
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRigid NON-donor pincer ligand complexes of lutetium and lanthanum: synthesis and hydroamination catalysis†
Kelly S. A. Motolkoa,
David J. H. Emslie *a and
James F. Brittenb
*a and
James F. Brittenb
aDepartment of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, Ontario L8S 4M1, Canada. E-mail: emslied@mcmaster.ca; Fax: +1-905-522-2509; Tel: +1-905-525-9140 extn 23307
bMcMaster Analytical X-Ray (MAX) Diffraction Facility, Department of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, Ontario L8S 4M1, Canada
First published on 26th May 2017
Abstract
Reaction of H2XN2 {4,5-bis(2,4,6-triisopropylanilino)-2,7-di-tert-butyl-9,9-dimethylxanthene} with [Lu(CH2SiMe3)3(THF)2], and crystallization from O(SiMe3)2, yielded [(XN2)Lu(CH2SiMe3)(THF)]·(O(SiMe3)2)1.5 (1·(O(SiMe3)2)1.5). Lanthanum complexes of the XN2 dianion were also prepared by salt metathesis; treatment of H2XN2 with excess KH in DME produced the dipotassium salt, [K2(XN2)(DME)x] (x = 2–2.5), and subsequent reaction with [LaCl3(THF)3] afforded [{(XN2)LaCl(THF)}x]·(O(SiMe3)2)0.25x (2·(O(SiMe3)2)0.25x; x = 1 or 2) after crystallization from O(SiMe3)2. Compound 2 reacted with two equivalents of LiCH2SiMe3, to form the dialkyl-‘ate’ complex, [Li(THF)x][(XN2)La(CH2SiMe3)2]·Toluene·LiCl (3·toluene·LiCl; x = 3). Both 1 and 3 (x = 4) were structurally characterized, and were evaluated as catalysts for intramolecular hydroamination; while 3 showed poor activity, 1 is highly active for both intramolecular hydroamination and more challenging intermolecular hydroamination. Reactions with unsymmetrical alkenes yielded Markovnikov products, and the activity of 1 surpassed that of the previously reported yttrium analogue in the reaction of diphenylacetylene with 4-tert-butylbenzylamine.
1 Introduction
A variety of rare earth alkyl and amido complexes have demonstrated high activity for alkene and alkyne hydroamination catalysis, typically exceeding that achievable with transition metal catalysts.1 However, the ease with which hydroamination reactions occur varies greatly as a function of the substrate. In particular, intramolecular hydroamination reactions are much more facile than intermolecular variants, and intramolecular hydroamination reactions are generally more favorable for (a) alkynes vs. alkenes, (b) substrates leading to 5- vs. 6- vs. 7-membered ring formation, (c) 1-aminoalkenes and 1-aminoalkynes di-substituted at the 2-position (Thorpe–Ingold effect; bulkier phenyl substituents are more effective than methyl substituents), and (d) aminoalkenes without additional alkene substitution (e.g. H2NCH2CPh2CH2CR![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR′ where R = R′ = H).1–5 Intermolecular reactions with unactivated alkenes are particularly challenging, and only a handful of effective catalysts have been reported for these substrates, including rare earth ansa-cyclopentadienyl complexes (a in Fig. 1) developed by Marks et. al.,6,7 yttrium complexes of both bidentate and tridentate 1,1′-binaphthalene-backbone ligands (b and c in Fig. 1) reported by Hultzsch,8,9 and an yttrium complex of a rigid xanthene-backbone NON-donor pincer ligand (d in Fig. 1) reported recently by the Emslie group.10
CHR′ where R = R′ = H).1–5 Intermolecular reactions with unactivated alkenes are particularly challenging, and only a handful of effective catalysts have been reported for these substrates, including rare earth ansa-cyclopentadienyl complexes (a in Fig. 1) developed by Marks et. al.,6,7 yttrium complexes of both bidentate and tridentate 1,1′-binaphthalene-backbone ligands (b and c in Fig. 1) reported by Hultzsch,8,9 and an yttrium complex of a rigid xanthene-backbone NON-donor pincer ligand (d in Fig. 1) reported recently by the Emslie group.10
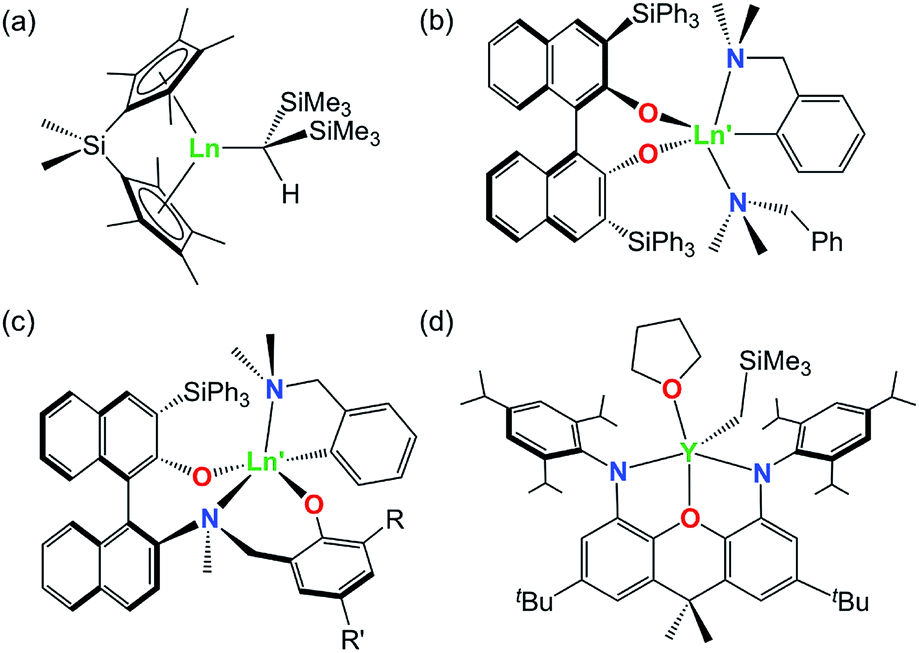 | ||
| Fig. 1 Highly active rare earth catalysts for intermolecular hydroamination of unactivated alkenes (Ln = Nd; Ln′ = Y). | ||
For trivalent rare earth catalyzed alkene hydroamination, catalytic activity typically increases in parallel with metal ionic radius. For example, in cyclization reactions with H2CCHCH2CMe2CH2NH2, the activity of [ Ln{CH(SiMe3)2}], [{Me2Si(C5Me4)2}Ln{CH(SiMe3)2}] (b in Fig. 1) and [(L)Ln{C6H4(CH2NMe2)-o}] (c in Fig. 1; R = SiPh3, R′ = Me) increased in the order (a) Lu < Sm < La,11,12 (b) Lu < Sm < Nd,13 and (c) Sc < Lu < Y,9 respectively. Similarly, with E-PhHCCHCH2CPh2CH2NH2 as the substrate, the activity of [{(Ind)(CH2)2N(o-C6H10)NMe2}Ln{N(SiMe3)2}] (Ind = 1-indenyl) increased in the order Sc < Lu < Y < Sm,14 and for H2CCHCH2CPh2CH2NH2 cyclization at 60 °C, [{OC6H3(o-tBu)(o-CHNAr)}2Ln(CH2SiMe2Ph)] (Ar = C6H3iPr2-2,6) was inactive for Sc, but active for Y.15,16 By contrast, for alkyne hydroamination, this trend is reversed, and higher activity is commonly observed for smaller rare earth ions. As an example, the activity of [
Ln{CH(SiMe3)2}], [{Me2Si(C5Me4)2}Ln{CH(SiMe3)2}] (b in Fig. 1) and [(L)Ln{C6H4(CH2NMe2)-o}] (c in Fig. 1; R = SiPh3, R′ = Me) increased in the order (a) Lu < Sm < La,11,12 (b) Lu < Sm < Nd,13 and (c) Sc < Lu < Y,9 respectively. Similarly, with E-PhHCCHCH2CPh2CH2NH2 as the substrate, the activity of [{(Ind)(CH2)2N(o-C6H10)NMe2}Ln{N(SiMe3)2}] (Ind = 1-indenyl) increased in the order Sc < Lu < Y < Sm,14 and for H2CCHCH2CPh2CH2NH2 cyclization at 60 °C, [{OC6H3(o-tBu)(o-CHNAr)}2Ln(CH2SiMe2Ph)] (Ar = C6H3iPr2-2,6) was inactive for Sc, but active for Y.15,16 By contrast, for alkyne hydroamination, this trend is reversed, and higher activity is commonly observed for smaller rare earth ions. As an example, the activity of [ Ln{CH(SiMe3)2}] for HC
Ln{CH(SiMe3)2}] for HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(CH2)3NH2 cyclization increased in the order La < Nd < Sm < Lu.17 However, these general trends are not always followed; for H2CCHCH2CMe2CH2NH2 cyclization, the activity of [Ln{N(SiMe3)2}3] grafted onto partially hydroxylated periodic mesoporous silica increased in the order Nd < La < Y.18 Additionally, for intermolecular hydroamination of Ph(CH2)2CHCH2 with benzylamine, yttrium and lutetium [(L)Ln{C6H4(CH2NMe2)-o}] (c in Fig. 1; R = R′ = tBu) catalysts9 showed comparable activity, while the lanthanum analogue was nearly inactive.19–29
C(CH2)3NH2 cyclization increased in the order La < Nd < Sm < Lu.17 However, these general trends are not always followed; for H2CCHCH2CMe2CH2NH2 cyclization, the activity of [Ln{N(SiMe3)2}3] grafted onto partially hydroxylated periodic mesoporous silica increased in the order Nd < La < Y.18 Additionally, for intermolecular hydroamination of Ph(CH2)2CHCH2 with benzylamine, yttrium and lutetium [(L)Ln{C6H4(CH2NMe2)-o}] (c in Fig. 1; R = R′ = tBu) catalysts9 showed comparable activity, while the lanthanum analogue was nearly inactive.19–29
As noted above, we previously described the synthesis and hydroamination activity of [(XN2)Y(CH2SiMe3)(THF)] (XN2 = 4,5-bis(2,4,6-triisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene; d in Fig. 1),10 and in this work we report the synthesis of lutetium and lanthanum XN2 complexes. These complexes were targeted in order to probe the effectiveness of the rigid XN2 pincer ligand to support robust alkyl derivatives of smaller and larger rare earth elements, and to assess the activity of the complexes for alkene and alkyne hydroamination.
2 Results and discussion
The alkane elimination reaction between H2XN2 and [Lu(CH2SiMe3)3(THF)2] yielded [(XN2)Lu(CH2SiMe3)(THF)]·(O(SiMe3)2)1.5 (1·(O(SiMe3)2)1.5) in 59% yield, after recrystallization from O(SiMe3)2 (Scheme 1). By contrast, the analogous reaction between H2XN2 and [Sc(CH2SiMe3)3(THF)2] in benzene at 24 °C was unsuccessful, resulting only in unreacted proligand and [Sc(CH2SiMe3)3(THF)2] thermal decomposition products. The 1H NMR spectrum of 1 is largely analogous to that of [XN2Y(CH2SiMe3)(THF)], which we previously reported,10 and is indicative of Cs symmetry in solution, with two Ar–H, ortho-CHMe2 and CMe2 peaks. Compound 1 was found to be fairly thermally stable, being only 25% decomposed after 24 h at 90 °C.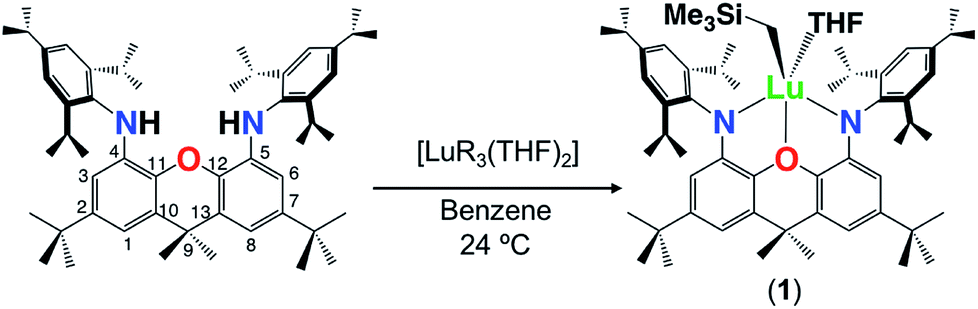 | ||
| Scheme 1 Synthesis of lutetium complex 1 (R = CH2SiMe3). | ||
Crystals of 1·(C6H6)0.5 (lattice solvent is residual benzene from the synthesis) were grown by cooling a concentrated O(SiMe3)2 solution to −30 °C (Fig. 2). In the solid state, the XN2 backbone is slightly bent with a 26° angle away from planarity, based on the orientation of the two xanthene aryl rings. This orientation is mirrored in [(XN2)Y(CH2SiMe3)(THF)], which displayed a backbone angle of 25° and a very similar geometry at the rare earth metal.10 Lutetium is 5-coordinate with the three anionic donors and coordinated THF arranged in an approximate tetrahedron around the metal center. The largest angle in this approximate tetrahedron is the N(1)–Lu–N(2) angle of 130°, and the smallest is the O(2)–Lu–C(54) angle of 97°, while the other angles are between 102° and 110°. The oxygen donor of the xanthene backbone is coordinated on the N(1)/N(2)/C(54) face of the tetrahedron closest to the nitrogen donors. Lutetium lies 0.74 Å out of the plane of the XN2 ligand donor atoms, leading to a 53° angle between the NON and the NLuN planes. The neutral oxygen donor on the xanthene backbone is located 0.5 Å out of the N(1)/C(4)/C(5)/N(2) plane in order to coordinate to lutetium, with N–Lu–O(1) angles of 69 and 70°. Additionally, the nitrogen donors on the XN2 ligand are bent towards lutetium, illustrated by the C(1)⋯C(8), C(4)⋯C(5) and N(1)⋯N(2) distances of 5.02 Å, 4.57 Å and 4.04 Å respectively, which are comparable with those in [(XN2)Y(CH2SiMe3)(THF)] (4.98 Å, 4.56 Å and 4.06 Å).10
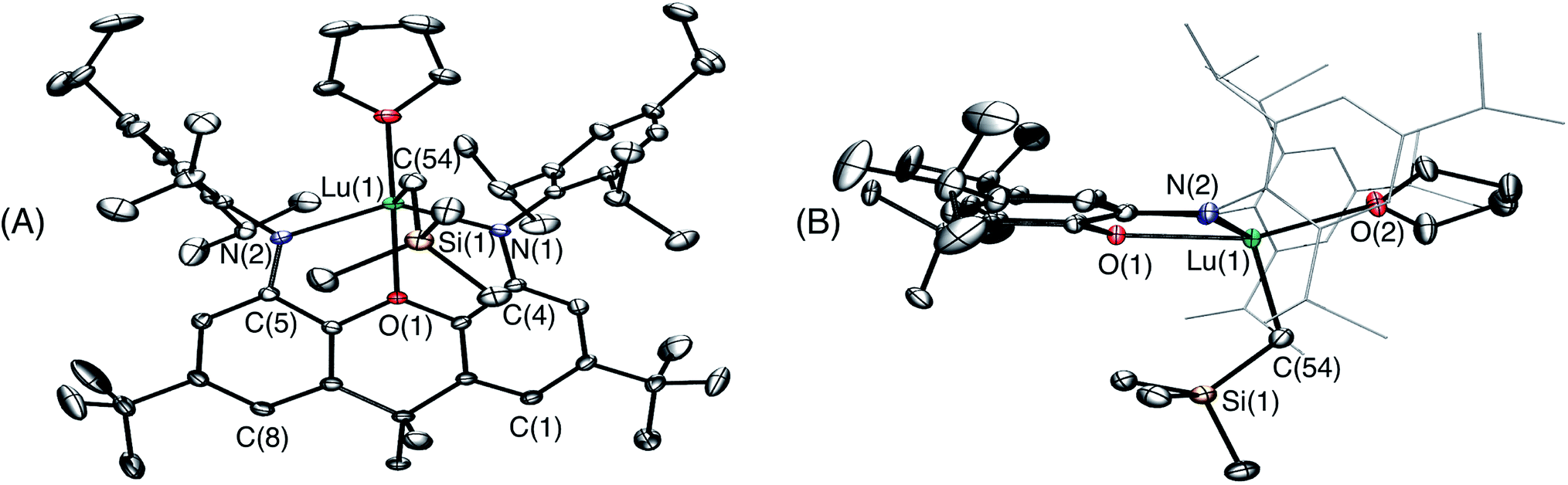 | ||
| Fig. 2 Two views of the X-ray crystal structure for 1·(C6H6)0.5. Ellipsoids are set to 50%. Hydrogen atoms and lattice solvent are omitted. The tert-butyl groups are rotationally disordered over two positions, and only one is shown for clarity. In view B the 2,4,6-triisopropylphenyl groups are depicted in wire-frame format. Selected bond lengths [Å] and angles [°]: Lu–N(1) 2.221(2), Lu–N(2) 2.228(2), Lu–C(54) 2.326(2), Lu–O(1) 2.299(1), Lu–O(2) 2.264(1), N(1)–Lu–N(2) 130.41(6), N(1)–Lu–C(54) 106.09(7), N(2)–Lu–C(54) 110.41(7), O(1)–Lu–C(54) 104.38(6), O(2)–Lu–C(54) 97.56(7), O(2)–Lu–N(2) 105.39(6), O(2)–Lu–N(1) 101.76(6). | ||
The Lu–N distances of 2.221(2) Å and 2.228(2) Å are slightly shorter than those in [(XN2)Y(CH2SiMe3)(THF)] (2.252(3) Å),10 consistent with the smaller ionic radius of LuIII compared to YIII (0.861 Å vs. 0.900 Å).30 Additionally, the Lu–N distances in 1 fall within the range reported for related compounds such as [{(2-ArNCMe)(6-ArNCMe2)C5H3N}Lu(CH2SiMe3)2] (Ar = C6H3iPr2-2,6; 2.188(4) Å)31 and [{1,8-(PziPr)2Cz}Lu(CH2SiMe3)2] (PziPr = 1-(3-isopropyl)pyrazolyl; Cz = 3,6-dimethylcarbazole; 2.231(3) Å).32 The Lu–C(54) distance of 2.326(2) Å is shorter compared to that in [(XN2)Y(CH2SiMe3)(THF)] (2.364(3) Å), also in keeping with the relative sizes of yttrium and lutetium (vide supra). Additionally, this distance falls within the range of Lu–C distances reported in the literature. For example, the Lu–C distances in the aforementioned alkyl complexes range from 2.329(6) Å to 2.374(3) Å,31,32 and Lu–C in [{(2-NAr)(6-Xyl)C5H3N}2Lu(CH2SiMe3)(THF)] (Ar = C6H3iPr2-2,6; Xyl = o-xylyl) is 2.323(14) Å.33
[(XN2)Lu(CH2SiMe3)(THF)]·(O(SiMe3)2)1.5 (1·(O(SiMe3)2)1.5) was tested as an ethylene polymerization catalyst at 24 °C and 80 °C (toluene, 1 atm ethylene, 1 h) but exhibited negligible activity. Compound 1 was also investigated as a catalyst for both intra- and inter-molecular hydroamination and the results are summarized in Tables 1 and 2. Compound 1 catalyzed intramolecular hydroamination of a range of substrates in benzene at 24 °C, proceeding to >99% completion in all cases. The time required to reach >99% completion was slightly increased compared to reactions catalyzed by the yttrium complex, [(XN2)Y(CH2SiMe3)(THF)], which is consistent with the majority of previous reports (vide supra), in which hydroamination activity increases with increasing rare earth metal size.30 This is particularly evident in entries 2 and 4 in Table 1, as [(XN2)Y(CH2SiMe3)(THF)] achieved >99% conversion after 1.5 h and 34 h,10 whereas 1 required 2.75 h and 48 h respectively. Nevertheless, the ability of 1 and the yttrium analogue to catalyze these more challenging intramolecular hydroamination reactions at room temperature stands these catalysts apart from most others.10
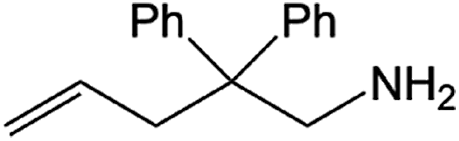
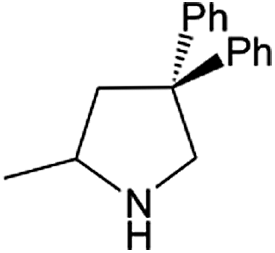
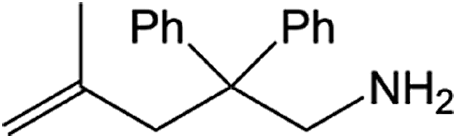
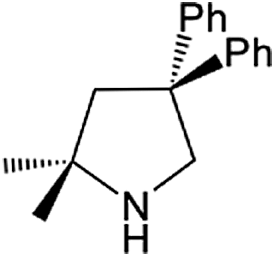
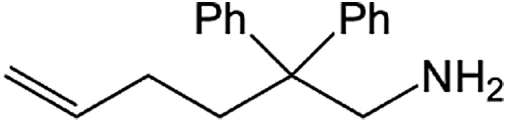
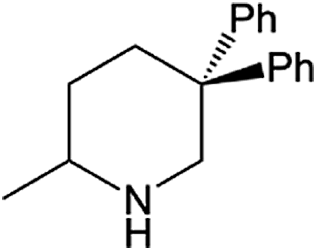

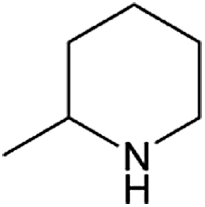
| Entry | Amine | Alkene or alkynea | Product | Time | Temp. (°C) | Product formationb,c | % Markovnikov productc | Ntd (h−1) |
|---|---|---|---|---|---|---|---|---|
a Alkene/alkyne present in 20 fold excess relative to the amine.b Conversion determined by product: unreacted amine ratio.c Determined by GC-MS.d Turnover frequency.e In entry 4 the product is formed as a single isomer, whereas in entries 5 and 6 the products are formed as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.35 and 1:0.24 mixtures of the E and Z isomers (based on literature assignments for similar compounds),35,36 respectively. :0.35 and 1:0.24 mixtures of the E and Z isomers (based on literature assignments for similar compounds),35,36 respectively. |
||||||||
| 1 | 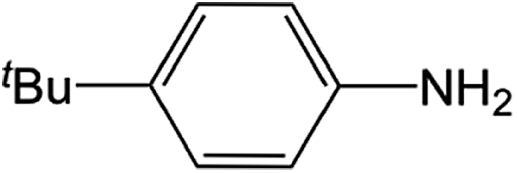 |
 |
 |
24 h | 110 | 11% | 97 | 0.04 |
| 2 | 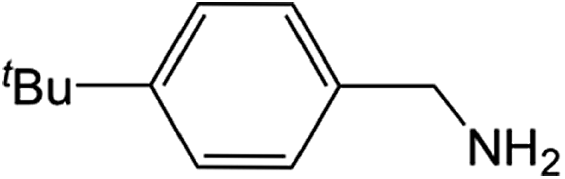 |
 |
 |
24 h | 110 | 83% | >99 | 0.35 |
| 3 | 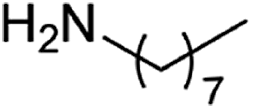 |
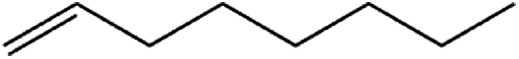 |
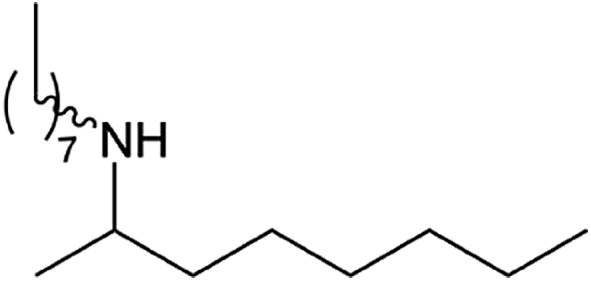 |
24 h | 110 | 98% | >99 | 0.41 |
| 4 | 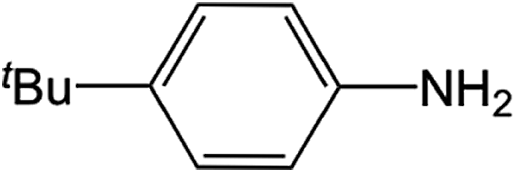 |
 |
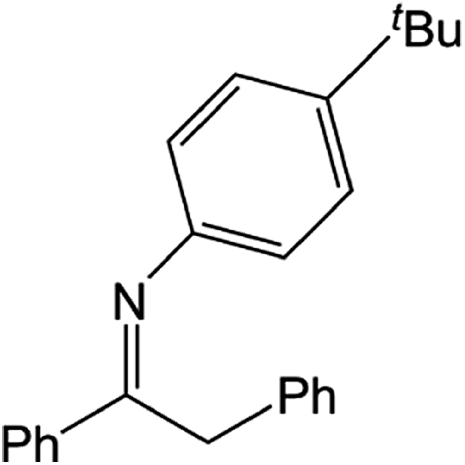 |
24 h | 110 | >99%e | N/A | 0.42 |
| 5 | 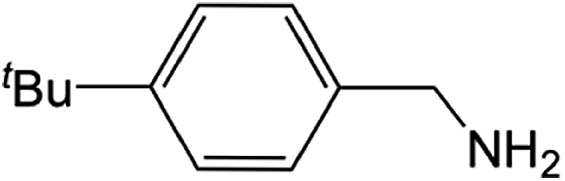 |
 |
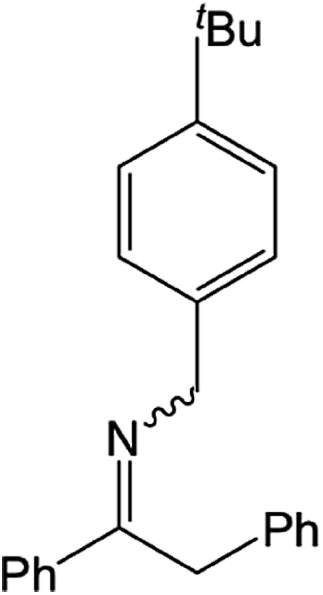 |
24 h | 110 | >99%e | N/A | 0.42 |
| 6 | 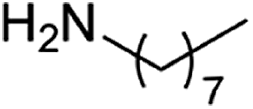 |
 |
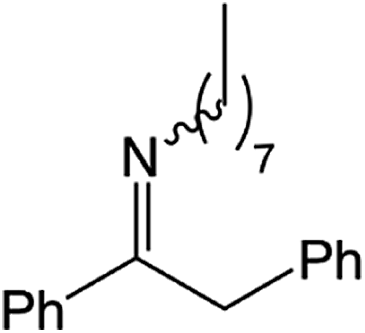 |
24 h | 110 | >99%e | N/A | 0.42 |
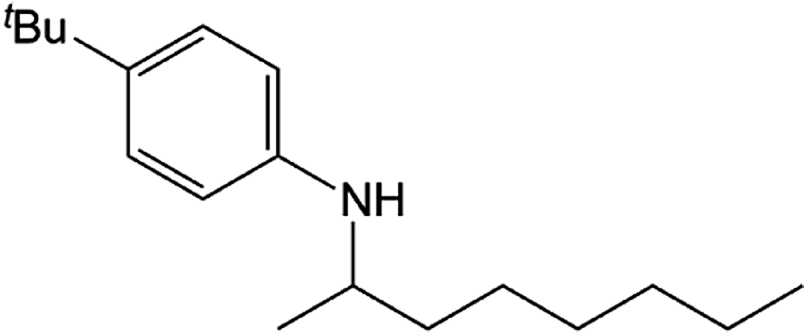
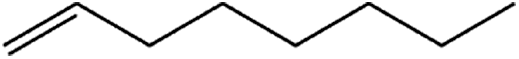
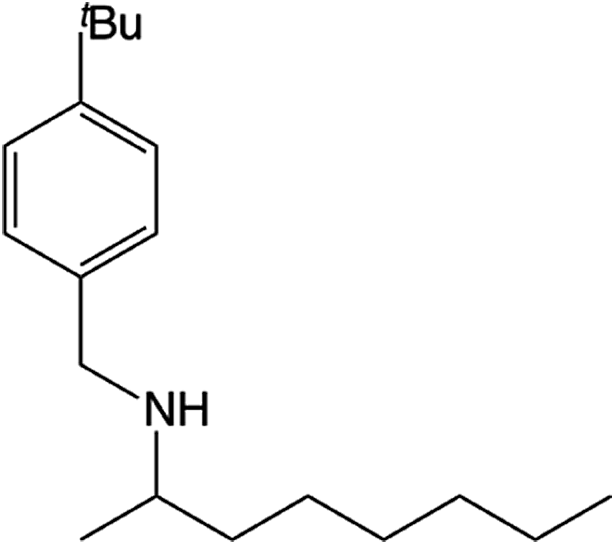
Compound 1 also catalyzed intermolecular hydroamination with 4-tert-butylaniline, 4-tert-butylbenzylamine and octylamine in combination with 1-octene and diphenylacetylene, and in all reactions with 1-octene, the Markovnikov product was formed selectively. These reactions were performed in toluene at 110 °C and the degree of conversion was determined by GC-MS (Table 2). Over a 24 h time period, the reaction of 1-octene with octylamine (entry 3) resulted in a turnover frequency (Nt) of 0.41 h−1, which is greater than that obtained for the reaction with 4-tert-butylbenzylamine (entry 2, 0.35 h−1), which in turn is significantly greater than that obtained for the reaction with 4-tert-butylaniline (entry 1, 0.04 h−1). These results are consistent with the increased donor ability and reduced steric bulk of the former amines. The same trend was previously observed for [(XN2)Y(CH2SiMe3)(THF)],10 and the ability of 1 to catalyze intermolecular hydroamination of 1-octene (an unactivated alkene) places it in a select group of catalysts with this capability (vide supra). The intermolecular hydroamination activity of 1 closely mirrors that of the yttrium analogue, although for entries 2 and 5 in Table 2, compound 1 afforded lower and higher activities, respectively (Nt = 0.35 vs. 0.40 and 0.42 vs. 0.33). The intermolecular reactions with the largest conversions after 24 h at 110 °C (10 mol% catalyst) were those utilizing diphenylacetylene (entries 4–6), as the amounts of unreacted 4-tert-butylaniline, 4-tert-butylbenzylamine and octylamine were below the detection limit of the GC instrument.
In order to further explore the effectiveness of the XN2 ligand for rare earth coordination, and the impact of metal ionic radius on hydroamination activity, the synthesis of lanthanum XN2 complexes was undertaken. As the lanthanum trialkyl compound, [La(CH2SiMe3)3(THF)x] is not readily accessible,34 salt metathesis was employed for ligand attachment in the place of alkane elimination. Stirring H2XN2 with excess KH in DME at 24 °C produced the dipotassium salt of the 4,5-bis(2,4,6-triisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene ligand, [K2(XN2)(DME)x] (x = 2–2.5), as a beige solid in 80% isolated yield. [K2(XN2)(DME)2] was reacted with [LaCl3(THF)3] in THF at 24 °C, and after recrystallization from O(SiMe3)2, [{(XN2)LaCl(THF)}x]·(O(SiMe3)2)0.25x (2·(O(SiMe3)2)0.25x; x = 1 or 2) was obtained in 51% yield as an off-white solid (Scheme 2).
 | ||
| Scheme 2 Synthesis of [K2(XN2)(DME)x] and complexes 2 and 3. | ||
Attempts to synthesize a lanthanum monoalkyl complex were undertaken via reactions of trimethylsilylmethyl lithium and methyl lithium with [{(XN2)LaCl(THF)}x]·(O(SiMe3)2)0.25x (2·(O(SiMe3)2)0.25x; x = 1 or 2). NMR-scale reactions were performed in d8-THF at 24 °C and both resulted in the formation of a dialkyl-‘ate’ complex, based on the integrations of the respective alkyl peaks, regardless of whether one equivalent (per La) or an excess of the alkali metal-alkyl reagent was added. The reaction utilizing trimethylsilylmethyl lithium was pursued further, as it provides a direct comparison with the lutetium and yttrium trimethylsilylmethyl complexes of the XN2 ligand. [{(XN2)LaCl(THF)}x]·(O(SiMe3)2)0.25x (2·(O(SiMe3)2)0.25x; x = 1 or 2) reacted with 2 equivalents of trimethylsilylmethyl lithium in THF at 24 °C, and after removal of the salts by centrifugation in toluene and layering with hexanes at −30 °C, [Li(THF)x][(XN2)La(CH2SiMe3)2]·toluene·LiCl (3·toluene·LiCl; x = 3) was isolated as a pale yellow solid in 55% yield (Scheme 2). Compound 3 is sparingly soluble in benzene and other non-polar solvents such as hexanes and pentane, so all characterization was carried out in d8-THF. The 1H NMR spectrum of 3 revealed the expected signals for the XN2 ligand backbone and only one set of signals for the two alkyl groups was observed between 25 and −80 °C (a singlet with an integration of 18 for LaCH2SiMe3 and a singlet with an integration of 4 for LaCH2SiMe3). Crystals of [Li(THF)4][(XN2)La(CH2SiMe3)2]·THF were grown from a concentrated THF solution of 3·toluene·LiCl (x = 3), layered with pentane and cooled to −30 °C (Fig. 3).
 | ||
| Fig. 3 Two views of the X-ray crystal structure for 3·THF (x = 4). Ellipsoids are set to 50%. Hydrogen atoms and lattice solvent are omitted. The tert-butyl groups are rotationally disordered over two positions, and only one is shown for clarity. In view A the [Li(THF)4]+ cation is omitted for clarity. In view B the 2,4,6-triisopropylphenyl groups and the THF molecules are depicted in wire-frame format. Selected bond lengths [Å] and angles [°]: La–N(1) 2.462(5), La–N(2) 2.445(5), La–C(54) 2.573(7), La–C(58) 2.613(7), La–O(1) 2.643(4), N(1)–La–N(2) 118.22(17), N(1)–La–C(54) 108.9(2), N(1)–La–C(58) 100.9(2), N(2)–La–C(54) 106.3(2), N(2)–La–C(58) 120.7(2), O(1)–La–C(54) 97.63(18), O(1)–La–C(58) 159.7(2), C(54)–La–C(58) 100.0(2), N(1)–La–O(1) 63.59(15), N(2)–La–O(1) 62.72(15). | ||
In the solid state, lanthanum is 5-coordinate with the two amido donors and two alkyl groups arranged in a distorted tetrahedron around the metal center. The largest angle in this approximate tetrahedron is the N(1)–La–N(2) angle of 118°, and the smallest is the C(54)–La–C(58) angle of 100°, while the other angles are between 101° and 106°. Lanthanum lies 0.96 Å out of the plane of the XN2 ligand donor atoms, leading to a 50° angle between the NON and NLaN planes. The XN2 backbone is bent with a 35° angle away from planarity, based on the orientation of the two aryl rings of the xanthene backbone. The neutral oxygen donor of the xanthene backbone is located 0.64 Å out of the N(1)/C(4)/C(5)/N(2) plane in order to coordinate to lanthanum, resulting in N–La–O(1) angles of 63–64°. In addition, it is of note that the nitrogen donors on the ligand are not bent towards lanthanum to the extent that they are in 1, illustrated by the C(1)⋯C(8), C(4)⋯C(5) and N(1)⋯N(2) distances in 3 of 4.90 Å, 4.57 Å and 4.21 Å respectively, compared to 5.02 Å, 4.57 Å and 4.04 Å in 1.
The La–N distances of 2.462(5) Å and 2.445(5) Å are substantially lengthened relative to those in [(XN2)Y(CH2SiMe3)(THF)] (2.252(3) Å)10 and [(XN2)Lu(CH2SiMe3)(THF)]·(O(SiMe3)2)1.5 (1) (2.221(2) Å and 2.228(2) Å), consistent with the large ionic radius of lanthanum compared to yttrium and lutetium, combined with increased steric hindrance and an overall negative charge in 3, resulting in a less electrophilic metal centre. For analogous reasons, the La–C(54) and La–C(58) distances of 2.573(7) Å and 2.613(7) Å are also greatly elongated compared to those in [(XN2)Y(CH2SiMe3)(THF)] (2.364(3) Å)10 and 1 (2.326(2) Å). However, both the La–N and La–C distances in 3 are significantly shorter than those previously reported for [{(R)-Binap(NCyp)2}La{(μ-Me)2Li(THF)}{(μ-Me)Li(THF)}2] (Binap = 2,2′-disubstituted-1,1′-binaphthyl; Cyp = cyclopentyl; La–N = 2.626(7)–2.677(8) Å; La–C = 2.704(8)–2.832(11) Å).29,37
Reaction of [Li(THF)3][(XN2)La(CH2SiMe3)2]·toluene·LiCl (3·toluene·LiCl) with [{(XN2)LaCl(THF)}x]·(O(SiMe3)2)0.25x (2·(O(SiMe3)2)0.25x; x = 1 or 2) did not provide access to the neutral XN2 lanthanum alkyl; at 24 °C no reaction was observed, and heating to 70 °C resulted only in thermal decomposition of 3·toluene·LiCl.
Rare earth alkyl-‘ate’ complexes have been reported to catalyze intramolecular hydroamination as well as intermolecular hydroamination. A few examples include, [{(R)-Binap(NCyp)2}La{(μ-Me)2Li(THF)}{(μ-Me)Li(THF)}2] which catalyzed asymmetric intramolecular hydroamination of amino-1,3-dienes,29 [Li(THF)4][{(R)-Binap(NCyp)2}Y(CH2SiMe3)2] (Binap = 2,2′-disubstituted-1,1′-binaphthyl; Cyp = cyclopentyl) which catalyzed intramolecular hydroamination of secondary aminoalkenes,38 [Li(THF)4][{ArNC(Me)C(Me)NAr}Y(CH2SiMe3)2] (Ar = C6H3iPr2-2,6) which catalyzed the intermolecular hydroamination reaction of styrene and pyrrolidine,39 and [{(R)-Binap(NCyp)2}Y{(μ-Me)2Li(THF)2}{(μ-Me)Li(THF)}] (Binap = 2,2′-disubstituted-1,1′-binaphthyl; Cyp = cyclopentyl) which catalyzed 1-amino-2,2-diphenyl-4-pentene cyclization, requiring 1.9 h at 25 °C with 6 mol% catalyst loading to reach 100% conversion.37 Complex 3·toluene·LiCl, (x = 3) was tested as a catalyst for intramolecular hydroamination with 1-amino-2,2-diphenyl-4-pentene in d8-THF at 24 °C. However, the time required to reach >99% completion (45 h) was significantly increased compared to that required for [(XN2)Y(CH2SiMe3)(THF)] and [(XN2)Lu(CH2SiMe3)(THF)] (1·(O(SiMe3)2)1.5) (<10 min) under analogous conditions in benzene (or in THF for the yttrium complex). Consequently, the catalytic activity of 3 was not further investigated.
3 Summary and conclusion
The rigid NON-donor pincer ligand, XN2, has been employed for the synthesis of a lutetium monoalkyl complex (1), a lanthanum chloride complex (2) and a lanthanum dialkyl-‘ate’ complex (3). Complex 3 was tested as a catalyst for intramolecular hydroamination, but showed low activity. By contrast, the neutral lutetium alkyl complex, 1, is highly active for both intra- and inter-molecular hydroamination with a variety of substrates. For intramolecular alkene hydroamination, the time required to reach >99% completion was slightly increased compared to the previously reported yttrium analogue. By contrast, the intermolecular hydroamination reaction between 4-tert-butylbenzylamine and diphenylacetylene afforded a higher turnover number than the yttrium analogue.4 Experimental
General details
An argon-filled MBraun UNIlab glove box equipped with a −30 °C freezer was employed for the manipulation and storage of all air-sensitive compounds, and reactions were performed on a double manifold high vacuum line using standard techniques.40 Residual oxygen and moisture was removed from the argon stream by passage through an Oxisorb-W scrubber from Matheson Gas Products. A Fisher Scientific Ultrasonic FS-30 bath was used to sonicate reaction mixtures where indicated. A VWR Clinical 200 Large Capacity Centrifuge (with 28° fixed-angle rotors that hold 12 × 15 mL or 6 × 50 mL tubes) in combination with 15 mL Kimble Chase glass centrifuge tubes was used when required (inside the glovebox). Diethyl ether, THF, toluene, benzene and hexanes were initially dried and distilled at atmospheric pressure from Na/Ph2CO. Hexamethyldisiloxane (O(SiMe3)2) was dried and distilled at atmospheric pressure from Na. Unless otherwise noted, all proteo solvents were stored over an appropriate drying agent (pentane, hexanes, hexamethyldisiloxane (O(TMS)2/O(SiMe3)2) = Na/Ph2CO/tetra-glyme; Et2O = Na/Ph2CO) and introduced to reactions via vacuum transfer with condensation at −78 °C. The deuterated solvents (ACP Chemicals) C6D6, THF-d8 and toluene-d8 were dried over Na/Ph2CO.The H2XN2,10 [Lu(CH2SiMe3)3(THF)2],41 and the commercially unavailable intramolecular hydroamination reagents10,42 were prepared according to literature procedures. 1-Amino-5-hexene was purchased from GFS Chemicals, dried over CaH2 and distilled prior to use. 1,3,5-Triisopropylbenzene, xanthone, KH (30 wt% in mineral oil), LiCH2SiMe3 (1.0 M in pentane), MeLi (1.6 M in Et2O), nBuLi (1.6 M in hexanes), Br2, NaH, NaOtBu, Pd(OAc)2, DPEPhos, [bis{2-(diphenylphosphino)phenyl}ether], diphenylacetylene and MgSO4 were purchased from Sigma-Aldrich. LuCl3 and LaCl3 were purchased from Strem Chemicals. Solid LiCH2SiMe3 and MeLi were obtained by removal of the solvent in vacuo, and solid KH was obtained by filtration and washing with hexanes. [LuCl3(THF)3] and [LaCl3(THF)3] were obtained by refluxing the anhydrous lutetium/lanthanum trihalide in THF for 24 h followed by removal of the solvent in vacuo. 4-tert-Butyl-aniline, 4-tert-butylbenzylamine, n-octylamine and 1-octene were purchased from Sigma-Aldrich, dried over molecular sieves and distilled prior to use. Argon (99.999% purity) and ethylene (99.999% purity) were purchased from Praxair.
Combustion elemental analyses were performed both at McMaster University on a Thermo EA1112 CHNS/O analyzer and by Midwest Microlab, LLC, Indianapolis, IN, USA. NMR spectroscopy (1H, 13C{1H}, DEPT-Q, COSY, HSQC, HMBC) was performed on Bruker DRX-500 and AV-600 spectrometers. All 1H NMR and 13C NMR spectra were referenced relative to SiMe4 through a resonance of the employed deuterated solvent or proteo impurity of the solvent; C6D6 (7.16 ppm), d8-Tol (2.08, 6.97, 7.01, 7.09 ppm), d8-THF (1.72, 3.58 ppm) for 1H NMR; and C6D6 (128.0 ppm), d8-Tol (20.43, 125.13, 127.96, 128.87, 137.48 ppm), d8-THF (25.31, 67.21 ppm) for 13C NMR. Herein, numbered proton and carbon atoms refer to the positions of the xanthene backbone, as shown in Scheme 1. Inequivalent ortho isopropyl protons are labeled A and B, while inequivalent aryl ring protons and inequivalent methyl protons are labeled ′ and ′′, so that the corresponding carbon resonances can be identified.
X-ray crystallographic analyses were performed on suitable crystals coated in Paratone oil and mounted on a SMART APEX II diffractometer with a 3 kW sealed tube Mo generator in the McMaster Analytical X-ray (MAX) Diffraction Facility. In all cases, non-hydrogen atoms were refined anisotropically and hydrogen atoms were generated in ideal positions and then updated with each cycle of refinement. GC-MS analyses were performed using an Agilent 6890N gas chromatograph (Santa Clara, CA, USA), equipped with a DB-17ht column (30 m × 0.25 mm i.d. × 0.15 μm film, J & W Scientific) and a retention gap (deactivated fused silica, 5 m × 0.53 mm i.d.), and coupled to an Agilent 5973 MSD single-quadrupole mass spectrometer. One microliter of sample was injected using Agilent 7683 autosampler in splitless mode. The injector temperature was 230 °C and carrier gas (helium) flow was 0.7 mL min−1. The transfer line was 280 °C and the MS source temperature was 230 °C. The column temperature started at 50 °C and was raised to 300 °C at 8 °C min−1. It was then held at 300 °C for 15 min to give a total run time of 46.25 min. Full scan mass spectra between m/z 50 and 800 were acquired after a five minute solvent delay.
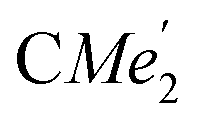 ), 1.72 (s, 3H,
), 1.72 (s, 3H, 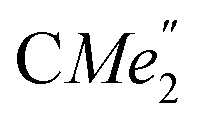 ), 1.49 (d, 6H, 3JH,H 6.73 Hz, A-ortho
), 1.49 (d, 6H, 3JH,H 6.73 Hz, A-ortho -), 1.47 (d, 6H, 3JH,H 6.73 Hz, A-ortho-
-), 1.47 (d, 6H, 3JH,H 6.73 Hz, A-ortho-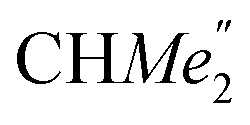 ), 1.26 (s, 18H, CMe3), 1.24 (d, 6H, 3JH,H 6.78 Hz, B-ortho-
), 1.26 (s, 18H, CMe3), 1.24 (d, 6H, 3JH,H 6.78 Hz, B-ortho-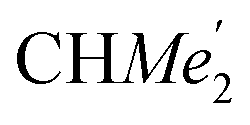 ), 1.21 (m, 12H, para-CHMe2), 1.01 (d, 6H, 3JH,H 6.78 Hz, B-ortho-
), 1.21 (m, 12H, para-CHMe2), 1.01 (d, 6H, 3JH,H 6.78 Hz, B-ortho-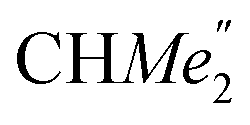 ), 0.83 (s, 4H, 1 equiv. THF-C3,4H2), 0.35 (s, 9H, LuCH2SiMe3), −0.40 (s, 2H, LuCH2SiMe3). 13C NMR (C6D6, 126 MHz): δ 147.83 (Xanth-C4), 147.72 (Xanth-C2) 147.38 (A-ortho-CCHMe2), 146.17 (B-ortho-CCHMe2), 145.33 (para-CCHMe2), 141.82 (Ar-Cipso), 141.30 (Xanth-C11), 130.56 (Xanth-C10), 122.20 (Ar-CH′), 121.85 (Ar-CH′′), 109.35 (Xanth-C3H), 106.47 (Xanth-C1H), 70.85 (THF-C2,5H2), 37.99 (LuCH2SiMe3), 35.57 (Xanth-C9Me2), 35.25 (
), 0.83 (s, 4H, 1 equiv. THF-C3,4H2), 0.35 (s, 9H, LuCH2SiMe3), −0.40 (s, 2H, LuCH2SiMe3). 13C NMR (C6D6, 126 MHz): δ 147.83 (Xanth-C4), 147.72 (Xanth-C2) 147.38 (A-ortho-CCHMe2), 146.17 (B-ortho-CCHMe2), 145.33 (para-CCHMe2), 141.82 (Ar-Cipso), 141.30 (Xanth-C11), 130.56 (Xanth-C10), 122.20 (Ar-CH′), 121.85 (Ar-CH′′), 109.35 (Xanth-C3H), 106.47 (Xanth-C1H), 70.85 (THF-C2,5H2), 37.99 (LuCH2SiMe3), 35.57 (Xanth-C9Me2), 35.25 (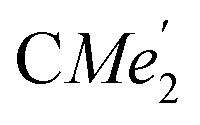 ), 35.05 (CMe3), 34.50 (para-CHMe2), 31.91 (CMe3), 28.39 (B-ortho-CHMe2), 27.67 (A-ortho-CHMe2), 27.12 (A-ortho-
), 35.05 (CMe3), 34.50 (para-CHMe2), 31.91 (CMe3), 28.39 (B-ortho-CHMe2), 27.67 (A-ortho-CHMe2), 27.12 (A-ortho-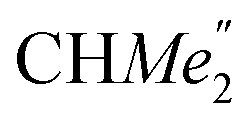 ), 25.85 (B-ortho-
), 25.85 (B-ortho-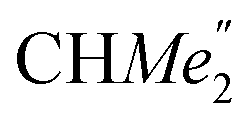 ), 25.48 (B-ortho-
), 25.48 (B-ortho- ), 25.04 (
), 25.04 (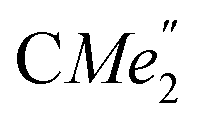 ), 24.85 (THF-C3,4H2), 24.62 (A-ortho-
), 24.85 (THF-C3,4H2), 24.62 (A-ortho-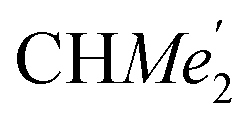 ), 24.51 (para-CHMe2), 4.13 (LuCH2SiMe3). Anal. calcd for C70H120N2O3.5LuSi4: C, 63.07; H, 9.07; N, 2.10%. Found: C, 63.03; H, 8.61; N, 2.30%.
), 24.51 (para-CHMe2), 4.13 (LuCH2SiMe3). Anal. calcd for C70H120N2O3.5LuSi4: C, 63.07; H, 9.07; N, 2.10%. Found: C, 63.03; H, 8.61; N, 2.30%.General procedure for intramolecular hydroamination
In the glove box, the appropriate amounts of 1 or 3 and the hydroamination substrate were weighed into separate vials, dissolved in C6D6 or d8-THF and placed in a teflon-valved J-young NMR tube. The reactions were monitored at 24 °C by 1H NMR spectroscopy and the expected products were confirmed by their agreement to reported literature spectra.42General procedure for intermolecular hydroamination
In the glove box, the appropriate amounts of 1, amine, and the alkene/alkyne were weighed into separate vials, dissolved in d8-toluene, placed in a Teflon-valved J-young NMR tube and then placed into a preheated oil bath at 110 °C. After heating for the designated amount of time, NMR spectra were obtained and the sample was submitted for analysis by GC-MS.Acknowledgements
D. J. H. E. thanks NSERC of Canada for a Discovery Grant and an NSERC Strategic Grant in collaboration with NOVA Chemicals. K. S. A. M. thanks the Government of Ontario for an OGS – Queen Elizabeth II Graduate Scholarship in Science and Technology (QEII GSST) scholarship. The authors would like to thank Dr Kirk Green and Dr Fan Fei from the McMaster Regional Centre of Mass Spectrometry (MRCMS) for acquisition and analysis of all the mass spectra.References
- A. L. Reznichenko and K. C. Hultzsch, Top. Organomet. Chem., 2011, 43, 51 CrossRef.
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed.
- A. L. Reznichenko, A. J. Nawara-Hultzsch and K. C. Hultzsch, Top. Curr. Chem., 2014, 343, 191 CrossRef CAS PubMed.
- A. A. Trifonov, I. V. Basalov and A. A. Kissel, Dalton Trans., 2016, 45, 19172 RSC.
- J.-S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584 CrossRef CAS PubMed.
- Y. Li and T. J. Marks, Organometallics, 1996, 15, 3770 CrossRef CAS.
- A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed.
- A. L. Reznichenko and K. C. Hultzsch, Organometallics, 2013, 32, 1394 CrossRef CAS.
- K. S. A. Motolko, D. J. H. Emslie and H. A. Jenkins, Organometallics, 2017, 36, 1601–1608 CrossRef CAS.
- M. R. Gagné and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 4109 CrossRef.
- M. R. Gagné, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275 CrossRef.
- S. Tian, V. M. Arredondo, C. L. Stern and T. J. Marks, Organometallics, 1999, 18, 2568 CrossRef CAS.
- Z. Chai, J. Chu, Y. Qi, M. Tang, J. Hou and G. Yang, RSC Adv., 2017, 7, 1759 RSC.
- F. Lauterwasser, P. G. Hayes, S. Bräse, W. E. Piers and L. L. Schafer, Organometallics, 2004, 23, 2234 CrossRef CAS.
- F. Lauterwasser, P. G. Hayes, W. E. Piers, L. L. Schafer and S. Bräse, Adv. Synth. Catal., 2011, 353, 1384 CrossRef CAS.
- Y. Li and T. J. Marks, J. Am. Chem. Soc., 1996, 118, 9295 CrossRef.
- E. L. Roux, Y. Liang, M. P. Storz and R. Anwander, J. Am. Chem. Soc., 2010, 132, 16368 CrossRef PubMed.
- For other recent examples of Lu and La hydroamination catalysts see 20–29.
- T. Spallek and R. Anwander, Dalton Trans., 2016, 45, 16393 RSC.
- T. S. Brunner, P. Benndorf, M. T. Gamer, N. Knöfel, K. Gugau and P. W. Roesky, Organometallics, 2016, 35, 3474 CrossRef CAS.
- A. Otero, A. Lara-Sánchez, J. A. Castro-Osma, I. Márquez-Segovia, C. Alonso-Moreno, J. Fernández-Baeza, L. F. Sánchez-Barba and A. M. Rodríguezc, New J. Chem., 2015, 39, 7672 RSC.
- K. Huynh, B. K. Anderson and T. Livinghouse, Tetrahedron Lett., 2015, 56, 3658 CrossRef CAS.
- K. Huynh, T. Livinghouse and H. M. Lovick, Synlett, 2014, 25, 1721 CrossRef.
- A. G. Trambitas, D. Melcher, L. Hartenstein, P. W. Roesky, C. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 6753 CrossRef CAS PubMed.
- A. Otero, A. Lara-Sánchez, C. Nájera, J. Fernández-Baeza, I. Márquez-Segovia, J. A. Castro-Osma, J. Martínez, L. F. Sánchez-Barba and A. M. Rodríguez, Organometallics, 2012, 31, 2244 CrossRef CAS.
- P. Benndorf, J. Jenter, L. Zielke and P. W. Roesky, Chem. Commun., 2011, 47, 2574 RSC.
- D. V. Vitanova, F. Hampel and K. C. Hultzsch, J. Organomet. Chem., 2011, 696, 321 CrossRef CAS.
- I. Aillaud, C. Olier, Y. Chapurina, J. Collin, E. Schulz, R. Guillot, J. Hannedouche and A. Trifonov, Organometallics, 2011, 30, 3378 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- T. M. Cameron, J. C. Gordon, R. Michalczyk and B. L. Scott, Chem. Commun., 2003, 2282 RSC.
- K. R. D. Johnson, B. L. Kamenz and P. G. Hayes, Organometallics, 2014, 33, 3005 CrossRef CAS.
- S. Qayyum, G. G. Skvortsov, G. K. Fukin, A. A. Trifonov, W. P. Kretschmer, C. Döring and R. Kempe, Eur. J. Inorg. Chem., 2010, 248 CrossRef CAS.
- S. Bambirra, F. Perazzolo, S. J. Boot, T. J. J. Sciarone, A. Meetsma and B. Hessen, Organometallics, 2008, 27, 704 CrossRef CAS.
- H. Ahlbrecht and S. Fischer, Tetrahedron, 1970, 26, 2837 CrossRef CAS.
- I. A. Tonks, J. C. Meier and J. E. Bercaw, Organometallics, 2013, 32, 3451 CrossRef CAS.
- I. Aillaud, D. Lyubov, J. Collin, R. G. J. Hannedouche, E. Schulz and A. Trifonov, Organometallics, 2008, 27, 5929 CrossRef CAS.
- C. Queffelec, F. Boeda, A. Pouilhès, A. Meddour, C. Kouklovsky, J. Hannedouche, J. Collin and E. Schulz, ChemCatChem, 2011, 3, 122 CrossRef CAS.
- A. A. Kissel, T. V. Mahrova, D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, I. D. Rosal and L. Maron, Dalton Trans., 2015, 44, 12137 RSC.
- B. J. Burger and J. E. Bercaw, Vacuum Line Techniques for Handling Air-Sensitive Organometallic Compounds, in Experimental Organometallic Chemistry: A Practicum in Synthesis and Characterization, American Chemical Society, Washington, D.C., 1987, vol. 357, p. 79 Search PubMed.
- F. Estler, G. Eickerling, E. Herdtweck and R. Anwander, Organometallics, 2003, 22, 1212 CrossRef CAS.
- M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for new compounds, and NMR and mass spectra for hydroamination reaction products. CCDC 1544724 and 1544725. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04432a |
| This journal is © The Royal Society of Chemistry 2017 |