
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ ATR-FTIR investigation and theoretical calculation of the interactions of chromate and citrate on the surface of haematite (α-Fe2O3)
W. G. Gao ac,
X. C. Liub and
M. F. Chen*a
ac,
X. C. Liub and
M. F. Chen*a
aKey Laboratory of Soil Environment and Pollution Remediation, Institute of Soil Science, Chinese Academy of Sciences, Nanjing 210008, China. E-mail: mfchen@issas.ac.cn
bState Key Laboratory for Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 130100, China
cUniversity of the Chinese Academy of Sciences, Beijing 100049, China
First published on 22nd August 2017
Abstract
In situ attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy was used to study the molecular kinetics of Cr(VI) reduction by citric acid at the α-Fe2O3–water interface. Both citrate and Cr(VI) were adsorbed to the surface of α-Fe2O3 through the formation of stable monodentate and bidentate complexes. Chromate was potentially reduced by citric acid hydroxyl groups (–COO−) on the α-Fe2O3 surface sites. Monodentate complexes were favoured at pH 4.0 due to strong electrostatic attraction forces between the ions and minerals. Density functional theory was used to calculate the adsorption and interaction processes of Cr(VI) and citrate on α-Fe2O3; the calculations indicated that the adsorption of Cr(VI) or citrate can occur spontaneously and result in the formation of stable complexation structures. However, the co-adsorption of Cr(VI) and citrate on α-Fe2O3 is energetically unstable and leads to the initiation of redox reactions on the α-Fe2O3 surface. These findings have important implications for the fate of chromate in the environment. Therefore, our work describes the molecular mechanism of Cr(VI) reduction by citrate on the surface of α-Fe2O3 for the first time. The results can be correlated to the fate of chromate in the environment in the presence of α-Fe2O3 and low molecular weight organic acids.
1 Introduction
Chromium(VI) (Cr(VI)) is one of the most frequently detected industrial groundwater contaminants. Cr(VI) is a carcinogen, and it exists in an oxyanionic form as chromate. The fate of Cr(VI) depends on its adsorption reactions on mineral surfaces in the absence of reducing conditions.1 Adsorption onto minerals is a common method for removing Cr(VI) from the environment.2 The reduction of Cr(VI) to Cr(III) is another important strategy for detoxifying Cr(VI)-contaminated sites, and it is an effective treatment method.Several studies have investigated the rapid removal of Cr(VI) from solutions using different types of adsorbents, such as metal oxides,3 and minerals.4 The molecular mechanisms of Cr(VI) adsorption on iron oxides were recently investigated by Johnston et al.,5 who used theoretical calculations and in situ attenuated total reflected Fourier transformed infrared spectroscopy (ATR-FTIR) to elucidate the adsorption structure of Cr(VI) on the surface of hematite. Cr(VI) can also be reduced to Cr(III) using natural organic matter (NOM); this process is greatly affected by the presence of coexisting metal ions6 and the properties of the minerals.7
The interactions between Cr(VI) and natural organic acids at mineral–water interfaces have become an increasingly important area of interest when considering the behaviour and toxicity of Cr(VI) in the environment. As one of the most ubiquitous natural organic acids, citric acid is an exudate of plants and can reduce Cr(VI) into Cr(III) under certain conditions. Earlier studies have confirmed that mineral surfaces can act as catalysts for redox reactions between Cr(VI) and citric acids to form nontoxic Cr(III).8–11 But the molecular mechanisms of their interactions at mineral–water interfaces remain unclear.
In this study, the molecular kinetics of Cr(VI) reduction by citrate on the surface of α-Fe2O3 was investigated in situ by using ATR-FTIR spectroscopy. Density functional theory (DFT) was used to calculate the electron densities and variations of the interactions between Cr(VI) and citrate at Fe2O3–water interfaces with different environmentally relevant pH values. We hypothesized that negatively charged Cr(VI) and citrate would adsorb to the positively charged α-Fe2O3 surface through the formation of monodentate complexes due to electrostatic attraction. Based on this assumption, the binding and free energies of the clusters were calculated to infer their possible reaction pathways. Our findings elucidate how these reactions occur at the molecular level.
2 Materials and methods
2.1 Chemicals
All chemicals and reagents were of analytical grade or higher purity and were used as received, and all solutions were prepared using Millipore filtered water (18 MΩ, Millipore Corp.). α-Fe2O3 was purchased from Sino Chemicals Corp., with a purity of more than 99.9%. Powder X-ray diffraction (XRD) was used to determine the bulk crystalline phase of the α-Fe2O3. The BET surface area of the studied α-Fe2O3 was 4.08 ± 0.02 m2 g−1. For in situ ATR-FTIR experiments, α-Fe2O3 was suspended in Millipore water by sonicating for 6 hours before use.2.2 In situ ATR-FTIR analysis
In situ flow cell ATR-FTIR spectroscopy was performed using a Bruker FTIR (Bruker Optics) spectrometer equipped with a multi-bounce PIKE flow cell part (PIKE Corp.). Data collection and analyses were performed using the OPUS software package (v6.5). Scans were recorded from 4000 to 400 cm−1 at a resolution of 8 cm−1. α-Fe2O3 film coatings (∼0.5 mg) were prepared by depositing 0.8 mL of the α-Fe2O3 suspension on a ZnSe internal reflection element (IRE) and were dried under a stream of nitrogen gas. Next, the IRE was mounted on a flow cell with a peristaltic pump to allow a specified volume of solution to pass over the film. The concentrations of Cr(VI) and citric acid were set at 0.1 mM each, and the solution pH was adjusted by titrating with 0.1 M HNO3 or NaOH. Before the adsorption experiment, the film was equilibrated with H2O (18 MΩ) at the specified pH for 20 min. The flow rate was set to approximately 1 mL min−1.2.3 DFT calculations
The theoretical frequencies of the possible surface complex structures were calculated using the approach described by Kubicki et al.12 The Fe2O3 surface was represented using an Fe2OxHy di-iron hydroxide cluster model that consisted of two octahedrally coordinated Fe atoms connected via two Fe–(OH)–Fe linkages. This model has been used in many other theoretical studies and has been proven valid. All calculations were performed using the Gaussian09 package and run on the Tianhe-2 supercomputer at the LvLiang Cloud Computing Center. DFT calculations were performed using the B3LYP method5,13 and the 6-311G++(d,p) basis set. The geometries of the clusters were optimized using the Berny algorithm and GEDIIS with redundant internal coordinates to a potential energy minimum without symmetry or geometrical constraints. Next, the clusters were analysed for harmonic frequencies. Short-range solvation effects were accounted for by using 3 explicit water molecules, and the long-range solvation effect was modelled using the integral equation formalism version of the polarizable continuum model (PCM).14 All atomic charges were calculated based on the natural bond orbital (NBO) population analysis code in Gaussian09.Because the Fe2O3 surface should be protonated and saturated with water molecules in aqueous solutions, the adsorption of either HCrO4− or citrate would not occur unless one or more of the saturating H2O molecules were replaced by the adsorbents. We defined the adsorption energy and free energy by using the following chemical equation:
| Ion + cluster·3H2O = ion_cluster·4H2O | (1) |
Thus we have:
| ΔEad = E(ion_cluster·4H2O) − E(ion) − E(cluster·3H2O) | (2) |
| ΔGad = G(ion_cluster·4H2O) − G(ion) − G(cluster·3H2O) | (3) |
In the case of co-adsorption,
| ΔEad = E(ionA_ionB_cluster·5H2O) − E(ionA) − E(ionB) − E(cluster·3H2O) | (4) |
| ΔGad = G(ionA_ionB_cluster·5H2O) − G(ionA) − G(ionB) − G(cluster·3H2O) | (5) |
3 Results and discussion
3.1 In situ ATR-FTIR studies of Cr(VI) and citrate on α-Fe2O3
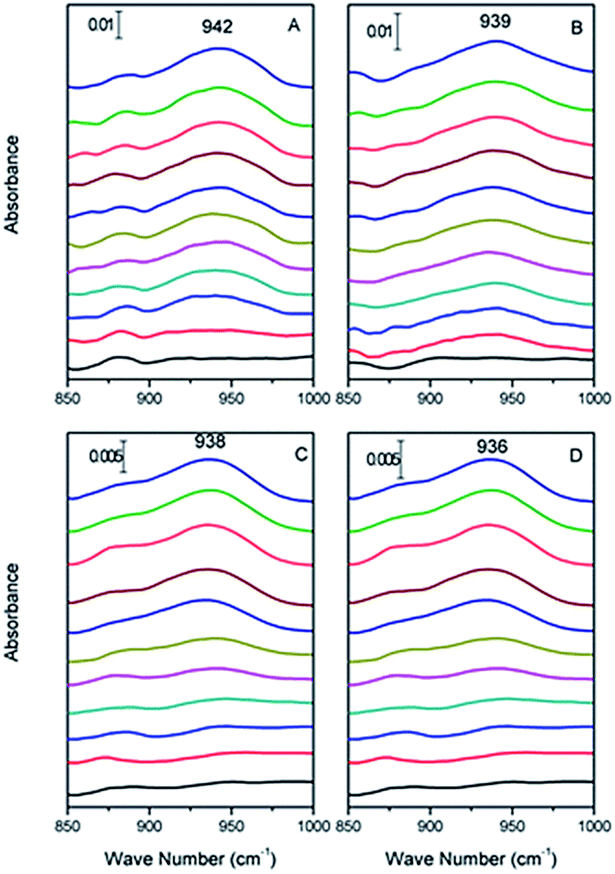 | ||
| Fig. 1 In situ ATR-FTIR spectrum of Cr(VI) adsorbed on haematite film at pH (A) 2.0, (B) 4.0, (C) 6.0, and (D) 8.0, respectively, from 0 to 30 min (from bottom to top). | ||
Specifically, the Cr(VI) adsorbed on α-Fe2O3 resulted in spectral features at 942, 939, 938 and 936 cm−1 at pH 2.0, 4.0, 6.0, and 8.0, respectively. In addition, the data obtained for pH 2.0, 4.0, 6.0 and 8.0 showed similar vibrational modes of Cr–O that increased with time and reached equilibrium after 30 min with no visible changes. We attributed the increased intensity of the spectral features representing associated Cr(VI) to its adsorption to the α-Fe2O3 surface. To qualify the interface absorption kinetics of Cr(VI), the peak areas of the Cr–O stretching vibrations at each specific pH were calculated as a function of time, as shown in Fig. 2, which is consistent with the expectation that chromate absorption on the α-Fe2O3 increased as the pH decreased, as previously overserved in batch studies.16,17
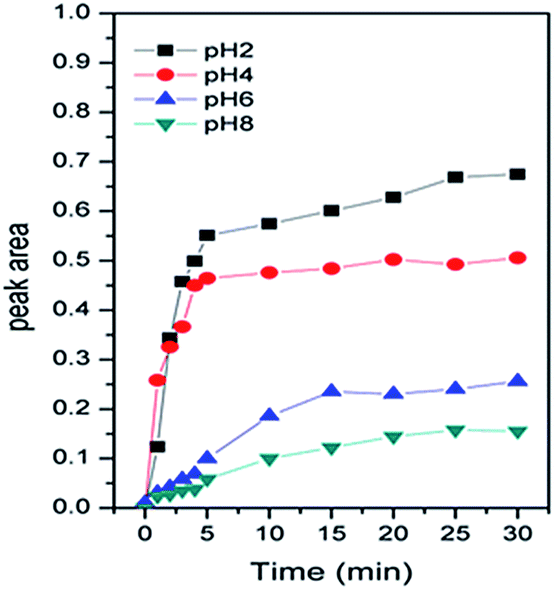 | ||
| Fig. 2 Areas of the IR peaks from Cr(VI) adsorbed on haematite at different pH values. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the –COOH radical resulted in a peak at 1740 cm−1, the single carboxyl radical resulted in a peak at 1740 cm−1, and the single carboxyl bond (νC–OH) position of the –COOH radical resulted in a peak at 1270 cm−1, as reported by Bhandari et al.18 and Max and Chapados.19 The peak at 1400 cm−1 was interpreted as the symmetric vibrations of ionized –COO− radicals (νs(COO−)), and the frequency range of 1570–1595 cm−1 was attributed to the asymmetric vibrations of the ionized –COO− radicals (νas(COO−)).19 The intensity of the peaks from the vibrations of these radicals slightly changed with pH, especially the intensities of νas(COO−) and νs(COO−) as the hydrolysis of protons from the citric acid occurred with increasing pH.
O) of the –COOH radical resulted in a peak at 1740 cm−1, the single carboxyl radical resulted in a peak at 1740 cm−1, and the single carboxyl bond (νC–OH) position of the –COOH radical resulted in a peak at 1270 cm−1, as reported by Bhandari et al.18 and Max and Chapados.19 The peak at 1400 cm−1 was interpreted as the symmetric vibrations of ionized –COO− radicals (νs(COO−)), and the frequency range of 1570–1595 cm−1 was attributed to the asymmetric vibrations of the ionized –COO− radicals (νas(COO−)).19 The intensity of the peaks from the vibrations of these radicals slightly changed with pH, especially the intensities of νas(COO−) and νs(COO−) as the hydrolysis of protons from the citric acid occurred with increasing pH.
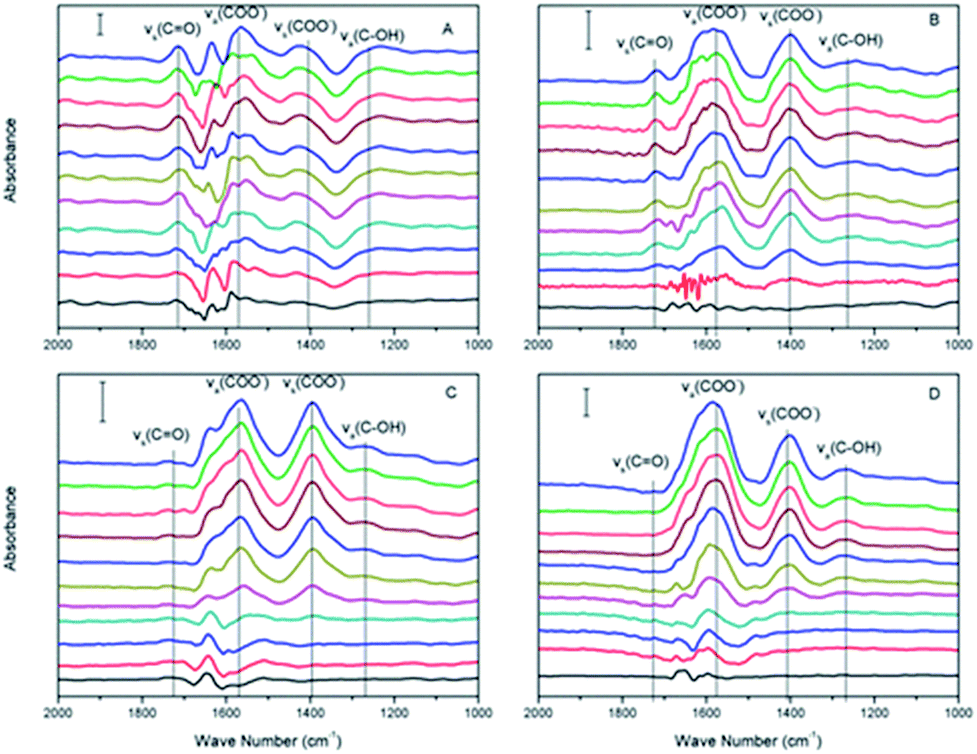 | ||
| Fig. 3 In situ ATR-FTIR spectra of citrate adsorbed on haematite film at pH (A) 2.0, (B) 4.0, (C) 6.0, and (D) 8.0, respectively, from 0 to 30 min (from bottom to top). | ||
Within the equilibrium pH range, citric acids were expected to gradually dissociate and remain deprotonated (negatively charged), and the surface of α-Fe2O3 was expected to remain positively charged (pHzeta ≈ 8.4). The –COO− radicals become adsorbed to the α-Fe2O3 surface via electrostatic attraction when the solution pH is greater than the pKa1 of citric acid. The areas of the νas(COO−) and νs(COO−) peaks were calculated to study their absorption kinetics on the α-Fe2O3 surface, as shown in Fig. 4A and B, respectively. As the pH increased, the intensities of νas(COO−) and νs(COO−) increased and were maximized at pH 6.0. Although the maximum electronegativity of citric acid should occur at pH > 8.0, the surface of α-Fe2O3 is negatively charged when the solution pH is greater than its isoelectric point, which would prevent the adsorption of more –COO− radicals on α-Fe2O3.
 | ||
| Fig. 4 Areas of the IR peaks from the carboxyl radicals (–COO−) of citrate adsorbed on haematite at different pH values. | ||
O) slightly decreased after flushing with water at pH 4 for 20 min until equilibrium was reached (Fig. 5A-f), at which time 0.1 mM citric acid at pH 4 was introduced into the ATR flow cell. The intensity of ν(CrO) gradually decreased and disappeared after 30 min. However, this result did not exclude the adsorbed Cr(VI) that was displaced by the citric acid via a competitive adsorption mechanism.
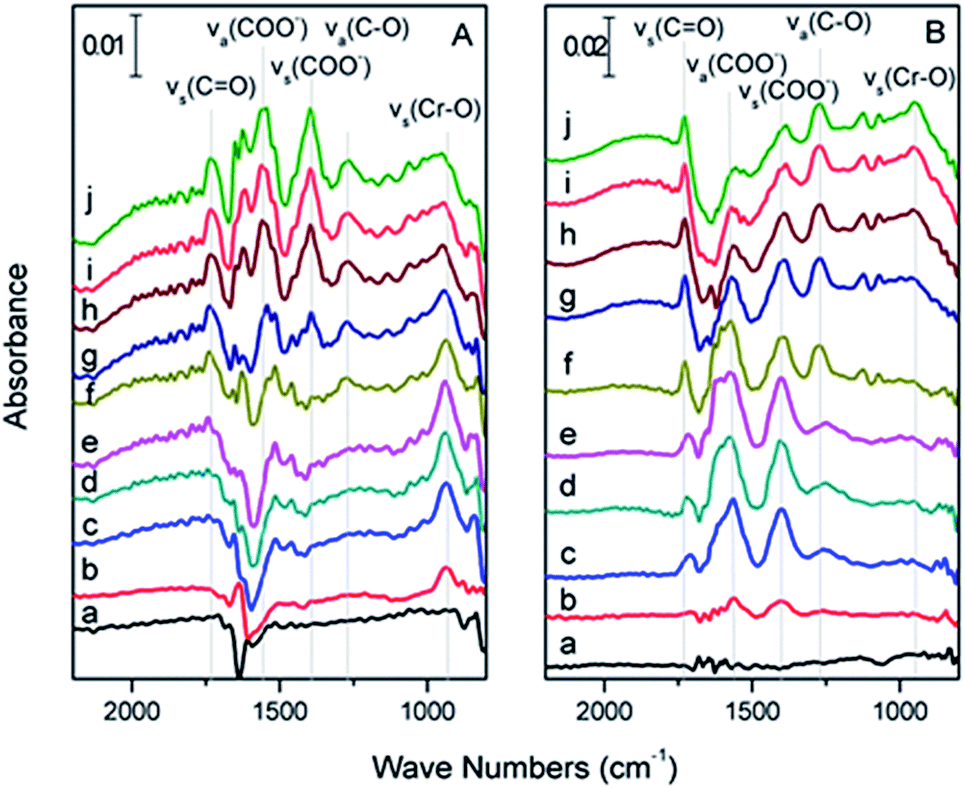 | ||
| Fig. 5 (A) In situ ATR-FTIR spectrum of haematite (a) flushed with pH 4.0 H2O for 15 min and after subsequent exposure to a flowing solution of 0.1 mM Cr(VI) at pH 4.0 for (b) 1, (c) 5, (d) 15 and (e) 30 min and (f) flushed with pH 4 H2O for 20 min and after subsequent exposure to a flowing solution of 0.1 mM citric acid at pH 4 for (g) 1, (h) 5, (i) 15, and (j) 30 min (B) in situ ATR-FTIR spectrum of haematite (a) flushed with pH 4.0 H2O for 15 min and after subsequent exposure to a flowing solution of 0.1 mM citric acid at pH 4.0 for (b) 1, (c) 5, (d) 15 and (e) 30 min; and (f) flushed with pH 4 H2O for 20 min and after subsequent exposure to a flowing solution of Cr(VI) 0.1 mM at pH 4 for (g) 1, (h) 5, (i) 15, and (j) 30 min. | ||
During the flow sequence, the characteristic peaks of citric acid grew with time (Fig. 5B(a, b, c, d, and e), respectively), which meant that the citric acid was adsorbed on the α-Fe2O3 film. Because –COOH and –COO− had different vibration modes, as mentioned above, we inferred that the –COOH and –COO− functional groups of citric acid were both adsorbed to the α-Fe2O3 surface. The vibration intensity of the citric acid remained stable when flushed with water at pH 4 water (Fig. 5B-f) for 20 min, at which time the pH 4.0 Cr(VI) solution was pumped into the ATR flow cell. Interestingly, as the intensity of the ν(CrO) peak increased, the intensities of the νs(COO−) and νas(COO−) peaks gradually decreased. However, the intensities of the νs(CO) and νas(C–O) peaks of the –COOH radicals remained the same throughout the experiment (Fig. 5B(g, h, i and j), respectively), which indicated that the adsorbed –COO− radical was the electron supplier rather than the –COOH radical of citric acid.
3.2 DFT calculations
 | ||
| Fig. 6 The optimized structures of the di-iron hydroxide cluster (A), with HCrO4− adsorption (B), citrate adsorption (C), and HCrO4− and citrate co-adsorption (D). Fe: orange, O: red, Cr: dark blue, C: grey, and H: white. | ||
| Energy | HCrO4− | Citrate | HCrO4− + citrate |
|---|---|---|---|
| ΔEad (kJ mol−1) | −73.06 | −79.99 | −106.25 |
| ΔGad (kJ mol−1) | −24.94 | −38.51 | −1.38 |
The co-adsorption state (−106.25 kJ mol−1) is energetically less favourable than the independent adsorption of both ions (153.05 kJ mol−1), presumably due to steric electronic effects.
As expected, adsorption causes monomers to lose some degree of freedom and is unfavourable in terms of entropy change. This effect is reflected by the difference in the adsorption energies and the adsorption free energies. The entropic effect results in a co-adsorption free energy of only −1.38 kJ mol−1; thus, the co-adsorption state is not very stable and may cause conformational changes, such as desorption or redox reactions. Redox reactions were favoured in this study.
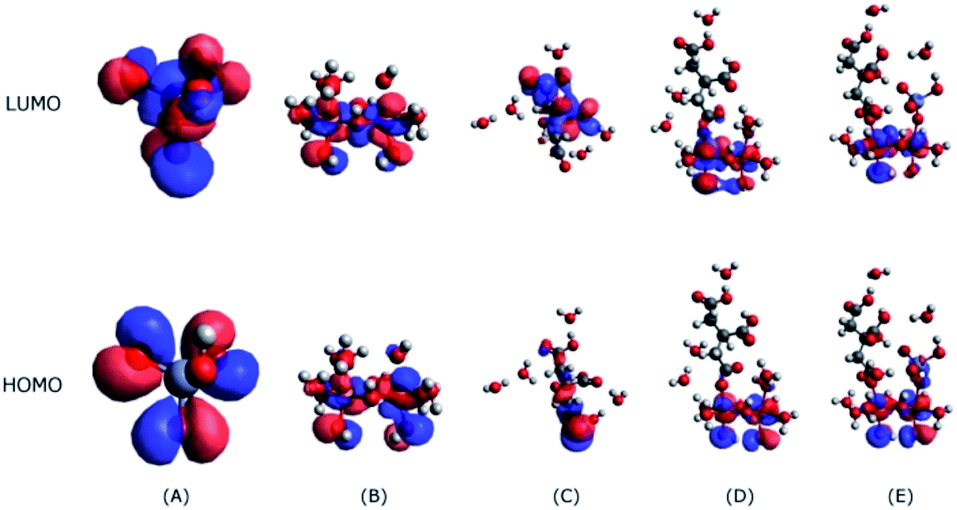 | ||
| Fig. 7 The HOMO and LUMO levels of HCrO4− (A), the Fe2O3 cluster (B), citrate (C), the citrate adsorption on the di-iron hydroxide cluster (D), and the citrate and HCrO4− co-adsorption on the di-iron hydroxide cluster (E). | ||
The interactions between citrate and Fe2O3 clusters involve electron flow from citrate to Fe2O3; hence, the HOMO orbitals of citrate will interact with the LUMO orbitals of Fe2O3 (Fig. 7B). As Fig. 7C shows, the HOMO orbital of citrate mostly resides on the bottom (carboxyl group), indicating that it will bind with Fe2O3 clusters through the carboxyl groups; this binding is evidenced by the interaction model in Fig. 7D. Despite the citrate adsorption, the LUMO orbital of the citrate–Fe2O3 complex is still located on the Fe2O3 part, showing that it still has the ability to attract HCrO4− through the interaction between its LUMO orbital and the HOMO orbital of HCrO4− (Fig. 7A). This observation proves that Fe2O3 is able to serve as a catalytic substrate for the co-adsorption and further reaction of citrate and HCrO4−, and this behaviour is in line with our experimental observations.
3.3 Reaction mechanisms
Johnston et al.5 indicated chromate mainly formed inner sphere complexes with α-Fe2O3. From a quantitative point of view, the absorbance signal of the adsorbed chromate increased as the solution pH decreased in previous large batch studies.21–25 Additionally, the spectra of adsorbed chromate on the α-Fe2O3 film were significantly different from the spectrum of aqueous chromate, which produced a single peak at 880 cm−1.26 Chromate is adsorbed to the α-Fe2O3 by electrostatic attraction and ligand exchange, resulting in the formation of monodentate complexes, with simultaneous desorption of surface hydroxyl groups from the iron surface sites. The complex geometry of the surface includes bidentate and monodentate complexes at low pH. Monodentate complexation is also a precursor for the formation of bidentate complexes.27
Citric acid can also adsorb onto α-Fe2O3 through electrostatic attraction and surface ligand exchange mechanisms.28 Citric acid formed innersphere complexes with the surface of α-Fe2O3 (ref. 29) and symmetrical monodentate and bidentate surface complexes.30 Protons generally dissociate from citric acid at three different pH values (pKa1 = 3.13, pKa2 = 4.76, and pKa3 = 6.40).29 As shown in Fig. 3, the first two prominent IR bands were assigned to the asymmetric (νas) and symmetric (νs) stretching vibrations of the carboxylate (–COO−) groups, which were located at 1570–1595 cm−1 and 1400 cm−1,29 respectively. The bands at 1270 cm−1 and 1740 cm−1 were interpreted as the bands of vibrational stretching and bending of CO and C–O in the –COOH groups of citric acid, respectively, because at least one carboxylic group likely remained in the protonated state under the experimental conditions (pH < pKa3).29 When the solution pH was greater than the pKa3, these two bands gradually disappeared, as shown in Fig. 3. Therefore, citric acid could form monodentate or bidentate ring complexes with the α-Fe2O3 surface.30
The binding energies of different complexing structures of HCrO4− and citric acid adsorbed on α-Fe2O3 at pH 4.0 were calculated by using DFT, as shown in Table 1. Both citrate and HCrO4− ions have similar adsorption energies of approximately −70 kJ mol−1. Co-adsorption could spontaneously occur because the free energy dropped significantly (−106.25 kJ mol−1); however, the binding energy of the co-adsorption state remained relatively unchanged, which indicated that co-adsorption only occurred as a transitional state and was unstable. Based on calculations of the HOMO, the most active electron in citrate is concentrated in the –COO− radicals. The active electron density shifted to the Fe–O bond after citrate was adsorbed to an iron cluster. After co-adsorption with Cr(VI), the most active electrons were shared by HCrO4− and the iron cluster, indicating that electrons were transferred from the carboxyl groups of citrate to Cr(VI) via the Fe–O bond of α-Fe2O3.
4 Conclusions
This study provides molecular level insights regarding co-adsorption and the interactions of Cr(VI) and citrate on the α-Fe2O3 surface. The in situ ATR-FTIR spectroscopy results show that Cr(VI) and citrate adsorbed to the α-Fe2O3 surface and formed inner-sphere complexes. Furthermore, the DFT calculations indicated that the adsorption of both Cr(VI) and citrate on the surface of α-Fe2O3 was energetically favourable. The co-adsorption of ions on the surface of α-Fe2O3 could lead to electron transfer from the carboxyl group of citrate to Cr(VI), which would drive the reduction of Cr(VI) to Cr(III). Collectively, the in situ ATR-FTIR spectroscopy and adsorption energy results and the IR spectrum predicted by DFT theory show that the interactions between Cr(VI) and citrate at the α-Fe2O3–water interface can be theoretically and experimentally explained. Given the ubiquitous co-existence of α-Fe2O3 and low molecular weight organic acids in natural environments, this study could be used to understand the fundamental mechanisms that determine the fate of Cr(VI) in the environment.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Science Foundation of China (Grants No. 51309214 & No. 21507138), the Natural Science Foundation of Jiangsu Province, China (Grants No. SBK2015041561), the National High Technology Research and Development Program (2013AA06A208), and the Science and Technology Service Network Initiative (STS, KFJ-EW-STS-091). We thank Prof. Cheng Gu at Nanjing University for performing the in situ ATR-FTIR analysis.References
- C. P. Johnston and M. Chrysochoou, Environ. Sci. Technol., 2016, 50, 3589–3596 CrossRef CAS PubMed.
- Z.-N. Huang, X.-L. Wang and D.-S. Yang, Water Sci. Eng., 2015, 8, 226–232 CrossRef.
- J. Hu, G. Chen and I. M. Lo, Water Res., 2005, 39, 4528–4536 CrossRef CAS PubMed.
- K. Matern and T. Mansfeldt, Environ. Chem., 2016, 13, 674–681 CrossRef CAS.
- C. P. Johnston and M. Chrysochoou, Geochim. Cosmochim. Acta, 2014, 138, 146–157 CrossRef CAS.
- C. Li, Y.-Q. Lan and B.-L. Deng, Pedosphere, 2007, 17, 318–323 CrossRef CAS.
- B. Sarkar, R. Naidu, G. S. R. Krishnamurti and M. Megharaj, Environ. Sci. Technol., 2013, 47, 13629–13636 CrossRef CAS PubMed.
- B. Deng and A. T. Stone, Environ. Sci. Technol., 1996, 30, 2484–2494 CrossRef CAS.
- J. W. Yang, Z. S. Tang, R. F. Guo and S. Q. Chen, Environ. Prog. Sustainable Energy, 2008, 27, 302–307 CAS.
- G. Yang, L. Tang, Y. Cai, G. Zeng, P. Guo, G. Chen, Y. Zhou, J. Tang, J. Chen and W. Xiong, RSC Adv., 2014, 4, 58362–58371 RSC.
- R. Dai, C. Yu, J. Liu, Y. Lan and B. Deng, Environ. Sci. Technol., 2010, 44, 6959–6964 CrossRef CAS PubMed.
- J. D. Kubicki, K. D. Kwon, K. W. Paul and D. L. Sparks, Eur. J. Soil Sci., 2007, 58, 932–944 CrossRef CAS.
- C. P. Johnston and M. Chrysochoou, Environ. Sci. Technol., 2012, 46, 5851–5858 CrossRef CAS PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Theory and Applications in Inorganic Chemistry, Wiley, Hoboken, NJ, 2008 Search PubMed.
- V. T. Bodunde, F. A. Adekola and N. Abdus-Salam, J. N. C. Acad. Sci., 2013, 129, 24–33 Search PubMed.
- S. Yin and D. E. Ellis, Surf. Sci., 2009, 603, 736–746 CrossRef CAS.
- N. Bhandari, D. B. Hausner, J. D. Kubicki and D. R. Strongin, Langmuir, 2010, 26, 16246–16253 CrossRef CAS PubMed.
- J.-J. Max and C. Chapados, J. Phys. Chem. A, 2004, 108, 3324–3337 CrossRef CAS.
- K. Axe and P. Persson, Geochim. Cosmochim. Acta, 2001, 65, 4481–4492 CrossRef CAS.
- D. Rai, L. Eary and J. Zachara, Sci. Total Environ., 1989, 86, 15–23 CrossRef CAS PubMed.
- O. Ajouyed, C. Hurel, M. Ammari, L. B. Allal and N. Marmier, J. Hazard. Mater., 2010, 174, 616–622 CrossRef CAS PubMed.
- D. D. Maksin, A. B. Nastasović, A. D. Milutinović-Nikolić, L. T. Suručić, Z. P. Sandić, R. V. Hercigonja and A. E. Onjia, J. Hazard. Mater., 2012, 209, 99–110 CrossRef PubMed.
- T. J. Reich and C. M. Koretsky, Geochim. Cosmochim. Acta, 2011, 75, 7006–7017 CrossRef CAS.
- G. Du, Z. Li, L. Liao, R. Hanson, S. Leick, N. Hoeppner and W.-T. Jiang, J. Hazard. Mater., 2012, 221, 118–123 CrossRef PubMed.
- M. M. Hoffmann, J. G. Darab and J. L. Fulton, J. Phys. Chem. A, 2001, 105, 1772–1782 CrossRef CAS.
- P. R. Grossl, M. Eick, D. L. Sparks, S. Goldberg and C. C. Ainsworth, Environ. Sci. Technol., 1997, 31, 321–326 CrossRef CAS.
- W. Stumm and E. Wieland, in Aquatic Chemical Kinetics: Reaction Rates of Processes in Natural Waters. Environmental Science and Technology Series, ed. W. Stumm, John Wiley & Sons, New York, 1990, pp. 367–400 Search PubMed.
- I. A. Mudunkotuwa and V. H. Grassian, J. Am. Chem. Soc., 2010, 132, 14986–14994 CrossRef CAS PubMed.
- O. W. Duckworth and S. T. Martin, Geochim. Cosmochim. Acta, 2001, 65, 4289–4301 CrossRef CAS.
- D. E. Latta, J. E. Bachman and M. M. Scherer, Environ. Sci. Technol., 2012, 46, 10614–10623 CrossRef CAS PubMed.
- B. Deng and A. T. Stone, Environ. Sci. Technol., 1996, 30, 463–472 CrossRef CAS.
- B. Flockhart, S. Uppal and R. Pink, Trans. Faraday Soc., 1971, 67, 513–525 RSC.
- Y. Lan, C. Li, J. Mao and J. Sun, Chemosphere, 2008, 71, 781–787 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |