
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A palladium-catalyzed oxidative cross-coupling reaction between aryl pinacol boronates and H-phosphonates in ethanol†
Te-Hsuan Chen‡
,
Daggula Mallikarjuna Reddy‡ and
Chin-Fa Lee *
*
Department of Chemistry, National Chung Hsing University, Taichung, Taiwan 402, Republic of China. E-mail: cfalee@dragon.nchu.edu.tw; Fax: +886-4-2286-2547; Tel: +886-4-2284-0411 ext. 810
First published on 14th June 2017
Abstract
The first successful oxidative coupling reaction of aryl pinacol boronic esters with H-phosphonates to deliver aryl phosphorous compounds is reported herein. These reactions between aryl boronic reagents and H-phosphonates were carried out synergistically using a Pd catalyst, additive and oxidant. Without using bases and ligands, phosphorylation was accomplished in an environmentally-friendly manner under mild conditions in ethanol.
Introduction
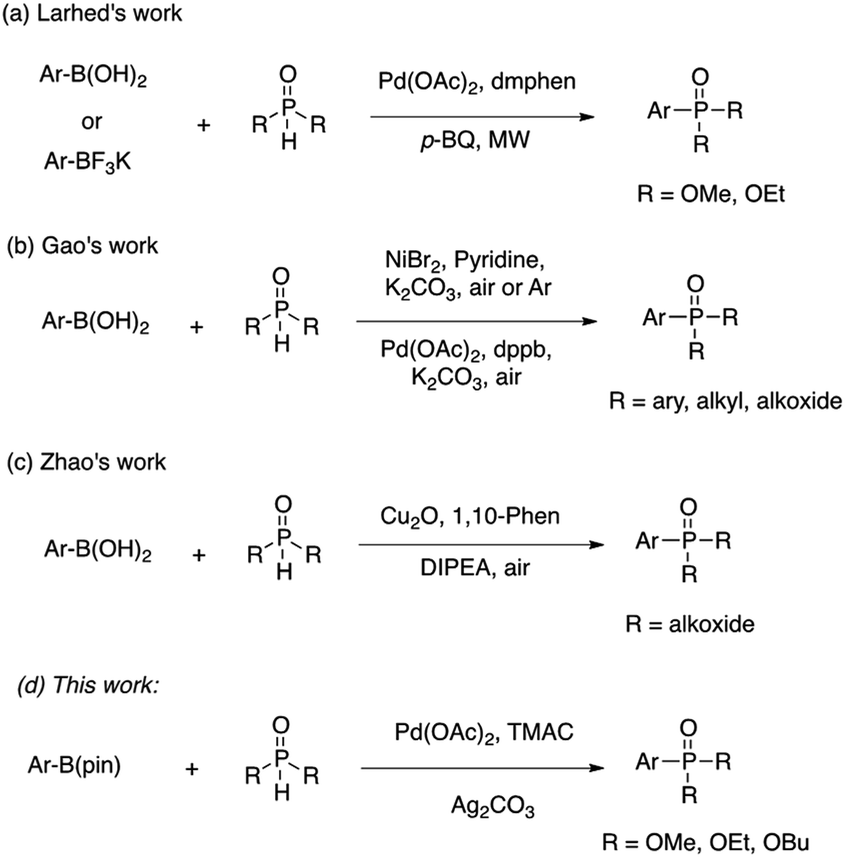
The development of versatile methods for the synthesis of arylphosphorous compounds1 has attracted much attention, owing to their broad applications in medicinal chemistry,2 materials chemistry,3 organic synthesis4 and catalysis.5 In 1981, Hirao and co-workers reported the first construction of C(sp2)–P bonds through palladium-catalyzed cross-coupling of aryl halides with H-phosphonates.6 During the past three decades, the scope of the reactions catalyzed by Pd, Cu and Ni or other metals reagents to form C(sp2)–P bonds employing functionalized arenes with phosphorus reagents has been significantly expanded to various aryl halides,7 triflates,8 imidazolylsulfonates,9 diazonium salts,10 o-aryl silyl triflates,11 arylhydrazines,12 arylnitriles,13 aryl pivalates,14 sodium arylsulfinates15 and diaryliodonium salts.16 Arylphosphorus compounds can also be produced by the palladium or copper-catalyzed directed C–H/P(O)–H coupling reactions.17Aryl boronic acids18 are extensively used as substrates in transition-metal catalyzed cross-coupling reactions including Suzuki–Miyaura cross-coupling reactions,19 Cu-catalyzed C–O, C–N, C–S and C–Se coupling reactions,20 and Rh-catalyzed conjugate additions to carbonyl compounds,21 owing to their commercial availability and structural diversity. There are only a few examples reported for C–P bond forming reactions using aryl boronic acids (Scheme 1a–c).22 Notably, additional ligands and bases are required in these works. Aryl boronic esters, especially aryl pinacol boronates, have received a great deal of attention in the catalysis community. In general, aryl boronic esters exhibit greater chemical stability and readily soluble in aprotic solvents. In most cases, they also exhibit stability towards column chromatography, which aids in their ease of isolation and purification. In addition, many are liquids at room temperature and can be easily distilled. However, to the best of our knowledge, the preparation of arylphosphorus compounds via oxidative cross coupling reactions of aryl boronic esters with H-phosphonates has never been developed. Herein, we report the first coupling reaction of aryl pinacol boronic esters with H-phosphonates through palladium catalysis (Scheme 1d). The features of this system include that the reactions are performed in ethanol and also declined the requirement of external ligand and base. The most important feature that we observed was that the reaction conditions used in Scheme 1a–c were not successful for the coupling of aryl pinacol boronate with H-phosphonate.
 | ||
| Scheme 1 Synthesis of aryl phosphorous compounds from arylboronic reagents. | ||
Results and discussion
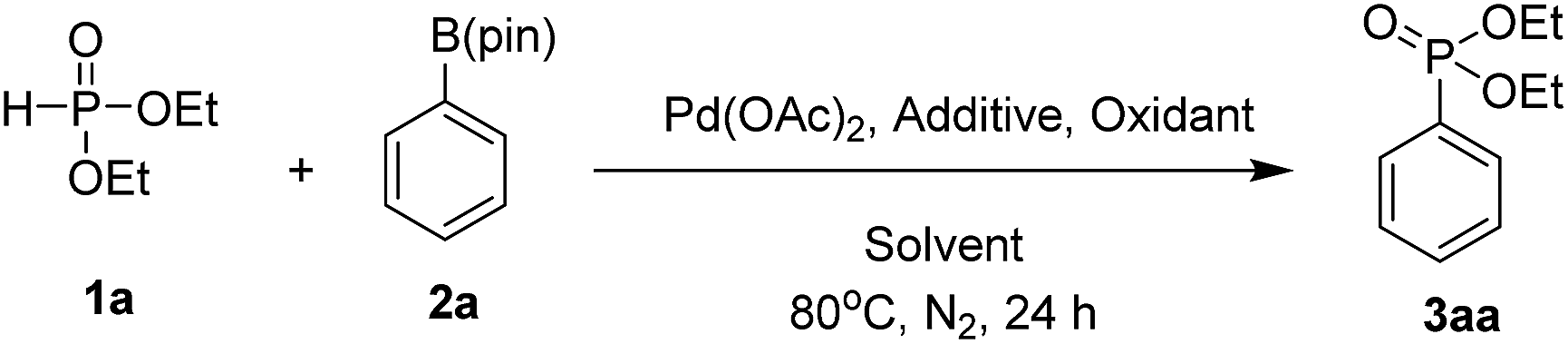
We selected diethyl phosphite (1a) and phenyl pinacol boronate (2a) as model substrates to optimize the reaction conditions (Table 1). Initially, the reaction was carried out with Pd(OAc)2 (5 mol%) as the catalyst and Ag2CO3 (1.5 equiv.) as the oxidant in DMF at 80 °C for 24 h; however, only a trace amount of the desired product diethyl phenylphosphonate 3aa was detected (entry 1). No product or only a trace amount of the product was observed when DMSO, CH3CN or toluene was used as the solvent (entries 2–4). To our delight, when the solvent was changed to ethanol, the coupling product 3aa was obtained at 22% yield (entry 5). Key role of ethanol in this coupling reaction to obtain the desired product can be explained by the possible coordination of ethanol at boron centre of aryl pinacol boronate. Due to the co-ordination of ethanol with aryl pinacol boronate the electron density at aryl system may be increased. When we chose tetramethylammonium bromide (TMAB), tetramethylammonium chloride (TMAC), LiCl, CsCl and TBACl as the additives, the coupling product 3aa with TMAC and LiCl obtained 76% and 75% yields, respectively (entries 7 and 9), whereas other additives gave inferior results. To our delight, we chose TMAC as additive for this reaction. Enhancement of the yield of product in the presence of TMAC was because of its phase transfer catalytic and inert counter ionic nature. The yield of the product was decreased to 32% when the reaction was conducted with tert-butanol as the solvent (entry 11). The choice of oxidant was crucial in this coupling reaction. When we replaced Ag2CO3 with AgOAc and Cu(OAc)2, both led to 3aa at lower yields (entries 12 and 14); meanwhile, no reaction occurred when K2S2O8 or oxygen (ballon) was used as the oxidant (entries 13 and 15). The superiority of Ag2CO3 over other oxidants in this reaction could explained by its additional basic nature that can promote the deprotonation of H-phosphonates. In the screening of the palladium source, Pd(PPh3)2Cl2, Pd(OCOCF3)2 and PdCl2 showed lower reactivity (entries 16–18) compared with Pd(OAc)2.
|
||||
|---|---|---|---|---|
| Entry | Additive | Oxidant | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1.5 eq.), Pd(OAc)2 (5 mol%), additive (1.5 eq.), oxidant (1.5 eq.), and solvent (3.0 mL) under an atmosphere of N2 for 24 h at 80 °C.b Isolated yield.c Pd(PPh3)2Cl2 as the catalyst.d Pd(OCOCF3)2 as the catalyst.e PdCl2 as the catalyst. | ||||
| 1 | — | Ag2CO3 | DMF | Trace |
| 2 | — | Ag2CO3 | DMSO | 0 |
| 3 | — | Ag2CO3 | CH3CN | Trace |
| 4 | — | Ag2CO3 | Toluene | Trace |
| 5 | — | Ag2CO3 | EtOH | 22 |
| 6 | TMAB | Ag2CO3 | EtOH | 25 |
| 7 | TMAC | Ag2CO3 | EtOH | 76 |
| 8 | CsCl | Ag2CO3 | EtOH | 50 |
| 9 | LiCl | Ag2CO3 | EtOH | 75 |
| 10 | TBAC | Ag2CO3 | EtOH | 35 |
| 11 | TMAC | Ag2CO3 | t-BuOH | 32 |
| 12 | TMAC | AgOAc | EtOH | 35 |
| 13 | TMAC | K2S2O8 | EtOH | N.R. |
| 14 | TMAC | Cu(OAc)2 | EtOH | 28 |
| 15 | TMAC | O2 | EtOH | N.R. |
| 16c | TMAC | Ag2CO3 | EtOH | 64 |
| 17d | TMAC | Ag2CO3 | EtOH | 62 |
| 18e | TMAC | Ag2CO3 | EtOH | 65 |
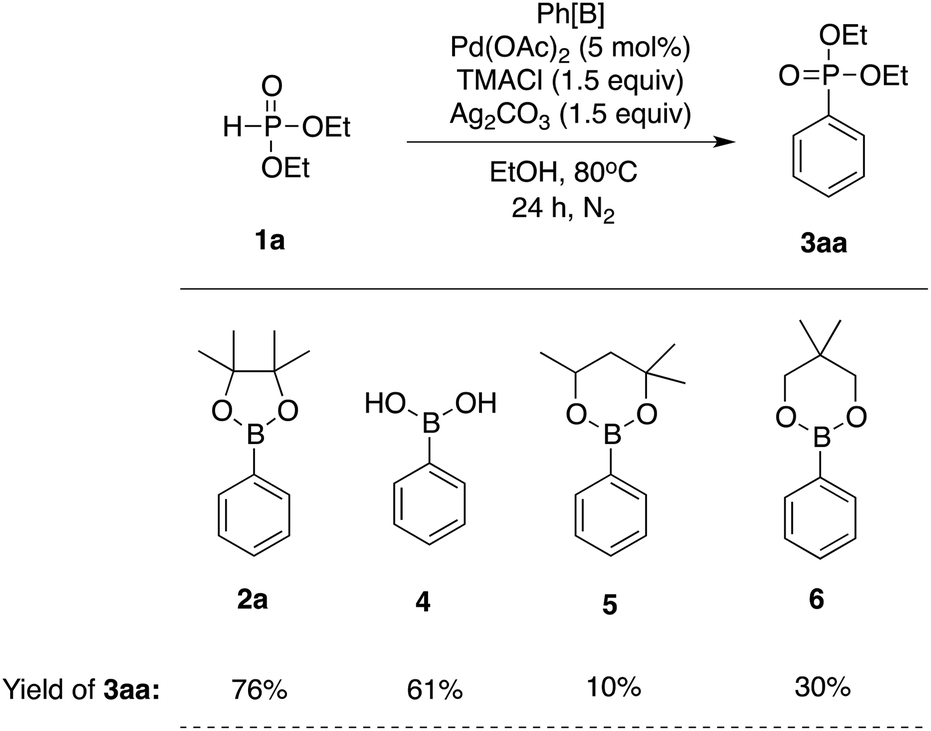
We next investigated the relative reactivity of phenyl boronic acid derivatives (Table 2). When phenyl boronic acid (4) was used as a boron reagent, the coupling product 3aa was obtained at 61% yield. Phenyl boronic esters, such as phenyl hexylene glycol boronate (5) and phenyl neopentyl glycol boronate (6), were also tested, but both gave the product at lower yields when compared with phenyl pinacol boronate (2a).
| a Reaction conditions: 0.5 mmol of 1a, 1.5 equivalent of Ph[B], 5 mol% Pd(OAc)2, 1.5 equivalent of TMACl, 1.5 equivalent of Ag2CO3, and 3 mL of ethanol were charged into a Schlenk tube under nitrogen atmosphere and heated at 80 °C for 24 h. |
|---|
 |
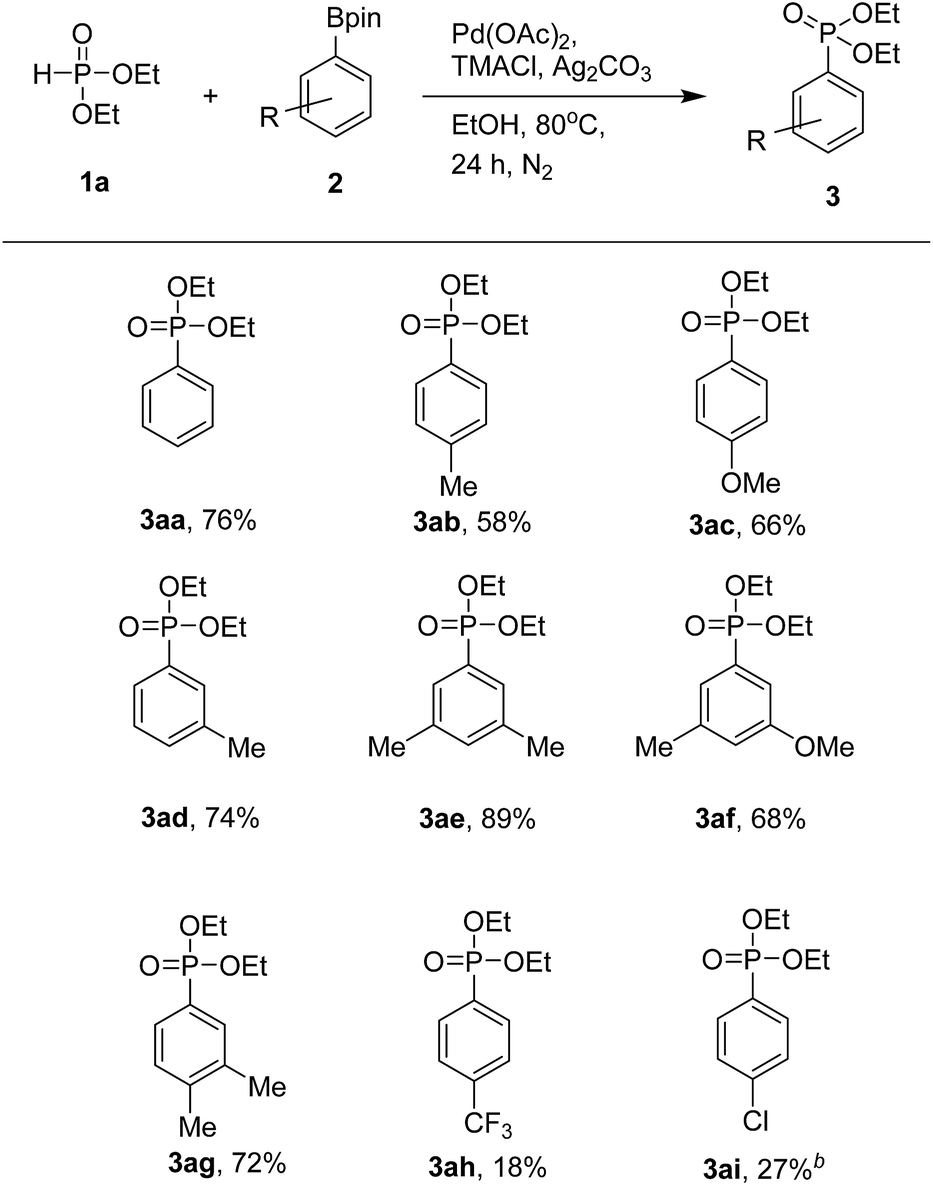
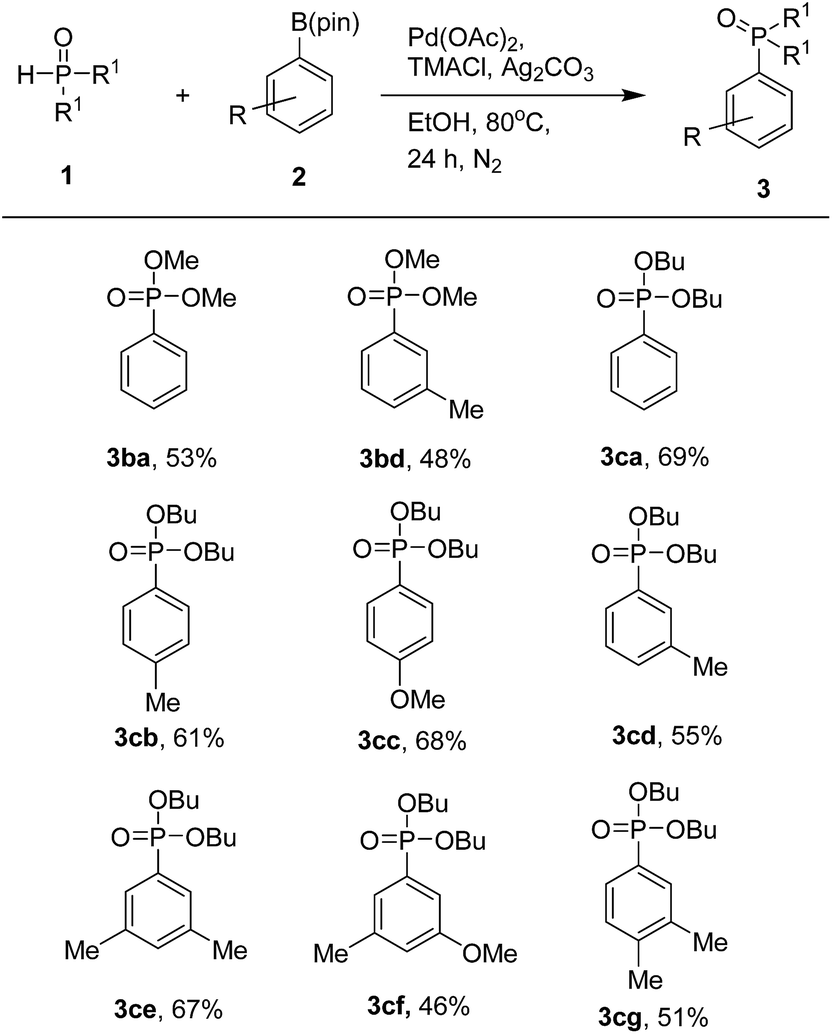
With the optimized protocol from previous works, we examined the scope of the substrates in this catalytic system (Table 3). Various aryl pinacol boronates 2 were found to couple with 1a to produce the corresponding diethyl arylphosphonates 3 at moderate to good yields. It is worth mentioning that higher yields of products were obtained with aryl pinacol boronates 2 bearing electron-donating groups. Thus, with electron-donating substituents such as methyl and methoxy groups at the C-3 or C-4 position of 2, one could obtain the corresponding products at good yields (3aa–3ag). In contrast, substrates with electron-withdrawing trifluoromethyl and chloro groups led to products at lower yields (3ah and 3ai). When we used 3-bromophenyl pinacol boronate as a coupling partner, the desired product was obtained in trace amount along with de-brominated arylphosphonate (3aa) in lower yield. Use of various aryl pinacol boronic esters such as 4-nitrophenyl, 4-methoxycarbonylphenyl, 4-cyanophenyl, naphthyl, 4-hydroxyphenyl pinacol boronates and heteroaryl pinacol boronates such as 3-thienyl, 5-indolyl pinacol boronates as substrates to couple with H-phosphonate, failed to the formation of desired products instead detected the de-borolyted products. Furthermore, we also tested the reaction by using ortho substituted aryl pinacol boronates such as 2-chlorophenyl and mesityl pinacol boronates. Under the reaction conditions, these two substrates also failed to react with H-phosphonate. The formation of de-borolyted product was detected by GC analysis. Another important factor that was responsible for lowering the yield of product was formation trialkylphosphates by the reaction of H-phosphonate with solvent (ethanol).
The generality of this coupling reaction was further demonstrated by using two other substituted phosphites, dimethyl phosphite (1b) and dibutyl phosphite (1c). When screening with different aryl pinacol boronates 2, both phosphites could produce the corresponding products at moderate to good yields (Table 4).
| a Reaction conditions: 0.5 mmol of 1, 1.5 equivalent of boronate 2, 5 mol% Pd(OAc)2, 1.5 equivalent of TMACl, 1.5 equivalent of Ag2CO3, and 3 mL of ethanol were charged into a Schlenk tube under nitrogen atmosphere and heated at 80 °C for 24 h. |
|---|
 |
Although the mechanism of this tranformation is not clear at this stage, based on experimental results, we propose that this Pd-catalyzed cross-coupling takes place via the catalytic cycle shown in Scheme 2. Firstly, the Pd(II) complex A reacted with the phosphorous nucleophile generated by deprotonation of the P(O)H compound in the presence of a Ag2CO3 to provide intermediate B.22a,d The resulting intermediate B associated with arylpinacol boronate (2) to give another intermediate C,22a,d,23 which upon reductive elimination, afforded the desired coupling product (3) and Pd(0) species. Finally, the Pd(0) species was oxidized by Ag2CO3, leading to the regeneration of Pd(II) complex A as a catalytically active species.
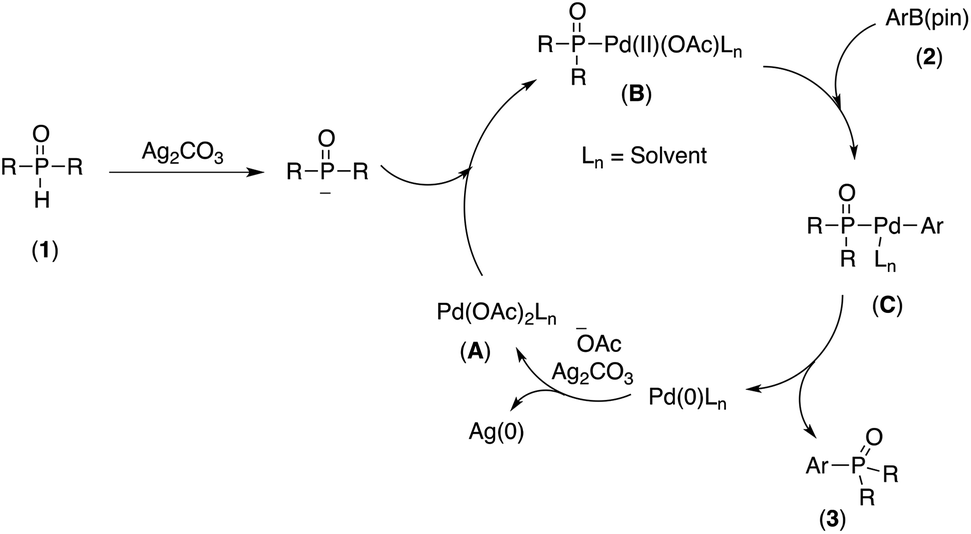 | ||
| Scheme 2 Plausible mechanism for the formation of 3. | ||
Conclusion
In conclusion, we demonstrated a novel Pd-catalyzed cross-coupling reaction of aryl pinacol boronates with H-phosphonates, which afforded good yields of arylphosphorus compounds under mild conditions in ethanol. A variety of readily available aryl pinacol boronates can be used in this coupling reaction. We believe that this is a new protocol for the construction of valuable C–P bonds from readily available, environmentally friendly chemical sources.Experimental
General information
All commercial chemicals were used as received except where noted. Aryl pinacol boronates were all prepared through literature procedures.24,25 Experiments were performed under a dinitrogen atmosphere using standard Schlenk techniques unless otherwise stated. Flash chromatography was performed on Merck silica gel 60 (230–400 mesh).Analysis
NMR spectra were recorded on a Varian Unity Inova-600 or a Varian Mercury-400 instrument using CDCl3 as a solvent. Chemical shifts are reported in parts per million (ppm) and referenced to the residual solvent resonance. Coupling constant (J) are reported in hertz (Hz). Standard abbreviations indicating multiplicity were used as follows: s = singlet, d = doublet, t = triplet, dd = double doublet, q = quartet, quin = quintet, m = multiplet, b = broad. GC-MS analysis were performed on a Agilent Technologies 5977A GC equipped with Agilent 7890B MS. High-resolution mass spectra were carried out on a Jeol JMS-HX 110 spectrometer by the services at the National Chung Hsing University.General procedure for synthesis of compound 3
To a mixture of aryl pinacol boronic ester (0.75 mmol, 1.5 equiv.), Pd(OAc)2 (5.6 mg, 5 mol%), Ag2CO3 (0.21 g, 1.5 equiv.) and tetramethylammonium chloride (82 mg, 1.5 equiv.) were added H-phosphonate (0.5 mmol) and ethanol (3.0 mL) under nitrogen atmosphere. The resulting suspension was heated at 80 °C for 24 h. After 24 h, the reaction mixture was cooled down to room temperature and diluted with ethyl acetate (20 mL). The resulting solution was directly filtered through a pad of celite and the filtrate was concentrated under vacuum. The crude product was purified by flash column chromatography (ethyl acetate/hexane = 30–80/20–70).Acknowledgements
The Ministry of Science and Technology, Taiwan (Most 105-2113-M-005-001-) and the National Chung Hsing University are gratefully acknowledged for financial support.References
- (a) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley Interscience, New York, 2000 Search PubMed; (b) Organophosphorus Reagents, ed. P. J. Murphy, Oxford University Press, Oxford, U.K., 2004 Search PubMed; (c) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed; (d) C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed.
- (a) Y. C. Kim, S. G. Brown, T. K. Harden, J. L. Boyer, G. Dubyak, B. F. King, G. Burnstock and K. A. Jacobson, J. Med. Chem., 2001, 44, 340–349 CrossRef CAS PubMed; (b) J. J. Shie, J. M. Fang, S. Y. Wang, K. C. Tsai, Y. S. Cheng, A. S. Yang, S. C. Hsiao, C. Y. Su and C. H. Wong, J. Am. Chem. Soc., 2007, 129, 11892–11893 CrossRef CAS PubMed; (c) T. S. Kumar, S. Y. Zhou, B. V. Joshi, R. Balasubramanian, T. H. Yang, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2010, 53, 2562–2576 CrossRef CAS PubMed.
- (a) H. H. Chou and C. H. Cheng, Adv. Mater., 2010, 22, 2468–2471 CrossRef CAS PubMed; (b) F. M. Hsu, C. H. Chien, C. F. Shu, C. H. Lai, C. C. Hsieh, K. W. Wang and P. T. Chou, Adv. Funct. Mater., 2009, 19, 2834–2843 CrossRef CAS.
- (a) T. Baumgartner and R. Reau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (b) R. Engel, Chem. Rev., 1977, 77, 349–367 CrossRef CAS.
- (a) H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151–4202 CrossRef CAS PubMed; (b) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and R. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS PubMed.
- (a) T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Tetrahedron Lett., 1980, 21, 3595–3598 CrossRef CAS; (b) T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Synthesis, 1981, 56–57 CrossRef CAS; (c) T. Hirao, T. Masunaga, N. Yamada, Y. Ohshiro and T. Agawa, Bull. Chem. Soc. Jpn., 1982, 55, 909–913 CrossRef CAS.
- (a) D. Gelman, L. Jiang and S. L. Buchwald, Org. Lett., 2003, 5, 2315–2318 CrossRef CAS PubMed; (b) H.-H. Rao, Y. Jin, H. Fu, Y.-Y. Jiang and Y.-F. Zhao, Chem.–Eur. J., 2006, 12, 3636–3646 CrossRef CAS PubMed; (c) C. Huang, X. Tang, H. Fu, Y.-Y. Jiang and Y.-F. Zhao, J. Org. Chem., 2006, 71, 5020–5022 CrossRef CAS PubMed; (d) M. Kalek, A. Ziadi and J. Stawinski, Org. Lett., 2008, 10, 4637–4640 CrossRef CAS PubMed; (e) M. Kalek and J. Stawinski, Organometallics, 2008, 27, 5876–5888 CrossRef CAS; (f) M. Kalek, M. Jezowska and J. Stawinski, Adv. Synth. Catal., 2009, 351, 3207–3206 CrossRef CAS; (g) N. T. McDougal, J. Streuff, H. Mukherjee, S. C. Virgil and B. M. Stoltz, Tetrahedron Lett., 2010, 51, 5550–5554 CrossRef CAS PubMed; (h) E. L. Deal, C. Petit and J. L. Montchamp, Org. Lett., 2011, 13, 3270–3273 CrossRef CAS PubMed; (i) X.-H. Zhang, H.-Z. Liu, X.-M. Hu, G. Tang, J. Zhu and Y.-F. Zhao, Org. Lett., 2011, 13, 3478–3481 CrossRef CAS PubMed; (j) H.-Y. Zhang, M. Sun, Y.-N. Ma, Q.-P. Tiana and S.-D. Yang, Org. Biomol. Chem., 2012, 10, 9627–9633 RSC; (k) M. Sun, Y.-S. Zang, L.-K. Hou, X.-X. Chen, W. Sun and S.-D. Yang, Eur. J. Org. Chem., 2014, 6796–6801 CrossRef CAS.
- (a) K. S. Petrakis and T. L. Nagabhushan, J. Am. Chem. Soc., 1987, 109, 2831–2833 CrossRef CAS; (b) X. Lu and J. Zhu, Synthesis, 1987, 726–727 CrossRef CAS.
- Y. Luo and J. Wu, Organometallics, 2009, 28, 6823–6826 CrossRef CAS.
- R. Berrino, S. Cacchi, G. Fabrizi, A. Goggiamani and P. Stabile, Org. Biomol. Chem., 2010, 8, 4518–4520 CAS.
- (a) R. A. Dhokale and S. B. Mhaske, Org. Lett., 2013, 15, 2218–2221 CrossRef CAS PubMed; (b) Q. Chen, X. Yan, Z. Du, K. Zhang and C. Wen, J. Org. Chem., 2016, 81, 276–281 CrossRef CAS PubMed; (c) G. Yang, C. Shen, M. Quan and W. Zhang, Tetrahedron, 2016, 72, 333–337 CrossRef CAS.
- (a) S.-Y. Chen, R.-S. Zeng, J.-P. Zou and O. T. Asekun, J. Org. Chem., 2014, 79, 1449–1453 CrossRef CAS PubMed; (b) W. Xu, G. Hu, P. Xu, Y. Gao, Y. Yin and Y. Zhao, Adv. Synth. Catal., 2014, 356, 2948–2954 CrossRef CAS.
- (a) M. Sun, H.-Y. Zhang, Q. Han, K. Yang and S.-D. Yang, Chem.–Eur. J., 2011, 17, 9566–9570 CrossRef CAS PubMed; (b) J.-S. Zhang, T. Chen, J. Yanga and L.-B. Han, Chem. Commun., 2015, 51, 7540–7542 RSC.
- J. Yang, T. Chen and L.-B. Han, J. Am. Chem. Soc., 2015, 137, 1782–1785 CrossRef CAS PubMed.
- T. Miao and L. Wang, Adv. Synth. Catal., 2014, 356, 967–971 CrossRef CAS.
- J. Xu, P. Zhang, Y. Gao, Y. Chen, G. Tang and Y. Zhao, J. Org. Chem., 2013, 78, 8176–8183 CrossRef CAS PubMed.
- (a) Y. Kuninobu, T. Yoshida and K. Takai, J. Org. Chem., 2011, 76, 7370–7376 CrossRef CAS PubMed; (b) C.-G. Feng, M. Ye, K.-J. Xiao, S. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 9322–9325 CrossRef CAS PubMed; (c) S. Wang, R. Guo, G. Wang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2014, 50, 12718–12721 RSC.
- (a) H. C. Brown, G. W. Kramer, A. B. Levy and M. M. Midland, Organic Syntheses via Boranes, Wiley-Interscience, New York, 1975 Search PubMed; (b) H. C. Brown and B. Singaram, Acc. Chem. Res., 1988, 21, 287–293 CrossRef CAS; (c) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) D. M. T. Chan, K. L. Monaco, R. Li, D. Bonne, C. G. Clark and P. Y. S. Lam, Tetrahedron Lett., 2003, 44, 3863–3865 CrossRef CAS; (b) C. C. Tzschucke, J. M. Murphy and J. F. Hartwig, Org. Lett., 2007, 9, 761–764 CrossRef CAS PubMed; (c) J.-H. Cheng, C.-L. Yi, T.-J. Liu and C.-F. Lee, Chem. Commun., 2012, 48, 8440–8442 RSC; (d) C.-L. Yi, T.-J. Liu, J.-H. Cheng and C.-F. Lee, Eur. J. Org. Chem., 2013, 2013, 3910–3918 CrossRef CAS.
- (a) M. P. Sibi, H. Tatamidani and K. Patil, Org. Lett., 2005, 7, 2571–2573 CrossRef CAS PubMed; (b) M. Ogasawara, H. L. Ngo, T. Sakamoto, T. Takahashi and W. Lin, Org. Lett., 2005, 7, 2881–2884 CrossRef CAS PubMed; (c) G. Chen, N. Tokunaga and T. Hayashi, Org. Lett., 2005, 7, 2285–2288 CrossRef CAS PubMed.
- (a) M. Andaloussi, J. Lindh, J. Savmarker, P. J. R. Sjoberg and M. Larhed, Chem.–Eur. J., 2009, 15, 13069–13074 CrossRef CAS PubMed; (b) R.-Q. Zhuang, J. Xu, Z.-S. Cai, G. Tang, M.-J. Fang and Y.-F. Zhao, Org. Lett., 2011, 13, 2110–2113 CrossRef CAS PubMed; (c) G. Hu, W. Chen, T. Fu, Z. Peng, H. Qiao, Y. Gao and Y. Zhao, Org. Lett., 2013, 15, 5362–5365 CrossRef CAS PubMed; (d) T. Fu, H. Qiao, Z. Peng, G. Hu, X. Wu, Y. Gao and Y. Zhao, Org. Biomol. Chem., 2014, 12, 2895–2902 RSC.
- M. C. Kohler, T. V. Grimes, X. Wang, T. R. Cundari and R. A. Stockland Jr, Organometallics, 2009, 28, 1193–1201 CrossRef CAS.
- J. Ratniyom, N. Dechnarong, S. Yotphan and S. Kiatisevi, Eur. J. Org. Chem., 2014, 2014, 1381–1385 CrossRef CAS.
- B. M. Partridge and J. F. Hartwig, Org. Lett., 2013, 15, 140–143 CrossRef CAS PubMed.
- X. Lu and J. Zhu, Synthesis, 1987, 726–727 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: For 1H and 13C spectra of componunds 3. See DOI: 10.1039/c7ra04619g |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |