DOI:
10.1039/C7RA04751G
(Paper)
RSC Adv., 2017,
7, 29927-29932
Dual role of polyphosphoric acid-activated nitroalkanes in oxidative peri-annulations: efficient synthesis of 1,3,6,8-tetraazapyrenes†
Received
27th April 2017
, Accepted 3rd June 2017
First published on 8th June 2017
Abstract
A highly efficient synthetic method for expeditious and selective assembly of tetraazopyrenes (TAPy) is reported, based on the novel reaction of electrophilically activated nitroalkanes with aromatic amines. Remarkably, the nitroalkanes play a dual role in this process, also serving as mild and efficient oxidants promoting aromatization of the final product and allowing for the exclusion of a poorly controllable aerobic treatment.
Introduction
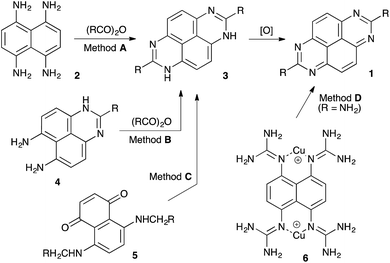
During the past decade, increasing attention of many research groups has been placed on the development of new π-conjugated organic materials for application in electronic and optoelectronic devices, such as light-emitting diodes (LEDs), field-effect transistors (FETs), and efficient and cost-effective solar cells.1,2 Among other fused polycyclic systems, tetraazapyrenes (TAPy, 1) have great promise as novel luminescent materials in sensors, lasers, and semi-conductor devices due to their favorable Red–Ox and photochemical properties.3 However, synthetic approaches to this heterocyclic core are limited to a handful of reliable methods. The most common synthetic route employs a double-fold cyclo-condensation of naphthalene-1,4,5,8-tetraamine (2) with carboxylic anhydrides and subsequent oxidation of the intermediate 1,8-dihydropyrimido[4,5,6-gh]perimidines 3 (Scheme 1, method A).3a,b Alternatively, this protocol could utilize 1H-perimidine-6,7-diamines 4 (method B)4 that are readily accessible via Pozharskii's electrophilic nitration approach.4 More unusual approaches involve thermally induced double-fold cyclo-condensation of 5,8-bis(methylamino)naphthalene-1,4-diones (5) (Scheme 1, method C),5 or oxidative decomposition of dinuclear copper-2,2′,2′′,2′′′-(naphthalene-1,4,5,8-tetrayl)tetraguanidine complexes 6 (method D).6 Notably, all of these methods rely on an oxidation step (usually aerobic) that could be substrate-dependent, providing inconsistent results upon scaling up. Herein we wish to report an innovative approach to the TAPy scaffolds, utilizing the nucleophilic reaction of substrates 2 and 4 with umpolung-activated electrophilic nitroalkanes. Remarkably, the latter compounds also serve as oxidizing agents for the aromatization of the target heterocyclic system.
 |
| | Scheme 1 | |
Results and discussion
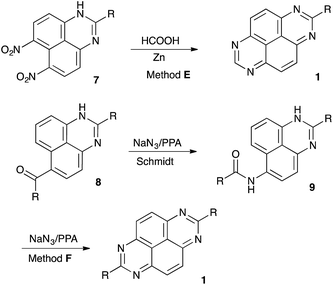
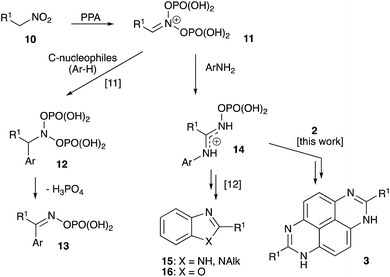
For several years our group had a great interest in development novel synthetic approaches towards fused aromatic polyheterocyclic cores targeting material science7 and medicinal chemistry applications.8 Several innovative approaches towards TAPy scaffolds were also developed (Scheme 2). These methods involved one-pot double nitration of perimidines followed by zinc-assisted reductive cyclo-condensation with formic acid (method E),9 and various peri-annulation cascades triggered by the Schmidt reaction of 6(7)-acylperimidines 8 (Scheme 2) (method F).10 We also demonstrated that nitroalkanes 10 react with polyphosphoric acid (PPA) to form highly stabilized aci form 11, which can serve as electrophilic component in highly selective Nef-like processes involving various carbon-11 and nitrogen-based nucleophiles12 (Scheme 3). Indeed, reactions of 11 with electron-rich aromatic compounds usually affords phosphorylated N-hydroxyhydroxylamine species 12, possessing a hydrogen atom at α-position, which undergoes facile elimination of H3PO4 to furnish phosphorylated oxime 10. The latter can undergo further Beckman or aza-Wagner–Meerwein rearrangements providing unusual avenues for selective installation of functional groups into aromatic compounds (Scheme 3).13 Reactions of 11 with nitrogen-based nucleophiles (typically anilines) provide stabilized amidinium ions 14. Installation of nucleophilic functionality in the strategic position could trigger intramolecular cyclo-condensation. This approach allowed us to develop expeditious synthetic route towards imidazoles 15 and oxazoles 16 (Scheme 3).14 We envisioned that similar strategy could potentially be employed for the synthesis of heteroaromatic core 3 (Scheme 3) via reaction of nitroalkanes with 1H-perimidine-6,7-diamines 4.
 |
| | Scheme 2 | |
 |
| | Scheme 3 | |
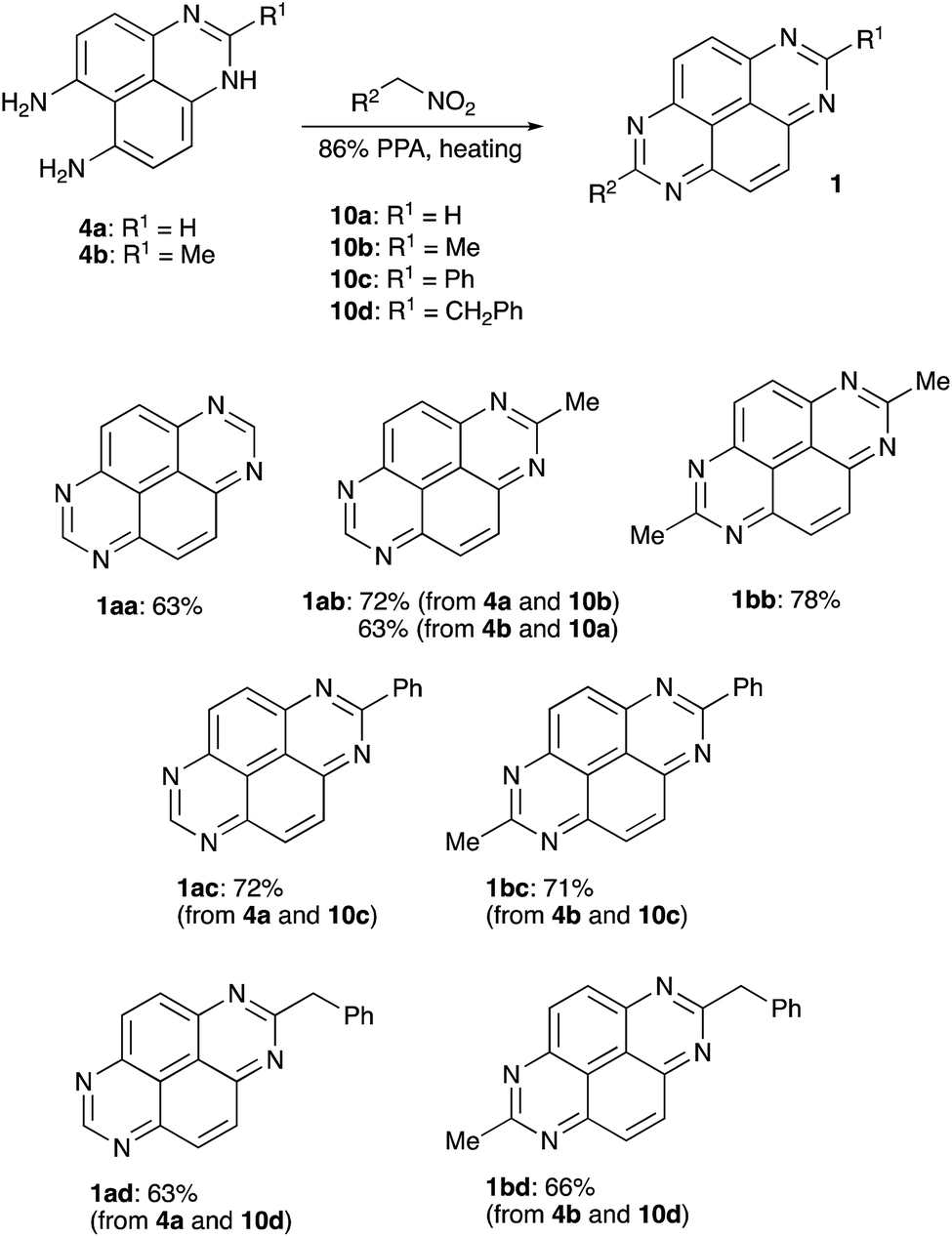
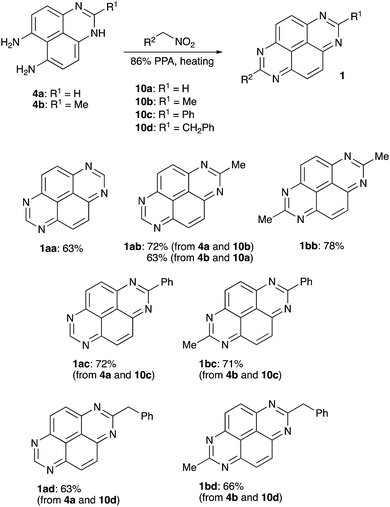
To test this idea we stirred 1H-perimidine-6,7-diamine (4a) in 86% PPA in the presence of nitromethane (10a). Interestingly, we did not observe formation of compound 3 (R1 = H). Instead, TAPy (1aa) formed as sole product in a yield of 63% (Scheme 4). Due to the symmetry of the scaffold, 2-Me-TAPy (1ab) can be assembled in two different way, either starting from 4a and nitroethane (10b) or by combining 2-methyl-1H-perimidine-6,7-diamine (4b) with nitromethane (10a). Both protocols work fine, affording the corresponding product 1ab in 72% and 63% yield, respectively (Scheme 4). 2,7-Me2-TAPy (1bb) was synthesized in a similar manner by treatment of 4b with nitroethane (10b). Evidently, a phenyl substituent could be introduced into the target scaffold via the reaction of an appropriate perimidine with α-nitrotoluene (10c). Indeed, 2-Ph-TAPy (1ac) and 2-Me-7-PhTAPy (1bc) were accessed via this mode very efficiently (Scheme 4). Finally, (2-nitroethyl)-benzene (10d) can be employed for the installation of benzylic substituents. Both tested perimidines 4a and 4b afforded the corresponding 7-benzyl-TAPy's 1ad and 1bd in good yields (Scheme 4).
 |
| | Scheme 4 | |
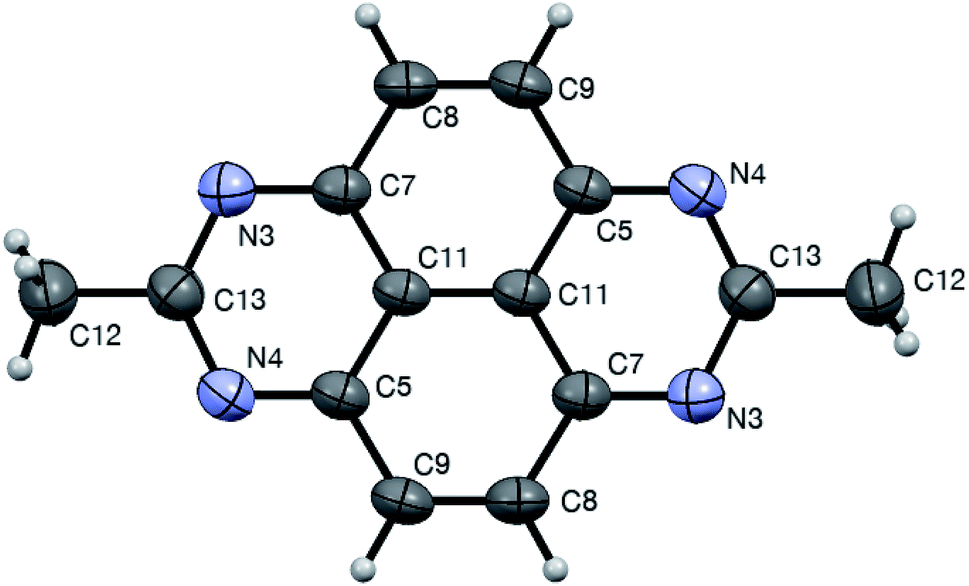
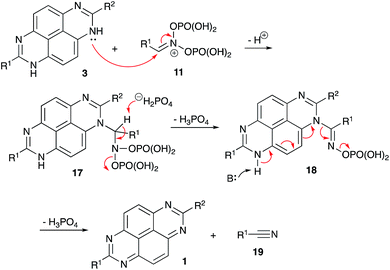
As mentioned above, the tetraazopyrenes 1 were obtained in the featured reaction with nitroalkanes directly, and reduced form 3 was never detected in the reaction mixtures. This also was the case even when the annulations were carried out under inert atmosphere to exclude the possibility of aerobic oxidation. We reasoned, that the nitroalkane aci formmay play here a dual role, serving both as an electrophilic reagent in cyclization and as an oxidant. Indeed, if we suppose that 1,8-dihydropyrimido[4,5,6-gh]perimidine core 3 was formed in the reaction, it also could be involved in nucleophilic attack at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of aci form 11. The resulting N-(imino-methyl)amidine 18 would then experience deprotonation of a distal N–H bond triggering elimination of phosphoric acid and nitrile 19 and leading to aromatization of the heterocyclic core of 1 (Scheme 5) (Fig. 1).
N bond of aci form 11. The resulting N-(imino-methyl)amidine 18 would then experience deprotonation of a distal N–H bond triggering elimination of phosphoric acid and nitrile 19 and leading to aromatization of the heterocyclic core of 1 (Scheme 5) (Fig. 1).
 |
| | Scheme 5 | |
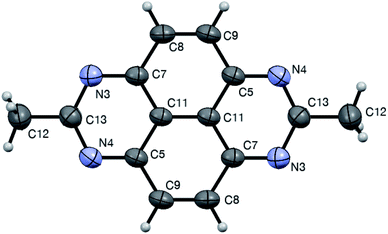 |
| | Fig. 1 ORTEP drawing od crystal structure of compound 1bb (CCDC 1544322†) showing atom numbering labels and 50% probability amplitude displacement ellipsoids. | |
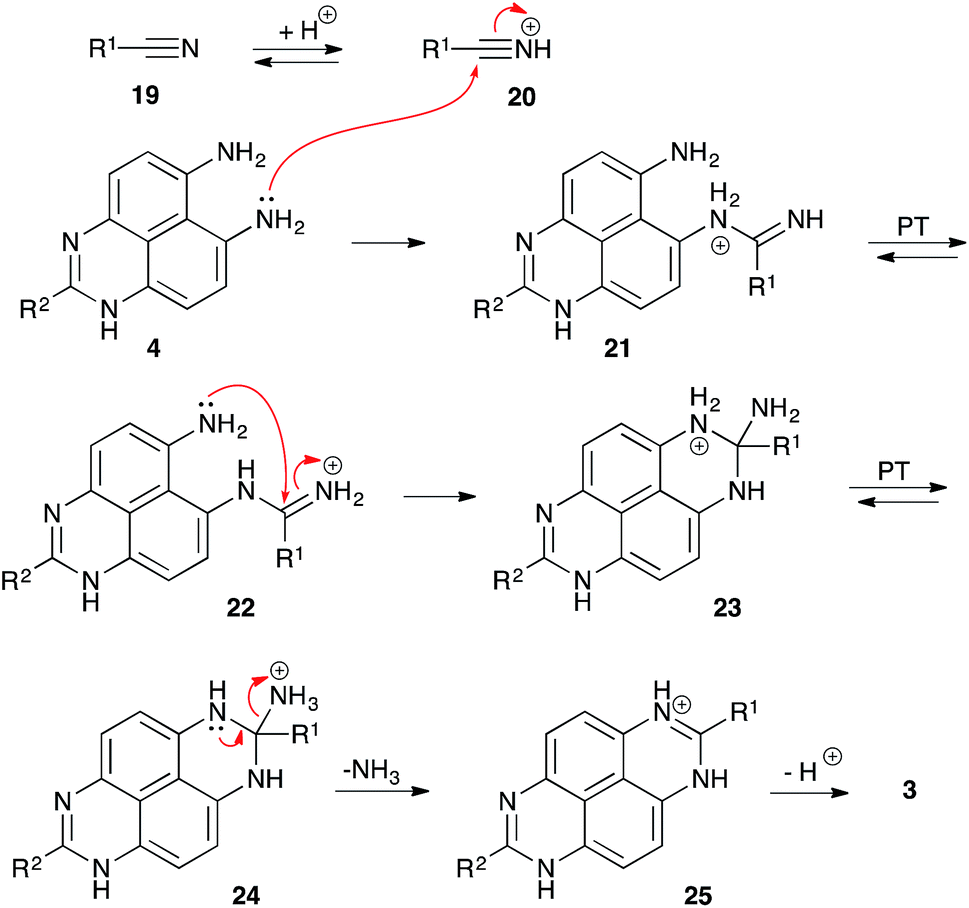
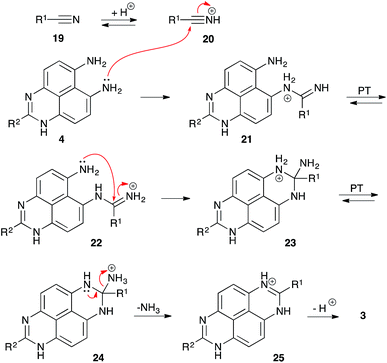
It should be pointed out that in acidic medium, nitrile 19 exists in equilibrium with highly electrophilic protonated form 20 (Scheme 6). We believe that this form might be responsible for the featured peri-annulation process according to the mechanism described in Scheme 6. Indeed, nucleophilic attack of the aniline moiety in perimidine 4 should afford amidinium species 21, which should exist in equilibrium with form 22. Subsequent 6-exo-trig nucleophilic ring closure should provide diaminomethanaminium moiety 23, that could experience a proton transfer to arrange an exo-cyclic ammonium leaving group in structure 24. Finally, elimination of ammonia would afford cyclic amidinium 25, which, after deprotonation, would provide 1,8-dihydropyrimido[4,5,6-gh]perimidine structure 3 (Scheme 6). The latter would be again oxidized by nitroalkanes into TAPy (1), as described in Scheme 5, and during this process nitrile 19 would be regenerated.
 |
| | Scheme 6 | |
The following empirical observation strongly supports this mechanistic hypothesis. Firstly, trace amounts of phenylacetonitrile were detected by GC and HRMS in reactions mixtures involving nitroalkane 10d. Secondly, peri-annulation of compound 4b with acetonitrile (instead of nitroethane) proceeded under similar reaction conditions (inert atmosphere was used to minimize aerobic oxidation) affording mixtures of 3bb and small amounts of 1bb (the latter was presumably formed due to oxidation during isolation). Thirdly, this mixture was quickly transformed into pure 1bb after treatment with nitroethane in 86% PPA.
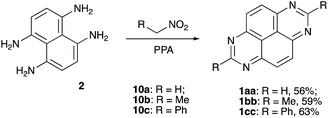
Having in hand such an efficient protocol for the synthesis of TAPy structures 1 from permidine precursors 4, we reserved the efforts to expand this methodology into a double-fold cyclocondensation; simultaneously installing two six-membered heterocyclic rings on the naphthalene-1,4,5,8-tetraamine (2). To test this idea we treated a solution of tetraamine 2 in 86% PPA with an excess of nitroalkane (10a, 10b, or 10c) at 100–150 °C (see Experimental part for details). Gratifyingly, the desired process proceeded smoothly, affording the corresponding symmetric TAPy structures (1aa, 1bb, or 1cc, respectively) as sole products, although only in medium yields (Scheme 7).
 |
| | Scheme 7 | |
Conclusions
In conclusion, we have developed highly efficient and chemoselective method for the synthesis of tetraazapyrenes (TAPy) based on the reaction of 1H-perimidine-6,7-diamines or naphthaene-1,4,5,8-tetraamine with nitroalkanes. This reaction demonstrates a very curious example of PPA-assisted activation of nitroalkane species towards nucleophilic attack. Interestingly, the electrophilically activated nitroalkane also plays a role of mild and efficient oxidant, providing direct access to the TAPy aromatic core and allowing excluding poorly controllable aerobic oxidation step.
Experimental part
1H and 13C NMR (DEPT-Q) spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with BBO probe in CDCl3, using TMS as an internal standard. The abbreviation “ψt” is used in listed spectra for doublets of doublets with non-resolved central lines. High-resolution mass spectra were registered with a Bruker Maxis spectrometer (electrospray ionization, in MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart smp30 apparatus. All reactions were performed in oven-dried 5 mL Erlenmeyer flasks (that can be open to the atmosphere), employing overhead stirring. Reaction progress and purity of isolated compounds were monitored by TLC on Silufol UV-254 plates. Perimidines 4a–b (in the form of trihydrochlorides),4 hexachlorostannate of tetraaminonaphthalene 2,3b and nitroalkenes 10b, c13 were prepared according to the literature procedures. All other reagents and solvents were purchased from commercial vendors and used as received.
General procedure for preparation of 1,3,6,8-tetraazapyrenes from 1H-perimidine-6,7-diamines (method A)
Reaction vessels charged with diaminoperimidine trihydrochloride (0.5 mmol), nitroalkane (1.0 mmol), and polyphosphoric acid (86% P2O5, 1.5 g) were vigorously stirred and heated at appropriate temperature (14 h at 100 °C for reactions involving nitromethane; 3 h at 110 °C for nitroethane, and 8 h 150 °C for phenylnitromethane and phenylnitroethane, respectively). Initially, intense foaming of the mixture was observed, which seized as the process progressed. Upon completion of the reaction, the mixture was poured into cold water (50 mL), neutralized with aqueous ammonia, extracted with ethyl acetate (5 × 70 mL), and concentrated in vacuum. Compounds 1aa, 1ab, 1bb can be obtained in pure form after trituation of the residue in dichloromethane (1 mL) and subsequent filtration. The filtrate was concentrated and purified by flash column chromatography eluting with ethyl acetate to obtain an additional crop of material (about 15–20%). For all other compounds, combined ethyl acetate extracts were concentrated and the residual material was purified by preparative column chromatography on silica gel eluting with ethyl acetate.
General procedure for preparation of 1,3,6,8-tetraazapyrenes from naphthalene-1,4,5,8-tetraamine (method B)
This protocol is identical to the one described above (method A), except for employing a mixture of naphthalene-1,4,5,8-tetraamine 2 (in the form of the stable hexachlorostannate that can be taken directly from reduction of the corresponding nitroarenes)3b,14 (0.261 g, 0.5 mmol), nitroalkane (2.0 mmol), and polyphosphoric acid (86% P2O5, 2.5 g).
Pyrimido[4,5,6-gh]perimidine (1aa)10,15
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.15 (10% EtOH/EtOAc); yield 65 mg (0.32 mmol, 63%) – via method A; 58 mg (0.28 mmol, 56%) – via method B; 1H NMR (CDCl3) δ, ppm: 10.08 (s, 2H), 8.71 (s, 4H); 13C NMR (CDCl3) δ, ppm: 158.0 (2C), 152.8 (4C), 136.5 (4C), 114.2 (2C); IR (NaCl, cm−1): 3050, 1603, 1437, 1240, 1180, 965, 843, 712; HRMS (ES TOF) calcd for C12H7N4 (M + H)+: 207.0665, found 207.0660 (2.4 ppm).
2-Methylpyrimido[4,5,6-gh]perimidine (1ab)10
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.21 (10% EtOH/EtOAc); yield 79 mg (0.36 mmol, 72%) – via method A in reaction of 4a with 10b; 69 mg (0.32 mmol, 63%) – in reaction of 4b with 10a; 1H NMR (CDCl3) δ, ppm: 9.92 (s, 1H), 8.51 (d, J = 9.5 Hz, 2H), 8.45 (d, J = 9.5 Hz, 2H), 3.17 (s, 3H); 13C NMR (CDCl3) δ, ppm: 168.6, 155.8, 154.7 (2C), 152.4 (2C), 137.1 (2C), 135.2 (2C), 113.7 (2C), 25.3; IR (NaCl, cm−1): 3057, 1562, 1551, 1478, 1491, 1355, 1342, 856; HRMS (ES TOF) calcd for C13H9N4 (M + H)+: 221.0822, found 221.0825 (1.4 ppm).
2,7-Dimethylpyrimido[4,5,6-gh]perimidine (1bb)11b
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.25 (10% EtOH/EtOAc); yield 91 mg (0.39 mmol, 78%) – via method A; 69 mg (0.30 mmol, 59%) – via method B; 1H NMR (CDCl3) δ, ppm: 8.35 (s, 4H), 3.03 (s, 6H); 13C NMR (CDCl3) δ, ppm: 166.9 (2C), 152.3 (4C), 135.6 (4C), 111.6 (2C), 26.8 (2C); IR (NaCl, cm−1): 3055, 1569, 1558, 1493, 1485, 1361, 1347, 858; HRMS (ES TOF) calcd for C14H11N4 (M + H)+: 235.0978, found 235.0983 (2.1 ppm).
2-Phenylpyrimido[4,5,6-gh]perimidine (1ac)11a
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.63 (10% EtOH/EtOAc); yield 102 mg (0.36 mmol, 72%) – via method A in reaction of 4c with 10a; 1H NMR (CDCl3) δ, ppm: 9.66 (s, 2H), 8.81 (d, J = 8.3 Hz, 2H), 8.57 (d, J = 9.5 Hz, 2H), 8.52 (d, J = 9.5 Hz, 2H), 7.64–759 (m, 3H); 13C NMR (CDCl3) δ, ppm: 165.9, 155.4, 154.7 (2C), 152.4 (2C), 138.4, 137.2 (2C), 135.2 (2C), 133.0, 131.2 (2C), 129.3 (2C), 114.2, 112.7; FT IR (NaCl, cm−1): 3052, 1577, 1555, 1499, 1483, 861, 723, 641; HRMS (ES TOF) calcd for C18H11N4 (M + H)+: 283.0978, found 283.0971 (2.5 ppm).
2-Methyl-7-phenylpyrimido[4,5,6-gh]perimidine (1bc)11b
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.75 (10% EtOH/EtOAc); yield 105 mg (0.36 mmol, 71%) – via method A in reaction of 4b with 10c; 1H NMR (CDCl3) δ, ppm: 8.84 (d, J = 8.3 Hz, 2H), 8.66 (d, J = 9.5 Hz, 2H), 8.56 (d, J = 9.5 Hz, 2H), 7.61 (ψt, 1H), 7.55 (d, J = 7.4 Hz, 2H), 3.24 (s, 3H); 13C NMR (CDCl3) δ, ppm: 166.8, 164.0, 153.0 (4C), 138.4, 136.8 (2C), 135.9 (2C), 133.0, 131.3 (2C), 129.2 (2C), 112.8 (2C), 21.2; FT IR (NaCl, cm−1): 3036, 1579, 1554, 1491, 1482, 858, 775, 637; HRMS (ES TOF) calcd for C19H13N4 (M + H)+: 297.1135, found 297.1129 (2.0 ppm).
2,7-Diphenylpyrimido[4,5,6-gh]perimidine (1cc)11b
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.85 (10% EtOH/EtOAc); yield 113 mg (0.32 mmol, 63%) – via method B; 1H NMR (CDCl3) δ, ppm: 8.84 (d, J = 8.5 Hz, 4H), 8.68 (s, 4H), 7.63–7.52 (m, 6H); 13C NMR (CDCl3) δ, ppm: 167.0 (2C), 152.3 (4C), 139.1 (2C), 135.2 (4C), 133.1 (2C), 131.3 (4C), 129.5 (4C), 113.0 (2C); FT IR (NaCl, cm−1): 3045, 1573, 1561, 1499, 1485, 852, 770, 641; HRMS (ES TOF) calcd for C24H15N4 (M + H)+: 359.1291, found 359.1301 (2.8 ppm).
2-Benzylpyrimido[4,5,6-gh]perimidine (1ad)
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.65 (10% EtOH/EtOAc); yield 93 mg (0.32 mmol, 63%) – via method A in reaction of 4a with 10d; 1H NMR (CDCl3) δ, ppm: 10.01 (s, 1H), 8.51 (d, J = 9.9 Hz, 2H), 8.47 (d, J = 9.9 Hz, 2H), 7.51 (d, J = 7.8 Hz, 2H), 7.35–7.32 (m, 2H), 7.21, 7.23–7.19 (m, 1H), 4.23 (s, 2H); 13C NMR (CDCl3) δ, ppm: 163.2, 157.9, 152.3 (2C), 151.6 (2C), 139.7, 136.4 (2C), 134.5 (2C), 129.4 (2C), 128.6 (2C), 126.7, 115.2 (2C), 29.8; FT IR (NaCl, cm−1): 3047, 1582, 1555, 1491, 1475, 863, 761, 633; HRMS (ES TOF) calcd for C19H13N4 (M + H)+: 297.1135, found 297.1143 (2.7 ppm).
2-Benzyl-7-methylpyrimido[4,5,6-gh]perimidine (1bd)
This material was obtained as a yellowish crystalline solid, mp over 300 °C, Rf 0.76 (10% EtOH/EtOAc); yield 102 mg (0.33 mmol, 66%) – via method A in reaction of 4b with 10d; 1H NMR (CDCl3) δ, ppm: 8.53 (s, 4H), 7.52 (d, J = 7.8 Hz, 2H), 7.35–7.32 (m, 2H), 7.21, 7.23–7.19 (m, 1H), 4.18 (s, 2H), 3.19 (s, 3H); 13C NMR (CDCl3) δ, ppm: 166.8 (2C), 152.1 (4C), 137.7, 135.4 (4C), 129.0 (2C), 128.2 (2C), 126.6, 111.4 (2C), 29.2, 26.6; FT IR (NaCl, cm−1): 3053, 1574, 1568, 1483, 1489, 1351, 1344, 866; HRMS calcd for C20H15N4 (M + H)+: 311.1291, found 311.1282 (2.9 ppm).
Reaction of 4b with acetonitrile to intercept 2,7-dimethyl-1,8-dihydropyrimido[4,5,6-gh]perimidine (3bb) (method C)
A mixture of 2-methyl-6,7-diaminoperimidine trihydrochloride (71 mg, 0.25 mmol) and polyphosphoric acid (86% P2O5, 1.0 g) was vigorously stirred under argon atmosphere and dry acetonitrile (52 μL, 41 mg, 1.0 mmol) was injected in a single portion. The mixture was heated for 3 h at 100 °C, and then poured into freshly distilled degassed water (50 mL), neutralized with aqueous ammonia, extracted with EtOAc (5 × 50 mL), and concentrated under argon. Crude compound 3b was isolated as brownish-red solid, Rf 0.10 (EtOAc/EtOH 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), relatively stable in solid form stored under inert atmosphere, but quickly decomposing in solutions or upon contact with air. GC detection as well as mass-spectral characterization of this compound proved impossible due to very rapid oxidation into 1bb upon heating. Our attempt to measure of 13C NMR spectrum also proved unfruitful, since this compound decomposes in solutions faster than it was necessary to accumulate sufficient number of scans for carbon spectrum. We were able to measure 1H NMR spectrum (see ESI† for spectral chart). This experiment demonstrated, that 3bb exists in solution in CDCl3 as a mixture of equilibrating 1,8-dihydro- and 1,6-dihydro-tautomers along with minute amount of oxidized product 1bb.
:1), relatively stable in solid form stored under inert atmosphere, but quickly decomposing in solutions or upon contact with air. GC detection as well as mass-spectral characterization of this compound proved impossible due to very rapid oxidation into 1bb upon heating. Our attempt to measure of 13C NMR spectrum also proved unfruitful, since this compound decomposes in solutions faster than it was necessary to accumulate sufficient number of scans for carbon spectrum. We were able to measure 1H NMR spectrum (see ESI† for spectral chart). This experiment demonstrated, that 3bb exists in solution in CDCl3 as a mixture of equilibrating 1,8-dihydro- and 1,6-dihydro-tautomers along with minute amount of oxidized product 1bb.
Reaction of 2,7-dimethyl-1,8-dihydropyrimido[4,5,6-gh]perimidine (3bb) with nitroethane in PPA
Compound 3bb (59 mg, 0.25 mmol) obtained in the reaction of 4b and acetonitrile as described above (method C) was stirred at 100 °C in 86% PPA (1.0 g) under argon atmosphere. Nitroethane (18 μL, 19 mg, 0.25 mmol, 1.00 equiv.) was injected in a single portion and the stirring was continued for 3 min, after which color of the reaction mixture changes from dark-red to green indicating the completion of the oxidation process. The post-reaction work up and isolation of the product was carried out in the same way as described for method A. Compound 1bb (47 mg, 0.20 mmol, 80%) was obtained as sole product. Physical and spectral properties of this material were identical to those described above for the sample obtained via method A.
Acknowledgements
This project received financial support from the Ministry of Education and Science of the Russian Federation (the Agreement number 02.a03.0008). Financial support from the Russian Foundation for Basic Research (grants #16-03-00177a and 16-3300483 mol_a) is also gratefully acknowledged.
Notes and references
- For reviews, see:
(a) F. Würthner and R. Schmidt, ChemPhysChem, 2006, 7, 793–797 CrossRef PubMed
 ;
(b) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed ;
(c) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed ;
(d) V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed ;
(e) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed ;
(f) H. E. Katz and J. Huang, Annu. Rev. Mater. Res., 2009, 39, 71–92 CrossRef CAS ;
(g) B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed ;
(h) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed ;
(i) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed .
;
(b) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed ;
(c) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed ;
(d) V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed ;
(e) Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed ;
(f) H. E. Katz and J. Huang, Annu. Rev. Mater. Res., 2009, 39, 71–92 CrossRef CAS ;
(g) B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed ;
(h) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed ;
(i) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed . - See, for example:
(a) P. Jonkheijm, N. Stutzmann, Z. Chen, D. M. de Leeuw, E. W. Meijer, A. P. H. J. Schenning and F. Würthner, J. Am. Chem. Soc., 2006, 128, 9535–9540 CrossRef CAS PubMed ;
(b) H. Z. Chen, M. M. Ling, X. Mo, M. M. Shi, M. Wang and Z. Bao, Chem. Mater., 2007, 19, 816–824 CrossRef CAS ;
(c) R. Schmidt, M. M. Ling, J. H. Oh, M. Winkler, M. Könemann, Z. Bao and F. Würthner, Adv. Mater., 2007, 19, 3692–3695 CrossRef CAS ;
(d) R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS PubMed ;
(e) H. Qian, W. Yue, Y. Zhen, S. Di Motta, E. Di Donato, F. Negri, J. Qu, W. Xu, D. Zhu and Z. Wang, J. Org. Chem., 2009, 74, 6275–6282 CrossRef CAS PubMed ;
(f) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed ;
(g) H. C. Hesse, J. Weickert, C. Hundschell, X. Feng, K. Müllen, B. Nickel, A. J. Mozer and L. Schmidt-Mende, Adv. Energy Mater., 2011, 1, 861–869 CrossRef CAS .
-
(a) S. C. Martens, L. Hahn, F. Lombeck, A. Rybina, H. Wadepohl and L. H. Gade, Eur. J. Org. Chem., 2013, 5295–5302 CrossRef CAS ;
(b) S. Geib, S. C. Martens, U. Zschieschang, F. Lombeck, H. Wadepohl, H. Klauk and L. H. Gade, J. Org. Chem., 2012, 77, 6107–6116 CrossRef CAS PubMed ;
(c) W.-C. Chen and I. Chao, J. Phys. Chem. C, 2014, 118, 20176–20183 CrossRef CAS .
- A. F. Pozharskii, V. N. Koroleva, I. V. Komissarov, I. T. Fillippov and I. V. Borovlev, Pharm. Chem. J., 1976, 10, 1613–1617 CrossRef .
- Z. D. Wang, J. Eilander, M. Yoshida and T. Wang, Eur. J. Org. Chem., 2014, 7664–7674 CrossRef CAS .
- A. Ziesak, T. Wesp, O. Huebner, E. Kaifer, H. Wadepohl and H.-J. Himmel, Dalton Trans., 2015, 44, 19111–19125 RSC .
- See, for recent examples:
(a) A. V. Aksenov, N. A. Aksenov, D. S. Ovcharov, D. A. Aksenov, L. G. Voskressensky and M. Rubin, RSC Adv., 2016, 6, 82425–82431 RSC ;
(b) A. V. Aksenov, M. H. Magamadova, D. A. Lobach, I. V. Aksenova, I. V. Malikova and M. Rubin, Chem. Heterocycl. Compd., 2014, 50, 1298–1304 CrossRef CAS S. V. Shcherbakov, D. A. Lobach, M. Rubin and A. V. Aksenov, Chem. Heterocycl. Compd., 2014, 50, 757–760 CrossRef CAS .
- See, for example:
(a) A. V. Aksenov, D. A. Aksenov, N. A. Orazova, N. A. Aksenov, G. D. Griaznov, A. De Carvalho, R. Kiss, V. Mathieu, A. Kornienko and M. Rubin, J. Org. Chem., 2017, 82, 3011–3018 CrossRef CAS PubMed ;
(b) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, J. P. Matheny and M. Rubin, RSC Adv., 2015, 5, 8647–8656 RSC ;
(c) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, A. S. Bijieva and M. Rubin, Org. Biomol. Chem., 2014, 12, 9786–9788 RSC ;
(d) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, L. V. Frolova, A. Kornienko, I. V. Magedov and M. Rubin, Chem. Commun., 2013, 49, 9305–9307 RSC .
- A. V. Aksenov, A. S. Lyakhovnenko, T. S. Perlova and I. V. Aksenova, Chem. Heterocycl. Compd., 2011, 47, 916–917 CrossRef CAS .
-
(a) A. V. Aksenov, N. A. Aksenov, A. S. Lyakhovnenko, A. N. Smirnov, I. I. Levina and I. V. Aksenova, Chem. Heterocycl. Compd., 2013, 49, 980–987 CrossRef CAS ;
(b) A. V. Aksenov, A. S. Lyakhovnenko, N. T. Karaivanov and I. V. Aksenova, Chem. Heterocycl. Compd., 2010, 46, 1146–1147 CrossRef CAS ;
(c) A. V. Aksenov, A. S. Lyakhovnenko, A. N. Spicin and I. V. Aksenova, Chem. Heterocycl. Compd., 2011, 47, 1180–1182 CrossRef CAS ;
(d) A. V. Aksenov, N. A. Aksenov, A. E. Tsys′, V. I. Goncharov and S. N. Ovcharov, Russ. Chem. Bull., 2013, 62, 1127–1128 CrossRef CAS .
-
(a) A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synth. Commun., 2012, 42, 541–547 CrossRef CAS ;
(b) A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Chem. Heterocycl. Compd., 2011, 46, 1405–1406 CrossRef CAS ;
(c) A. V. Aksenov, N. A. Aksenov, O. N. Nadein and A. E. Tsys, Chem. Heterocycl. Compd., 2010, 46, 1025–1026 CrossRef CAS ;
(d) A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synlett, 2010, 2628–2630 CrossRef CAS ;
(e) A. V. Aksenov, N. A. Aksenov, N. A. Orazova, D. A. Aksenov, M. V. Dmitriev and M. Rubin, RSC Adv., 2015, 5, 84849–84855 RSC ;
(f) N. A. Aksenov, A. V. Aksenov, O. N. Nadein, D. A. Aksenov, A. N. Smirnov and M. Rubin, RSC Adv., 2015, 5, 71620–71626 RSC .
- A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenova and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289–4295 CAS .
-
(a) R. Sarkar and S. Mukherjee, Org. Lett., 2016, 18, 6160–6163 CrossRef CAS PubMed ;
(b) M. S. Manna and S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 130–133 CrossRef CAS PubMed .
- W. Will, Chem. Ber., 1895, 28, 2234–2235 CrossRef CAS .
- O. Dimroth and H. Roos, Liebigs Ann. Chem., 1927, 456, 177–192 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Spectral and crystallographic data. CCDC 1544322. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04751g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Dmitrii S. Ovcharova,
Nicolai A. Aksenovab,
Dmitrii A. Aksenova,
Oleg N. Nadeina and
Michael Rubin
*a,
Dmitrii S. Ovcharova,
Nicolai A. Aksenovab,
Dmitrii A. Aksenova,
Oleg N. Nadeina and
Michael Rubin