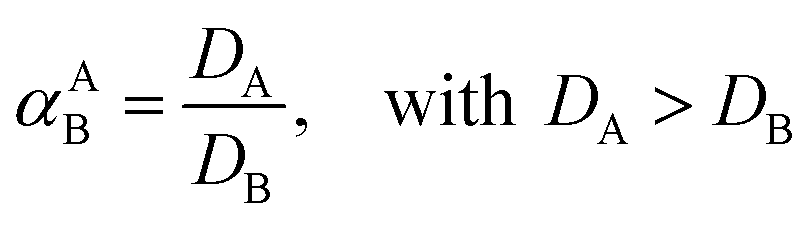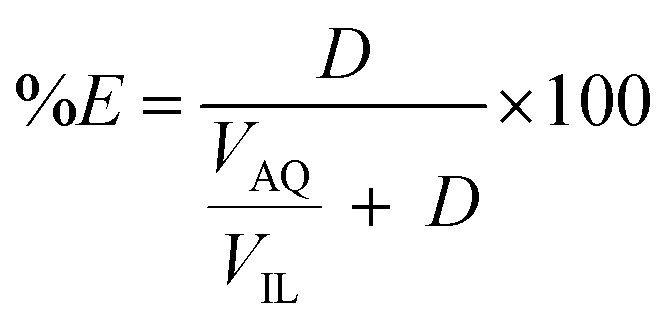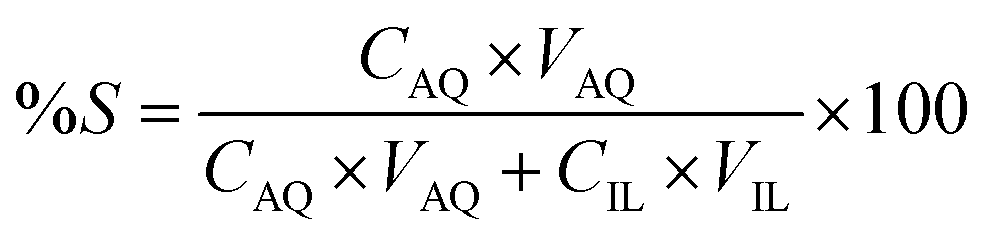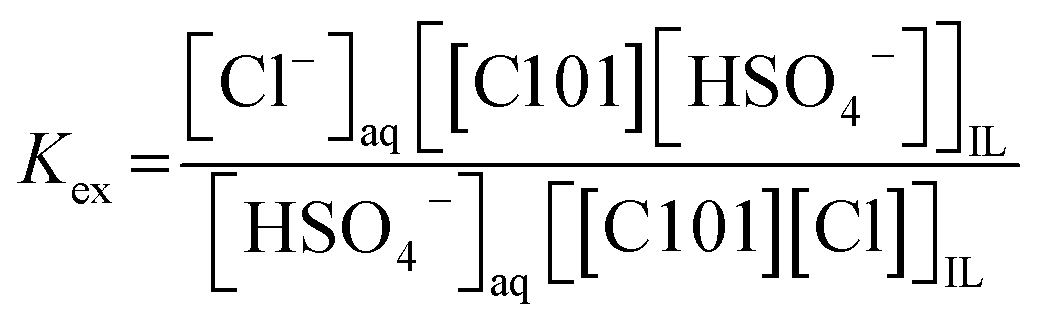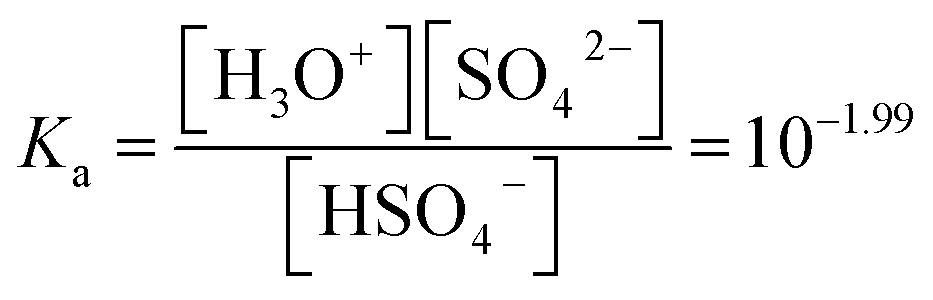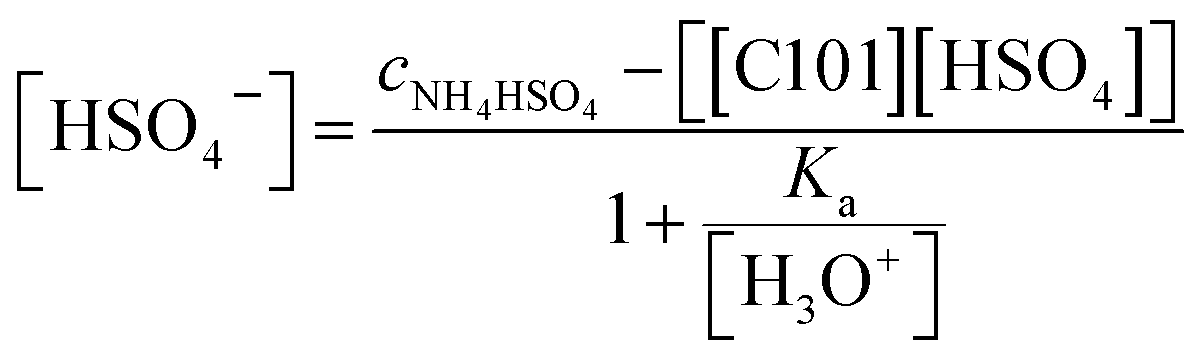DOI:
10.1039/C7RA04753C
(Paper)
RSC Adv., 2017,
7, 35992-35999
Cobalt(II)/nickel(II) separation from sulfate media by solvent extraction with an undiluted quaternary phosphonium ionic liquid†
Received
27th April 2017
, Accepted 9th July 2017
First published on 19th July 2017
Abstract
Leaching of cobalt and nickel from ores, urban waste and industrial residues is in general performed by sulfuric acid rather than hydrochloric acid, because sulfuric acid is cheaper and much less corrosive than hydrochloric acid. The choice of the acidic lixiviant has a large influence on the down-stream separation of Co and Ni, since the anion in the leachate determines the choice of solvent extraction system that can be applied. Co/Ni separations from sulfate media are traditionally performed by acidic extractants. In this paper, the extraction of Co(II) from sulfate media and its separation from Ni(II) is investigated using the quaternary phosphonium ionic liquid trihexyl(tetradecyl)phosphonium chloride (Cyphos IL 101, [C101][Cl]). Several extraction parameters have been optimized: acid concentration, salt concentration, time, temperature and the initial metal concentration in the aqueous feed solution. Furthermore, the extraction mechanism was investigated. The separation factors of Co(II)/Ni(II) are very high, in the order of 105. Stripping of the extracted Co(II) ions from the loaded ionic liquid phase was achieved by mixing with water. The ionic liquid phase was afterwards regenerated by washing with a CaCl2 solution. The extraction of Co(II) and Ni(II) with [C101][Cl] was compared to the extraction with the thiocyanate form of Cyphos IL 101, [C101][SCN]. It was found that extraction of Co(II) with [C101][SCN] occurred according to a split-anion mechanism, whereas a regular anion-exchange mechanism was observed for [C101][Cl].
1 Introduction
The separation of cobalt from nickel is a challenging task in extractive metallurgy because of the similarities in the chemical properties of these two elements.1 The Co/Ni separation is important for the production of these transition metals and corresponding salts from their primary ores, including nickel–copper sulfides and nickel laterite ores, and for the recycling of these metals from end-of-life consumer goods such as spent nickel-metal hydride (NiMH) batteries and lithium-ion batteries.2–10 In general, a combination of leaching, solvent extraction, precipitation and electrochemical techniques is applied to recover cobalt and nickel from these ores and waste fractions. Sulfuric acid is preferred over hydrochloric acid as the lixiviant, since the former acid is cheaper and less corrosive. The choice of the lixiviant has a substantial impact on the down-stream processing of the leachate because the effectiveness of the extractant in the subsequent solvent extraction process depends on the type of anion (sulfate or chloride). Co/Ni separations are traditionally performed with acidic extractants such as organophosphorus acids, including D2EHPA, PC88A, Cyanex 272 and Cyanex 302. These systems can be applied to both chloride and sulfate solutions, but the selectivities are rather low, a strict pH control is required and stripping must be performed by strong acids.1,10–12 On the other hand, quaternary ammonium and phosphonium extractants have been shown to be very efficient extractants for the separation of Co(II) and Ni(II) from chloride media. These extractants preferentially extract Co(II) as its tetrachlorocobaltate(II) complex, [CoCl4]2−, via an anion-exchange mechanism.13–15 However, few studies report the use of these anion-exchange extractants for extraction from sulfate media.16 Quaternary ammonium and phosphonium extractants with long aliphatic chains, such as Cyphos IL 101, the commercial name for trihexyl(tetradecyl)phosphonium chloride ([C101][Cl]), and Aliquat 336, a commercial mixture of C8–C10 trialkylmethylammonium chlorides, both belong to the class of solvents called ionic liquids (ILs). These solvents consist entirely of ions and show general characteristic properties including a broad liquidus range, a negligible vapor pressure and a low flammability.17–21 Since Cyphos IL 101 and Aliquat 336 are liquid at room temperature, they can be employed in undiluted form as the organic phase in solvent extraction, avoiding the use of volatile organic solvents as diluents.13,22,23
Recently, Larsson and Binnemans introduced a new approach to solvent extraction with ILs, called split-anion extraction. This approach allowed the extraction of trivalent rare-earth ions from a chloride solution by quaternary phosphonium thiocyanate and nitrate ILs.24 The basis of this technique is the use of basic extractants (anion exchangers), with anions of the type A−, for the extraction of metal ions from a solution containing a different type of anion, indicated by B−. The IL anions A− should have a higher tendency for the formation of anionic complexes with the targeted metal ion than the aqueous anions B−. Furthermore, the anions B− in the aqueous phase should have a lower affinity for the IL phase than the anions A− in the IL phase so that no anion-exchange occurs upon contact of the IL phase with the aqueous phase. The general extraction reaction is described by eqn (1):
| | |
Maqx+ + xBaq− + y[Q][A]IL ⇄ [Q(y−x)][MAy]IL + x[Q][B]IL
| (1) |
where Q denotes the cation of the IL, and (aq) and (IL) denote species in the aqueous and IL phase, respectively. Extraction is achieved by addition of B
− anions to the aqueous phase, which are co-extracted to the IL phase in order to maintain charge neutrality.
In principle, split-anion extraction could be applied to the extraction and separation of Co(II) and Ni(II) from sulfate media for two main reasons. First of all, the aqueous phase anions, namely sulfate SO42− and hydrogen sulfate HSO4−, are generally more hydrated than the chloride anions in the IL phase, as described by the Hofmeister series.25,26 Secondly, Co(II) has a high tendency to form tetrachlorocobaltate(II) complexes, [CoCl4]2− in chloride ILs.13
In this paper, we describe the extraction of Co(II) from sulfate solutions and the separation from Ni(II) using the undiluted ionic liquid Cyphos IL 101, abbreviated as [C101][Cl]. The results are compared to Co(II) and Ni(II) extraction with the thiocyanate form of the ionic liquid, abbreviated as [C101][SCN]. The influence of several extraction parameters is investigated. Furthermore, it is examined whether the extraction occurs via the split-anion mechanism or the classical anion-exchange mechanism.
2 Experimental
2.1 Chemicals
(NH4)2SO4 was purchased from VWR Chemicals (Leuven, Belgium), CoSO4·7H2O from Merck (Overijse, Belgium) and NiSO4·6H2O (99%) from JT Baker (Geel, Belgium). H2SO4 (96% solution in water) and K2CrO4 (99.5%) were ordered from Acros (Geel, Belgium), HCl (37% solution in water) from Fisher Scientific (Aalst, Belgium), ethanol absolute from VWR Chemicals (Leuven, Belgium), Triton X-100 and NH4HSO4 (≥99.0%) from Sigma-Aldrich (Diegem, Belgium), the silicone solution in isopropanol from SERVA Electrophoresis (Heidelberg, Germany) and the metal standard solutions of Co, Ni and Tm for TXRF (1000 μg mL−1 in 2–5% HNO3) from Chem-Lab (Zedelgem, Belgium). Cyphos IL 101 was obtained from Cytec Industries (Ontario, Canada). All products were used as received, without any further purification.
2.2 Instrumentation
Elemental analyses (Co, Ni, S, Cl, P) of both the aqueous and organic phases of the extraction experiments were performed by total-reflection X-ray fluorescence (TXRF) spectroscopy on a Bruker Picofox S2 TXRF spectrometer.27 UV-VIS absorption spectra were recorded in dual-beam mode on a Varian Cary 5000 spectrophotometer in a 0.2 cm quartz cuvette, with pure [C101][Cl] as the blank. Ultrapure water (18.2 MΩ cm) was used for all dilutions and prepared using a Sartorius Arium Pro ultrapure water system. pH measurements were performed using a Mettler-Toledo SevenCompact pH meter combined with a Hamilton Slimtrode pH-electrode. Extractions in 4 mL vials were performed in a Nemus Life TMS-200 thermoshaker with temperature control, operating at 2000 rpm, and centrifuged in a Heraeus Labofuge 200. Extractions in 50 mL centrifuge tubes were performed in a ThermoFisher MaxQ 2000 orbital shaker at 300 rpm and room temperature for 30 min, unless stated otherwise. Stability of the ionic liquid phase was tested with 1H and 31P NMR on a Bruker Ascend 400 spectrometer operating at 400 and 162 MHz respectively. NMR samples were prepared by dilution in acetonitrile-d3.
2.3 Elemental analysis with TXRF
TXRF samples of the aqueous solutions were prepared by first adding 50 μL of a Tm standard solution (1000 μg mL−1) to 100 μL of the aqueous solution and diluting with 850 μL of a 5 wt% Triton-X 100 solution in water. Secondly, this mixture was again diluted 10 times with a 5 wt% Triton-X 100 solution. Samples of IL solutions were prepared in a similar fashion as the aqueous solutions, but in this case a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of ethanol and 5% Triton-X 100 was used for dilution. A droplet (3 μL) of the prepared sample solution was placed on a quartz glass sample carrier, pretreated with 30 μL of a silicone solution in isopropanol to make the surface more hydrophobic, and dried at 60 °C for 30 min. The samples carriers were measured with the TXRF spectrometer for 500 s and the results were analyzed with the Bruker Spectra Picofox software (version 7.5.3.0). All measurements were performed in triplicate and the measurement error was determined as the standard deviation on the measurements.
:1 solution of ethanol and 5% Triton-X 100 was used for dilution. A droplet (3 μL) of the prepared sample solution was placed on a quartz glass sample carrier, pretreated with 30 μL of a silicone solution in isopropanol to make the surface more hydrophobic, and dried at 60 °C for 30 min. The samples carriers were measured with the TXRF spectrometer for 500 s and the results were analyzed with the Bruker Spectra Picofox software (version 7.5.3.0). All measurements were performed in triplicate and the measurement error was determined as the standard deviation on the measurements.
2.4 Titration
The chloride or thiocyanate concentrations in the aqueous phase were determined by precipitation titration with AgNO3, with potassium chromate, K2CrO4, as an indicator. An aliquot of the aqueous phase (0.1 mL) was mixed with 0.5 mL of a 100 g L−1 K2CrO4 solution and further diluted to 10 mL with ultrapure water. A 0.1 M AgNO3 solution was then added dropwise and a white/yellow-colored suspension was formed. The endpoint of the titration was reached when the suspension turned dark brown. The remaining concentration of chloride or thiocyanate in solution after titration is negligible.
2.5 Preparation of [C101][SCN]
[C101][Cl] was converted to its thiocyanate form, [C101][SCN], by mixing with a 100 g L−1 solution of potassium thiocyanate. The mixture was stirred for 3 h at room temperature and allowed to settle afterwards. The IL phase was separated and the procedure was repeated two more times to ensure full conversion to the thiocyanate form, as verified by TXRF.
2.6 Extraction experiments
Extractions were generally performed by mixing of a metal sulfate solution with water-saturated [C101][Cl], in its chloride form or thiocyanate form, for 30 min, in a 1:1 AQ/IL phase volume ratio, either in a 4 mL glass vial (14.75 × 45 mm), closed off with a screw cap for the parameter study or in 50 mL centrifuge tubes for the recycling experiment. The temperature was controlled at 25 °C, as set by the thermomixer, for most experiments (unless otherwise stated). Afterwards, the mixtures were centrifuged to accelerate the phase disengagement and the aqueous and IL phase were separated for further analysis by TXRF. The metal extraction and separation between two metals A and B were evaluated by calculating the distribution ratios D, separation factor αAB, and the percentage extraction %E, using eqn (2)–(4):| |
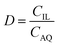 | (2) |
| |
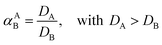 | (3) |
| |
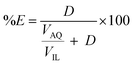 | (4) |
where CIL, VIL and CAQ, VAQ are the equilibrium metal concentration and volume of the IL and aqueous phase, respectively.
2.7 Stripping experiments
Stripping was performed by mixing the loaded IL phase, with the strip solution, namely water, for 30 min, in a 1:1 AQ/IL phase volume ratio. The mixtures were centrifuged to accelerate the phase disengagement and the aqueous and IL phase were separated for further analysis with TXRF. The stripping was evaluated by calculating the percentage stripping %S, using eqn (5):| |
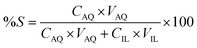 | (5) |
where CIL, VIL and CAQ, VAQ are the equilibrium metal concentration and volume of the IL and aqueous phase, respectively.
2.8 Regeneration of the IL
[C101][Cl] was regenerated from the stripped IL phase by contacting the IL phase with a 4 M CaCl2 solution in an AQ/IL 1:1 phase ratio, and shaken for 15 minutes. The mixture was centrifuged to separate the CaSO4 precipitate from the IL and the aqueous phase, after which the IL phase was neutralized by titration with NaOH in an AQ/IL 1:1 ratio until a pH between 4 and 7 was obtained.
3 Results and discussion
3.1 Influence of time and temperature
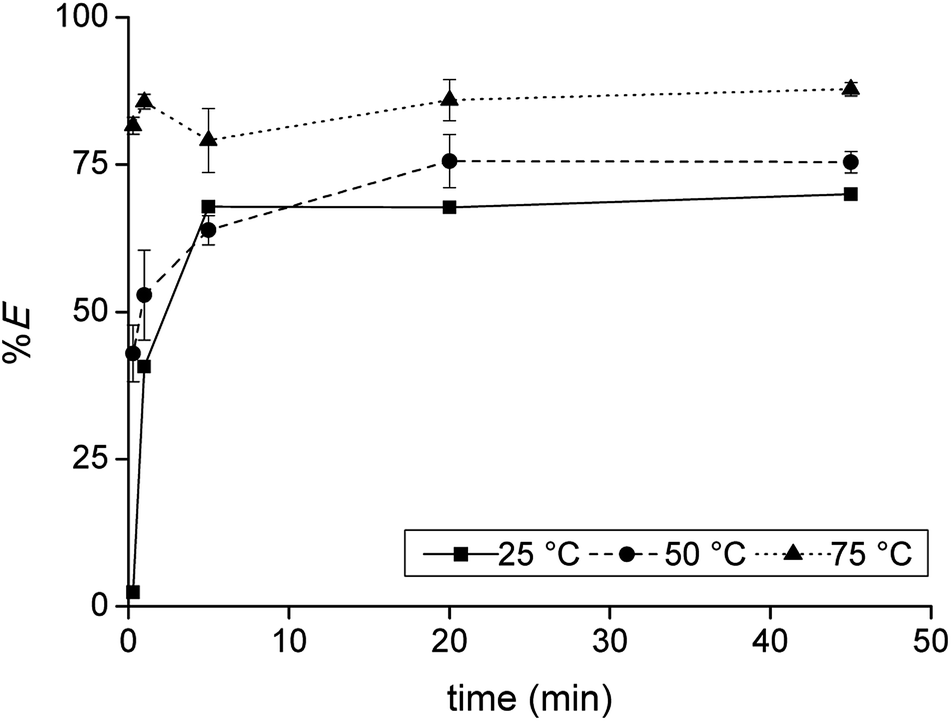
Firstly, the extraction of Co(II) and Ni(II) was investigated as a function of shaking time and temperature to determine the optimal shaking conditions to reach extraction equilibrium (Fig. 1). The results show that the equilibrium for Co(II) extraction is reached within approximately 30 min at room temperature. With increasing temperature, the kinetics are improved and extraction occurs faster, which is clear from the data points at lower shaking times. At 75 °C, equilibrium is reached almost instantaneously. This increase in extraction speed is mostly caused by a decrease in viscosity of [C101][Cl] with increasing temperatures. Furthermore, the position of the extraction equilibrium is influenced by the temperature, since %E at equilibrium increased with increasing temperatures. Consequently, the extraction of Co(II) is an endothermic process.28 The Ni(II) concentration in the IL phase after extraction at equilibrium conditions was below the detection limit of the TXRF (estimated to be <0.2 mg L−1), which means that the extraction of Ni(II) was negligible. The separation factor αCoNi for the separation of Co(II) and Ni(II) with [C101][Cl] was estimated to be larger than 105.
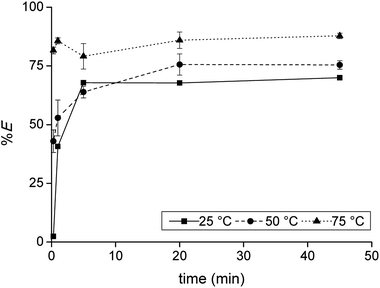 |
| | Fig. 1 Extraction of Co(II) with [C101][Cl] from sulfate solution containing Co(II) and Ni(II) as a function of shaking temperature and time. [Co]aq,in = 0.5 g L−1, [Ni]aq,in = 0.5 g L−1, [H2SO4]aq,in = 4 M, [(NH4)2SO4]aq,in = 3 M, AQ/IL = 1:1. Extraction of Ni(II) was found to be negligible and is therefore omitted from the plot. | |
3.2 Influence of acid and salt concentration
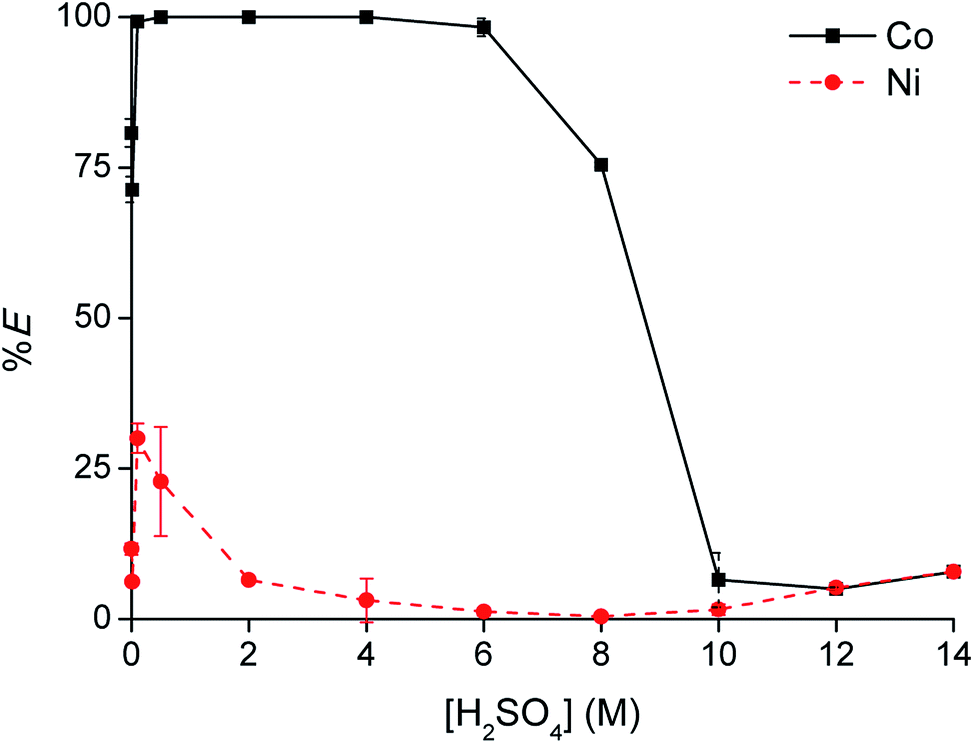
To optimize the position of the extraction equilibrium, the influence of the composition of the matrix in the aqueous phase was investigated by varying the (initial) concentration of H2SO4 and (NH4)2SO4 in the aqueous phase (Fig. 2a and b). The addition of only (NH4)2SO4 did not result in any extraction of Co(II) or Ni(II) to [C101][Cl]. This is an important finding, since this indicates that salting-out with the double-negatively charged sulfate anion SO42− to the IL phase via a split-anion mechanism is not possible for Co(II) and Ni(II). There are two possible explanations for this observation: (1) SO42− is too hydrophilic to be co-extracted to the IL phase via a split-anion mechanism,25 (2) SO42− coordinates to Co(II), forming hydrated sulfato complexes in the aqueous phase which prevent extraction of Co(II). Extraction was increased by addition of H2SO4 to the aqueous phase. H2SO4 is a strong acid and fully dissociated in the aqueous phase to the hydrogen sulfate ion, HSO4−. The pKa of HSO4− is 1.99, so an equilibrium will be set between HSO4− and SO42−. The actual concentration of HSO4− can be calculated from the pKa. The efficiency of the extraction process, expressed either as %E or logD, was linearly correlated to the calculated HSO4− concentration. This gives an indication for the role of HSO4− in the extraction process.
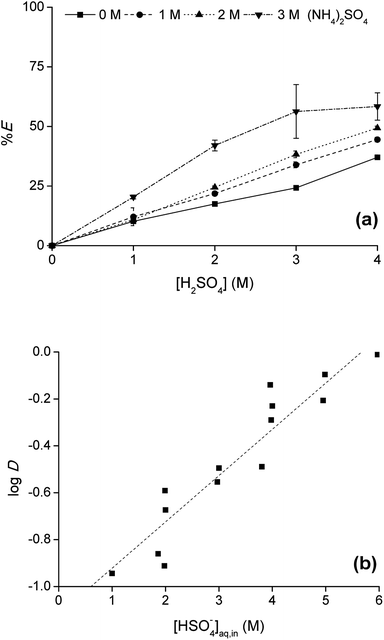 |
| | Fig. 2 Extraction of Co(II) with [C101][Cl] from a sulfate solution containing Co(II) and Ni(II) (a) as a function of the concentration [H2SO4]aq,in and [(NH4)2SO4]aq,in added to the aqueous phase and (b) as a function of the calculated concentration [HSO4−]aq,in. [Co]aq,in = 0.5 g L−1, [Ni]aq,in = 0.5 g L−1, AQ/IL = 1:1. Extraction of Ni(II) was found to be negligible and is therefore omitted from the plot. | |
Since the extraction increased with increasing hydrogen sulfate concentration in the aqueous phase, the addition of H2SO4 in high concentrations was investigated as well. The Co(II) extraction increased, reaching a maximum at a H2SO4 concentration of 11 M. Higher acid concentrations led to a decrease in %E of Co(II).
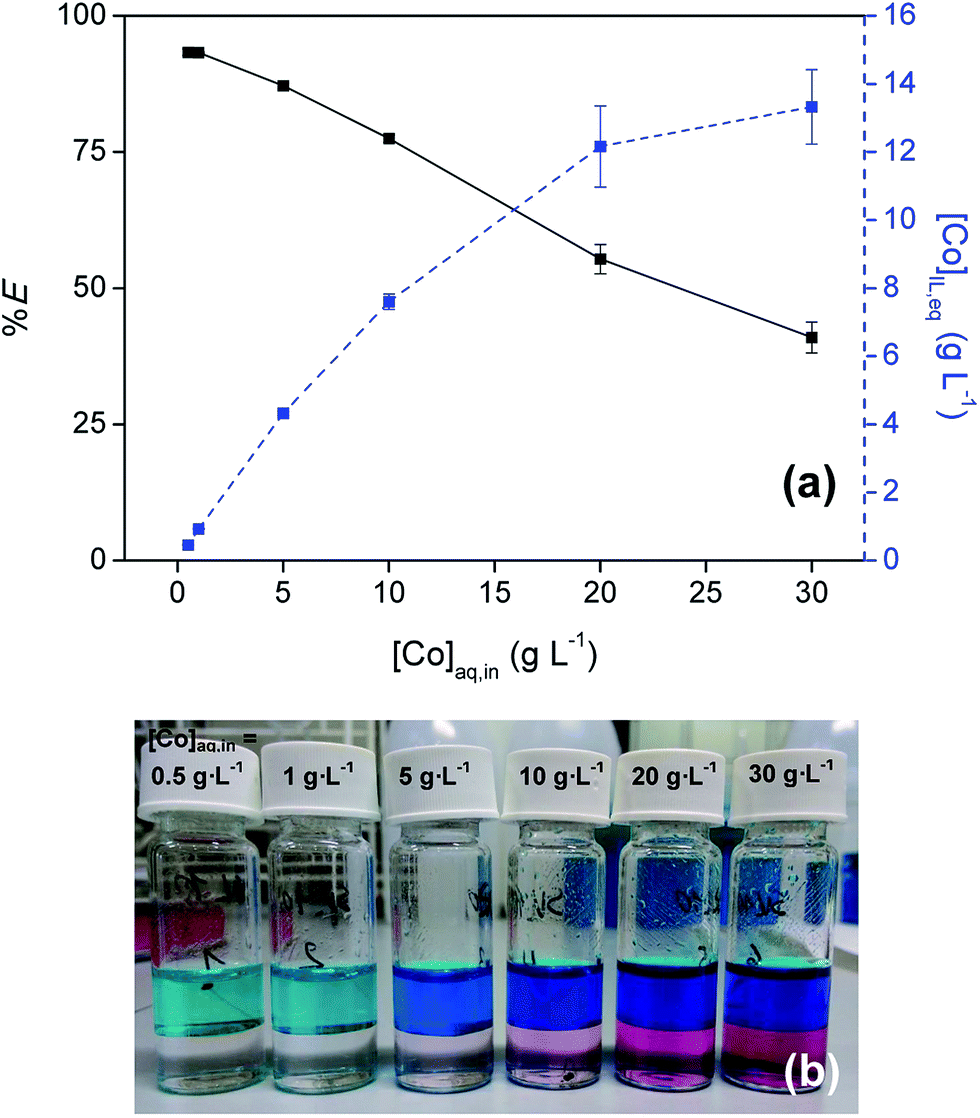
3.3 Influence of the metal concentration
The influence of the Co(II) concentration on the extraction was examined by performing extractions with varying initial Co(II) concentration in the aqueous phase (0.1–30 g L−1) at an initial H2SO4 concentration of 9 M (Fig. 3). Cobalt concentrations up to 13 g L−1 were reached in the IL phase at the highest initial concentration. At these high concentrations in the feed solution the %E dropped below 50% due to loading effects.
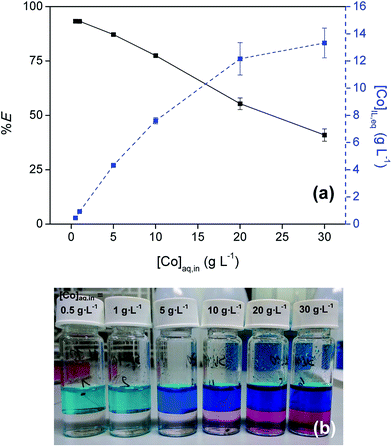 |
| | Fig. 3 (a) Extraction of Co(II) with [C101][Cl] from sulfate solution as a function of [Co]aq,in (0.1–30 g L−1). [(NH4)2SO4]aq,in = 0 M, [H2SO4]aq,in = 9 M, AQ/IL = 1:1. (b) Picture of the corresponding extraction mixtures. | |
3.4 Phase disengagement
Centrifugation was used to accelerate the phase disengagement in all shaking experiments. However, from an industrial point of view, it is also important to gain information on the settling duration without centrifugation, since gravitational forces are employed for the settling stage in for instance industrial mixer-settlers. The settling was tested in a centrifuge tube of 50 mL, by mixing 20 mL of aqueous phase and 20 mL of water-saturated [C101][Cl]. Phase disengagement was fast, with two separately identifiable phases appearing instantly and full settling after less than one minute. Thus, the settling speed is estimated at 0.2 m h−1.
3.5 Extraction mechanism
First, it was studied whether the extraction of Co(II) from sulfate media with [C101][Cl] occurred through a split-anion mechanism, as described by eqn (6) and (7) for sulfate and hydrogen sulfate, respectively.| | |
Coaq2+ + SO4,aq2− + 4[C101][Cl]IL ⇄ [C101]2[CoCl4]IL + [C101]2[SO4]IL
| (6) |
| | |
Coaq2+ + 2HSO4,aq− + 4[C101][Cl]IL ⇄ [C101]2[CoCl4]IL + 2[C101][HSO4]IL
| (7) |
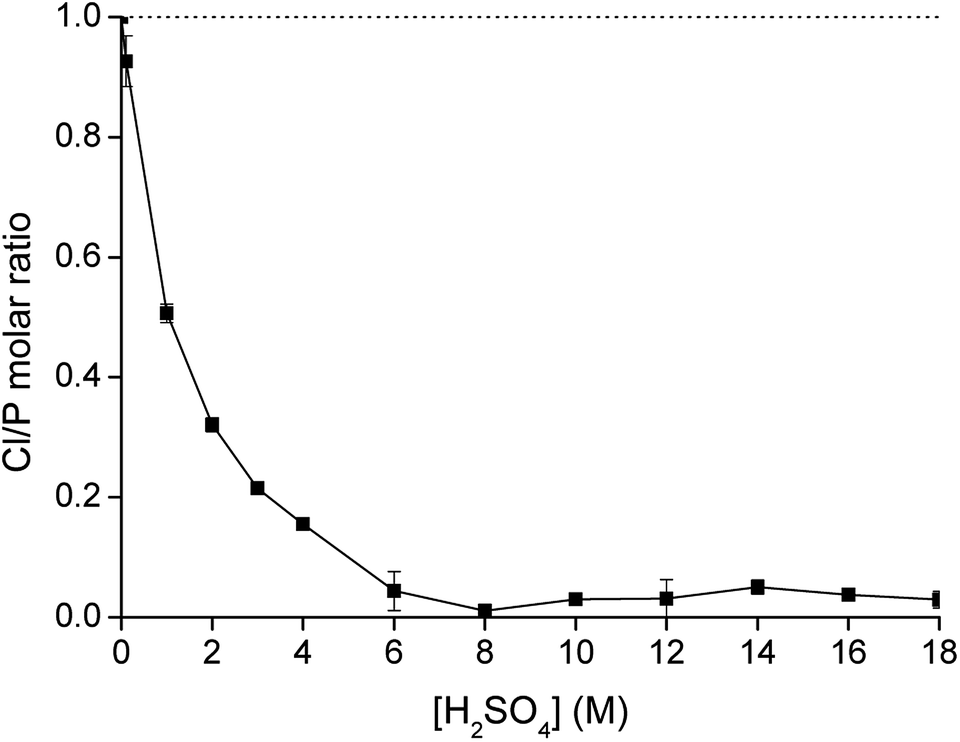
From Fig. 2, it was observed that no extraction occurred upon increasing concentration of (NH4)2SO4 without the presence of the acid H2SO4. Therefore, eqn (6) is not relevant to describe the extraction of Co(II). On the other hand, upon the addition of H2SO4, the sulfate ions are converted to hydrogen sulfate ions and Co(II) is extracted, with a direct relation between extraction and the hydrogen sulfate concentration. If the extraction takes place via the mechanism shown in eqn (7), hydrogen sulfate is co-extracted to the IL phase upon extraction of Co(II). To verify this experimentally, the relative S, Cl and P concentrations in the IL phase were measured with TXRF. The results showed that the S concentration increased after extraction, but also that the Cl concentration decreased, which does not correspond to what would be expected from eqn (7). To further investigate this observation, [C101][Cl] was mixed with different concentrations of H2SO4 in a 1:1 AQ/IL phase ratio (without the presence of metal ions) and it was seen that the Cl concentration drastically decreased with increasing H2SO4 concentration, while the S concentration increased (Fig. 4). This indicates that upon mixing of [C101][Cl] with a H2SO4 solution, the chloride anions are exchanged by hydrogen sulfate anions. At high hydrogen sulfate concentrations, the IL [C101][Cl] is converted to the hydrogen sulfate form [C101][HSO4]. The exchange equilibrium constant Kex is defined by eqn (8):
| |
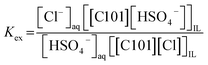 | (8) |
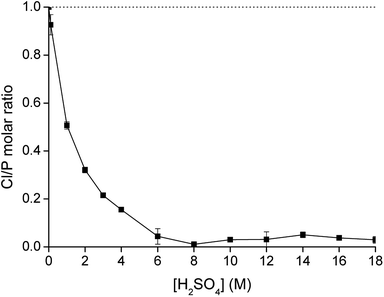 |
| | Fig. 4 Exchange of chloride by hydrogen sulfate upon contacting water-saturated [C101][Cl] with a H2SO4 solution as a function of the sulfuric acid concentration (AQ/IL = 1:1), expressed as the molar ratio Cl/P measured in the IL phase. | |
The value of Kex was calculated after equilibrating 1 mL of water-saturated [C101][Cl] with 1 mL of a 100 g L−1 (0.87 M) solution of NH4HSO4 and analyzing both aqueous and organic phase. The equilibrium chloride concentration in the aqueous phase [Cl−] was determined experimentally by titration with AgNO3 and was found to be 0.54 M. The ratio of the IL in the hydrogen sulfate form over the chloride form [[C101][HSO4]]IL/[[C101][Cl]]IL was determined by measuring the relative concentrations of P, S and Cl in the IL phase by TXRF and was found to be 0.79. The equilibrium concentration of hydrogen sulfate [HSO4−]aq was calculated using the mass balance (eqn (9)):
| | |
cNH4HSO4 = [HSO−4] + [SO42−] + [[C101][HSO4]]
| (9) |
The initial analytical concentration of NH4HSO4 in the aqueous phase was equal to 0.87 M, and the concentration of the IL in the hydrogen sulfate form in the IL phase was measured to be equal to 0.7 M. The equilibrium sulfate concentration can be written as a function of the hydrogen sulfate concentration using the acid dissociation constant Ka (eqn (10)).29
| |
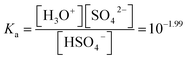 | (10) |
The pH was 0.67. The combination of eqn (9) and (10) gives eqn (11):
| |
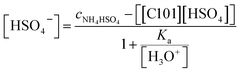 | (11) |
By inserting all calculated equilibrium concentrations in eqn (8) and (11), the exchange equilibrium constant Kex was calculated to be equal to 100.3. Note that latter calculation is an approximation, since the contribution of the ionic strength to the activity of ionic species and the extraction of acid to the IL phase were assumed to be negligible,30 and merely indicates that the position of hydrogen sulfate in the Hofmeister series is approximately the same as chloride for anion exchange in the undiluted trihexyl(tetradecyl)phosphonium system:25
| SO42− ≪ HSO4− ∼ Cl− < NO3− < SCN− < Tf2N− |
Thus, upon mixing of the IL phase with the sulfate aqueous feed solution, the chloride ions from the IL are relatively easily exchanged by hydrogen sulfate ions from the aqueous phase. The exact influence of this exchange on the extraction mechanism is not completely clear. Probably, these exchanged chloride anions readily coordinate to Co(II) in the aqueous phase and Co(II) is most likely extracted to the IL phase through a classical anion-exchange mechanism. It can be concluded that not all aqueous-IL anion combinations A−/B− that theoretically apply to the rules of split-anion extraction, are also practically applicable. For instance, no extraction occurred from solutions containing only sulfate anions, SO42−, probably indicating that sulfate is too hydrophilic and too highly hydrated to be co-extracted to the IL phase.
The speciation of Co(II) in the IL phase was studied by optical absorption spectroscopy. A comparison was made between extraction of Co(II) with [C101][Cl] from an aqueous solution containing 4 M of H2SO4, 3.5 M H2SO4 + 3 M (NH4)2SO4 and 6 M HCl as a reference (Fig. 5). Earlier work on the extraction of Co(II) with [C101][Cl] from chloride media showed that the tetrachloro species, [CoCl4]2−, is formed in the IL phase. The spectra of the IL phase of Co(II) extracted from 4 M of H2SO4 and 3.5 M H2SO4 + 3 M (NH4)2SO4 showed the same absorption bands in the region 600–750 nm as the reference spectrum of extraction from 6 M HCl, with the same molar absorptivity ε, as calculated from the absorbance with the analytical Co concentration in the IL phase. Thus, for the extraction from sulfate media by [C101][Cl], Co(II) is also mostly present as a tetrachloro species, [CoCl4]2−. Furthermore, the matrix of the aqueous phase does not have a large influence on the speciation in the IL phase since the spectrum of extraction from 4 M of H2SO4 is very similar to the spectrum of extraction from 3.5 M H2SO4 + 3 M (NH4)2SO4.
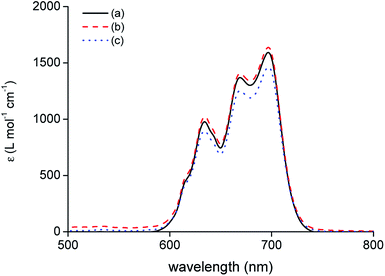 |
| | Fig. 5 Absorption spectra of the IL phase after extraction of Co(II) by [C101][Cl] from: (a) 4 M of H2SO4, (b) 3.5 M H2SO4 + 3 M (NH4)2SO4 and (c) 6 M HCl. [Co]aq,in = 0.5 g L−1, AQ/IL = 1:1, optical path length 0.2 cm. | |
3.6 Stripping of the loaded IL phase
Stripping of metal ions extracted with basic anion exchangers is generally performed with water or a dilute salt solution.31 Therefore, water stripping was tested to remove the extracted Co(II) from the loaded IL phase obtained after extraction from an aqueous solution containing Co(II) and Ni(II), 0.5 g L−1 each, with 4 M H2SO4 and 3 M (NH4)2SO4 as the matrix. The IL phase was mixed with water in a AQ/IL 1:1 phase ratio. The stripping percentage %S was calculated to be >99% since approximately all extracted Co(II) was recovered in the strip solution and the remaining Co(II) concentration in the IL phase was below the detection limit of the TXRF (<0.2 mg L−1).
3.7 Reusability of the IL
In order to have a feasible process, the IL phase should be recovered and reused in a next extraction cycle. However, as was stated before, the chloride ions of [C101][Cl] phase are exchanged for hydrogen sulfate ions. Therefore, an extra regeneration step was required to convert the IL back to the chloride form. This was done by contacting the IL phase obtained after the stripping described in the previous section, with a 4 M CaCl2 solution. The hydrogen sulfate ions present in the IL phase were exchanged by the chloride ions, after which CaSO4 is precipitated from the aqueous phase, pushing the exchange equilibrium to the right, as described by eqn (12).| | |
[C101][HSO4]IL + CaCl2,aq ⇄ [C101][Cl]IL + CaSO4,aq + HClaq
| (12) |
During the regeneration, HCl is formed, which is known to be efficiently extracted to quaternary ammonium and phosphonium chloride salts.30 Therefore, the regenerated IL was mixed with water and the extracted HCl was neutralized by titration with NaOH until an aqueous pH between 4 and 7 was obtained. The final AQ/IL phase ratio was approximately 1:1. The obtained IL phase was checked by TXRF. The relative S content compared to P was below the detection limit (<0.5%), which means that the regeneration of the chloride form of the IL was successful.
Furthermore, the stability of the ionic liquid phase was checked by comparing the 1H and 31P NMR spectra the ionic liquid phase after extraction and regeneration, with the spectra of the commercial Cyphos IL 101 before use (ESI, Fig. S1–S6†). The spectra showed the same peaks before and after extraction, indicating that structural decomposition of the ionic liquid in the extraction process is negligible.
The water content of the IL phase during the extraction process and after regeneration was monitored. Water-saturated [C101][Cl] contained 13.8 wt% water and the IL phase after extraction contained only 3.1 wt% water. However, the volume of the phases remained almost constant before and after extraction. The exchange of chloride by hydrogen sulfate likely resulted in an increase in volume of the IL phase, which was cancelled out by the decrease in water content due to the salting-out effect of the hydrogen sulfate matrix in the aqueous phase. The regenerated IL phase before washing contained 9.0 wt% water, which was slightly lower than water-saturated [C101][Cl], due to the CaCl2 which was present in the aqueous phase in the regeneration step. However, after the neutralization step, the water content was again increased to 13.5 wt%.
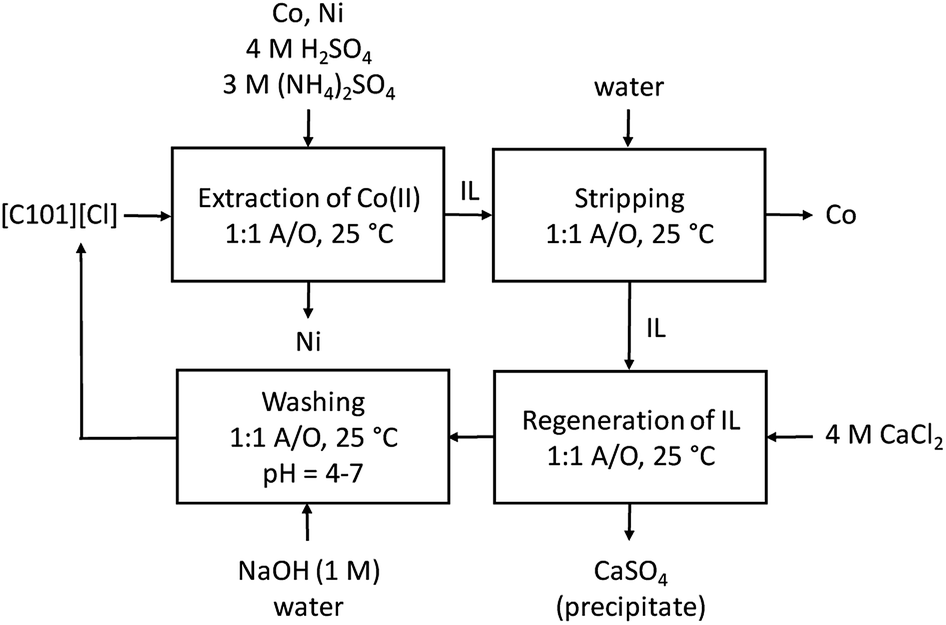
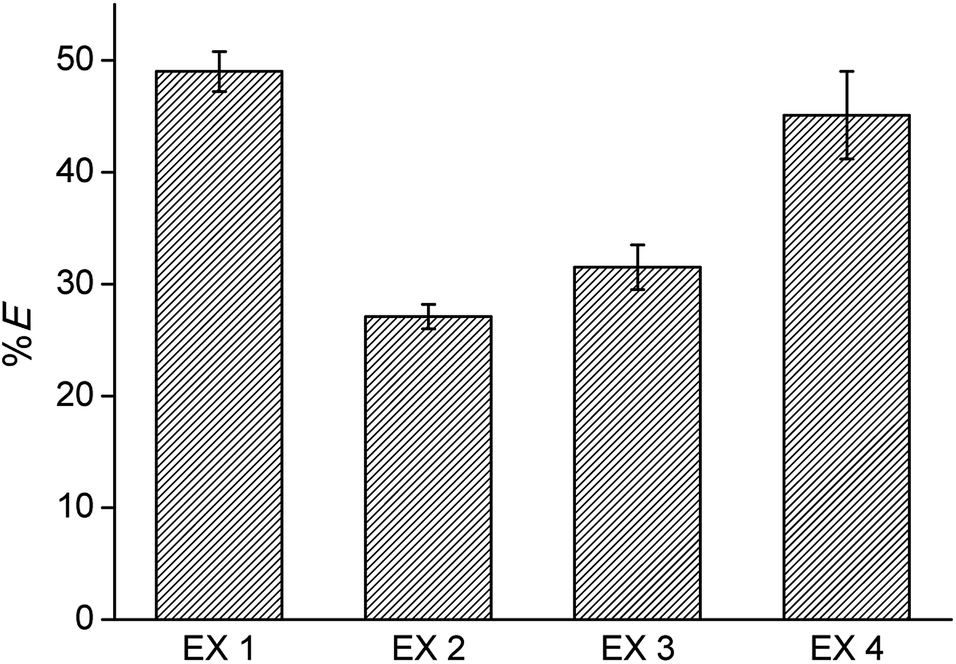
The reusability of the regenerated IL was investigated by recycling the IL three times. Each time, extraction was performed from an aqueous solution containing Co(II) and Ni(II), 0.5 g L−1 each, with 4 M H2SO4 and 3 M (NH4)2SO4 as the matrix, in a AQ/IL 1:1 phase ratio. Stripping was performed with water, and regeneration with 4 M CaCl2. The cycle was each time ended with a washing step during which the HCl formed and extracted during the regeneration step, is removed by neutralization with NaOH until a pH between 4 and 7 is obtained. An overview of the optimized process is given in Fig. 6 and the extraction results are summarized in Fig. 7. The %E of Co(II) did not remain the same in the four regeneration steps. This is not due to chemical or structural changes in the [C101][Cl] since NMR studies showed that the ionic liquid did not decompose over the process (ESI, Fig. S1–S6†). Most likely, the extraction conditions in the four extractions were not exactly the same, possibly because of the lack of good control during the neutralization at a laboratory scale. Nevertheless, the results show that it is possible to reuse the ionic liquid.
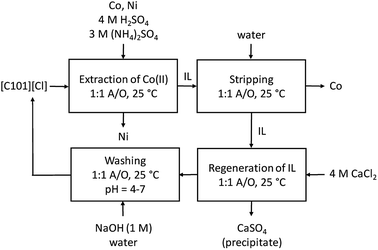 |
| | Fig. 6 Overview of the extraction process for the separation of Co(II) and Ni(II) from a sulfuric solution with [C101][Cl]. | |
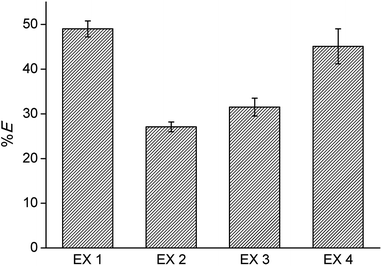 |
| | Fig. 7 Reusability of the ionic liquid phase: extraction of Co(II) using fresh [C101][Cl] (EX1), compared to extraction with one, two and three times regenerated [C101][Cl] (EX2, EX3, EX4), following the optimized extraction process. | |
3.8 Extraction with [C101][SCN]
[C101][Cl] was unable to extract Co(II) from sulfate solution by the split-anion mechanism, due to the anion-exchange between the chloride ions of the IL and hydrogen sulfate ions from the aqueous phase. Therefore, [C101][Cl] was converted to its thiocyanate form and tested for Co(II) extraction. Thiocyanate ions are more hydrophobic than chloride ions,25 so the thiocyanate anion in the IL phase should be less prone to exchange with hydrogen sulfate. Furthermore, thiocyanate is known to be even stronger coordinating to Co(II) than chloride.32 Conversion of [C101][Cl] to [C101][SCN] was performed via a metathesis reaction with KSCN. Extraction of Co(II) and Ni(II) was investigated as a function of the H2SO4 concentration in the aqueous phase (Fig. 8). Extraction of Co(II) was efficient, with 75% extraction at 0 M H2SO4 up to over 99% extraction between 0.5 and 4 M H2SO4. In latter concentration range, the Co(II) concentration in the aqueous phase was even below the detection limit of the TXRF (<0.2 mg L−1). At high H2SO4 concentrations (>8 M), extraction efficiency decreased again, similarly as in the case of [C101][Cl]. Also, Ni(II) showed an increase in extraction, reaching a maximum extraction of 30% at 0.5 M H2SO4. The separation factors αCoNi between 0.5 and 4 M H2SO4 were higher than 105.
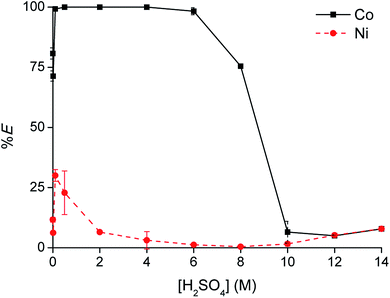 |
| | Fig. 8 Extraction of Co(II) with [C101][SCN] from sulfate solution containing Co(II) and Ni(II) as a function of [H2SO4]aq,in. [Co]aq,in = 0.5 g L−1, [Ni]aq,in = 0.5 g L−1, [(NH4)2SO4]aq,in = 0 M, AQ/IL = 1:1. | |
To investigate whether the thiocyanate anions from the IL phase might have been exchanged by hydrogen sulfate anions from the aqueous phase, the thiocyanate content in the aqueous phase was tested by titration with AgNO3 in the presence of K2CrO4 as an indicator. Immediately upon addition of the first drop of AgNO3 solution, a brown precipitate of Ag2CrO4 was formed, which indicated that the thiocyanate concentration in the aqueous phase after extraction was negligible. This observation proved that no exchange of thiocyanate for hydrogen sulfate had occurred during extraction. Thus, the only possible way to extract Co(II) and maintain charge neutrality in both phases is by co-extracting hydrogen sulfate ions according to the split-anion mechanism. The extraction can be described by eqn (13):
| | |
Coaq2+ + 2HSO4,aq− + 4[C101][SCN]IL ⇄ [C101]2[Co(SCN)4]IL + 2[C101][HSO4]IL
| (13) |
4 Conclusions
The separation of cobalt(II) and nickel(II) with [C101][Cl] and [C101][SCN] was investigated by extraction from sulfate media. This is relevant since extraction from sulfate media is currently mainly performed with less selective acidic extractants, which require strict pH control and stripping with concentrated acid. First, Co(II)/Ni(II) separation with the chloride IL was investigated and seemed to be strongly correlated to the hydrogen sulfate concentration. Co(II) extraction was close to 100% while no extracted Ni(II) could be detected, indicating a very good separation. Extraction did not follow the split-anion mechanism, but occurred through a regular anion-exchange mechanism. The IL chloride anions exchanged with the hydrogen sulfate anions from the aqueous phase. Optical absorption spectroscopy confirmed the presence of the tetrachlorocobaltate(II) complex, [CoCl4]2−, in the IL phase. Stripping was easily performed by mixing the loaded IL phase with water. After stripping, the IL was mixed with a CaCl2 solution to regenerate the IL in its chloride form. Finally, a comparison was made with [C101][SCN]. Thiocyanate is a more hydrophobic anion than chloride and it was confirmed that no exchange occurred with the hydrogen sulfate anions from the aqueous phase. Co(II) was extracted efficiently, even at low H2SO4 concentrations, and also some co-extraction of Ni(II) was detected. Nevertheless, the separation factors were still very high (>105). Furthermore, it was confirmed that Co(II) extraction with [C101][SCN] occurred according to a split-anion mechanism.
Acknowledgements
This research was financially supported by VLAIO Flanders (PhD fellowship to BO) and by the FWO Flanders (postdoctoral fellowship to TVH). Financial support by the KU Leuven (IOF-KP RARE3) is gratefully acknowledged.
Notes and references
- D. S. Flett, Chem. Sustainable Dev., 2004, 12, 81–91 Search PubMed.
- T. Müller and B. Friedrich, J. Power Sources, 2006, 158, 1498–1509 CrossRef.
- A. Fernandes, J. C. Afonso and A. J. B. Dutra, Hydrometallurgy, 2013, 133, 37–43 CrossRef CAS.
- K. Larsson and K. Binnemans, Green Chem., 2014, 16, 4595–4603 RSC.
- P. Zhang, T. Yokoyama, O. Itabashi, Y. Wakui, T. M. Suzuki and K. Inoue, Hydrometallurgy, 1998, 50, 61–75 CrossRef CAS.
- N. Tzanetakis and K. Scott, J. Chem. Technol. Biotechnol., 2004, 79, 919–926 CrossRef CAS.
- V. T. Nguyen, J.-C. Lee, J. Jeong, B.-S. Kim and B. D. Pandey, Met. Mater. Int., 2014, 20, 357–365 CrossRef CAS.
- B. I. Whittington and D. Muir, Miner. Process. Extr. Metall. Rev., 2000, 21, 527–599 CrossRef.
- USGS, Mineral Commodity Summaries, 2016 Search PubMed.
- Z. Zhu, Y. Pranolo, W. Zhang and C. Y. Cheng, J. Chem. Technol. Biotechnol., 2011, 86, 75–81 CrossRef CAS.
- N. B. Devi, K. C. Nathsarma and V. Chakravortty, Hydrometallurgy, 1998, 49, 47–61 CrossRef CAS.
- I. Y. Fleitlikh, G. L. Pashkov, N. A. Grigorieva, L. K. Nikiforova, M. A. Pleshkov and Y. M. Shneerson, Solvent Extr. Ion Exch., 2011, 29, 782–799 CrossRef CAS.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC.
- P. Rybka and M. Regel-Rosocka, Sep. Sci. Technol., 2012, 47, 1296–1302 CrossRef CAS.
- B. Wassink, D. Dreisinger and J. Howard, Hydrometallurgy, 2000, 57, 235–252 CrossRef CAS.
- A. A. Nayl, J. Hazard. Mater., 2010, 173, 223–230 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- P. Wasserscheid and T. Welton, Ionic liquids in synthesis, Wiley-VCH, Weinheim, Germany, 2nd edn, 2008 Search PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- K. R. Seddon, Nat. Mater., 2003, 2, 363–365 CrossRef CAS PubMed.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- S. Wellens, R. Goovaerts, C. Moller, J. Luyten, B. Thijs and K. Binnemans, Green Chem., 2013, 15, 3160–3164 RSC.
- K. Larsson and K. Binnemans, Hydrometallurgy, 2015, 156, 206–214 CrossRef CAS.
- D. Dupont, D. Depuydt and K. Binnemans, J. Phys. Chem. B, 2015, 119, 6747–6757 CrossRef CAS PubMed.
- F. Hofmeister, Arch. Exp. Pathol. Phar., 1888, 24, 247–260 CrossRef.
- S. Riaño, M. Regadío, K. Binnemans and T. Vander Hoogerstraete, Spectrochim. Acta, Part B, 2016, 124, 109–115 CrossRef.
- G. R. Choppin and A. Morgenstern, Solvent Extr. Ion Exch., 2000, 18, 1029–1049 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Taylor & Francis, 85th edn, 2004 Search PubMed.
- T. Sato, H. Watanabe and S. Kikuchi, J. Appl. Chem. Biotechnol., 1975, 25, 63–72 CAS.
- J. Rydberg, M. Cox, C. Musikas and G. R. Choppin, Solvent extraction principles and practice, Marcel Dekker, Inc., New York, 2nd edn, 2004 Search PubMed.
- E. Högfeldt, Stability constants of metal-ion complexes: part A: inorganic ligands, Pergamon Press, 1982 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04753c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Koen Binnemans
and
Koen Binnemans