
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrogenation of p-chloronitrobenzene over an Fe promoted Pt/AC catalyst
Hu Chen,
Daiping He *,
Qingqing He,
Ping Jiang,
Gongbing Zhou and
Wensheng Fu
*,
Qingqing He,
Ping Jiang,
Gongbing Zhou and
Wensheng Fu
Key Lab of Green Synthesis and Applications, College of Chemistry, Chongqing Normal University, Chongqing 401331, China. E-mail: hedaiping@126.com; Fax: +86-23-6536-2777; Tel: +86-23-6591-0309
First published on 5th June 2017
Abstract
Activated carbon supported Pt (Pt/AC) and Fe modified Pt (Pt–Fe/AC) catalysts have been investigated for selective hydrogenation of p-chloronitrobenzene to p-chloroaniline. It was found that the incorporation of an appropriate amount of Fe (4 wt%) resulted in a remarkable increase in activity and selectivity to p-chloroaniline, and the catalytic hydrodechlorination over the catalyst was fully suppressed at complete conversion of p-chloronitrobenzene. X-ray photoelectron spectroscopy (XPS) revealed the electron transfer from Pt nanoparticles to Fe2O3, leading to the electron-deficient state of the Pt nanoparticles in the present catalyst, which is responsible for improved performance of the Pt–Fe/AC catalyst for p-chloronitrobenzene hydrogenation to p-chloroaniline.
1 Introduction
The reduction of chloronitrobenzenes to chloroanilines is significant in organic synthesis because the chloroanilines are the key intermediates of pharmaceuticals, polymers, pesticides, explosives, fibers, dyes and cosmetics.1 The traditional methods that use either the Fe/HCl system (Bechamp reaction) or metal sulfides inevitably generate large amounts of waste, which requires costly and laborious separation and disposal.2 Synthesis of chloroanilines using catalytic hydrogenation starting with chloronitrobenzenes offers several advantages over chemical reduction, including cheap hydrogen, easy product separation and a nonpolluting process. A severe problem related to the catalytic hydrogenation has been the co-occurrence of the catalytic hydrodechlorination reaction of the substrates,3–5 which significantly limits the selectivity to chloroanilines and leads to a lot of wastes. Furthermore, hydrogen chloride produced from the hydrodechlorination is also corrosive to the reactor. It is highly desirable to develop heterogeneous catalysts that would ensure the selective reduction of the nitro groups while leave intact of the carbon–chlorine bond in the same molecule. Many efforts have been devoted to performing selective hydrogenation of chloronitrobenzenes to corresponding chloroanilines over a plentiful of metal catalysts,6–11 including Pt, Pd, Au, Ru, Ni and Ag. The gold catalyst offers the highest selectivity of chloroanilines for this hydrogenation,12,13 but platinum seems to be the best catalyst for minimizing dechlorination combined with a fast rate of reduction of the nitro group and being highly resistant to deactivation.14 In most cases, high selectivity is always achieved at the expense of activity by introducing either transition-metal salts or additives that partly poison the active sites.15–17 The choice of components is critical in developing an efficient and viable catalyst, and due attention should be paid to the interaction between active metal and promoter, since the nature of this interaction plays an important role in shaping the activity, selectivity and stability of the catalyst. Mahata N et al.18 studied the influence of Cu on the catalytic properties of Pt/C for the hydrogenation of chloronitrobenzenes. It is found that the strong interactions between Pt and Cu result in partial coverage of the Pt surface by Cu. The superficial Cu species activate the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of chloronitrobenzene and also inhibit the adsorption of the desired product chloroaniline. Thus, the bimetallic catalysts exhibited excellent performance in producing selectively chloroanilines (>99%) in comparison to the Pt/AC catalyst. Moreover, the high catalytic activity of the Pt nanoclusters was maintained.
O bond of chloronitrobenzene and also inhibit the adsorption of the desired product chloroaniline. Thus, the bimetallic catalysts exhibited excellent performance in producing selectively chloroanilines (>99%) in comparison to the Pt/AC catalyst. Moreover, the high catalytic activity of the Pt nanoclusters was maintained.
In this paper, we report the promotional effect of Fe on the catalytic performance of Pt/C catalyst. The possible function of Fe in Pt–Fe/AC catalysts is discussed on the basis of catalyst characterization and catalytic results.
2 Experimental
2.1 Catalyst preparation
An activated carbon (AC) sample was obtained from Chongqing Chuandong Chemical Company and was crushed into 220–250 mesh powder. It was washed with distilled water, then dried at 383 K overnight and calcined in flowing air at 523 K for 3 h. The Pt–Fe/AC catalyst was prepared by a wet impregnation technique. Typically, 2.0 g of activated carbon with BET surface area of 411 m2 g−1 was added to 50 mL aqueous Fe(NO3)3 (0.03 mol L−1) and the suspension was stirred for 24 h. Then, 1.58 mL aqueous H2PtCl6 (3.8 × 10−3 mol L−1) was added dropwise to the mixture for 24 h upon continuous stirring. The deposition of Pt and Fe nanoparticles from the aqueous solution occurred when a stoichiometric excess of aqueous NaBH4 (0.5 mol L−1) was added dropwise to the mixture. Stirring was continued for a further 5 h. The solids were filtered and washed thoroughly with distilled water until free of chloride ions. The obtained sample was dried at 383 K for 10 h and calcined in flowing air at different temperature for 5 h. The platinum, iron in the catalyst was 0.29 wt% and 3.86 wt% according to ICP-AES analysis, respectively.2.2 Catalyst characterization
The platinum and iron contents in the 0.3% Pt–4% Fe/AC catalyst were analyzed by means of inductively coupled plasma-atomic emission spectroscopy (ICP-AES) after the sample was dissolved in aqua regia. X-ray powder diffraction (XRD) patterns were recorded on a Rigaku D/Max Ultima IV X-ray diffractometer using Cu Kα radiation with accelerating voltage of 40 kV and current of 30 mA. Transmission electron microscopy (TEM) measurement was performed on a JEOL JEM-2000EX microscope operated at an accelerating voltage of 120 kV. An energy-dispersive X-ray (EDX) spectroscopic detecting unit was used to collect the EDX spectra for elemental analysis. X-ray photoelectron spectra (XPS) were obtained on an ESCALAB 250 X-ray photoelectron spectroscopy using Mg-Kα X-ray as the excitation source. XPS spectra were calibrated based on the graphite C 1s peak at 284.6 eV.2.3 Catalyst testing
The catalytic hydrogenation was carried out with magnetic stirring (900 rpm) in a 60 mL Teflon-lined stainless steel autoclave containing 1.0 g of p-chloronitrobenzene (p-CNB) in 8.0 mL ethanol (solvent). The catalyst used in each run of the reaction was 20.0 mg. The reactor was flushed one time with 0.5 MPa N2 and then four times with 0.5 MPa H2 before it was pressurized to the desired H2 pressure and placed in a heat installation maintained at the reaction temperature. The reaction was stopped at a selected time by cooling the reactor in a ice water bath. The products were analyzed by GC (GC9890) with an SE-30 capillary column and an FID as detector, using o-xylene as an internal standard.3 Results and discussion
3.1 Catalyst characterization
The XRD patterns of the AC support and 0.3% Pt–4% Fe/AC catalyst are shown in Fig. 1. The AC support shows the characteristic diffraction peaks of graphitized activated carbon at 2θ = 24.3°, 35.4° and 43.2°.19 The minor peaks at 20.7°, 26.5°, 39.4°, 50.1° and 68.1° are attributed to the presence of trace quantity of silica.20 The XRD pattern of the 0.3% Pt–4% Fe/AC catalyst is similar to that of the AC support as we can see from Fig. 1b. No characteristic diffraction peaks at 39.9°, 46.4°, 67.5° index to (111), (200), (220) planes of metallic Pt21 and 44.4° assigned to Fe (110) plane22 were detected in the 0.3% Pt–4% Fe/AC catalyst, probably because of the low percentage of metals on the catalyst and/or the small particle size.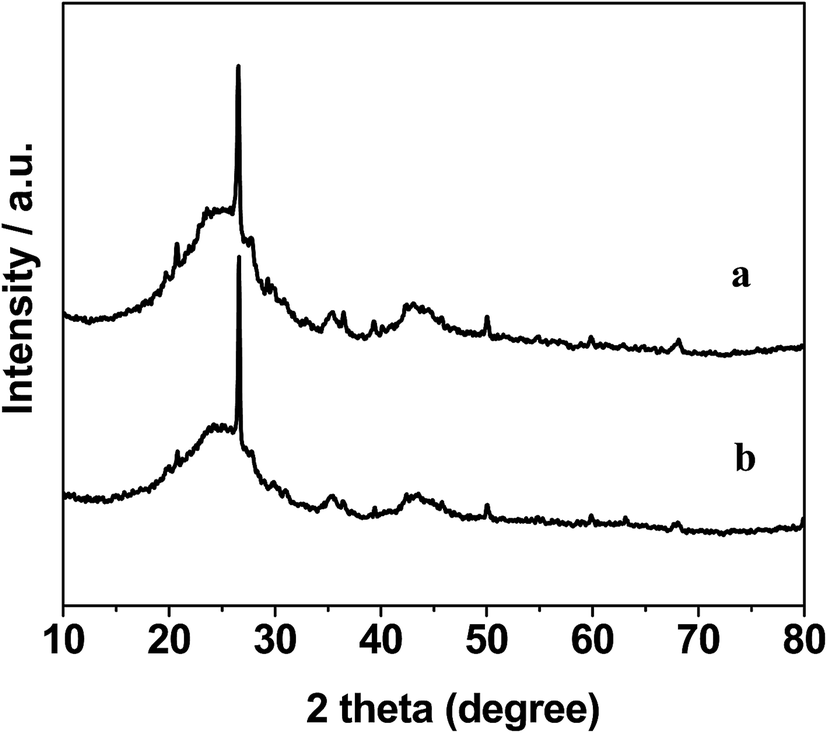 | ||
| Fig. 1 XRD patterns of AC (a) and 0.3% Pt–4% Fe/AC (b). | ||
Fig. 2 shows the representative TEM images of the 0.3% Pt–4% Fe/AC catalyst. As can be seen from the TEM image (Fig. 2a), small black spots can be discerned from the background over the 0.3% Pt–4% Fe/AC catalyst. A combined EDX analysis showed that the signal of Pt and Fe could be detected in the small black spots (Fig. 2b). Therefore, we conclude that the small black spots in Fig. 2a are the images of the Pt and Fe species. The Fe and Pt particles in the 0.3% Pt–4% Fe/AC catalyst have an average diameter of 4.5 nm, with a size distribution from 3 to 7 nm (Fig. 2c), which is consistent with the absence of characteristic diffraction peaks of Fe and Pt in the XRD pattern of the 0.3% Pt–4% Fe/AC, suggesting that the Fe and Pt species on AC support are in a homogenous dispersion without any obvious aggregation.
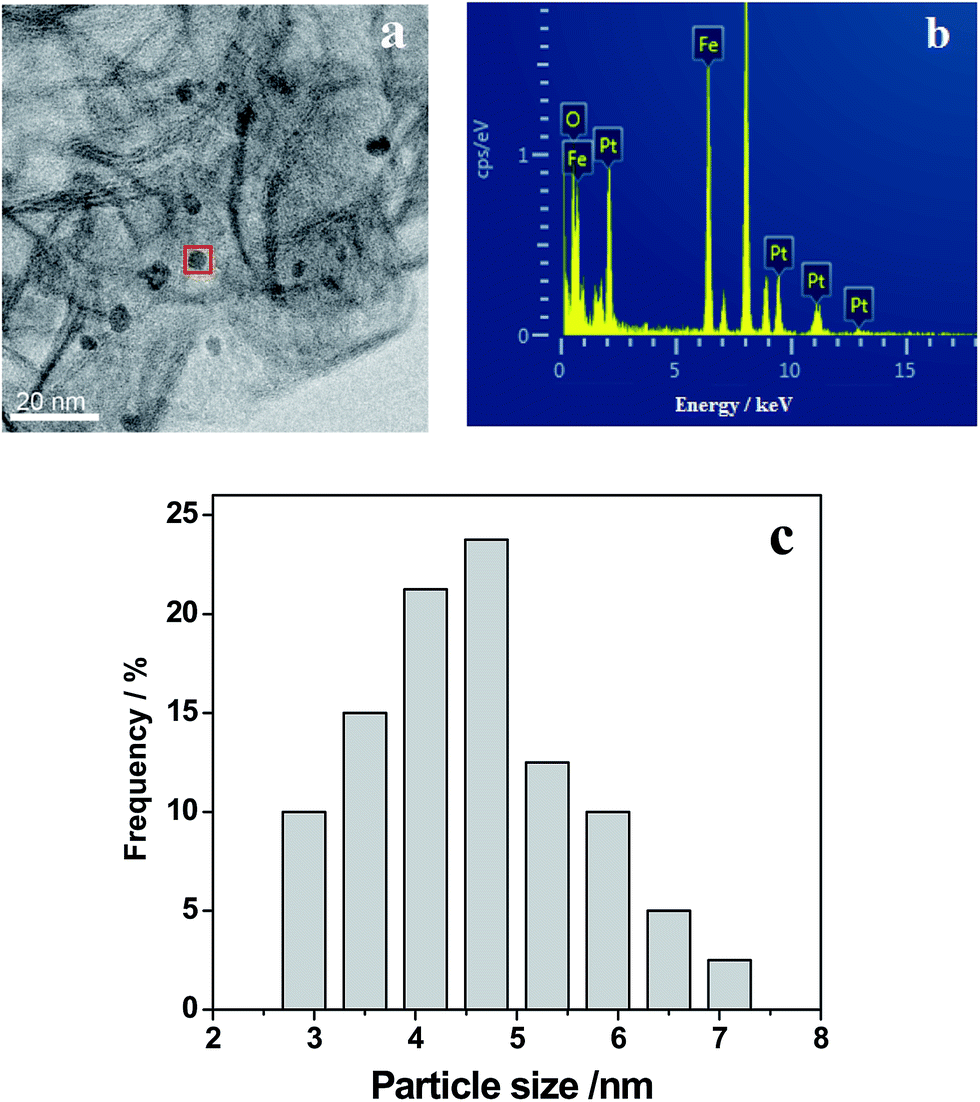 | ||
| Fig. 2 TEM image of 0.3% Pt–4% Fe/AC (a), EDX spectra of the selected area (b) and size distributions of Pt–Fe particles in the catalyst (c). | ||
XPS analyses of the 0.3% Pt/AC and 0.3% Pt–4% Fe/AC catalysts were performed. The Fe 2p spectrum of the 0.3% Pt–4% Fe/AC can be deconvoluted three peaks (Fig. 3). The peaks can be assigned to Fe3+ in Fe2O3, Fe8/3+ in Fe3O4 and Fe2+ in FeO, respectively.23 It is evident that the calcination of the 0.3% Pt–4% Fe/AC at 473 K in air leads to the formation of Fe2O3 on AC. Fig. 4 presents the Pt 4f spectra of the 0.3% Pt/AC and 0.3% Pt–4% Fe/AC catalysts. The Pt 4f7/2 photoelectron peak of the 0.3% Pt/AC catalyst is observed at 71.2 eV (Fig. 4a), which is consistent with the characteristic peak of Pt 4f7/2 for metallic Pt at 71.1 eV.24 However, the binding energy of the Pt 4f7/2 level in the 0.3% Pt–4% Fe/AC catalyst (Fig. 4b) is 0.9 eV higher than that of metallic Pt. This reveals the electron transfer and the electron-deficient state of the Pt nanoparticles in the 0.3% Pt–4% Fe/AC catalyst. Wang et al.25 also observed the same phenomenon in the Pt/γ-Fe2O3 catalyst. Based on these results, we believe that the electron transfer may occur from the Pt particles to the Fe2O3 in the 0.3% Pt–4% Fe/AC catalyst.
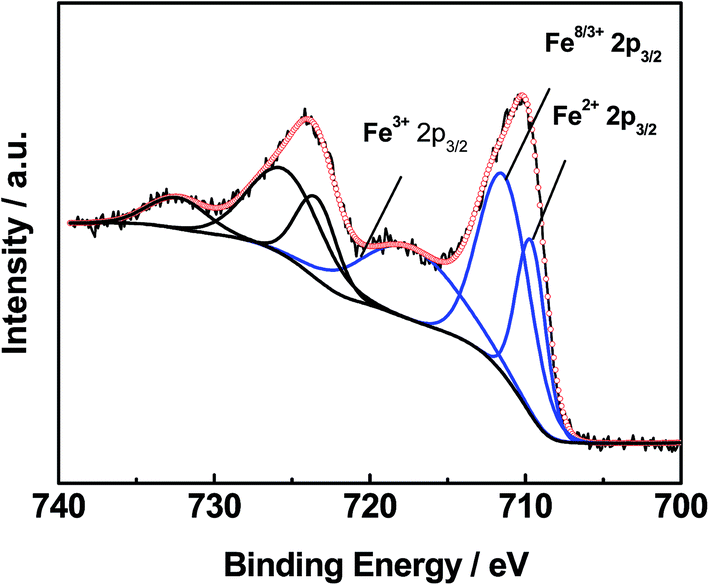 | ||
| Fig. 3 XPS spectrum of Fe 2p3/2 for 0.3% Pt–4% Fe/AC. | ||
 | ||
| Fig. 4 XPS spectra of Pt 4f5/2 for 0.3% Pt/AC (a) and 0.3% Pt–4% Fe/AC (b). | ||
3.2 Catalytic performance
Table 1 shows the catalytic properties of the 4% Fe/AC, 0.3% Pt/AC and 0.3% Pt–Fe/AC catalysts for hydrogenation of p-CNB at 303 K, 1.0 MPa H2. No reaction of p-CNB was observed in the absence of the catalyst and over the 4% Fe/AC catalyst. The conversion of p-CNB over the 0.3% Pt/AC catalyst was 37% for 40 minutes. When the same 0.3% Pt/AC was loaded with 4% Fe, however, a dramatically high p-CNB conversion (65%) was achieved under the same reaction conditions, and the selectivity to the desired product p-CAN also increased from 84.0% to 94.2%, which may be attributed to FexOy species activated the NO bond which becomes highly susceptible to hydrogen attack.26 When Fe loading amount further increases from 4 wt% to 6 wt%, the conversion of p-CNB decreases from 65% to 33%, which may be caused by partial coverage of the Pt surface by excess FexOy. Interestingly, the undesired hydrodechlorination reaction observed over the Pt/AC was not detected over the 0.3% Pt–4% Fe/AC catalyst. Special attention was paid to distinguish byproducts of the p-CNB hydrogenation reactions with GC-MS. Any byproduct that amounted to 0.1% in the reaction mixture can be unambiguously detected in our method of product analysis. We did not detect any by-products derived from the hydrodechlorination over the 0.3% Pt–4% Fe/AC catalyst, even when the reaction time was extended by 30 minutes after p-CNB was exhausted, which indicates that the undesired hydrodechlorination reaction usually observed over other metal catalysts27–29 can be successfully avoided over the catalyst. XPS data indicate that the electron transfer occurred from Pt to Fe2O3 make the Pt nanoparticles in the 0.3% Pt–4% Fe/AC catalyst an electron-deficient state. The electron-deficient state of the Pt particles could weaken the extent of electron feedback from the Pt particles to the aromatic ring in chloroanilines and suppressed the hydrodechlorination reaction.25 Therefore, the high selectivity of p-CAN over the 0.3% Pt–4% Fe/AC catalyst may attribute to the electron-deficient state of the Pt nanoparticles.
| Catalyst | p-CNB conv./% (min) | Sel. (%) | ||
|---|---|---|---|---|
| p-CAN | ANb | Othersc | ||
| a n.d.: not detected.b AN refers to aniline.c Other products were p-chloronitrosobenzene, 4,4′-dichloroazoxybenzene and 4,4′-dichloroazobenzene. Reaction conditions: 20.0 mg catalyst, 1.0 g p-CNB in 8.0 mL ethanol (solvent), 303 K, 1.0 MPa H2. | ||||
| 4% Fe/AC | n.d. (40) | n.d. | n.d. | n.d. |
| 0.3% Pt/AC | 37 (40) | 84.0 | n.d. | 16.0 |
| 0.3% Pt–2% Fe/AC | 50 (40) | 92.6 | n.d. | 7.4 |
| 0.3% Pt–4% Fe/AC | 65 (40) | 94.2 | n.d. | 5.8 |
| 0.3% Pt–4% Fe/AC | 100 (90) | 100 | n.d. | n.d. |
| 0.3% Pt–4% Fe/AC | 100 (120) | 100 | n.d. | n.d. |
| 0.3% Pt–6% Fe/AC | 33 (40) | 92.7 | n.d. | 7.3 |
3.3 Effect of calcination temperature
The hydrogenation properties of the 0.3% Pt–4% Fe/AC calcined at different temperature are also investigated, and the results are listed in Table 2. From Table 2, we can see that different calcination temperature of the 0.3% Pt–4% Fe/AC has significant influence on the hydrogenation of p-CNB. The 0.3% Pt–4% Fe/AC calcined at 473 K shows the best catalytic activity and selectivity towards p-CAN, which could can be the result of the favorable formation of Fe2O3 on AC at 473 K, and the sintering of the Pt nanoparticles at higher calcination temperature (>473 K).3.4 Effect of reaction conditions
The reaction temperature also plays a significant role in the p-CNB hydrogenation reaction (Table 3). The conversion of p-CNB and selectivity to p-CAN was 65% and 94.2% at 303 K, respectively. The conversion increased significantly to 100% and the selectivity reached 100% without formation of any decholorination product when the reaction temperature increased from 303 K to 333 K. The byproducts produced in the reaction can be further hydrogenated to the desired product p-CAN. These data demonstrate that the undesired further hydrogenation of p-CAN can be avoided over the 0.3% Pt–4% Fe/AC catalyst in a broad window (303–333 K).This advantage of the catalyst is clearly shown by the catalytic properties under the elevated H2 pressure (Table 4). The conversion of p-CNB and selectivity to p-CAN was 30% and 92.9% at 0.5 MPa H2, respectively. The conversion increased to 100% and the selectivity reached 98.5% at 2.0 MPa H2. It is interesting to note that any by-products derived from the hydrodechlorination were not detected, even when all of the p-CNB was exhausted under 3.0 MPa, which demonstrates a remarkable feature of the 0.3% Pt–4% Fe/AC catalyst for avoiding successfully any occurrence of the further hydrogenation of p-CAN even when very high H2 pressure is used for the reaction.
3.5 Catalyst stability
The recycling procedure of the 0.3% Pt–4% Fe/AC catalyst was performed for examining the stability of the catalyst. The catalyst after reaction was separated by centrifugation, washed with ethanol, dried under vacuum and then reused for the same reaction. As shown in Table 5, both conversion and selectivity did not exhibit a considerable decrease for at least 4 cycles. We checked the Pt, Fe content in the recovered catalyst after 4 cycles and found that there was only a slight loss of Pt (0.02 wt%) and Fe (0.35 wt%) in the recovered catalyst. TEM analysis showed good dispersion of Pt–Fe particles in the recovered 0.3% Pt–4% Fe/AC (Fig. 5a). The average Pt–Fe particle size was 4.6 nm, with a size distribution from 3 to 7 nm (Fig. 5b), which is similar to that in the as-prepared fresh 0.3% Pt–4% Fe/AC catalyst (4.5 nm) (Fig. 2c). The as-prepared 0.3% Pt–4% Fe/AC catalyst showed high stability.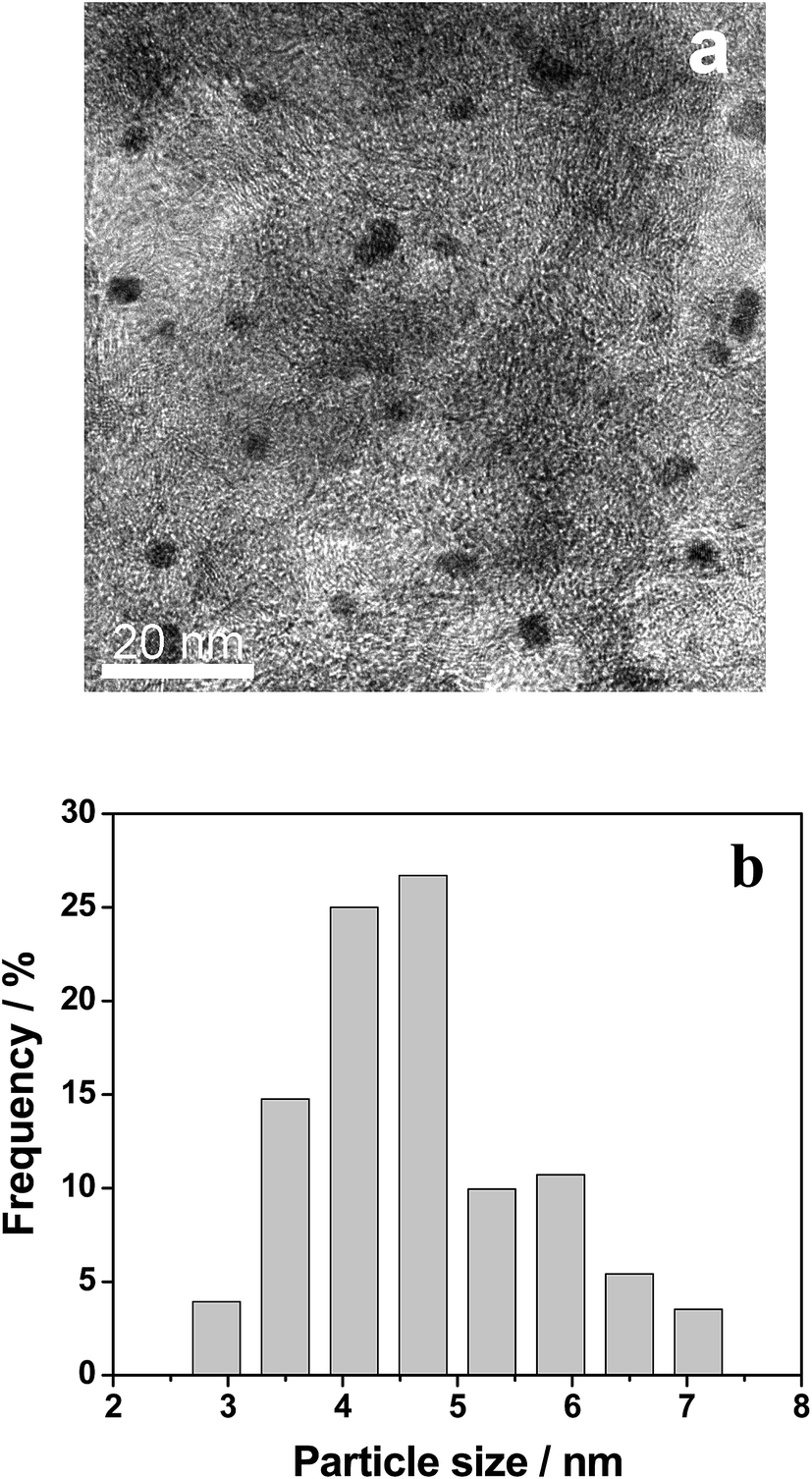 | ||
| Fig. 5 TEM image of the 0.3% Pt–4% Fe/AC after catalysis (a) and size distributions of Pt–Fe particles in the catalyst (b). | ||
4 Conclusion
In this study, we have prepared Fe modified Pt nanoparticles over activated carbon and investigated their catalytic performance on the chemoselective hydrogenation of p-chloronitrobenzene. It was found that the incorporation of an appropriate amount of Fe (4 wt%) resulted in a remarkable increase in activity, and the selectivity to p-chloronitrobenzene could be up to 100% at complete conversion of p-chloronitrobenzene over 0.3% Pt–4% Fe/AC catalyst even at high temperature and H2 pressure. X-ray photoelectron spectroscopy revealed the formation of Fe2O3 and the electron transfer from Pt nanoparticles to Fe2O3, leading to the electron-deficient state of the Pt nanoparticles in the present catalyst, which are responsible for the improved performance of 0.3% Pt–4% Fe/AC catalyst for p-chloronitrobenzene hydrogenation to p-chloroaniline. High reaction temperature and H2 pressure are favorable for p-CNB hydrogenation to p-CAN over the 0.3% Pt–4% Fe/AC catalyst.Acknowledgements
This work was supported by the Science and Technology Innovation Project of Chongqing (cstc2016shmszx20004) and Project of International Science and Technology Cooperation in Chongqing (cstc2014gjhz20002).References
- R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121 CrossRef CAS.
- H. U. Blaser, U. Siegrist, H. Steiner and M. Studer, in Fine Chemicals through Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2001, pp. 389–406 Search PubMed.
- M. H. Liu, X. X. Mo, Y. Y. Liu, H. L. Xiao, Y. Zhang, J. Y. Jing, V. Colvin and W. W. Yu, Appl. Catal., A, 2012, 439–440, 192 CrossRef CAS.
- S. Ichikawa, T. Seki and T. Ikariya, Adv. Synth. Catal., 2014, 356, 2643 CrossRef CAS.
- A. B. Dongil, L. Pastor-Pérez, J. L. G. Fierro, N. Escalona and A. Sepúlveda-Escriban, Catal. Commun., 2016, 75, 55 CrossRef CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero and M. A. Keane, Appl. Catal., A, 2008, 334, 199 CrossRef.
- L. N. Liu, L. N. Xing, C. X. Cheng, L. X. Xia and H. Y. Liu, RSC Adv., 2016, 6, 31871 RSC.
- Y. C. Liu, C. Y. Huang and Y. W. Chen, Ind. Eng. Chem. Res., 2006, 45, 62 CrossRef CAS.
- Y. Y. Fan, H. L. Xiao, N. Sui, M. H. Liu and W. W. Yu, RSC Adv., 2014, 4, 11788 RSC.
- E. Bertolucci, R. Bacsa, A. Benyounes, A. M. Raspolli-Gallett, M. R. Axet and P. Serp, ChemCatChem, 2015, 7, 2971 CrossRef CAS.
- F. Cardenas-Lizana, D. Lamey, N. Perret, S. Gomez-Quero, L. Kiwi-Minsker and M. A. Keane, Catal. Commun., 2015, 21, 46 CrossRef.
- C. H. Camposa, M. Jofréa, C. C. Torresa, B. Pawelecb, J. L. G. Fierrob and P. Reyesa, Appl. Catal., A, 2014, 482, 127 CrossRef.
- D. P. He, X. D. Jiao, P. Jiang, J. Wang and B. Q. Xu, Green Chem., 2012, 14, 111 RSC.
- M. H. Liu, W. Y. Yu and H. F. Liu, J. Mol. Catal. A: Chem., 1999, 138, 295 CrossRef CAS.
- M. Makosch, W. I. Lin, V. Bumbálek, J. Sá, J. W. Medlin, K. Hungerbühler and J. A. van Bokhoven, ACS Catal., 2012, 2, 2079 CrossRef CAS.
- S. Furukawa, Y. Yoshida and T. Komatsu, ACS Catal., 2014, 4, 1441 CrossRef CAS.
- K. Möbus, D. Wolf, H. Benischke, U. Dittmeier, K. Simon, U. Packruhn, R. Jantke, S. Weidlich, C. Weber and B. S. Chen, Top. Catal., 2010, 53, 1126 CrossRef.
- N. Mahata, O. S. G. P. Soaresa, I. Rodríguez-Ramos, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., A, 2013, 464–465, 28 CrossRef CAS.
- H. Zhu, W. F. Han, H. F. Chai and H. Z. Liu, Chin. J. Catal., 2007, 28, 196 CAS.
- P. R. Shukla, S. B. Wang, H. Q. Sun, H. M. Ang and M. Tadé, Appl. Catal., B, 2010, 100, 529 CrossRef CAS.
- A. Shukla, R. K. Singha, T. Sasaki and R. Bal, Green Chem., 2015, 17, 785 RSC.
- W. J. Li, L. M. Ye, P. Long, J. Chen, H. Ariga, K. Asakurab and Y. Z. Yuan, RSC Adv., 2014, 4, 29072 RSC.
- J. Gong, K. Yao, J. Liu, Z. W. Jiang, X. C. Chen, X. Wen, E. Mijowska, N. N. Tian and T. Tang, J. Mater. Chem. A, 2013, 1, 5247 CAS.
- L. Olsson and E. Fridell, J. Catal., 2002, 210, 340 CrossRef CAS.
- J. L. Zhang, Y. Wang, H. Ji, Y. G. Wei, N. Z. Wu, B. J. Zuo and Q. L. Wang, J. Catal., 2005, 229, 114 CrossRef CAS.
- X. X. Han, R. X. Zhou, G. H. Lai and X. M. Zheng, Catal. Today, 2004, 93–95, 433 CrossRef CAS.
- C. Lian, H. Q. Liu, C. Xiao, W. Yang, K. Zhang, Y. Liu and Y. Wang, Chem. Commun., 2012, 48, 3124 RSC.
- H. M. Liu, H. B. Yu, C. R. Xiong and S. H. Zhou, RSC Adv., 2015, 5, 20238 RSC.
- C. S. Lu, M. J. Wang, Z. L. Feng, Y. N. Qi, F. Feng, L. Ma, Q. F. Zhang and X. N. Li, Catal. Sci. Technol., 2017, 7, 1581 CAS.
| This journal is © The Royal Society of Chemistry 2017 |