
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe photoprocess effects of an amino group located at different positions along the polymethine chain in indodicarbocyanine dyes†
Jianfang Cao *a,
Jiangli Fanb,
Wen Sunb,
Yu Guoa,
Hongmei Wua and
Xiaojun Peng*b
*a,
Jiangli Fanb,
Wen Sunb,
Yu Guoa,
Hongmei Wua and
Xiaojun Peng*b
aSchool of Chemical and Environmental Engineering, Liaoning University of Technology, No. 169 Shiying Road, Guta District, Jinzhou 121001, China. E-mail: caojf@lnut.edu.cn
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, No. 2 Linggong Road, High-tech District, Dalian 116024, China
First published on 14th June 2017
Abstract
A systematic spectral analysis is presented for Cy3, Cy5, Cy7 and Cy9 aminocyanine dyes. The amino group in different positions of the polymethinic chain causes different background spectral properties. Computational studies on these dyes reveal that the length of C–N bond of amino group in the even position is close to that of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond, which prevents the conjugation of the whole polymethine chain. The amino group located in the even position acts as the electron acceptor at excited state (a-ICT) of aminocyanine dyes. While, the amino group in the odd position is the electron donor at excited state (ICT) of aminocyanine dyes. The rotations of amino groups are more difficult for aminocyanine dyes with the amino group in the even position than that of the amino group in the odd postion. The results demonstrate a foundation for interpretation of the behavior of the dyes, thus providing guidelines for future design of new aminocyanine fluorophores.
N double bond, which prevents the conjugation of the whole polymethine chain. The amino group located in the even position acts as the electron acceptor at excited state (a-ICT) of aminocyanine dyes. While, the amino group in the odd position is the electron donor at excited state (ICT) of aminocyanine dyes. The rotations of amino groups are more difficult for aminocyanine dyes with the amino group in the even position than that of the amino group in the odd postion. The results demonstrate a foundation for interpretation of the behavior of the dyes, thus providing guidelines for future design of new aminocyanine fluorophores.
1. Introduction
The polymethine carbocyanine dyes have been widely used as biological labeling reagents, fluorescence probes, and the imaging of biological targets in vivo, targeting in living cells and discriminating the dead cells from the living ones, due to their excellent photophysical properties.1–13 However, the small Stokes shift is a fatal fault for traditional cyanine dyes (<20 nm), which inevitably causes an inner filter effect, resulting in low fluorescence quantum yield and detection sensitivity.14 In addition, in order to avoid the above problem, Peng et al.15 developed a kind of aminocyanine with the amino substitution at the central position of Cy7. Surprisingly, the Stokes shift was enlarged from about 20 nm to more than 140 nm, which well avoided the shortcoming from scattering light and excited light. Furthermore, Peng et al.16 developed a trimethine cyanine dye with amino substitution in the meso position of the polymethine chain (Stokes shift 74 nm). Nagano and coworkers17 synthesized a series of IR-786 amine-substituted tricarbocyanines with a large Stokes' shift. They studied the photophysical perperties of these dyes and found that the electron-donating ability of the amine substituent affected the wavelength of the absorption maximum. Gallay et al.18 reported an N,N-dimethylamino substituted at β-position of Cy5 with the Stokes shift as 70 nm. Symmetric cyanine dyes that employ a (substituted) amino group along the polymethine chain show pronounced Stokes shifts, which is expected to be advantageous for many applications such as labels and probes.19–24 The mechanism for a large Stokes shift of amino substitution at the central position of Cy7 has been investigated by time-dependent density functional theory (TDDFT) at B3LYP/6-31G(d,p) level using Gaussian 09 software suite.25–27 However, the mechanism to enlarge the Stokes shift through the amino substitution at different positions of such dyes are still unknown. It is important to note that the recent literature on amino-substituted cyanine dyes neglects to discuss the role of amino group located at different positions along the polymethine chain in these compounds.The aim of the present work is to elucidate the photoprocesses effects of amino group located at different positions along the polymethine chain in indodicarbocyanine dyes. Theoretical simulations are recognized as a powerful tool which not only provide a complementary spectroscopic view on the measured data but also can guide the synthesis of the promising derivatives.28–31 We describe a series of aminocyanine dyes (Scheme 1) and proceed the system research on the influence of the amino group on the spectral properties, geometry optimizations, orbital transition and the potential energy curves by DFT and TDDFT method. Moreover, understanding these features is not only understand the effects of amino group on large Stokes shift and charge transfer process, but can even lead to the development of future aminocyanine design.
 | ||
| Scheme 1 Geometric structures for a series of aminocyanine dyes (see text for further details). | ||
2. Computational details
To gain a good theoretical understanding of the photoprocesses effects of amino group located at different positions along the polymethine chain, optical properties, geometry optimization, molecular orbital and potential energy curves with reactive coordinates are necessary. The molecular geometries were investigated using density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations, respectively.32 Becke's three-parameter hybrid exchange functions with Lee–Yang–Parr gradient-corrected correlation functional (B3LYP functional) is more suited to be in good accordance with the experimental results, and used in both the DFT and TD-DFT methods in the sequential work.33 The 6-31G(d,p) basis set was chosen as the basis set throughout, which is an appropriate basis set for such organic compounds. Moreover, electronic excitation energies of low-lying electronically excited states were also computed by the TDDFT method.34 A continuum solvation model, PCM, was used for the consideration of solvent effects in aqueous solution.35 In addition, the S0 potential energy curves were qualitatively scanned by constrained optimizations with keeping the dihedral angels fixed at a series of values. The excited-state potential energy curves were obtained by calculating the Franck–Condon transition energies for the ground-state optimized structures at fixed dihedral angles using TDDFT method. All calculations on electronic structures were carried out using the Gaussian 09 program suite.363. Results and discussion
3.1. Optical properties
To study the amino effect at different positions along the polymethine chain in indodicarbocyanine dyes, we design some new aminocyanine dyes including Cy3-NH2-1, Cy3-NH2-2, Cy5-NH2-1, Cy5-NH2-2, Cy5-NH2-3, Cy7-NH2-1, Cy7-NH2-2, Cy7-NH2-3, Cy7-NH2-4, Cy9-NH2-1, Cy9-NH2-2, Cy9-NH2-3, Cy9-NH2-4 and Cy9-NH2-5 (Scheme 1). Firstly, we optimized ground state and the first excited state geometric structures and calculated the absorption and emission spectra of all the molecules using the TD-DFT/B3LYP/6-31G(d,p) method with the PCM solvation model (data in Table 1). The local minima only have real frequencies. As shown in Table 1, all the molecules have large Stokes shifts. Based on absorption maxima and emission maxima, the effects of amino group located at different positions on spectral properties of the dyes were presented and discussed. The introduction of the amino group in the odd position of the polymethinic chain causes a redshift of the absorption maximum by 60–90 nm with respect to of the amino group in the even position of the polymethinic chain (Table 1). The aminocyanine dyes with the amino group in the odd position display larger Stokes shifts than the even position. For instance, the absorption maximum, the emission maxima and Stokes shifts of Cy7-NH2-1 and Cy7-NH2-3 are larger than Cy7-NH2-2 and Cy7-NH2-4. Similar results have also been obtained from other molecules including amino-substituted Cy3, Cy5, and Cy9. Our attention was eventually attracted by the amino group in different position with different background spectral properties. This prompted additional calculations to better understand the effects of the amino group on the geometry optimizations, orbital transition and the potential energy curves in the ground state and the first excited state.| abs (nm) | em (nm) | Stokes shift (nm) | |
|---|---|---|---|
| Cy3-NH2-1 | 507 | Dark state | — |
| Cy3-NH2-2 | 446 | 543 | 97 |
| Cy5-NH2-1 | 565 | 765 | 200 |
| Cy5-NH2-2 | 503 | 603 | 100 |
| Cy5-NH2-3 | 576 | 720 | 144 |
| Cy7-NH2-1 | 625 | 833 | 208 |
| Cy7-NH2-2 | 564 | 678 | 114 |
| Cy7-NH2-3 | 639 | 817 | 178 |
| Cy7-NH2-4 | 556 | 670 | 114 |
| Cy9-NH2-1 | 684 | 906 | 222 |
| Cy9-NH2-2 | 621 | 758 | 137 |
| Cy9-NH2-3 | 699 | 908 | 209 |
| Cy9-NH2-4 | 613 | 745 | 132 |
| Cy9-NH2-5 | 705 | 917 | 212 |
3.2. Optimized structures in the ground state and the first excited state
Some of the most important structural parameters are shown in Fig. 1. For Cy3-NH2-1, the length of C–N bond of amino group is 1.418 Å which is close to the normal C–N single bond. However, in the structure Cy3-NH2-2, the length of C–N bond of amino group is 1.346 Å and is shorter than normal C–N single bond but similar to that of CN double bond. For Cy5-NH2-1, the length of C–N bond of amino group is 1.419 Å, which is close to the bond length of Cy3-NH2-1. For Cy5-NH2-2, the length of C–N bond of amino group is 1.346 Å and is similar to the bond length of Cy3-NH2-2. For Cy5-NH2-3, the length of C–N bond of amino group is 1.402 Å and is between the length of C–N bond of Cy5-NH2-1 and Cy5-NH2-2. For Cy7-NH2-1, the length of C–N bond of amino group is 1.418 Å. For Cy7-NH2-2, the length of C–N bond of amino group is 1.350 Å. For Cy7-NH2-3, the length of C–N bond of amino group is 1.401 Å. For Cy7-NH2-4, the length of C–N bond of amino group is 1.349 Å. We found the length of C–N bond of amino group in the odd position of the polymethinic chain is longer than in the even position. Cy9 aminocyanine dyes have similar results. For Cy9-NH2-1, the length of C–N bond of amino group is 1.418 Å. For Cy9-NH2-2, the length of C–N bond of amino group is 1.349 Å. For Cy9-NH2-3, the length of C–N bond of amino group is 1.400 Å. For Cy9-NH2-4, the length of C–N bond of amino group is 1.350 Å. For Cy9-NH2-5, the length of C–N bond of amino group is 1.399 Å.
 | ||
| Fig. 1 The stable structures in the ground and calculated bond lengths of C–N of the aminocyanine dyes based on DFT//B3LYP/6-31G(d,p) basis set. | ||
Finally, based on optimized structures in the ground state, we found that the length of C–N bond of amino group in the even position is shorter than normal C–N single bond but close to that of CN double bond to prevent the conjugation of whole polymethine chain. However, the length of C–N bond of amino group in the odd position is close to than normal C–N single bond. The shorten length of C–N bond of amino group reduces the effective length of whole polymethine chain and further affects the maximum absorption and emission peak.
We further optimized the structures of the first excited state. The dyes transferred from ground state to the vertical excited state by excitation. Normally, the vertical excited state does not go back to ground state directly. The vertical excited state changes their configuration to get a new configuration with low energy as geometrical optimization of the first excited state S1, and then returns to the ground state through emitting long wavelength fluorescence. During this process, the configuration of amino group located at different positions along the polymethine chain takes evidently changes involved bond lengths, bond angles and dihedral angles of amino groups. So the comparisons about the bond lengths, bond angles and dihedral angles of amino groups between the ground state and the first excited state have been given in Table 2. About the structure of the first excited state Cy5-NH2-1, the bond length (C1–N1 bond) becomes obviously shorter and the bonding angles (C1–N1–H1, C1–N1–H2 and H1–N1–H2) get a little larger (three angles are increased by 5.78°, 5.31° and 5.50°, respectively) and the dihedral angles (H1–N1–C1–C2 and H2–N1–C1–C3) are decreased by 29.66° and 6.96°, respectively. And as respect to the structure of the first excited state Cy5-NH2-2, the bond length (C1–N1 bond) becomes obviously longer and the bonding angles (C1–N1–C2, H1–N1–C2 and H1–N1–C1) get slightly smaller (three angles are diminished by 5.22°, 5.69° and 5.16°, respectively) and the dihedral angles (H1–N1–C1–C2 and H2–N1–C1–C3) are increased by 29.27° and 21.50°, respectively. And for the structure of the first excited state Cy5-NH2-3, the bond length (C1–N1 bond) becomes obviously shorter and the bonding angles (C1–N1–C2, H1–N1–C2 and H1–N1–C1) get a little larger and both dihedral angles (C1–N1–C3–C4 and C2–N1–C3–C5) become shorter simultaneously (the two dihedral angles are decreased by 10.89° and 10.88°, respectively). The calculated bond lengths, bond angles and dihedral angles of Cy3, Cy7 and Cy9 aminocyanine dyes have the same varying trend to the computed results of Cy5 aminocyanine dyes, as shown in ESI part.†
| Dyes | Atomic number | S0 state | S1 state | |
|---|---|---|---|---|
| Cy5-NH2-1 | Bond length (Å) | N1–H1 | 1.016 | 1.011 |
| N1–H2 | 1.012 | 1.009 | ||
| N1–C1 | 1.419 | 1.382 | ||
| Bond angle (°) | H1–N1–C1 | 113.15 | 118.94 | |
| H2–N1–C1 | 112.89 | 118.21 | ||
| H1–N1–H2 | 109.19 | 114.68 | ||
| Dihedral angle (torsional angle) (°) | H1–N1–C1–C2 | 67.96 | 38.30 | |
| H2–N1–C1–C3 | 11.38 | 4.43 | ||
| Cy5-NH2-2 | Bond length (Å) | N1–H1 | 1.003 | 1.007 |
| N1–H2 | 1.007 | 1.012 | ||
| N1–C1 | 1.347 | 1.382 | ||
| Bond angle (°) | H1–N1–C1 | 122.50 | 117.28 | |
| H2–N1–C1 | 121.15 | 115.46 | ||
| H1–N1–H2 | 116.34 | 111.18 | ||
| Dihedral angle (torsional angle) (°) | H1–N1–C1–C2 | −8.54 | 20.74 | |
| H2–N1–C1–C3 | −7.76 | −29.26 | ||
| Cy5-NH2-3 | Bond length (Å) | N1–H1 | 1.013 | 1.009 |
| N1–H2 | 1.013 | 1.009 | ||
| N1–C1 | 1.402 | 1.372 | ||
| Bond angle (°) | C1–N1–C2 | 113.49 | 118.59 | |
| H1–N1–C2 | 113.49 | 118.59 | ||
| H1–N1–C1 | 108.42 | 113.13 | ||
| Dihedral angle (torsional angle) (°) | H1–N1–C1–C2 | 30.01 | 19.13 | |
| H2–N1–C1–C3 | −30.06 | −19.18 |
From the first excited-state structures of the three dyes, the amino groups in the polymethine chains are twisted out of the main plane owing to steric interaction, thus disturbing the planarity of the backbones of the main skeleton and creating a relatively distorted skeleton for the aminocyanine dyes. The first excited-state conformations were different from the ground-state structures, which would be significant in the study of large Stokes' shift.
3.3. Molecular orbital analysis
On the basis of calculation on Cy5 aminocyanine dyes shown in Fig. 2, for Cy5-NH2-2, the highest occupied molecular orbital (HOMO) is localized on the entire pentamethine chromophore including the end groups, but excluding the amino group substituent. The lowest unoccupied molecular orbital (LUMO) is localized predominantly on the actual pentamethine chain, including the amino group substituent. Alternatively, when dye molecule was excited, electron transfers from both side phenyl groups of the dye molecule Cy5-NH2-2 to the conjugated chain upon excitation, and the electron cloud density of LUMO on the amino groups substituent was increased corresponding to that of HOMO. It is demonstrated that the amino group acts as the electron acceptor at excited state, which is inverse to that for amino groups as electron donor in typical ICT process. This process can be called acceptor-ICT (photo-induced a-ICT). | ||
| Fig. 2 Frontier molecular orbitals corresponding to the ground-state S0 geometry (left), the first excited state S1 geometry (right) of the Cy5 aminocyanine dyes. The HOMO–LUMO gaps, absorption maxima, emission maxima and oscillator strengths for molecular orbitals are given. | ||
However, for Cy5-NH2-1 and Cy5-NH2-3, the highest occupied molecular orbital (HOMO) is localized on the entire pentamethine chromophore including the end groups and the amino group substituent. The lowest unoccupied molecular orbital (LUMO) is localized predominantly on the actual pentamethine chain, but excluding the amino group substituent. When the dye molecule was excited, the S0 → S1 transition is from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). The electron cloud density of LUMO on the amino groups substituent was decreased corresponding to that of HOMO. It is demonstrated that the amino group acts as the electron donor at excited state, which is accordance with that for amino groups as electron donor in typical ICT process.
Besides, geometrical optimization of the first excited state S1 was also performed. The S1 → S0 transition with the maximum oscillator strength is from the lowest unoccupied molecular orbital (LUMO) to the highest occupied molecular orbital (HOMO), corresponding to the maximum emission (in Fig. 2). For Cy5-NH2-2, the LUMO energy level is lowered 0.23 eV for the molecule in the S1 state compared with that in the S0 state; however the HOMO energy level is no change for the molecule in the S1 state compared with that in the S0 state. The amino group acted as the electron acceptor primarily destabilizes the LUMO energy level, leading to decrease the energy of the LUMO energy level. For Cy5-NH2-1, the LUMO energy level is lowered 0.15 eV for the molecules in the S1 state compared with that in the S0 state; however the HOMO energy level is raised 0.22 eV for the molecules in the S1 state compared with that in the S0 state. For Cy5-NH2-3, the LUMO energy level is lowered 0.08 eV for the molecules in the S1 state compared with that in the S0 state; however the HOMO energy level is raised 0.12 eV for the molecules in the S1 state compared with that in the S0 state. The amino group acted as the electron donor thus destabilizes the HOMO energy level, resulting in the increase of the HOMO energy level is greater than the decrease of the LUMO energy level. The HOMO is raised and the LUMO is lowered for the molecules in the S1 state compared with those in the S0 state, which leads to a clear decrease in the HOMO–LUMO gaps in the S1 state than in the S0 state, in agreement with the large Stokes shift.
We also get the same or similar results for Cy3, Cy7 and Cy9 aminocyanine dyes (see ESI part†). An important conclusion from this research is that the amino group acted as the electron acceptor at excited state (a-ICT) for aminocyanine dyes with amino group in the even position. While, on the contrary, the amino group is the electron donnor at excited state (ICT) for aminocyanine dyes with amino group in the odd position. The phenomena have not previously reported in the literature.
3.4. Nature of the different large Stokes shift
As above analysis demonstrated that, the difference between the first excited state structures and ground state structures and the electron transfers for the aminocyanine dyes both contribute to an enhanced Stokes shift, which is expected to exert a direct effect on the photophysical performance.It is noted that the aminocyanine dyes have different structures and the electron transfers of amino group located at different positions along the polymethine chain in indodicarbocyanine dyes. These features will produce different spectral characteristics; especially with different large Stokes shifts. Until recently, no reference has been published for the different large Stokes shifts of aminocyanine dyes, and the in-depth mechanism of the nature of the difference large Stokes shifts for these aminocyanine dyes are still unknown. Our previous research measured that the geometric distortion due to the rotation of the amino group and the skeleton of the polymethine chain produced large Stokes shift.37 To get a better understanding the nature of the different large Stokes shifts for the these dyes, we present a detailed study of the potential energy curves as a function of the amino group twisting angles of the these dyes.
Fig. 3 presents the calculated potential energy curves for rotation about amino group in the ground states and low-lying electronic excited states of Cy5-NH2-1, Cy5-NH2-2 and Cy5-NH2-3. For Cy5-NH2-1, the smallest energy gap (48.79 kcal mol−1 in Table 3) between ground state and the first excited state is ca. 0° twisted about the dihedral angle rotation, which provides the major nonradiative transition from an excited state to a ground state. The potential energy curves suggest that the rotation about the amino group is very easy, since there is lower energy barrier to rotation in the S1 state, especially from the angle of 70–0° with a very small energy barrier about 0.05 kcal mol−1. For Cy5-NH2-2, the smallest energy gap (53.16 kcal mol−1) between ground state and the first excited state is ca. 90° twisted about the dihedral angle rotation, which provides the major nonradiative transition from the first excited state to the ground state. The potential energy curves suggest that rotation about the central double bond is more difficult than Cy5-NH2-1, since there are higher energy barriers to rotation in the S1 state about 6.25 kcal mol−1. The barrier to rotation and the smallest energy gap between the first excited state and ground state for amino group of Cy5-NH2-3 is similar to these of the Cy5-NH2-1. Thus, for Cy5-NH2-1 and Cy5-NH2-3, the rotations of amino group are very easy by 0–70° with small energy barriers, which plays a major role in retuning the conforms of the first excited state and providing lager Stokes shift for aminocyanine dyes. For Cy5-NH2-2, the rotation of amino group is relatively difficult and is not conducive to the change of the conforms of the first excited state, resulting relatively smaller stokes shift than Cy5-NH2-1 and Cy5-NH2-3.
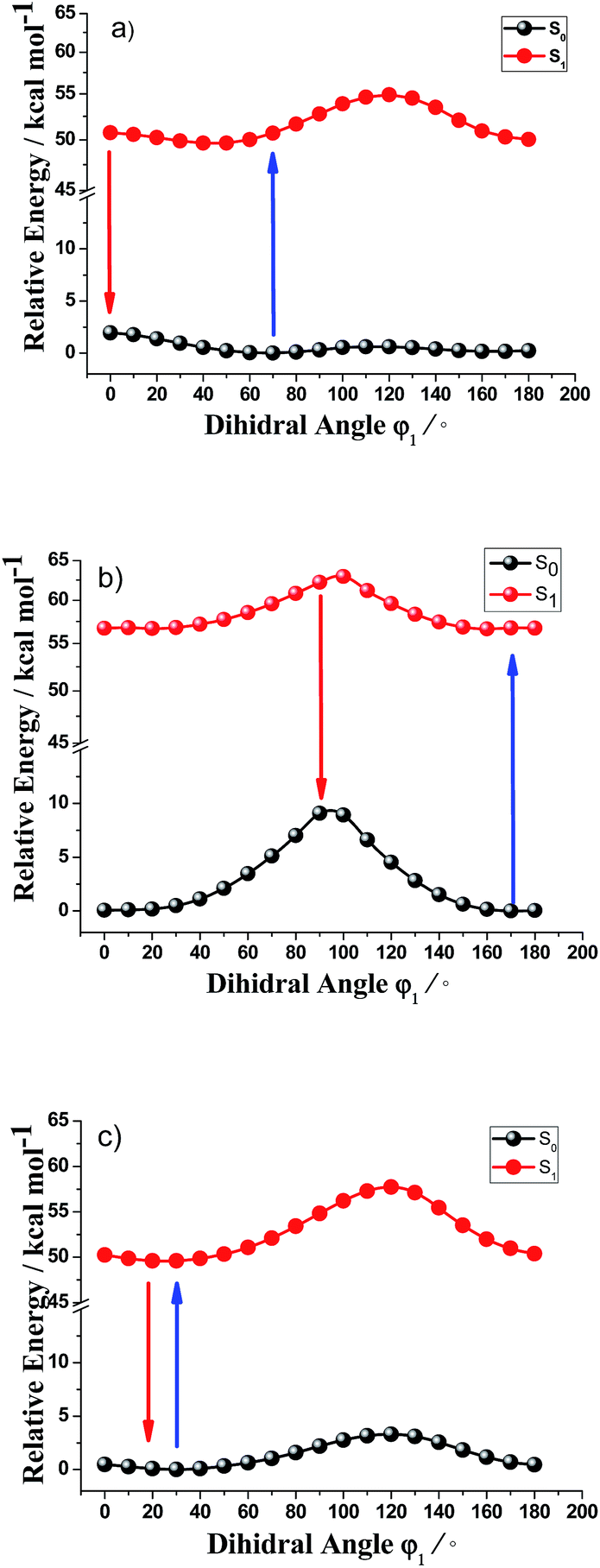 | ||
| Fig. 3 Energy levels of the S0 (black) and S1 (red) states of the amino group rotation around chemical bond C1–N1 (dihedral angle φ1) for Cy5-NH2-1 (a), Cy5-NH2-2 (b) and Cy5-NH2-3 (c) calculated at the TD-DFT/B3LYP/6-31G(d,p) level with PCM solvation model. | ||
| Cy5-NH2-1 φ1 | Cy5-NH2-2 φ1 | Cy5-NH2-3 φ1 | |
|---|---|---|---|
| Ea (kcal mol−1) | 0.05 | 6.20 | 0.01 |
| Egap (kcal mol−1) | 48.79 | 53.16 | 49.52 |
We calculated potential energy curves for different dihedral angles in the ground state and the first electronic excited states for Cy3, Cy7 and Cy9 aminocyanine dyes (see ESI part†). The energy barrier is 2.95 kcal mol−1 for Cy3-NH2-1 and 7.30 kcal mol−1 for Cy3-NH2-2. The energy barrier is 0.06 kcal mol−1 for Cy7-NH2-1, 7.30 kcal mol−1 for Cy7-NH2-2, 0.10 kcal mol−1 for Cy7-NH2-3 and 7.09 kcal mol−1 for Cy7-NH2-4. The energy barrier is 0.95 kcal mol−1 for Cy9-NH2-1, 7.65 kcal mol−1 for Cy9-NH2-2, 0.07 kcal mol−1 for Cy9-NH2-3, 6.56 kcal mol−1 for Cy9-NH2-4 and 0.01 kcal mol−1 for Cy3-NH2-5. Those calculations demonstrate that the rotations of amino group are very easy with small energy barriers for aminocyanine dyes with amino group in the odd position; nevertheless, the rotations of amino group are relatively difficult with higher energy barriers for aminocyanine dyes with amino group in the even position.
The excited-state barriers calculated in this work are slightly overestimated, due to the fact that the molecule was kept rigid except for rotation about a particular bond. But this strategy allows us to separate the effect of a single rotation from other rotations. It was found that allowing the molecule to relax along the remaining internal coordinates made little difference to the qualitative conclusions.38,39
4. Conclusions
In the framework of optimising the properties of aminocyanine dyes, the results showed that aminocyanine dyes with amino group in the odd position has commonality regarding the spectral properties, geometry optimizations, orbital transition and the potential energy curves. Similar results were found in the even position of such dyes. It was concluded that the spectral properties, geometry optimizations, orbital transition and the potential energy curves of aminocyanine dyes were highly depended on the amino group location along the polymethine chain. Such molecules being challenging targets for synthetic chemists, it is indeed of prime importance to guide the experimental efforts towards the most promising targets.Acknowledgements
This work was financially supported by the National Science Foundation of China (21606118 and 21601075), Natural Science Foundation of Liaoning Province (2015020249) and supported by the State Key Laboratory of Fine Chemicals (KF1614).Notes and references
- E. Sasaki, H. Kojima, H. Nishimatsu, Y. Urano, K. Kikuchi, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2005, 127, 3684–3685 CrossRef CAS PubMed.
- W. M. Leevy, S. T. Gammon, H. Jiang, J. R. Johnson, D. J. Maxwell, E. N. Jackson, M. Marquez, D. Piwnica-Worms and B. D. Smith, J. Am. Chem. Soc., 2006, 128, 16476–16477 CrossRef CAS PubMed.
- K. Kiyose, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2006, 128, 6548–6549 CrossRef CAS PubMed.
- C. Li, T. R. Greenwood, Z. M. Bhujwalla and K. Glunde, Org. Lett., 2006, 8, 3623–3626 CrossRef CAS PubMed.
- M. Zhu, M. Yuan, X. Liu, J. Xu, J. Lv, C. Huang, H. Liu, Y. Li, S. Wang and D. Zhu, Org. Lett., 2008, 10, 1481–1484 CrossRef CAS PubMed.
- B. Tang, H. Huang, K. Xu, L. Tong, G. Yang, X. Liu and L. An, Chem. Commun., 2006, 34, 3609–3611 RSC.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- Z. G. Yang, J. F. Cao, Y. X. He, J. H. Yang, T. Kim, X. J. Peng and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4563–4601 RSC.
- F. Yu, X. Han and L. Chen, Chem. Commun., 2014, 50, 12234–12249 RSC.
- F. Yu, P. Song, P. Li, B. Wang and K. Han, Analyst, 2012, 137, 3740–3749 RSC.
- F. Yu, P. Li, G. Li, G. Zhao, T. Chu and K. Han, J. Am. Chem. Soc., 2011, 133, 11030–11033 CrossRef CAS PubMed.
- S. Pascal, A. Haefele, C. Monnereau, A. Charaf-Eddin, D. Jacquemin, B. Le Guennic, C. Andraud and O. Maury, J. Phys. Chem. A, 2014, 118, 4038–4047 CrossRef CAS PubMed.
- V. Khimenko, A. K. Chibisov and H. Görner, J. Phys. Chem. A, 1997, 101, 7304–7310 CrossRef CAS.
- Z. Zhang and S. Achilefu, Org. Lett., 2004, 6, 2067–2070 CrossRef CAS PubMed.
- X. Peng, F. Song, E. Lu, Y. Wang, W. Zhou, J. Fan and Y. Gao, J. Am. Chem. Soc., 2005, 127, 4170–4171 CrossRef CAS PubMed.
- G. Chen, F. Song, X. Wang, S. Sun, J. Fan and X. Peng, Dyes Pigm., 2012, 93, 1532–1537 CrossRef CAS.
- K. Kiyose, S. Aizawa, E. Sasaki, H. Kojima, K. Hanaoka, T. Terai, Y. Urano and T. Nagano, Chem.–Eur. J., 2009, 15, 9191–9200 CrossRef CAS PubMed.
- O. Przhonska, M. Bondar, J. Gallay, M. Vincent, Y. Slominsky, A. Kachkovski and A. P. Demchenko, J. Photochem. Photobiol., B, 1999, 52, 19–29 CrossRef CAS.
- K. Kiyose, S. Aizawa, E. Sasaki, H. Kojima, K. Hanaoka, T. Terai, Y. Urano and T. Nagano, Chem.–Eur. J., 2009, 15, 9191–9200 CrossRef CAS PubMed.
- T. Myochin, K. Kiyose, K. Hanaoka, H. Kojima, T. Terai and T. Nagano, J. Am. Chem. Soc., 2011, 133, 3401–3409 CrossRef CAS PubMed.
- L. Yu, T. Li, Q. Wang, L. Li and L. Chen, Res. Chem. Intermed., 2014, 40, 1469–1481 CrossRef CAS.
- R. Gray, D. Walton, J. Bickerton, P. Richards and J. Heptinstall, Dyes Pigm., 1996, 30, 321–332 CrossRef CAS.
- R. Gray, D. Walton, J. Bickerton, P. Richards and J. Heptinstall, Dyes Pigm., 1998, 38, 97–105 CrossRef CAS.
- C. Ornelas, R. Lodescar, A. Durandin, J. W. Canary, R. Pennell, L. F. Liebes and M. Weck, Chem.–Eur. J., 2011, 17, 3619–3629 CrossRef CAS PubMed.
- E. K. U. Gross and W. Kohn, Phys. Rev. Lett., 1985, 55, 2850–2852 CrossRef CAS PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- A. Upadhyay, T. Bhatt, H. B. Tripathi and D. D. Pant, J. Photochem. Photobiol., A, 1995, 89, 201–207 CrossRef CAS.
- J. Cao, C. Hu, F. Liu, W. Sun, J. Fan, F. Song, S. Sun and X. Peng, ChemPhysChem, 2013, 14, 1601–1608 CrossRef CAS PubMed.
- J. F. Cao, C. Hu, W. Sun, Q. L. Xu, J. L. Fan, F. L. Song, S. G. Sun and X. J. Peng, RSC Adv., 2014, 4, 13385–13394 RSC.
- J. F. Cao, T. Wu, C. Hu, T. Liu, W. Sun, J. L. Fan and X. J. Peng, Phys. Chem. Chem. Phys., 2012, 14, 13702–13708 RSC.
- F. Liu, T. Wu, J. F. Cao, S. Cui, Z. G. Yang, X. X. Qiang, S. G. Sun, F. L. Song, J. L. Fan, J. Y. Wang and X. J. Peng, Chem.–Eur. J., 2013, 19, 1548–1553 CrossRef CAS PubMed.
- B. C. Popere, A. M. Della Pelle, A. Poe, G. Balaji and S. Thayumanavan, Chem. Sci., 2012, 3, 3093–3102 RSC.
- D. T. Nhan, N. K. Hien, H. Van Duc, N. T. Ai Nhung, N. T. Trung, D. U. Van, W. S. Shin, J. S. Kim and D. T. Quang, Dyes Pigm., 2016, 131, 301–306 CrossRef.
- M. Zhu, C. Shi, X. Xu, Z. Guo and W. Zhu, RSC Adv., 2016, 6, 100759–100764 RSC.
- H. Zhu, J. L. Fan, M. Li, J. F. Cao, J. Y. Wang and X. J. Peng, Chem.–Eur. J., 2014, 20, 4691–4696 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Nor-mand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, The Gaussian 09 package refer to Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. Cao, J. Fan, W. Sun, Z. Yang, C. Hu and X. Peng, Dyes Pigm., 2017, 141, 379–387 CrossRef CAS.
- X. Cao, R. W. Tolbert, J. L. McHale and W. D. Edwards, J. Phys. Chem. A, 1998, 102, 2739–2748 CrossRef CAS.
- G. L. Silva, V. Ediz, D. Yaron and B. A. Armitage, J. Am. Chem. Soc., 2007, 129, 5710–5718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04556e |
| This journal is © The Royal Society of Chemistry 2017 |