
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrotropic polymer-based paclitaxel-loaded self-assembled nanoparticles: preparation and biological evaluation
Lipeng Gaoa,
Liefang Gaoa,
Mingxue Fana,
Qilong Lia,
Jiyu Jina,
Jing Wanga,
Weiyue Lub,
Lei Yua,
Zhiqiang Yan *a and
Yiting Wang*a
*a and
Yiting Wang*a
aInstitute of Biomedical Engineering and Technology, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: zqyan@sat.ecnu.edu.cn; ytwang@nbic.ecnu.edu.cn
bDepartment of Pharmaceutics, School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery, Ministry of Education, Shanghai 201203, China
First published on 30th June 2017
Abstract
The poor compatibility of carrier materials with drugs is one of the main obstacles in the drug encapsulation of nano-drug delivery system (NDDS), hindering the clinical translation of NDDS. In this study, using paclitaxel (PTX) as the insoluble model drug, we conjugated N,N-diethylniacinamide (DENA), a hydrotropic agent of PTX, to the backbone of poly(L-γ-glutamyl-glutamine) (PGG), a water-soluble polymer, to prepare the “hydrotropic polymer” PGG–DENA to improve its compatibility with PTX. By virtue of the hydrotropic effect of the DENA group, PTX was encapsulated by PGG–DENA to obtain the hydrotropic polymeric nanoparticles (PGG–DENA/PTX NPs). PTX-conjugated poly(L-γ-glutamyl-glutamine) acid (PGG–PTX) NPs previously reported were used as the control in the study. The PGG–DENA/PTX NPs showed a z-average hydrodynamic diameter of about 70 nm, and good long-term stability in PBS solution at 4 °C. The cumulative release rate of PTX from PGG–DENA/PTX NPs reached 79.10% at 96 h, while that of PGG–PTX NPs was 22.96%. PGG–DENA/PTX NPs showed significantly increased in vitro cytotoxicity on NCI-H460 lung cancer cells compared with PGG–PTX NPs. The hemolysis study proved that the PGG–DENA/PTX NPs has good biocompatibility. These results indicated that by introducing the hydrotropic agent DENA, the hydrotropic polymer PGG–DENA becomes an effective carrier material of PTX. This study provides a solution to increase the compatibility of carrier materials with insoluble drugs, and also may provide an effective way to develop a series of personalized carrier materials suitable for different insoluble drugs.
1. Introduction
Micelles are widely studied because of their ability to increase drug solubility and stability, and their tumor targeting ability. Paclitaxel (PTX) encapsulated polyethylene glycol-b-polylactic acid (PEG-b-PLA) micelles have been approved by the FDA.1 The traditional micelles are formed by self-assembly of amphiphilic polymer materials (e.g., PEG-b-PLA, polyethylene glycol–phosphatidylethanolamine (PEG–PE)) via hydrophobic interaction. However, the drug encapsulation in the traditional micelles mainly depends on the non-specific hydrophobic interaction between the hydrophobic end of the polymer and the drug. Thus the traditional micelles usually only have a good encapsulating effect for a small amount of extremely hydrophobic drugs. But in fact most of drugs are often mildly hydrophobic, resulting in most of the reported micelles having low drug loading capacity and poor stability.2Accordingly, researchers introduced hydrotropic agents into the hydrophobic side of the polymeric materials to increase the drug encapsulation in micelles.3–5 Hydrotropic agents are a kind of small molecules that can increase the water solubility of insoluble drugs by forming a complex, association or complex salt.6 The micelles developed by combining the hydrotropic agents with the traditional micelles are so-called hydrotropic polymer micelles. Up to now, there have been several hydrotropic agents reported, such as N-methylpyridinium nicotinamide (PNA),7 N,N-dimethylbenzamide (DMBA),4 N,N-diethylniacinamide (DENA)3,4 and fluorenylmethoxycarbonyl (α-Fmoc).8 For example, Kim group developed a PEG-b-(poly-N,N-diethylnicotinamide) (PEG-b-PDENA), which can self-assemble to form micelles that showed good encapsulating effect for PTX.4 The solubility of PTX was increased by 6000 times, which is higher than the equivalent concentration of free DENA or PEG-b-PLA does. The PTX encapsulated PEG-b-PDENA micelles can be stably stored for at least 4 weeks.9 Because DENA has a special solubilization mechanism for PTX (aromatic ring buildup and hydrogen bonding), it showed higher solubilization effect on PTX than other cosolvents (DMBA or α-Fmoc) did.4 More importantly, different form the free hydrotropic agents, the polymerized hydrotropic agent cannot be easily taken up in vivo, thereby avoiding the potential toxicity to body. In addition, the stability and drug release behavior of micelles can be altered by adjusting the molecular weight of the hydrotropic end.4 In spite of this, the hydrotropic polymer micelles are also deficient: the hydrophobicity of the polymerized hydrotropic agents is weaker than that of the hydrophobic end of traditional micellar materials (such as PLA), especially for DENA. This resulted in a high critical micelle concentration (CMC) of these hydrotropic polymers, which is disadvantageous for the formation and further use of micelles.2 This problem hinders the further development of the hydrotropic polymer micelles.
PTX-conjugated poly(L-glutamic acid) (PGA–PTX, also known as Xyotax™), a polymer drug conjugate, has entered the clinical trials due to its high water solubility and antitumor effect.10,11 Poly(L-γ-glutamyl-glutamine)–paclitaxel (PGG–PTX) is another polymer drug conjugate we previously developed on the basis of PGA–PTX,12–14 which further increased the water solubility, increased the tolerable dose, reduced the side effects and improved the antitumor effects.15–18 The results showed that PGG–PTX could be directly dissolved in water and had a solubility of 50 mg mL−1, which was much higher than that of PGA–PTX (7 mg mL−1). It can self-assemble to form nano core–shell structure about 30 nm in water, with PTX as hydrophobic nucleus and PGG skeleton as hydrophilic shell. However, PGG–PTX is also deficient: since PTX is covalently coupled to the PGG backbone, its drug release performance is poor. The cumulative release rate of PTX from PGG–PTX nanoparticles (NPs) in vitro is only about 23% at 96 h.12
Based on the above considerations, we here conjugated DENA to the backbone of PGG to prepare the “hydrotropic polymer” PGG–DENA. Then by virtue of the hydrotropic effect of DENA group, PTX was encapsulated by PGG–DENA to obtain the hydrotropic polymeric nanoparticles (PGG–DENA/PTX NPs) (Fig. 1). We characterized the NPs by DLS, TEM, HPLC and evaluated the PTX release, cytotoxicity, long-term stability and cellular uptake by human NCI-H460 cancer cells in vitro and hemolysis study in vivo. PGG–DENA/PTX NPs exhibited a desirable drug release profile, and good biocompatibility and long-term stability.
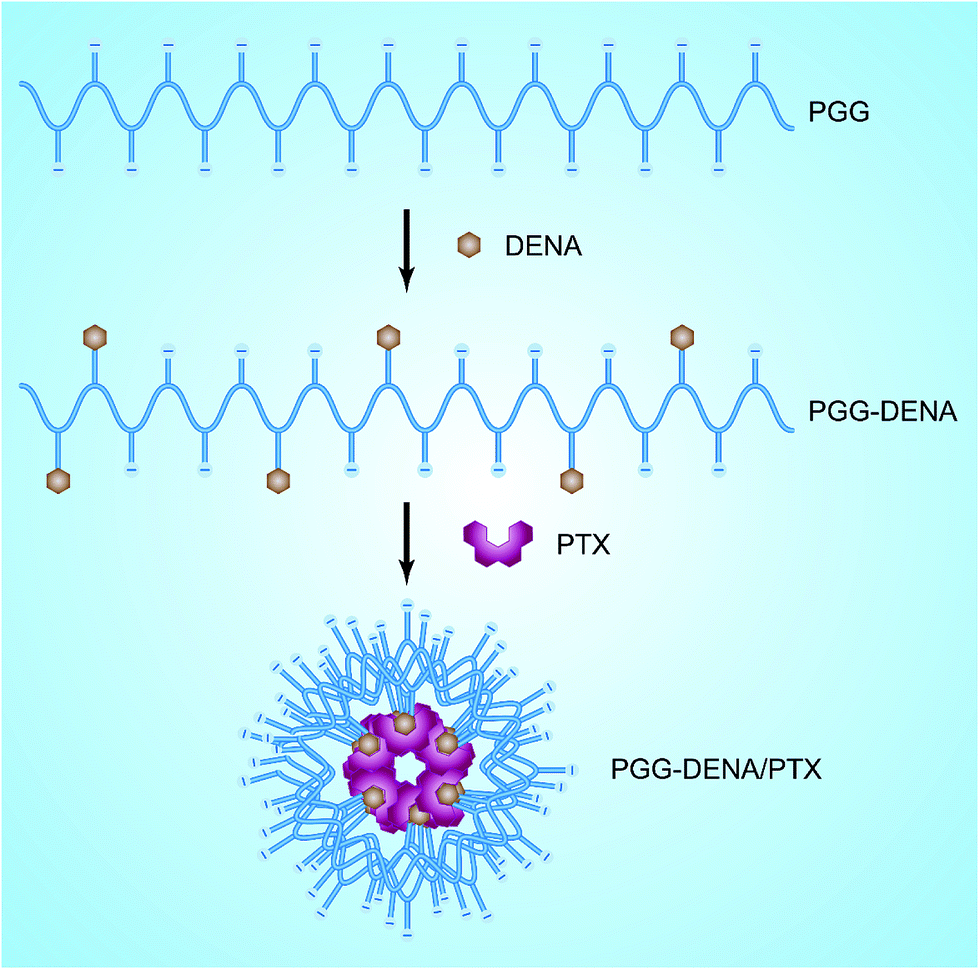 | ||
| Fig. 1 Schematics of the preparation process of the PGG–DENA/PTX. | ||
2. Materials and methods
2.1. Materials
PGG (poly-(L-γ-glutamyl-glutamine)) was synthesized by our laboratory.12 4-(Chloromethyl)benzoyl chloride, tert-butyldimethylsilyl chloride (TBSCL), 2-hydroxynicotinic acid, N,N′-carbonyldiimidazole (CDI) and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich, Inc. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) was purchased from EMD Chemicals Inc. (Darmstadt, Germany). All other chemicals and reagents were commercially available and directly used.Human NCI-H460 carcinoma cell line was obtained from Shanghai Institute of Cell Biology. This cell was cultured in RPMI 1640 supplemented with 10% FBS at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. DiO (3,3′-dioctadecyloxacarbocyanine, perchlorate) was purchased from Tianjin Biolite Biotech Co., LTD. Hoechst 33342 was purchased from Beyotime Institute of Biotechnology.
All experiments involving animals were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of East China Normal University. All experimental protocols were approved by the IACUC of East China Normal University. Male SD rats (6–8 weeks old) were obtained from SLAC Ltd (Shanghai, China) and maintained under SPF conditions. All efforts were made to minimize the number of animals used and their suffering. Animal experiments were reported in accordance with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
2.2. Synthesis of PGG–DENA
The synthesis of PGG–DENA was illustrated in Fig. 2. The starting material 4-(chloromethyl)benzoyl chloride (1) was reacted with boron hydride to prepare compound 2, which was protected with TBSCl to afford compound 3. The starting material 2-hydroxynicotinic acid (4) was reacted with diethylamine to produce compound 5. The compound 3 was reacted with compound 5 to obtain compound 6, which was deprotected to afford compound 7. Then the compound 7 was conjugated to PGG in the presence of EDC and DMAP to obtain the final “hydrotropic polymer” PGG–DENA, which was dialyzed with a tangential flow filtration system followed by lyophilization. | ||
| Fig. 2 Synthesis of PGG–DENA. Reagents and conditions: (a) NaBH4, THF/EtOH, rt; (b) TBSCl, DMAP, DCM, rt; (c) DEA, CDI, THF, 60 °C; (d) K2CO3, anhydrous acetone, 60 °C; (e) 2 M HCl (aq), rt; (f) PGG–COOH, EDC, DMAP, DMF, rt. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Then NaBH4 (0.908 g, 24 mmol) was added and stirred at room temperature under N2 for 2 h. The solvent was removed by rotary evaporation at 37 °C. Ethyl acetate was added and washed with 0.3 M NaHCO3 (aq). The organic layer was dried over Na2SO4, filtrated and concentrated to give (4-(chloromethyl)phenyl)methanol (2) (867 mg, 92%) as white solid. 1H-NMR (400 MHz, CDCl3) δ 4.50 (d, J = 4.8 Hz, 2H), 4.75 (s, 2H), 5.21 (t, J = 4.8 Hz, 1H), 7.32 (d, J = 7.6 Hz, 2H), 7.39 (d, J = 7.6 Hz, 2H).
:1, v/v). Then NaBH4 (0.908 g, 24 mmol) was added and stirred at room temperature under N2 for 2 h. The solvent was removed by rotary evaporation at 37 °C. Ethyl acetate was added and washed with 0.3 M NaHCO3 (aq). The organic layer was dried over Na2SO4, filtrated and concentrated to give (4-(chloromethyl)phenyl)methanol (2) (867 mg, 92%) as white solid. 1H-NMR (400 MHz, CDCl3) δ 4.50 (d, J = 4.8 Hz, 2H), 4.75 (s, 2H), 5.21 (t, J = 4.8 Hz, 1H), 7.32 (d, J = 7.6 Hz, 2H), 7.39 (d, J = 7.6 Hz, 2H).2.3. Preparation of PGG–DENA/PTX NPs
PGG–DENA/PTX NPs were prepared by the emulsification-solvent evaporation method.19,20 Briefly, 12 mg of PTX was dissolved in 2 mL of mixture of methylene chloride and acetone (3:1, v/v) as the organic phase, and 40 mg of PGG–DENA were suspended in 4 mL of sodium cholate solution as the water phase. The mixture was emulsified by ultrasonic method in ice bath and gently stirred at room temperature. The solution was then opened to air overnight to allow slow evaporation of organic solvent and formation of the PGG–DENA/PTX NPs, which were purified by G50 gel column connecting AKTA purifier (GE Healthcare) to remove the free PTX.
2.4. PTX solubility
The solubility of PTX was determined by high performance liquid chromatography (HPLC, Agilent 1260 series). DENA was dissolved in deionized water and packed in centrifuge tube (2 mL). Excess PTX was added to each tube. The mixture was stirred using a magnetic stirring bar for 24 h at 37 °C. The sample was mixed with 1 mL ethyl acetate to extract PTX. The ethyl acetate solution was dried with nitrogen blowing instrument. Then the samples were resuspended with acetonitrile and filtered through a 0.22 μm pore-sized filtration membrane. The amount of PTX in each tube was measured by HPLC with a UV detector at 228 nm.2.5. Characterization of PGG–DENA/PTX NPs
The particle sizes of PGG–DENA/PTX NPs were measured by dynamic light scattering (DLS) using a Mastersizer2000 (Malvern Instruments Inc) equipped with He–Ne laser (4 mW, 633 nm) light source and 90° angle scattered-light collection configuration.The particle charge was quantified as zeta potential using a Mastersizer2000 (Malvern Instruments Inc).
The morphology of PGG–DENA/PTX NPs were observed with transmission electron microscopy (TEM). The TEM study was carried out using a JEM-2100 (Hitachi, Tokyo, Japan) electron microscope operating at an accelerating voltage of 75 kV.
The drug loading capacity of PGG–DENA/PTX was determined by HPLC (Agilent 1260 series). PGG–DENA/PTX (4 mg) was placed in a 5 mL centrifuge tube. Then, 2 mL of acetonitrile was added and the solution was shaken horizontally at 120 min−1 with an incubator shaker (HZ-8812S, Scientific and Educational Equipment plant, China) at 37 °C. The supernatant solution was taken at 0.5 h, 3 h, 6 h, 9 h and 20 h, respectively, and filtered through a 0.22 μm pore-sized filtration membrane for HPLC determination.
2.6. Long-term stability of PGG–DENA/PTX NPs
PGG–DENA/PTX NPs solution (2.0 mg mL−1) was prepared in PBS. The particle size and polydispersity index (PDI) of the sample was monitored by DLS for 28 days.2.7. In vitro release of PTX from PGG–DENA/PTX NPs
To investigate the in vitro release profile of PTX from PGG–DENA/PTX NPs, we used sodium salicylate solution (0.8 M, pH 6.5) as the release medium as reported previously.21 The PGG–DENA/PTX NPs solution with the final concentration of 2 mg mL−1 was packed in 50 mL centrifuge tube and shaken horizontally at 120 min−1 at 37 °C. The sample was withdrawn at predetermined time points and mixed with 1 mL ethyl acetate to extract PTX. The ethyl acetate solution was dried with nitrogen blowing instrument. Then the samples were resuspended with acetonitrile and filtered through a 0.22 μm pore-sized filtration membrane. The medium was refreshed at various time intervals. The concentration of PTX was determined by HPLC analysis.2.8. In vitro cytotoxicity assays
The in vitro cytotoxicity of NPs was investigated by the CCK-8 assay according to the published protocols with modifications.22 NCI-H460 cells (5 × 103) were seeded in 96-well plates and incubated for 24 h in a humidified atmosphere with 5% CO2. Then serial dilutions of PTX, PGG–PTX NPs, PGG–DENA/PTX NPs and PGG–DENA were added to the plate (100 μL per well), respectively. After further incubation for up to 48 h, the cells were treated with 10 μL of CCK-8 solution and cultured for 4 h. The absorbance was measured with a microplate reader (SpectraMax M5, Molecular Devices, USA) at 450 nm. The survival rate was calculated using the following formula: viability rate = [(ODtest group − ODblank)/(ODcontrol group − ODblank)] × 100%, where ODtest group is the optical density (OD) of experiment group, ODcontrol group is the OD of control group, and ODblank is the OD of blank group.2.9. In vitro cellular uptake
:1, v/v) as the organic phase, and PGG–DENA/PTX NPs were suspended in sodium cholate solution as the water phase. The mixture was emulsified by ultrasonic method in ice bath and rotary evaporated to remove organic solvent and obtain the PGG–DENA/PTX/DiO NPs. PGG–DENA/PTX/DiO NPs were then purified by G50 gel column connecting AKTA purifier to remove free DiO.In order to observe the cellular uptake of NPs qualitatively, the treated cells were washed three times with PBS, then fixed with 4% paraformaldehyde, stained with Hoechst 33342 and observed using a confocal laser scanning microscope (CLSM, TCS SP5, Leica).
To quantitatively analyze the cellular uptake of NPs, the treated cells were washed with PBS, trypsinized and harvested by centrifugation at 1200 rpm for 5 min. The cells were resuspended in 200 μL PBS and filtered through a 40 mm nylon mesh to remove cell aggregates. The cell suspensions were then analyzed by flow cytometry (Guava easyCyte, USA).
2.10. Hemolysis study
Hemolysis study was carried out according to the published procedure.23 Briefly, freshly collected rat blood was washed three times with saline by centrifugation at 1500 rpm for 15 minutes. The red blood cell suspension was diluted with saline to obtain a 2% suspension (v/v). Various concentrations of PGG–DENA/PTX NPs, Cremophor EL, Tween 80, saline were added into the suspension, respectively. These samples were incubated at 37 °C for 1 hour, and centrifuged at 3000 rpm for 10 minutes. The supernatants were collected and analyzed for hemoglobin content by spectrophotometric detection at 545 nm. Analysis of each sample was performed in triplicate.2.11. Statistical analysis
Statistical differences were evaluated by two-tailed student's t-test for two groups of data and one-way ANOVA for over three groups of data. The differences were considered to be significant at P < 0.05 and very significant at P < 0.01.3. Results and discussion
3.1. PTX solubility
The solubilization of PTX by DENA was determined by HPLC. The results (Fig. 3) showed that the solubility of PTX increased gradually as the concentration of DENA increased. When the concentration of DENA reached 6 M, the solubility of PTX was increased to about 523 mg mL−1 in water (the intrinsic solubility of PTX is 0.0003 mg mL−1).24 The results indicating that DENA increased the solubility of PTX by 1.7 × 106-fold. | ||
| Fig. 3 The solubility of PTX as a function of the molar concentration of DENA. The solubility of PTX reached about 523 mg mL−1 at 6 M of DENA. Data are means ± SD from three measurements. | ||
3.2. Characterization of PGG–DENA
 | ||
| Fig. 4 Characterization of the PGG–DENA. (A) The chemical structure of the PGG–DENA. The green part is PGG and the red is DENA. (B) 1H-NMR spectra of PGG (I) and PGG–DENA (II). The characteristic peaks aromatic hydrogens of DENA at 7.80 (s, 1H), 7.59 (s, 1H), 7.21–7.30 (m, 4H) and 6.53 (s, 1H) ppm (the black arrow) showed that DENA and PGG were successfully linked together. | ||
:1. And the results also suggested the successful conjugated of DENA.
| Sample | Mw (kDa) | Mw/Mn (PDI) |
|---|---|---|
| a All data are expressed as the mean Mw of the samples (n = 3). | ||
| PGG | 51.29 | 1.31 |
| PGG–DENA | 65.52 | 1.43 |
3.3. Characterization of PGG–DENA/PTX NPs
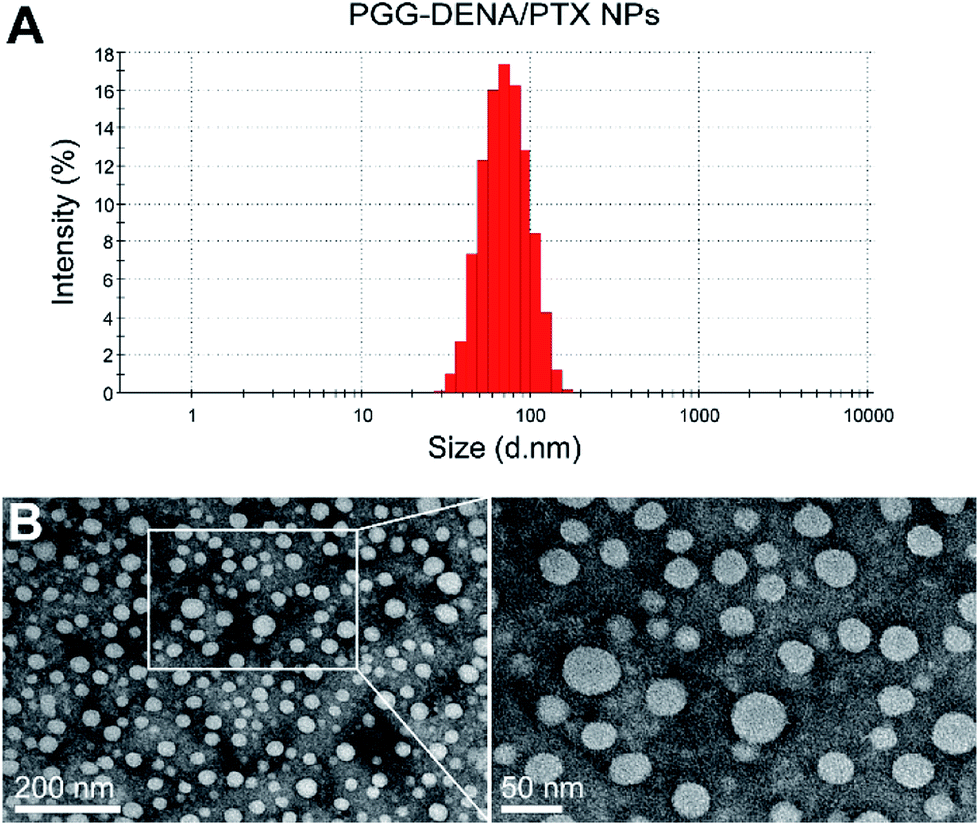 | ||
| Fig. 5 Particle size and morphology characterization of PGG–DENA/PTX NPs. DLS analysis (A) and TEM images (B) of PGG–DENA/PTX NPs. The NPs exhibited uniform spherical morphology as displayed in the TEM image. | ||
 | ||
| Fig. 6 Stability of PGG–DENA/PTX NPs in PBS solution at 4 °C. PGG–DENA/PTX NPs exhibited long-term stability. | ||
3.4. In vitro release of PTX from PGG–DENA/PTX NPs
The PTX release from PGG–DENA/PTX NPs was performed in sodium salicylate (0.8 M) at 37 °C for 96 h. As shown in Fig. 7, the results showed that the release of PTX from PGG–PTX NPs and PGG–DENA/PTX NPs gradually increased with time. Compared to PGG–PTX NPs (22.62%), the cumulative release rate of PTX from PGG–DENA/PTX NPs reached 79.10% at 96 h. PTX release from PGG–DENA/PTX NPs was remarkably higher than that from PGG–PTX NPs. This results indicated that the encapsulated PTX in PGG–DENA/PTX NPs is easier to be released than chemically bonded PTX in PGG–PTX NPs. | ||
| Fig. 7 The kinetics of PTX release from PGG–DENA/PTX NPs and PGG–PTX NPs in sodium salicylate at 37 °C (n = 3, bars represent means ± SD). | ||
3.5. In vitro cytotoxicity assays
The cell viability of NCI-H460 cells were evaluated following incubation with PTX, PGG–PTX NPs, PGG–DENA/PTX NPs and PGG–DENA. As shown in Fig. 8, the results showed that the cell viability of PTX, PGG–DENA/PTX NPs and PGG–PTX NPs decreased gradually as the drug concentration increased. PGG–DENA had no obvious cytotoxicity. The IC50 value for PTX, PGG–DENA/PTX NPs and PGG–PTX NPs was 0.2304 μg mL−1, 0.7356 μg mL−1 and 2.0253 μg mL−1, respectively. PTX had the strongest cytotoxicity. PGG–DENA/PTX NPs showed significantly increased cytotoxicity than PGG–PTX NPs, which should be resulted from the increased PTX release from PGG–DENA/PTX NPs compared with PGG–PTX NPs (Fig. 7).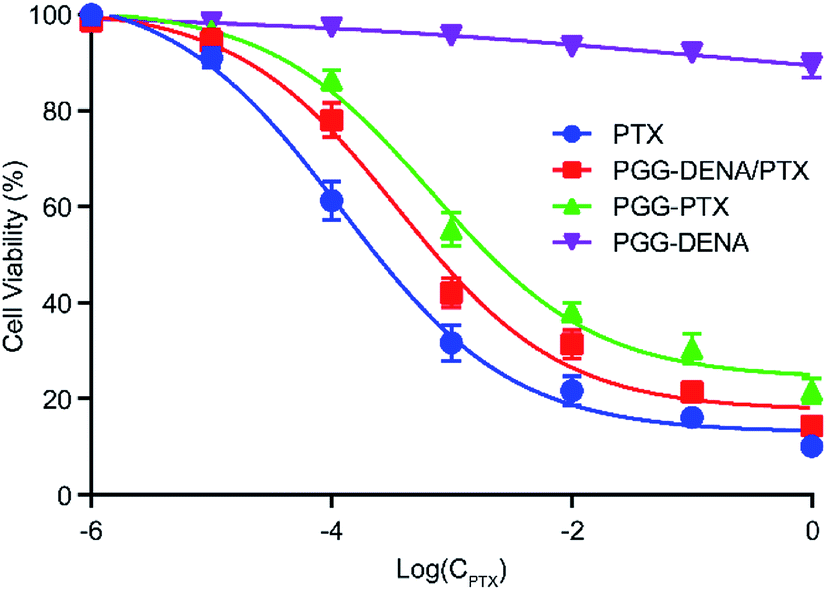 | ||
| Fig. 8 The cytotoxicity of PTX, PGG–PTX NPs, PGG–DENA/PTX NPs and PGG–DENA on NCI-H460 cells as measured by CCK-8 Kit. PGG–DENA/PTX NPs showed significantly increased cytotoxicity than PGG–PTX NPs. | ||
3.6. In vitro cellular uptake
In order to determine the interaction of PGG–DENA/PTX NPs with the tumor cells, we observed the cellular uptake of NPs by NCI-H460 cell line using CLSM and flow cytometer. As shown in Fig. 9, the percentages of fluorescent cells in PGG–PTX/DiO NPs and PGG–DENA/PTX/DiO NPs were 97.36% and 96.74%, and the mean fluorescent intensities for them were 183.40 and 177.03, respectively. These data showed that the PGG–PTX/DiO NPs and PGG–DENA/PTX/DiO NPs have similar cellular uptake by NCI-H460 cells, which may be attributed to their similar endocytic pathways. | ||
| Fig. 9 The CLSM images of cellular uptake and flow cytometry for PGG–PTX/DiO NPs (A) and PGG–DENA/PTX/DiO NPs (B). The numbers in flow cytometry pictures represent percentages of DiO-positive cells and mean of fluorescence intensity, respectively. These data showed that the PGG–PTX/DiO NPs and PGG–DENA/PTX/DiO NPs have similar cellular uptake by NCI-H460 cells. | ||
3.7. Hemolysis study
To determine the biocompatibility of PGG–DENA/PTX NPs, a hemolysis study was carried out. Our previous study showed that the PGG polymer have good biocompatibility.13 As shown in Fig. 10, the surfactants Tween 80 caused significant damage of red blood cells at 2 mg mL−1, and Cremophor EL did not induce substantial hemolysis until reaching 4 mg mL−1. By contrast, PGG–DENA/PTX NPs had no hemolytic activity even at high concentrations of 8 mg mL−1. These data suggested that PGG–DENA/PTX has good compatibility with erythrocytes and can be administered intravenously. | ||
| Fig. 10 Hemolysis of red blood cells after incubation with PGG–DENA/PTX NPs, Cremophor EL and Tween 80. Compared with Cremophor EL and Tween 80, PGG–DENA/PTX NPs showed almost no hemolytic activity. | ||
4. Conclusions
In this work, using PTX as the model drug, we conjugated DENA, the hydrotropic agents of PTX, to the backbone of a water-soluble polymer PGG to prepare the “hydrotropic polymer” PGG–DENA. Then by virtue of the hydrotropic effect of DENA group, PTX was successfully encapsulated by PGG–DENA to obtain the hydrotropic polymeric nanoparticles PGG–DENA/PTX NPs. The results showed that the cumulative release rate of PTX from PGG–DENA/PTX NPs reached 79.10% at 96 h. In vitro cytotoxicity assays, PGG–DENA/PTX NPs showed significantly increased cytotoxicity than PGG–PTX NPs. The NCI-H460 lung cancer cells have similar cell uptake of PGG–PTX/DiO NPs and PGG–DENA/PTX/DiO NPs. Furthermore, the hemolysis study proved that the PGG–DENA/PTX NPs has good compatibility with erythrocytes. The results indicated that the hydrotropic polymer PGG–DENA was an effective carrier material for PTX.The molecular structure of the hydrotropic agent is generally composed of an aromatic ring system and an anionic group, in which the aromatic ring moiety interacts with the drug and the anionic group brings a high water solubility. The mechanism of solubilization is much complicated, and the consensus on this can be summarized as the four points: hydrophobic interaction, aromatic ring accumulation, hydrogen bond formation and specific interaction.26 Application of hydrotropic agent is a very effective means for the solubilization of insoluble drugs, which can result in the increase of drug solubility by several orders of magnitude.27 However, it requires a high concentration of hydrotropic agents for effective solubilization, making it impossible for them to be used in vivo due to their potential toxicity. Thus the current application of hydrotropic agents are mostly limited in the drug solubilization in vitro, such as in drug dissolution test.28 Therefore, how to make full use of the solubilization effect of hydrotropic agents, as well as avoid their potential toxicity in vivo, becomes an important scientific issue. Our study here would provide an idea for this issue and expand the use of hydrotropic agents, making them a more effective and practical tool in pharmaceutical science.
The timely and adequate drug release in tumor tissues is a key step to exert the antitumor effect of NDDS. Accordingly, researchers have made great progress in the tumor microenvironment responsive NDDS in recent years, including pH-sensitive,29,30 temperature-sensitive,31 redox-sensitive32 and enzyme-sensitive,33 to improve the drug release behavior. In our study, the cumulative release rate of PTX from PGG–DENA/PTX NPs reached 79.10% at 96 h, while that of PGG–PTX NPs was only 22.96% (Fig. 7). This results indicated that the physical encapsulation of drugs might produce more adequate drug release than chemical conjugation does. What is more, PGG–DENA/PTX NPs can avoid the side effects of free DENA after administration. We also found that the PGG–DENA/PTX NPs showed significantly increased cytotoxicity than PGG–PTX NPs (Fig. 8), which should be resulted from the increased PTX release from PGG–DENA/PTX NPs compared with PGG–PTX NPs.
In summary, we have successfully developed a PGG–DENA/PTX NPs delivery system which exhibited a high drug release rate, good biocompatibility and long-term stability. This study provide a solution to increase the compatibility of carrier materials with insoluble drugs, and also may provide an effective way to develop a series of personalized carrier materials suitable for different insoluble drugs.
Conflict of interest
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.Acknowledgements
This work was supported by National Basic Research Program of China (2013CB932500), National Natural Science Foundation of China (60976004), “985” grants of East China Normal University (ECNU).References
- H. K. Ahn, M. Jung, S. J. Sym, D. B. Shin, S. M. Kang, S. Y. Kyung, J. W. Park, S. H. Jeong and E. K. Cho, Cancer Chemother. Pharmacol., 2014, 74, 277–282 CrossRef CAS PubMed.
- X. L. Zhang, Y. X. Huang and S. Li, Ther. Delivery, 2014, 5, 53–68 CrossRef CAS PubMed.
- H. Koo, K. H. Min, S. C. Lee, J. H. Park, K. Park, S. Y. Jeong, K. Choi, I. C. Kwon and K. Kim, J. Controlled Release, 2013, 172, 823–831 CrossRef CAS PubMed.
- J. Y. Kim, S. Kim, R. Pinal and K. Park, J. Controlled Release, 2011, 152, 13–20 CrossRef CAS PubMed.
- G. Saravanakumar, K. H. Min, D. S. Min, A. Y. Kim, C. M. Lee, Y. W. Cho, S. C. Lee, K. Kim, S. Y. Jeong, K. Park, J. H. Park and I. C. Kwon, J. Controlled Release, 2009, 140, 210–217 CrossRef CAS PubMed.
- J. J. Booth, M. Omar, S. Abbott and S. Shimizu, Phys. Chem. Chem. Phys., 2015, 17, 8028–8037 RSC.
- K. M. Huh, H. S. Min, S. C. Lee, H. J. Lee, S. Kim and K. Park, J. Controlled Release, 2008, 126, 122–129 CrossRef CAS PubMed.
- X. Gao, Y. Huang, A. M. Makhov, M. Epperly, J. Lu, S. Grab, P. Zhang, L. Rohan, X. Q. Xie, P. Wipf, J. Greenberger and S. Li, Mol. Pharm., 2013, 10, 187–198 CrossRef CAS PubMed.
- S. C. Lee, K. M. Huh, J. Lee, Y. W. Cho, R. E. Galinsky and K. Park, Biomacromolecules, 2007, 8, 202–208 CrossRef CAS PubMed.
- S. Sharma, J. Singh, A. Verma, B. V. Teja, R. P. Shukla, S. K. Singh, V. Sharma, R. Konwar and P. R. Mishra, RSC Adv., 2016, 6, 73083–73095 RSC.
- M. H. Han, H. Zheng, Y. F. Guo, Y. H. Wang, X. Y. Qi and X. T. Wang, RSC Adv., 2016, 6, 45664–45672 RSC.
- D. Yang, X. Liu, X. Jiang, Y. Liu, W. Ying, H. Wang, H. Bai, W. D. Taylor, Y. Wang, J. P. Clamme, E. Co, P. Chivukula, K. Y. Tsang, Y. Jin and L. Yu, J. Controlled Release, 2012, 161, 124–131 CrossRef CAS PubMed.
- S. Van, S. K. Das, X. Wang, Z. Feng, Y. Jin, Z. Hou, F. Chen, A. Pham, N. Jiang, S. B. Howell and L. Yu, Int. J. Nanomed., 2010, 5, 825–837 CrossRef CAS PubMed.
- D. Yang, L. Yu and S. Van, Cancers, 2010, 3, 17–42 CrossRef PubMed.
- Z. Feng, G. Zhao, L. Yu, D. Gough and S. B. Howell, Cancer Chemother. Pharmacol., 2010, 65, 923–930 CrossRef CAS PubMed.
- D. Yang, S. Van, X. Jiang and L. Yu, Int. J. Nanomed., 2011, 6, 85–91 CAS.
- D. Yang, S. Van, J. Liu, J. Wang, X. Jiang, Y. Wang and L. Yu, Int. J. Nanomed., 2011, 6, 2557–2566 CAS.
- D. Stirland, J. Nichols, S. Miura and Y. Bae, J. Controlled Release, 2013, 172, 1045–1064 CrossRef CAS PubMed.
- X. Li, Y. Ma, X. Zhang and G. Chen, Chin. JMAP, 2011, 28, 740–743 CAS.
- J. H. Kim, Y. S. Kim, K. Park, S. Lee, H. Y. Nam, K. H. Min, H. G. Jo, J. H. Park, K. Choi, S. Y. Jeong, R. W. Park, I. S. Kim, K. Kim and I. C. Kwon, J. Controlled Release, 2008, 127, 41–49 CrossRef CAS PubMed.
- S. Kim, J. Y. Kim, K. M. Huh, G. Acharya and K. Park, J. Controlled Release, 2008, 132, 222–229 CrossRef CAS PubMed.
- Y. Song, R. Guan, F. Lyu, T. Kang, Y. Wu and X. Chen, Mutat. Res., 2014, 769, 113–118 CrossRef CAS PubMed.
- X. Liu, Y. Xie, W. Li, W. Sheng, Y. Li, Z. Tong, H. Ni, C. Huselstein, X. Wang and Y. Chen, Bio-Med. Mater. Eng., 2015, 25, 47–55 Search PubMed.
- J. Lee, S. C. Lee, G. Acharya, C. J. Chang and K. Park, Pharm. Res., 2003, 20, 1022–1030 CrossRef CAS.
- D. Yang, S. Van, Y. Shu, X. Liu, Y. Ge, X. Jiang, Y. Jin and L. Yu, Int. J. Nanomed., 2012, 7, 581–589 CAS.
- J. Y. Kim, S. Kim, M. Papp, K. Park and R. Pinal, J. Pharm. Sci., 2010, 99, 3953–3965 CrossRef CAS PubMed.
- C. Som, B. Nowack, H. F. Krug and P. Wick, Acc. Chem. Res., 2013, 46, 863–872 CrossRef CAS PubMed.
- B. M. El-Houssieny, E. Z. El-Dein and H. M. El-Messiry, Drug Discoveries Ther., 2014, 8, 178–184 CrossRef CAS.
- M. Dalela, T. G. Shrivastav, S. Kharbanda and H. Singh, ACS Appl. Mater. Interfaces, 2015, 7, 26530–26548 CAS.
- S. Yin, L. Chang, T. Li, G. Wang, X. Gu and J. Li, RSC Adv., 2016, 6, 105957–105968 RSC.
- Z. Y. Wang, H. Zhang, Y. Yang, X. Y. Xie, Y. F. Yang, Z. Li, Y. Li, W. Gong, F. L. Yu, Z. Yang, M. Y. Li and X. G. Mei, Drug Delivery, 2016, 23, 1222–1231 CrossRef CAS PubMed.
- J. Li, T. Yin, L. Wang, L. Yin, J. Zhou and M. Huo, Int. J. Pharm., 2015, 483, 38–48 CrossRef CAS PubMed.
- N. Li, H. Cai, L. Jiang, J. Hu, A. Bains, J. Hu, Q. Gong, K. Luo and Z. Gu, ACS Appl. Mater. Interfaces, 2017, 9, 6865–6877 CAS.
| This journal is © The Royal Society of Chemistry 2017 |