
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, in vitro and in vivo evaluation of tacrine–cinnamic acid hybrids as multi-target acetyl- and butyrylcholinesterase inhibitors against Alzheimer's disease†
Yao Chenabc,
Hongzhi Line,
Jie Zhue,
Kai Gue,
Qi Lie,
Siyu Hee,
Xin Lue,
Renxiang Tanb,
Yuqiong Peia,
Liang Wub,
Yaoyao Bian*d and
Haopeng Sun *e
*e
aSchool of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, 210023, China
bState Key Laboratory Cultivation Base for TCM Quality and Efficacy, Nanjing University of Chinese Medicine, Nanjing, 210023, China
cJiangsu Key Laboratory for Functional Substance of Chinese Medicine, Nanjing University of Chinese Medicine, Nanjing, 210023, China
dSchool of Nursing, Nanjing University of Chinese Medicine, Nanjing, 210023, China. E-mail: 1691@163.com; Tel: +86-15952007562
eDepartment of Medicinal Chemistry, China Pharmaceutical University, Nanjing, 210009, China. E-mail: sunhaopeng@163.com; Tel: +86-25-85863169
First published on 5th July 2017
Abstract
Previously, we have reported tacrine–ferulic acid hybrids as multi-target cholinesterase inhibitors against Alzheimer's disease. However, the detailed structure–activity relationship (SAR), especially regarding the ferulic acid moiety, has yet to be elucidated. Herein we report the structural modification of the ferulic acid moiety, which is replaced by cinnamic acid with different substitutions. The target compounds are synthesized and evaluated for their in vitro cholinesterase inhibitory activities, inhibition of amyloid β-protein self-aggregation, cyto-protective effects against hydrogen peroxide and antiproliferative activity in PC-12 cells. The optimal compounds 35 and 36 are subsequently selected for in vivo assays. 36 shows much better performance in ameliorating the scopolamine-induced cognition impairment and less hepatotoxicity than tacrine. The compound serves as a good lead compound for further optimization.
Introduction
Alzheimer's disease (AD) is the most prevalent form of late-life mental failure in humans and it affects about 6% of the population aged over 65.1 It is estimated that more than 18 million people presently suffer from AD, and the number is predicted to sharply increase to 70 million by 2050.2 The cardinal features of AD include progressive memory impairment, disordered cognitive function, altered behavior such as depression, hallucination, delusion, and agitation, and a progressive decline in language function.3 So far, it is well accepted that AD is a multifactorial syndrome deriving from a complex array of neurochemical factors. During the process of AD, cholinergic neurons and synapses of the basal forebrain are selectively lost, causing cognitive impairment.4 These findings inspired several theories about AD pathogenesis, including cholinergic dysfunction,5 amyloid cascade,6 hyperphosphorylation of τ-protein,7 cell cycle hypothesis,8 and brain-derived neurotrophic factor hypothesis.9 Additionally, oxidative stress,10 free radical formation,11 metal dyshomeostasis,12 and mitochondrial dysfunction,13 are also reported to be tightly correlated to the development of AD by supplying an inflammatory micro-environmental condition. These theories increase understanding of the basic mechanism of AD, and, also depict a more complex AD scenario.The cholinergic hypothesis of the pathogenesis of AD asserts that dysfunction of cholinergic system, mainly decline of acetylcholine (ACh) level, results in the cognitive and memory deficits. Therefore, recovering cholinergic function is believed to be beneficial for the treatment of AD.14 Generally, ACh can be hydrolyzed by two types of cholinesterases (ChEs), namely acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). Although the elucidation of the pathophysiology of AD provides multiple potential drug targets for designing effective drugs, acetylcholinesterase inhibitors (AChEIs) still serve as the main therapeutic agents applied clinically for AD.
The enzymatic site of human AChE is a narrow gorge with a length of approximate 20 Å, which contains two binding sites: the catalytic active site (CAS) at the bottom and the peripheral anionic site (PAS) near the entrance of the gorge.15,16 CAS is in charge of the hydrolysis of ACh and is consisted of key residues including Ser203, Glu334, and His447, which are referred to as the catalytic triad.17 PAS is composed of several aromatic residues such as Trp86, Trp286.18 It has been proved that PAS is closely related to both hydrolysis of ACh and neurotoxic cascade of AD through AChE-induced β-amyloid (Aβ) aggregation.19 Under normal condition, AChE is more active and can hydrolyze about 80% of ACh in human brains.20 However, both the level and the activity of AChE in AD patients are found to be remarkably reduced, leading to the compensative upregulation of BuChE, which further modulates the ACh levels.21 Therefore, inhibitors of both AChE and BuChE, such as tacrine and rivastigmine, are expected to exert potent therapeutic effect on AD. Unfortunately, instead of curing or preventing the neurodegeneration, these drugs can only enable a palliative treatment.22 Considering the multifactorial nature of AD, the traditional agents designed by one-molecule one-target approach is insufficient to provide enough benefits. Thus, designing compounds that can simultaneously regulate multiple significant targets in the development of AD, has emerged as a new strategy. These compounds, which are referred to as multi-target-directed ligands (MTDLs),23 are considered to offer additional properties other than cholinesterase inhibition. Substantial studies have been performed to achieve different types of MTDLs, many of which have been proved to show promising pharmacological effects on AD.24–39 These results encourage medicinal chemists to continue this work. In recent years, designing MTDLs based on tacrine has attracted the attentions of medicinal chemists throughout the world and numerous related publications are disclosed to describe the efforts on this field. Compared to other AChEIs, tacrine is a good scaffold for the design of MTDLs due to its simple structure and high ligand efficiency (LE), which means tacrine can potently inhibit AChE with small number of non-hydrogen atoms. Moreover, tacrine has a good endurance against substantial structural modification while retaining the target-based activity, further provides a sound basis for the design of MTDLs. However, there is no newly approved small molecular agent for the treatment of AD in recent years, and most of the MTDLs remain at the stage of preclinical study. Therefore, it is still urgently needed for us to design new MTDLs and to fully understand the structural requirement through detailed structure–activity relationship (SAR) study.
Our group has been dedicated to the discovery of new MTDLs for nearly a decade. Previously, Fang L. et al.40 and Chen Y. et al.41 have disclosed a series of tacrine–ferulic acid hybrids as multifunctional potent ChEs inhibitors, most of which effectively inhibited ChEs in vitro in nanomolar range. These compounds were also proved to exert multiple functions, including antioxidant activities, vasorelaxation effects, and NO-donating behavior. In vivo studies by using the scopolamine-induced cognition impairment mouse model confirmed that these compounds can ameliorate the cognitive impairment and reduce the hepatotoxicity compared to the reference compound tacrine. These studies provided us promising lead compounds for further research. However, structural modification, especially on the ferulic acid moiety, is still limited and needs to be further elucidated. To deepen the understanding of the structural requirement for tacrine–ferulic acid hybrids, here we report the structural modification of ferulic acid moiety, which is replaced by cinnamic acid with different substitutions. The target compounds are synthesized and evaluated for their in vitro and in vivo activities related to the treatment of AD, including in vitro assays for cholinesterase catalytic activity, Aβ1–42 self-aggregation, cyto-protective effects against hydrogen peroxide and antiproliferative activity in PC-12 cells. Additionally, we also report the in vivo behavioral and hepatotoxic evaluations for the optimal compound selected from in vitro assays. Based on these results, we hope to supply more useful information of structure–activity relationship (SAR) that can guide further discovery of new MTDLs against AD.
Results and discussions
Compound design and chemistry
Compound CY-1, which was previously reported by our group, was used as the lead compound for structural modification.41 The ferulic acid moiety of CY-1 was replaced by cinnamic acid with various substitutions at different positions of the phenyl ring (Fig. 1).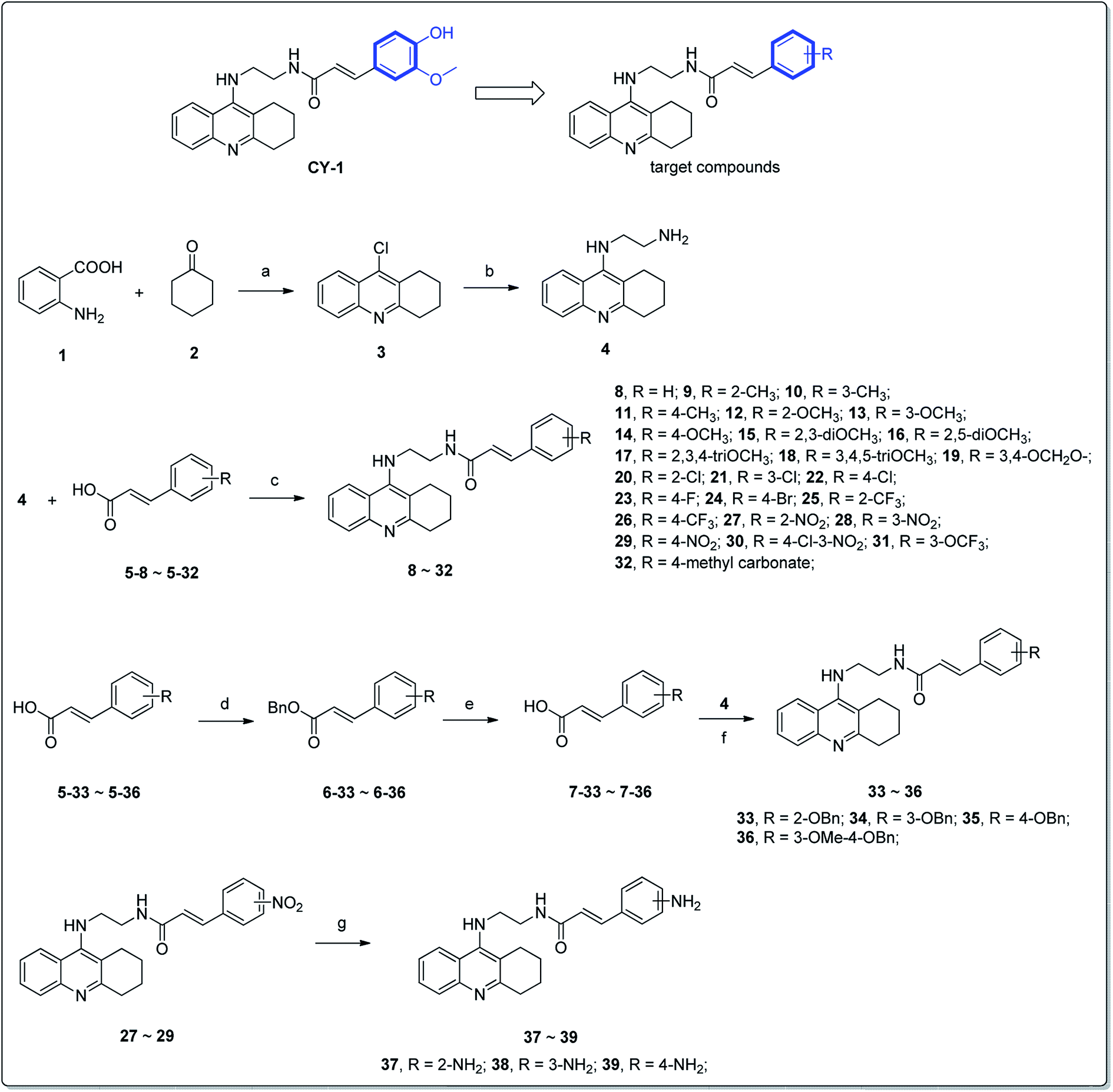 | ||
| Fig. 1 Design strategy and synthetic route of the target compounds. Reagents and conditions: (a) POCl3, reflux, 3 h; (b) pentanol, NH2(CH2)2NH2, NaI, reflux, 18 h; (c) PyBop, DIPEA, anhydrous CH2Cl2, room temp, 24 h; (d) benzyl bromide, K2CO3, DMF; (e) NaOH, MeOH, H2O, 5 h; (f) SnCl2·2H2O, EtOH, reflux, 8 h. | ||
The synthesis of the tacrine–cinnamic acid hybrids is described in Fig. 1. Anthranilic acid (1) was condensed with cyclohexanone (2) to yield chloro acridine 3.42 Treatment of 3 with ethane-1,2-diamine led to 4, which was condensed with different cinnamic acid analogs (5-8–5-32) to result in the target compounds 8–32. Hydroxyl substituted cinnamic acids (5-33–5-36) were first protected with benzyl to obtain intermediates 6-33–6-36, whose benzyl ester moiety was subsequently hydrolyzed to result in 7-33–7-36. They were condensed with 4 to acquire target compounds 33–36. Reduction of the nitro group of 27–29 resulted in 37–39.
Cholinesterase inhibitory activity and SAR analysis
The inhibitory effects of the synthesized compounds against AChE from electrophorus electricus (eeAChE) and BuChE from equine serum (eqBuChE) were determined, following Ellman's method.43 The data were expressed as IC50 values (Table 1). Most of the compounds were proved to be potent inhibitors of ChEs, with IC50 values lower than 100 nM. The AChE IC50 value of 8, an analog without any substitution at the cinnamic acid moiety, was higher than most of the substituted ones. Methyl substitution (9–11) led to an increase of AChE inhibitory activity. The position of the methyl group was also considered, showing that the activity was para- > meta- > ortho-. Interestingly, 11 showed much improved activity on AChE (IC50 = 34.3 ± 1.8 nM), while its inhibitory effect on BuChE remarkably reduced (IC50 = 86.9 ± 6.6 nM). When the methyl was replaced by a 4-methyl carbonate substitution (32), the compound exhibited a higher selectivity on AChE (AChE IC50 = 71.2 ± 2.4 nM, BuChE IC50 = 342.0 ± 61.5 nM). The results suggested that bulky functional groups were well tolerated by AChE, while they were restricted by BuChE. Therefore, bulky groups at para-position can be a good choice to enhance the target selectivity on AChE. Next, we designed a series of methoxy group analogs to evaluate the impact of this group on activity. For mono-substituted compounds (12–19), the activity on AChE was para- > meta- > ortho-, the same to that of methyl analogs. Substitution of methoxy group at meta-position (13) seemed have no impact on the target selectivity (AChE IC50 = 47.4 ± 2.0 nM, BuChE IC50 = 57.4 ± 5.6 nM). It was noticeable when methoxy group was at ortho-position (12), it showed a 7.99-fold selectivity on BuChE (AChE IC50 = 123.8 ± 7.6 nM, BuChE IC50 = 15.5 ± 1.5 nM). Similar results were also observed on 9 with 2-methyl substitution (AChE IC50 = 80.6 ± 9.8 nM, BuChE IC50 = 37.3 ± 3.3 nM). We inferred that the shape difference of the binding site between AChE and BuChE led to such phenomenon. The binding site of AChE is narrow and long, while BuChE is broad and short. As a result, substitution at the ortho-position can lead to steric hindrance to the narrow binding site of AChE but was tolerated by BuChE. Oppositely, para-substitution resulted in the elongation of the molecular shape of the inhibitors, and was more suitable for the binding site of AChE. For multi-substituted compounds (15–19), we could also observe a trend that ortho-substitution (15 and 16) was preferred by BuChE, while para-substitution was better for AChE (19). Interestingly, for triOCH3 analogs, 17 was very potent on both AChE and BuChE (IC50 = 17.3 ± 0.6 and 23.3 ± 2.3 nM, respectively), while 18 showed considerably high selectivity on AChE.
|
||||
|---|---|---|---|---|
| Cpd. | R | IC50 (nM) ± SEMa | SIf | |
| AChEb | BuChEc | |||
| a Concentration of the compound required for 50% inactivation of ChEs, data were shown in mean ± SEM of three experiments.b AChE (EC 3.1.1.7) from electric eel.c BuChE (EC 3.1.1.8) from horse serum.d AChE (EC 3.1.1.7) from human.e BuChE (EC 3.1.1.8) from human.f Selectivity index (SI) = AChE IC50/BuChE IC50. | ||||
| 8 | H | 92.5 ± 6.9 | 23.3 ± 2.7 | 4.0 |
| 9 | 2-CH3 | 80.6 ± 9.8 | 37.3 ± 3.3 | 2.2 |
| 10 | 3-CH3 | 78.9 ± 2.3 | 28.3 ± 2.0 | 2.8 |
| 11 | 4-CH3 | 34.3 ± 1.8 | 86.9 ± 6.6 | 0.4 |
| 12 | 2-OCH3 | 123.8 ± 7.6 | 15.5 ± 1.5 | 8.0 |
| 13 | 3-OCH3 | 47.4 ± 2.0 | 57.4 ± 5.6 | 0.8 |
| 14 | 4-OCH3 | 22.5 ± 1.1 | 61.3 ± 4.3 | 0.4 |
| 15 | 2,3-diOCH3 | 47.8 ± 2.5 | 23.2 ± 3.1 | 2.1 |
| 16 | 2,5-diOCH3 | 72.3 ± 6.2 | 26.1 ± 4.5 | 2.8 |
| 17 | 2,3,4-triOCH3 | 17.3 ± 0.6 | 23.3 ± 2.3 | 0.7 |
| 18 | 3,4,5-triOCH3 | 20.3 ± 0.5 | 270.6 ± 21.9 | 0.1 |
| 19 | 3,4-OCH2O– | 49.3 ± 2.5 | 80.7 ± 9.3 | 0.6 |
| 20 | 2-Cl | 50.4 ± 4.1 | 19.4 ± 1.4 | 2.6 |
| 21 | 3-Cl | 34.9 ± 4.3 | 21.8 ± 1.4 | 1.6 |
| 22 | 4-Cl | 21.3 ± 1.6 | 39.1 ± 3.7 | 0.5 |
| 23 | 4-F | 68.6 ± 5.7 | 125.8 ± 11.5 | 0.6 |
| 24 | 4-Br | 27.4 ± 1.7 | 92.5 ± 4.1 | 0.3 |
| 25 | 2-CF3 | 52.1 ± 1.5 | 28.4 ± 2.1 | 1.8 |
| 26 | 4-CF3 | 33.0 ± 1.3 | 170.6 ± 15.2 | 0.2 |
| 27 | 2-NO2 | 7.1 ± 0.5 | 48.2 ± 2.8 | 0.2 |
| 28 | 3-NO2 | 3.8 ± 0.1 | 34.7 ± 2.2 | 0.1 |
| 29 | 4-NO2 | 6.8 ± 0.7 | 154.3 ± 20.3 | 0.04 |
| 30 | 4-Cl-3-NO2 | 5.5 ± 0.2 | 64.4 ± 5.4 | 0.1 |
| 31 | 3-OCF3 | 56.5 ± 1.7 | 33.4 ± 4.9 | 1.7 |
| 32 | 4-Methyl carbonate | 71.2 ± 2.4 | 342.0 ± 61.5 | 0.2 |
| 33 | 2-OBn | 103.2 ± 9.9 | 13.6 ± 1.5 | 7.6 |
| 34 | 3-OBn | 40.1 ± 1.2 | 15.0 ± 1.1 | 2.7 |
| 35 | 4-OBn | 29.5 ± 1.0 | 42.6 ± 3.0 | 0.7 |
| 60.6 ± 5.7d | 86.1 ± 15.5e | 0.7 | ||
| 36 | 3-OMe-4-OBn | 15.8 ± 0.7 | 52.6 ± 5.6 | 0.3 |
| 55.1 ± 4.9d | 55.9 ± 3.3e | 1.0 | ||
| 37 | 2-NH2 | 28.7 ± 2.7 | 18.7 ± 2.2 | 1.5 |
| 38 | 3-NH2 | 54.7 ± 8.8 | 115.2 ± 12.5 | 0.5 |
| 39 | 4-NH2 | 173.3 ± 41.2 | 68.5 ± 7.9 | 2.5 |
| CY-1 | — | 62.0 ± 10.5 | 37.5 ± 9.7 | 1.7 |
| Tacrine | — | 69.8 ± 11.1 | 10.6 ± 1.1 | 6.6 |
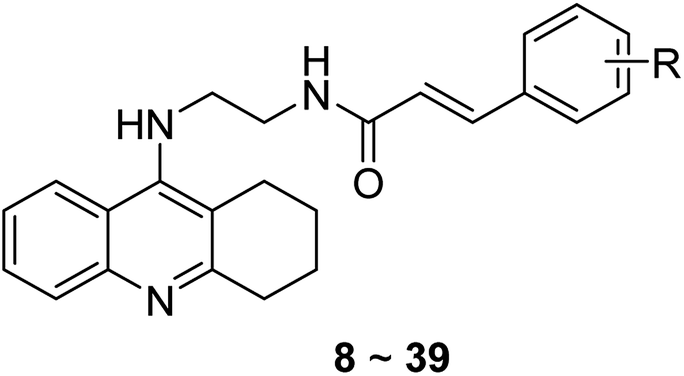
Next, we evaluated the impact of halogen atoms on the ChEs activity. When substituted by Cl (20–22), the activity on AChE was para- > meta- > ortho-, while it showed an opposite manner on BuChE. The results were in accordance with those from methyl and methoxy group substitution. When substituted by different halogen atoms, the activity on AChE was –Cl (22) ≈ –Br (24) > –F (23). Meanwhile, the three compounds with para-substitution were more selective on AChE than BuChE, a similar manner as mentioned above.
Then –NO2 (27–29) and –CF3 (25–26) and –OCF3 (31), three electron-withdrawing groups, were introduced as the R group. It was noteworthy that analogs with –NO2 substitution were the most active compounds on AChE among all the derivatives, with IC50 values in a single-digit nanomolar range. Meanwhile, they all exhibited a remarkable selectivity toward AChE than BuChE (SI = 0.04–0.15). The impact of –CF3 and –OCF3 was lower than –NO2, however, they also showed the same target selective rule to other groups mentioned above.
We subsequently introduced amino (37–39) as hydrogen-bond donating group. We found that such groups resulted in a remarkably reduced activity on AChE (IC50 = 54.7 ± 8.8–173.3 ± 41.2 nM) except 37, which was active toward both AChE and BuChE (IC50 = 28.7 ± 2.7, 18.7 ± 2.2 nM, respectively). Considering the hydrophobic nature of the binding site, especially for AChE, introduction of polar substitutions may lead to the improper intermolecular recognition, thus reducing the activity. Inspired by this, we replaced the hydroxyl to benzyloxyl. The ortho-substitution (33) exhibited high selectivity on BuChE, while meta- (34) and para-substitution (35 and 36) were preferable to AChE. These results further suggested the steric hindrance of the groups on the target selectivity.
To further validated the inhibitory activities of synthesized compounds on human ChEs, the representative compounds, 35 and 36 were selected for determination (Table 1). 35 exhibited huAChE IC50 = 60.6 ± 5.6 nM, huBuChE IC50 = 86.1 ± 15.5 nM; 36 exhibited huAChE IC50 = 55.1 ± 4.9 nM, huBuChE IC50 = 55.9 ± 3.3 nM. The results showed that the synthesized compounds efficiently inhibited the activities of human ChEs, further confirmed their activities as ChEs inhibitors.
Kinetic study of AChE inhibition
To further analyze the binding manner of the synthesized compounds to huAChE, the potent inhibitor 36 was selected as representative compound to perform the kinetic studies as described previously.34 Lineweaver–Burk reciprocal plots was applied to elucidate the type of inhibition of the test compounds. Briefly, Lineweaver–Burk plot can be described by reciprocal rates versus reciprocal substrate concentrations for the different inhibitor concentrations resulting from the substrate–velocity curves for ChEs. According to the results (Fig. 2), increasing concentration of the compounds (20, 60, 100, and 200 nM) resulted in both increased slopes (decreased Vmax) and intercepts (higher Km), indicating a mixed-type inhibition of 36. The detailed values of Vmax and Km at different concentrations are shown in Table 2.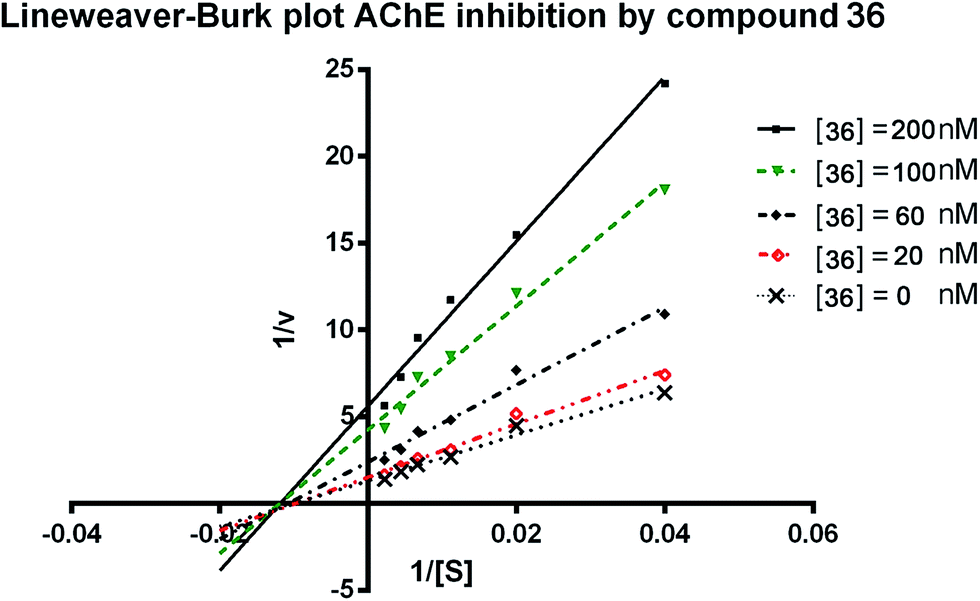 | ||
| Fig. 2 Lineweaver–Burk plots resulting from subvelocity curves of huAChE activity with different substrate concentrations (25–450 μM) in the absence and presence of 20, 60, 100, 200 nM 36. | ||
| Concentration (nM) | Vmax (μM min−1) | Km (μM) | R square |
|---|---|---|---|
| 0 | 0.9 ± 0.1 | 147.8 ± 18.8 | 0.99 |
| 5 | 0.8 ± 0.0 | 139.7 ± 16.3 | 0.99 |
| 10 | 0.5 ± 0.0 | 145.9 ± 22.4 | 0.99 |
| 15 | 0.3 ± 0.0 | 141.4 ± 25.2 | 0.99 |
| 40 | 0.2 ± 0.0 | 148.7 ± 27.5 | 0.99 |
Molecular modeling studies
To investigate the binding pattern of the synthesized compounds with huAChE, molecular docking studies were performed using Discovery Studio (DS). 35, and 36 were selected as representative compounds. As shown in Fig. 3A and B, both the compounds bound to AChE in a dual-site manner by occupying both CAS and PAS. The 1,2,3,4-tetrahydroacridin moiety of the two compounds inserted into the CAS. The tetrahydroacridin moiety formed multiple π–π stacking contacts with the aromatic sidechains of Trp86 and Tyr124. These hydrophobic contacts provided driving force for the binding of the compounds to CAS of huAChE. The substituted phenyl ring of cinnamic acid moiety was located at the PAS of huAChE binding groove.44 It formed π–π stacking contacts with the sidechain of Trp286, Tyr341. It was noticeable that the methoxy group at meta-position of the phenyl ring (36) formed an additional π–alkyl interaction with the sidechain of Trp286. The binding difference may explain the slightly better huAChE inhibitory activity of 35 than 36.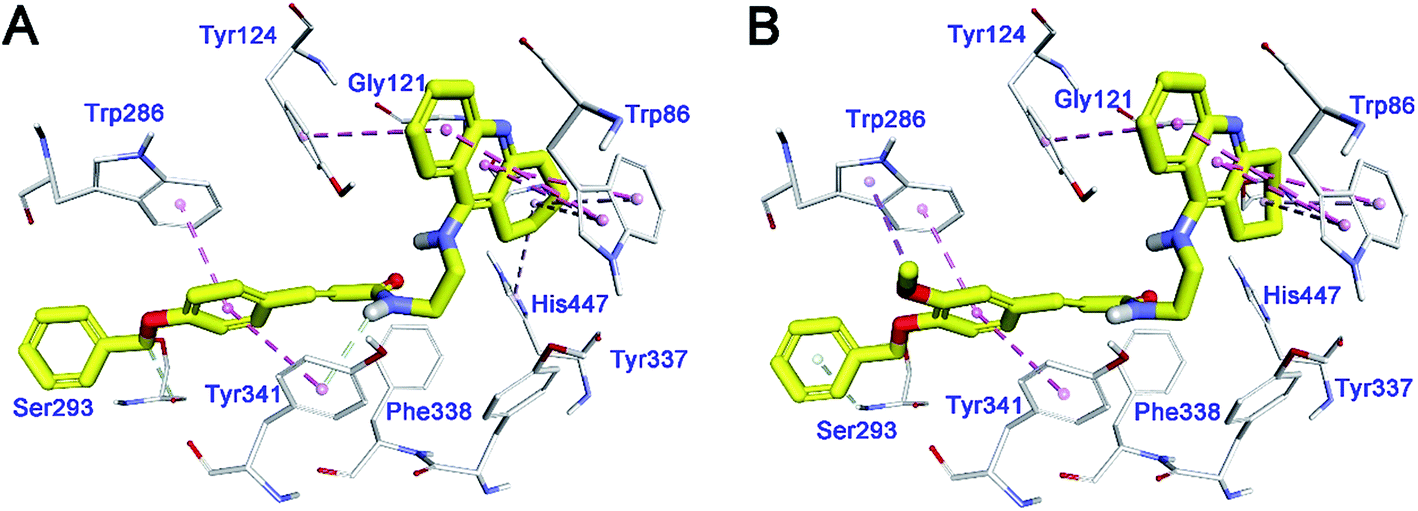 | ||
| Fig. 3 Binding mode prediction of 35 (A), and 36 (B) with huAChE (PDB id: 4EY7). Compounds were shown in stick mode colored in yellow. Key residues were labeled as thin stick mode colored in white. Intermolecular interactions were shown as dot lines with different colors according to the type of the interaction: light green, hydrophobic contact; pink, π–π stacking; purple, π–alkyl contact. | ||
In summary, the binding mode of the selected compounds supported the mixed-type of binding manner revealed by the kinetic study.
Inhibition of self-induced Aβ1–42 aggregation
All compounds were evaluated for their inhibitory capacity on self-induced Aβ1–42 aggregation based on a thioflavin T-based fluorometric assay. Curcumin, a natural product that is known to inhibit the Aβ1–42 self-aggregation, was used as the reference compound. Most of the analogs only showed poor or moderate inhibition on Aβ1–42 self-aggregation (ranging from 4.2 ± 0.1–37.8 ± 3.9%, Table 3). Two compounds, 35, and 36, exhibited inhibitory rate over 40% (40.7 ± 1.9, 42.2 ± 2.6%, respectively). Interestingly, 36 was potent on both AChE and self-induced Aβ1–42 aggregation, indicating its potential for acting as a multi-target compound. It seemed that a methyl group was preferred in inhibiting Aβ1–42 self-aggregation, especially when substituted at the para-position. Larger groups such as t-Bu led to a greater than 2-fold decrease in activity. Mono-substitution of methoxy group led to the completely loss of the inhibitory effect, however, di- or tri-substitution of methoxy groups (16, 18, 19) supplied moderate activity. Electron-withdrawing group, such as –NO2 and –CF3, remarkably reduced the activity. Substituent groups such as halogen, hydroxyl or amino group had no impact on the inhibition of Aβ1–42 self-aggregation, no matter what position they were at. It was noticeable that benzyl substituted compounds (33–36) exhibited the best activity among all the derivatives, suggesting this benzyl group was not only important for AChE inhibition, but also acted as a preferred moiety for designing new MTDLs.| Cpd. | Aβ IRa % | IC50b (μM) | Cpd. | Aβ IRa % | IC50b (μM) |
|---|---|---|---|---|---|
| a IR stands for inhibitory rate. Inhibition of self-induced Aβ1–42 aggregation was determined by thioflavin-T fluorescence method, and the data were shown in mean ± SEM of three independent experiments. The experiments were performed in the presence of 20 μM target compounds.b The antiproliferative activities of the target compounds on PC12 cells. The IC50 values were calculated based on three independent experiments and were shown in mean ± SEM. | |||||
| 8 | 7.0 ± 0.6 | 55.7 ± 5.8 | 24 | 21.6 ± 3.6 | 34.2 ± 4.7 |
| 9 | 33.2 ± 0.8 | 71.7 ± 6.8 | 25 | 7.0 ± 0.1 | 19.8 ± 2.4 |
| 10 | 32.9 ± 2.1 | 39.9 ± 4.0 | 26 | 22.4 ± 1.7 | 10.1 ± 1.0 |
| 11 | 40.9 ± 1.6 | 27.1 ± 3.2 | 27 | 6.0 ± 0.8 | 34.5 ± 2.5 |
| 12 | 4.2 ± 0.1 | 63.0 ± 9.2 | 28 | 5.6 ± 0.2 | 23.7 ± 1.2 |
| 13 | 8.4 ± 0.6 | 56.3 ± 5.1 | 29 | 13.6 ± 1.2 | 55.0 ± 6.5 |
| 14 | 7.9 ± 0.9 | 51.6 ± 9.4 | 30 | 19.4 ± 2.8 | 13.5 ± 1.7 |
| 15 | 5.1 ± 0.2 | 79.6 ± 11.5 | 31 | 30.0 ± 5.1 | 10.0 ± 1.0 |
| 16 | 34.5 ± 2.3 | 55.2 ± 4.4 | 32 | 17.6 ± 1.3 | 59.6 ± 8.0 |
| 17 | 21.4 ± 3.8 | 26.3 ± 2.9 | 33 | 40.7 ± 1.9 | 14.5 ± 1.9 |
| 18 | 28.8 ± 4.0 | 18.7 ± 1.4 | 34 | 35.9 ± 2.1 | 43.8 ± 3.9 |
| 19 | 31.3 ± 4.0 | 34.9 ± 4.0 | 35 | 37.8 ± 3.9 | 92.2 ± 8.8 |
| 20 | 30.1 ± 2.7 | 29.4 ± 1.9 | 36 | 42.2 ± 2.6 | 84.6 ± 7.3 |
| 21 | 28.9 ± 2.1 | 30.9 ± 4.2 | 37 | 19.9 ± 0.3 | 34.8 ± 2.6 |
| 22 | 35.3 ± 3.8 | 20.1 ± 3.0 | 38 | 14.4 ± 1.0 | 41.8 ± 3.9 |
| 23 | 22.7 ± 2.9 | 28.7 ± 2.1 | 39 | 25.1 ± 3.1 | 37.3 ± 4.1 |
| Curcumin | 46.2 ± 3.2 | — | |||
Cell toxicity and cyto-protection effects of the compounds in PC-12 neuroblastoma cells
We next focused on the cell toxicities of the synthesized compounds. They were evaluated for the anti-proliferative effects against neuroblastoma PC-12 cell line. Most of the synthesized compounds exhibited IC50 values above 30 μM (Table 3). 35 and 36 showed the best safety on PC-12 cells (IC50 = 92.2 ± 8.8 μM and 84.6 ± 7.3, respectively). Considering their good inhibitory activity on ChEs and self-induced Aβ1–42 aggregation, especially 36, they were selected for further in vivo evaluations. It was noteworthy that the nitro-substituted compounds 27, 28, and 30 showed much stronger anti-proliferative activity against PC-12 cells than most of the derivatives (IC50 = 34.5 ± 2.5, 23.7 ± 1.2, 13.5 ± 1.7 μM, respectively), indicating their potential cytotoxicity. Although these nitro-substituted compounds showed the best inhibitory effects on AChE, they only exerted poor activity on self-induced Aβ1–42 aggregation. Taken together, these compounds were not investigated for their in vivo activity. Besides, CF3-substituted compounds 25 and 26 also potently inhibited the proliferation of PC-12 cells (IC50 = 19.8 ± 2.4 and 10.1 ± 1.0 μM, respectively). Although strong electron-withdrawing groups were preferred for AChE inhibition, it seemed that they were prone to cause a strong cytotoxicity. Similar manner was observed in 3-OCF3 substituted analog 31 (IC50 = 10.0 ± 1.0 μM). Highly toxic compounds also included 18, 22, and 33 (IC50 = 18.7 ± 1.4, 20.1 ± 3.0, and 14.5 ± 1.9 μM, respectively).Then, we evaluated the cytoprotective effects of 35 and 36 on H2O2-induced cell damage. Treatment with 500 μM H2O2 for 24 h caused over 60% death rate of PC12 cells compared with the control group (Fig. 4). When pretreated with 35 and 36 for 24 h, the mortality rate of PC-12 cells caused by H2O2 was significantly attenuated. Such protective effect exhibited dose-dependent manner for both the two compounds. 36 showed a better cytoprotective effect than 35. It increased the cell viability to 63.1 ± 2.1% and 72.5 ± 2.1% at the concentration of 20 and 40 μM, respectively. These results indicated that 36 had a potential in antagonizing the oxidative stress.
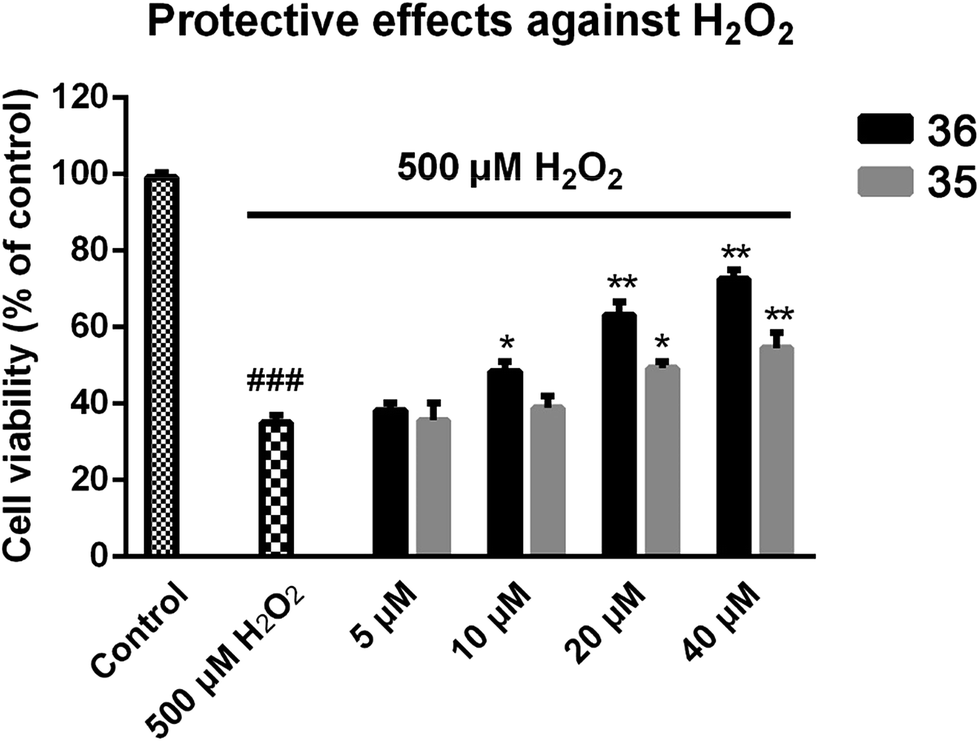 | ||
| Fig. 4 Protective effects of compound 35 or 36 against H2O2-induced cell death in PC-12 cells. PC12 cells were plated in a 96-well plate for 12 h and then treated with various concentrations of 35 or 36 for another 24 h. After replacing the medium containing 500 μM H2O2, incubation of the cells was continued for 12 h, then the cell viability was measured by MTT assay. All data represent the means ± SD of three independent experiments. Data were shown as mean ± SD (n = 3). p### < 0.001 compared to control, p* < 0.05, p** < 0.01 compared to H2O2-treated cells. | ||
Behavioral studies
Improvement of cognitive ability is the most significant profile of anti-AD agents. Based on the multiple evaluations mentioned above, compound 36 with the best multipotent activity profile was selected for in vivo behavioral study by using a Morris water maze test. The animal model was built on the basis of scopolamine-induced cognition-impaired adult ICR mice and was applied for the cognitive improvement effects of 36. Compound 35 was also evaluated with the aim to understand the importance of the methoxy group. Tacrine (20 μmol kg−1 body weight) was used as positive control. 35, 36, and tacrine were orally administered to the ICR mice 30 min before intraperitoneal (ip) administration of scopolamine (1 mg kg−1) or saline solution for 10 consecutive days to adapt the apparatus. The test included 5 days of learning and memory training and a probe trial on the sixth day. The mean escape latency values of all the groups on the sixth day were shown in Table 4 and Fig. 5A. Compared to the control group, scopolamine led to a remarkable delay of the latency to target (8.9 ± 4.0 seconds vs. 45.2 ± 11.6 seconds), indicating that the cognitive impairment mouse model was successfully built. Treatment of tacrine ameliorated the impairment and the latency to target reduced to 36.3 ± 11.6 seconds (*p < 0.05). 35 exhibited a comparable activity to tacrine (36.4 ± 14.3 seconds, *p < 0.05). Compared to tacrine and 35, 36 significantly reduced the latency to target (13.2 ± 7.6 seconds, ***p < 0.001), indicating that 36 considerably ameliorated the cognitive impairment of the treated mice and was much better than tacrine. The results also suggested the critical role of the methoxy group of ferulic acid moiety of compound 36. Removal of this group led to the markedly decrease of the in vivo activity as compared to 35. We confer there may be two reasons for the results: (1) the methoxy group may enhance the ability of 36 to penetrate the blood–brain barrier (BBB) and target the central nervous system (CNS); (2) the methoxy group may prevent the metabolism at meta-position of the phenyl ring, thus enhance the concentration of the compound to target CNS.| Group | Latency to target (s) | Distance to target (cm) |
|---|---|---|
| Control | 8.9 ± 4.0 | 292.8 ± 206.4**** |
| Model | 45.2 ± 11.6 | 1637.3 ± 517.1 |
| Tacrine | 36.3 ± 11.6* | 1125.3 ± 367.1 |
| 36 | 13.2 ± 7.6*** | 469.5 ± 278.8**** |
| 35 | 36.4 ± 14.3* | 1274.9 ± 452.6 |
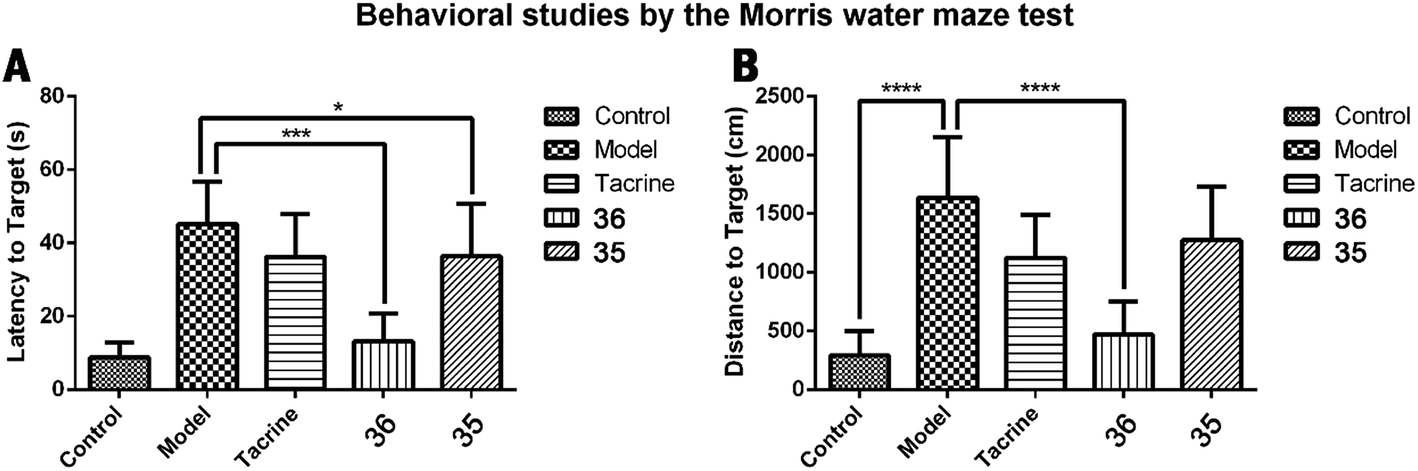 | ||
| Fig. 5 Effects of oral administration of tacrine (15 mg kg−1), 35 (15 mg kg−1), and 36 (15 mg kg−1) on scopolamine-induced cognitive impairment in ICR mice determined by the Morris water maze test. (A) The latency to target; (B) the distance to target. Data are presented as the mean ± SEM (n = 6; *p < 0.05, ***p < 0.001, ****p < 0.0001 vs. scopolamine group). | ||
The distance to target (Table 4 and Fig. 6B) and the trajectories of the mice in each group were also analyzed. Compared to the control group, administration of scopolamine remarkably led to the extended distance to target (1637.3 ± 517.1 cm vs. 292.8 ± 206.4 cm, ****p < 0.0001). Tacrine and 35 reduced the distance to target (1125.3 ± 367.1 cm, 1274.9 ± 452.6 cm, respectively). When treated with 36, the distance to target was significantly shortened (469.5 ± 278.8, ****p < 0.0001). These results were supported by trajectory analysis. As shown in Fig. 6B, the trajectory of the mice in scopolamine model group was very long and disordered, while tacrine and 35 groups (Fig. 6C and E) showed shortened distances, but still much longer than the control group (Fig. 6A). Mice treated with 36 almost recovered to the normal cognition (Fig. 6D), with a similar orientation and distance to that of the normal mice. Taken together, these results supported that 36 remarkably ameliorated the cognition impairment caused by scopolamine.
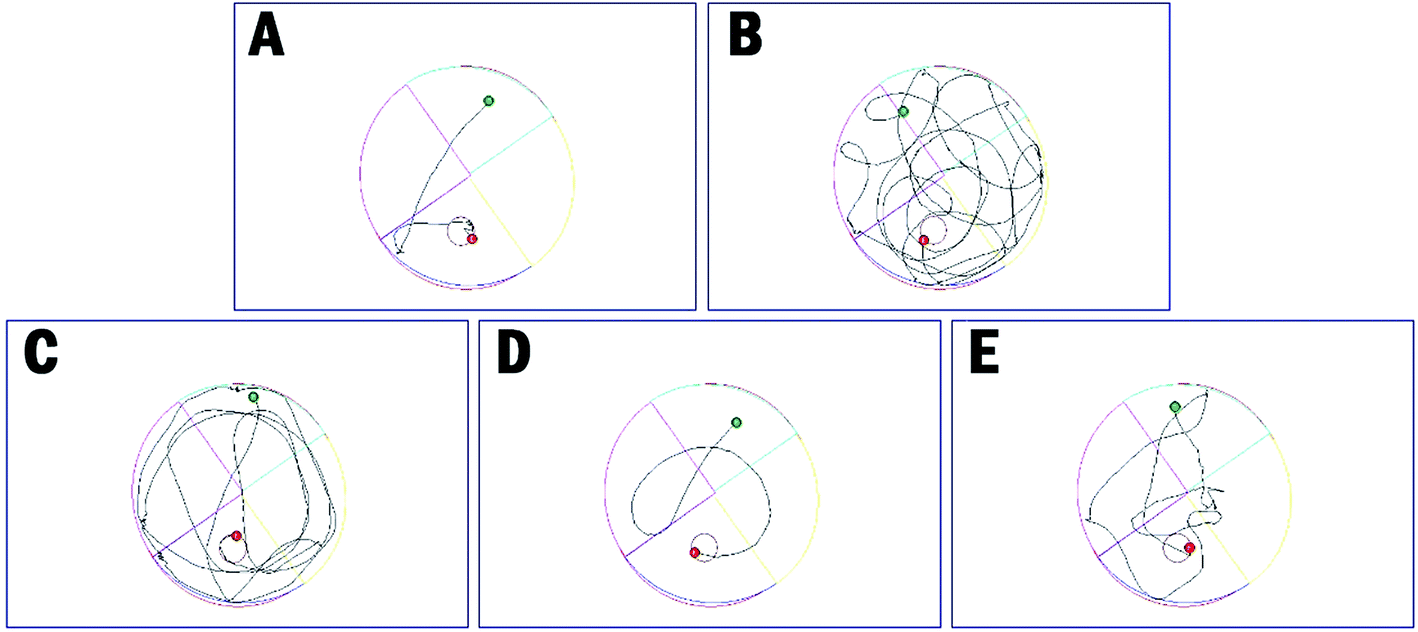 | ||
| Fig. 6 The trajectories of mice in control (A), model (B), tacrine (C), 35 (D), and 36 (E) group in the Morris water maze test. | ||
Hepatotoxicity studies
Given that the serious hepatotoxicity of tacrine has been the primary limitation for its clinical use, to ensure the safety of 35 and 36 for further development, we next investigated the possible drug-induced hepatotoxicity by comparing their toxic profile to tacrine. Heparinized serum was collected 8 h, 22 h, 36 h, 72 h, and 96 h after the administration of tacrine, 35 and 36 for the evaluation of the levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), two known biomarkers of live damage (Table 5 and Fig. 7). Compared to the control group, after the treatment of tacrine, the levels of ALT and AST were slightly induced at 22 h, but in general, no remarkable damage was observed. As expected, 36 did not showed any hepatotoxicity at all the time points, the level of ALT and AST even slightly reduced at 22 h, 36 h, and 96 h for ALT and 36 h and 96 h for AST, compared to those of tacrine group. The results indicated the hepatic safety profile of 36. Interestingly, 35 exhibited an induction of the ALT (36.3 ± 6.9 vs. 31.4 ± 2.3 at 8 h, 40.9 ± 6.9 vs. 32.9 ± 5.3 at 22 h) and AST level (102.6 ± 14.3 vs. 91.1 ± 16.0 at 8 h) compared to the control group, indicating a potential toxic effect of this compound. When the time extended to 72 h and 96 h, the levels of ALT and AST were comparable to those of control group. Considering the structural difference between 35 and 36, we can speculate that the methoxy group was beneficial to the hepatic safety. The results were also in accordance with those from the behavioral studies. Therefore, it is important to introduce proper groups at this position in order to avoid undesired metabolism.| Group | 8 h | 22 h | 36 h | 72 h | 96 h |
| ALT (U L−1) | |||||
| Control | 31.4 ± 2.3 | 32.9 ± 5.3 | 30.5 ± 3.0 | 28.2 ± 3.8 | 29.1 ± 6.0 |
| Tacrine | 28.3 ± 5.4 | 34.9 ± 8.6 | 32.4 ± 4.8 | 30.5 ± 3.4 | 30.5 ± 6.0 |
| 35 | 36.3 ± 6.9 | 40.9 ± 6.9 | 29.0 ± 8.0 | 29.7 ± 3.4 | 30.7 ± 4.0 |
| 36 | 32.0 ± 7.7 | 27.8 ± 5.0 | 27.8 ± 7.5 | 30.7 ± 4.0 | 27.5 ± 5.7 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| AST (U L−1) | |||||
| Control | 91.1 ± 16.0 | 91.5 ± 21.4 | 95.6 ± 9.3 | 83.9 ± 16.9 | 86.4 ± 18.2 |
| Tacrine | 89.3 ± 22.1 | 105.4 ± 13.6 | 87.8 ± 22.5 | 91.6 ± 18.5 | 87.7 ± 14.4 |
| 35 | 102.6 ± 14.3 | 93.7 ± 12.0 | 73.0 ± 10.0 | 78.1 ± 10.3 | 82.1 ± 16.7 |
| 36 | 91.2 ± 19.8 | 96.7 ± 15.6 | 78.0 ± 14.6 | 88.9 ± 24.1 | 77.9 ± 16.2 |
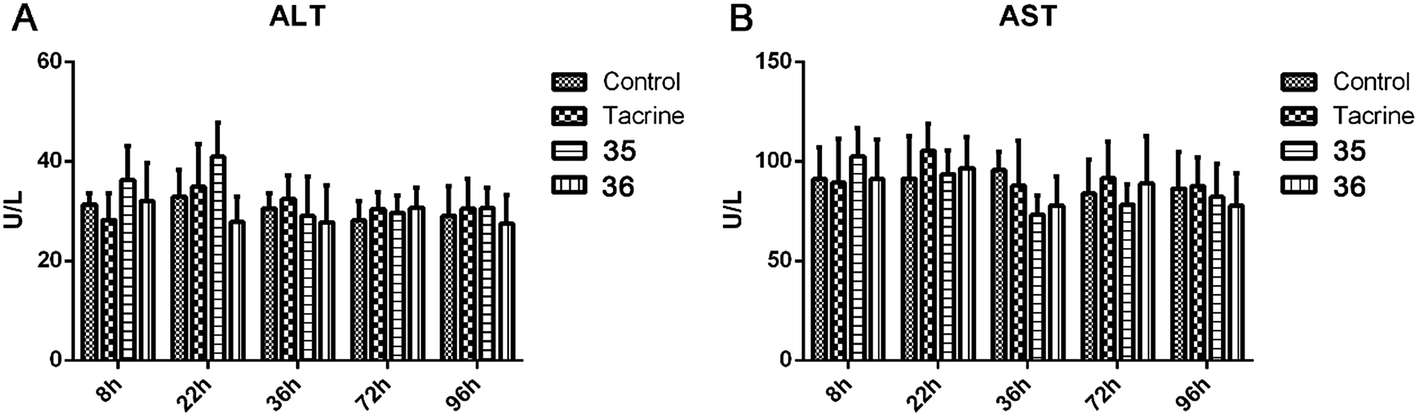 | ||
| Fig. 7 ALT (A) and AST (B) activity after the administration of tacrine, 35, and 36. Values are expressed as mean ± SEM (n = 6; t test, compared to the control of the same time after administration). | ||
To further analyze the hepatotoxicity of 35 and 36, morphologic studies by immunohistochemical staining were applied. Treatment of tacrine (Fig. 8B), 35 (Fig. 8C) or 36 (Fig. 8D) did not result in remarkable morphologic changes in liver compared to the control group (Fig. 8A). Taken together, 36 exhibited the highest safety among all the test compounds, ensuring its further development.
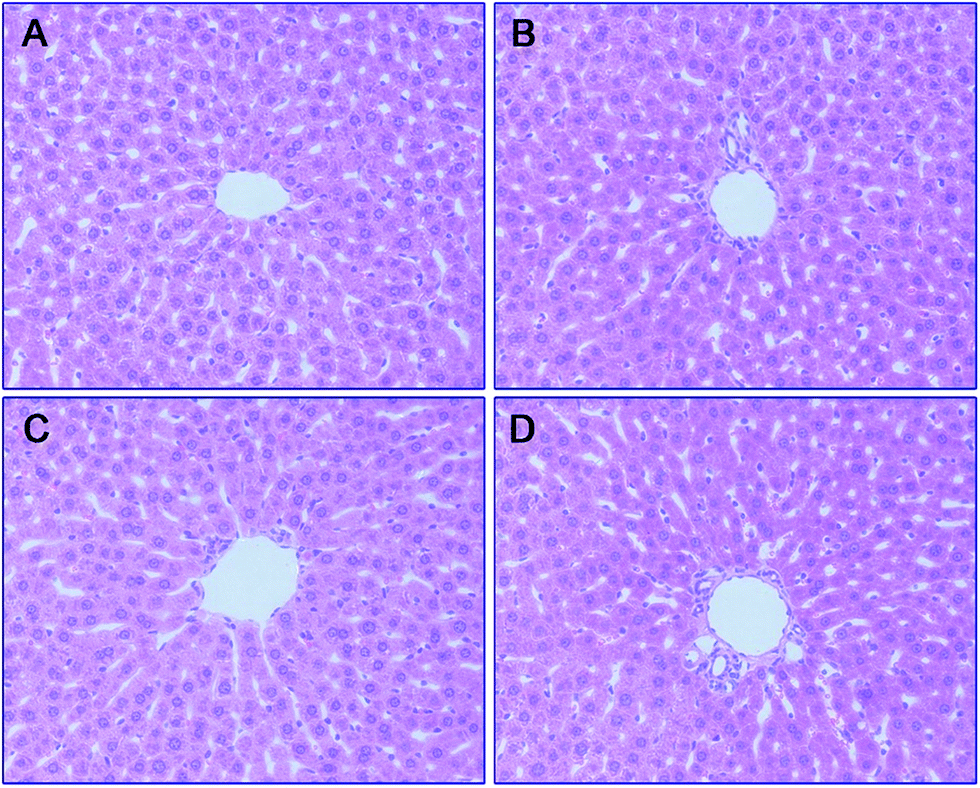 | ||
| Fig. 8 Histomorphological appearance of livers of male mice after treatment with the solvent only (control, A), or 22 h after administration of tacrine (B), 35 (C), or 36 (D). HE staining, original magnification ×200. | ||
Conclusions
CY-1 was a tacrine–ferulic acid hybrid reported by our group previously. Guided by this compound, in the present studies, a series of tacrine–cinnamic acid hybrids were designed and synthesized so as to identify the optimal substitution on the phenyl ring of the cinnamic acid moiety. Although there are several publications about tacrine–ferulic acid hybrid, as far as we concerned, this is the first medicinal chemistry study on the ferulic acid moiety. In vitro assays proved that most of the compounds effectively inhibited ChEs in the nanomolar range. Additionally, some interesting information was summarized from the SAR study and can guide the further optimization of this series of compounds. 36 was one of the most potent analogs, which was about 4-fold more active than the parent compound CY-1 against AChE. Kinetic studies and molecular docking indicated that 36 inhibited AChE in a mixed-type manner by simultaneously binding to CAS and PAS of AChE. This compound effectively inhibited the self-induced Aβ1–42 aggregation, and exhibited cytoprotective effects against H2O2 induced cell damage. Meanwhile, it was proved to be non-toxic to PC-12 cells when it exerted its biological functions, indicating its good safety. The in vitro assays confirmed the multifunctional potent manner of 36 as potential anti-AD agent. Therefore, it was subjected to in vivo evaluation including Morris water maze test and hepatotoxicity studies. 36 remarkably reduced the scopolamine-induced cognitive impairment in animal model and showed very low hepatotoxicity under the therapeutic concentration. Altogether, 36 can be considered as a promising lead compound for further identification of new anti-AD agents.Experimental sections
Chemistry
N-(1,2,3,4-Tetrahydroacridin-9-yl)ethane-1,2-diamine (4). To a solution of 3 (9-chlorotetrahydroacridine) (2 g, 9.2 mmol) in 10 mL of pentanol, diaminoethane (2.76 g, 45.94 mmol) were added. After refluxing for 24 h, the solution was cooled to room temperature and then acidified with hydrochloric acid. The solution was extracted with acid water (4 × 10 mL). The combined aqueous phase was basified by NaOH and then extracted with CH2Cl2 (4 × 15 mL). The water phase was combined and washed with brine, dried over anhydrous Na2SO4 overnight, and evaporated in vacuo. The residue was purified by column chromatography (CH2Cl2/MeOH = 20
:1, v/v, with 0.1% triethylamine). 1.5 g yellow oil was given (67.6%). 1H NMR (CD3OD): δ 8.13–8.10 (m, 1H, ArH), 7.78–7.75 (m, 1H, ArH), 7.60–7.54 (m, 1H, ArH), 7.42–7.36 (m, 1H, ArH), 3.61 (t, J = 7.50 Hz, 2H, NH–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 2.96–2.91 (m, 4H, C4–H2, C2–NH2), 2.76 (br, 2H, C1–H2), 1.91–1.88 (m, 4H, C3–H2, C2–H2); MS (GC) m/z (% rel. Int.) 241 (M+, 45), 197 (100) (found C, 74.99; H, 8.18; N, 16.98; C15H19N3 requires C, 74.65; H, 7.94; N, 17.41).
2), 2.96–2.91 (m, 4H, C4–H2, C2–NH2), 2.76 (br, 2H, C1–H2), 1.91–1.88 (m, 4H, C3–H2, C2–H2); MS (GC) m/z (% rel. Int.) 241 (M+, 45), 197 (100) (found C, 74.99; H, 8.18; N, 16.98; C15H19N3 requires C, 74.65; H, 7.94; N, 17.41).
N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)cinnamamide (8)45. Cinnamic acid (0.123 g, 0.83 mmol), pyBOP (0.44 g, 0.99 mmol), and DIPEA (0.14 g, 1.08 mmol) were dissolved in 3 mL of CH2Cl2, and the mixture was stirred at room temperature for 40 min. Then a solution of 4 (0.2 g, 0.83 mmol) in 2 mL CH2Cl2 was added. After being stirred for overnight at room temperature. The solvent was removed under vacuo, 20 mL ethyl acetate was added, the solid was filtered. Then the solid was purified by column chromatography (CH2Cl2/MeOH = 50
:1, v/v, with 0.1% triethylamine) to give a white powder 0.12 g (39.1%). Mp 186–187 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.53 (t, J = 6.10 Hz, 1H, ArH), 8.47 (d, J = 8.64 Hz, 1H, ArH), 7.96 (s, 1H, CONH), 7.86 (t, J = 7.58 Hz, ArH), 7.60–7.75 (m, 3H, ArH and NH), 7.40 (d, J = 6.84 Hz, 3H, ArH and COCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 6.57 (d, J = 15.81 Hz, 1H, COCCH), 4.02 (d, J = 5.40 Hz, 2H, NHC2), 3.57 (d, J = 5.40 Hz, 2H, C2NHCO), 2.94 (br, 2H, C4–H2), 2.68 (br, 2H, C1–H2), 1.82 (br, 4H, C2–H2 and C3–H2). 13C NMR (125 MHz, CDCl3): δ 167.61, 157.94, 151.06, 146.59, 141.52, 134.68, 129.85, 128.86, 128.66, 127.86, 127.67, 123.86, 122.85, 120.37, 119.73, 115.85, 63.96, 49.84, 40.68, 33.43, 25.01, 22.95, 22.53. HRMS (ESI): calcd for C24H25N3O [M + H]+ 372.207, found 372.2074. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.56 min, 97.077%.
C), 6.57 (d, J = 15.81 Hz, 1H, COCCH), 4.02 (d, J = 5.40 Hz, 2H, NHC2), 3.57 (d, J = 5.40 Hz, 2H, C2NHCO), 2.94 (br, 2H, C4–H2), 2.68 (br, 2H, C1–H2), 1.82 (br, 4H, C2–H2 and C3–H2). 13C NMR (125 MHz, CDCl3): δ 167.61, 157.94, 151.06, 146.59, 141.52, 134.68, 129.85, 128.86, 128.66, 127.86, 127.67, 123.86, 122.85, 120.37, 119.73, 115.85, 63.96, 49.84, 40.68, 33.43, 25.01, 22.95, 22.53. HRMS (ESI): calcd for C24H25N3O [M + H]+ 372.207, found 372.2074. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.56 min, 97.077%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(o-tolyl)acrylamide (9). According to the procedure used to synthesize 8, 9 was synthesized from (E)-3-(o-tolyl)acrylic acid (0.14 g, 0.83 mmol) to give a white powder 0.14 g (43.89%). Mp 202–204 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.83 (s, 1H, ArH), 8.53 (d, J = 8.53 Hz, 1H, ArH), 8.00 (d, J = 8.19 Hz, 2H, ArH and CONH), 7.84 (t, J = 7.59 Hz, 1H, ArH), 7.65 (d, J = 15.72 Hz, 1H, COCH
C), 7.56 (t, J = 7.68 Hz, 1H, ArH), 7.45 (d, J = 6.93 Hz, 1H, ArH), 7.24 (br, 2H, ArH and NH), 6.53 (d, J = 15.66 Hz, COCCH), 4.02 (d, J = 6.18 Hz, 2H, 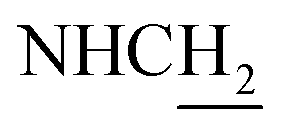 ), 3.56 (d, J = 5.22 Hz, 2H,
), 3.56 (d, J = 5.22 Hz, 2H, 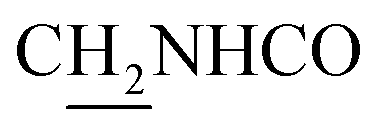 ), 3.00 (s, 3H, CH3) 2.71 (s, 2H, C4–
), 3.00 (s, 3H, CH3) 2.71 (s, 2H, C4–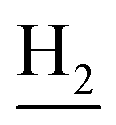 ), 2.35 (s, 2H, C1–
), 2.35 (s, 2H, C1– ), 1.82 (s, 4H, C2–
), 1.82 (s, 4H, C2– , C3–
, C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.78, 156.12, 148.29, 137.22, 137.00, 134.12, 132.79, 131.15, 129.76, 126.87, 126.45, 125.50, 123.47, 119.56, 115.95, 111.91, 48.70, 28.44, 24.31, 21.98, 21.44, 20.81. HRMS (ESI): calcd for C25H27N3O [M + H]+ 386.2227, found 386.2226. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.53 min, 95.828%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.78, 156.12, 148.29, 137.22, 137.00, 134.12, 132.79, 131.15, 129.76, 126.87, 126.45, 125.50, 123.47, 119.56, 115.95, 111.91, 48.70, 28.44, 24.31, 21.98, 21.44, 20.81. HRMS (ESI): calcd for C25H27N3O [M + H]+ 386.2227, found 386.2226. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.53 min, 95.828%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(m-tolyl)acrylamide (10). According to the procedure used to synthesize 8, 10 was synthesized from (E)-3-(m-tolyl)acrylic acid (0.14 g, 0.83 mmol) to give a white powder 0.14 g (43.89%). Mp 200–202 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.51–8.46 (m, 2H, ArH), 7.97 (br, 1H, CONH), 7.87 (t, J = 7.56 Hz, 1H, ArH), 7.78 (d, J = 7.65 Hz, 1H, ArH), 7.58 (t, J = 7.49 Hz, 1H, ArH), 7.40 (d, J = 15.84 Hz, 1H, COCH
C), 7.35–7.26 (m, 3H, ArH and NH), 7.19 (d, J = 7.26 Hz, 1H, ArH) 6.56 (d, J = 15.81 Hz, 1H, COCCH), 4.02 (d, J = 5.46, 2H, 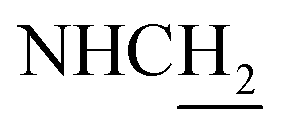 ), 3.57 (d, J = 5.61 Hz, 2H,
), 3.57 (d, J = 5.61 Hz, 2H, 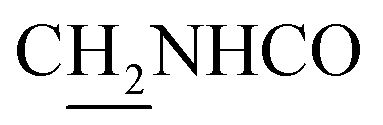 ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4–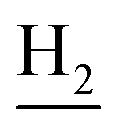 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–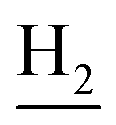 ), 2.31 (s, 3H, CH3), 1.83 (br, 4H, C2–
), 2.31 (s, 3H, CH3), 1.83 (br, 4H, C2–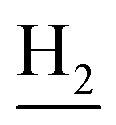 and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 167.06, 156.66, 151.00, 139.99, 138.64, 138.33, 135.08, 133.31, 130.86, 129.35, 128.61, 125.79, 125.60, 125.26, 121.68, 119.54, 116.00, 111.92, 48.82, 28.42, 24.21, 21.92, 21.38, 20.79. HRMS (ESI): calcd for C25H27N3O [M + H]+ 386.2227, found 386.2215. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.97 min, 99.188%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.06, 156.66, 151.00, 139.99, 138.64, 138.33, 135.08, 133.31, 130.86, 129.35, 128.61, 125.79, 125.60, 125.26, 121.68, 119.54, 116.00, 111.92, 48.82, 28.42, 24.21, 21.92, 21.38, 20.79. HRMS (ESI): calcd for C25H27N3O [M + H]+ 386.2227, found 386.2215. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.97 min, 99.188%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(p-tolyl)acrylamide (11). According to the procedure used to synthesize 8, 11 was synthesized from (E)-3-(p-tolyl)acrylic acid (0.14 g, 0.83 mmol) to give a white powder 0.17 g (53.29%). Mp 212–214 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.47 (d, J = 7.41 Hz, 2H, ArH), 7.95 (br, 1H, CONH), 7.86 (t, J = 7.32 Hz, 1H, ArH), 7.78 (d, J = 7.37 Hz, 1H, ArH), 7.57 (t, J = 7.56 Hz, 1H, ArH), 7.45–7.42 (m, 3H ArH and NH and COCH
C), 7.22 (d, J = 7.08 Hz, 1H, ArH), 6.81 (d, J = 15.69 Hz, 1H, COCCH), 4.02 (br, 2H,  ), 3.57 (br, 2H,
), 3.57 (br, 2H,  ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4– ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1– ), 2.31 (s, 3H, CH3), 1.83 (br, 4H, C2–
), 2.31 (s, 3H, CH3), 1.83 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 167.19, 156.69, 150.99, 139.98, 139.88, 138.32, 133.32, 132.38, 130.06, 128.07, 125.80, 125.60, 120.78, 119.52, 116.00, 111.91, 48.88, 28.41, 24.21, 21.92, 21.43, 20.79. HRMS (ESI): calcd for C25H27N3O [M + H]+ 386.2227, found 386.2219. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.75 min, 98.085%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.19, 156.69, 150.99, 139.98, 139.88, 138.32, 133.32, 132.38, 130.06, 128.07, 125.80, 125.60, 120.78, 119.52, 116.00, 111.91, 48.88, 28.41, 24.21, 21.92, 21.43, 20.79. HRMS (ESI): calcd for C25H27N3O [M + H]+ 386.2227, found 386.2219. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.75 min, 98.085%.
(E)-3-(2-Methoxyphenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (12). According to the procedure used to synthesize 8, 12 was synthesized from (E)-3-(2-methoxyphenyl)acrylic acid (0.15 g, 0.83 mmol) to give a white powder 0.15 g (45.05%). Mp 173–175 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.48–8.45 (m, 2H, ArH), 7.85 (t, J = 7.02 Hz, 2H, ArH and CONH), 7.78 (d, J = 7.77 Hz, 1H, ArH), 7.66 (d, J = 15.96 Hz, 1H, COCH
C), 7.57 (t, J = 6.93 Hz, 1H, ArH), 7.49 (d, J = 7.23, 1H, ArH), 7.36 (t, J = 7.62 Hz, 1H, ArH), 7.07 (d, J = 8.25 Hz, 1H, ArH), 6.97 (t, J = 7.35 Hz, ArH), 6.60 (d, J = 15.96 Hz, 1H, COCCH), 4.00 (d, J = 5.56 Hz, 2H,  ), 3.85 (s, 3H, OCH3), 3.55 (d, J = 5.71 Hz, 2H,
), 3.85 (s, 3H, OCH3), 3.55 (d, J = 5.71 Hz, 2H, 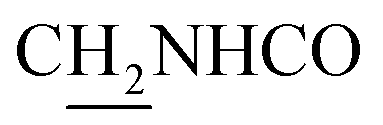 ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–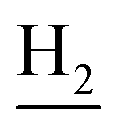 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–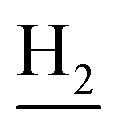 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–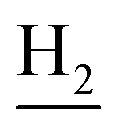 and C3–
and C3–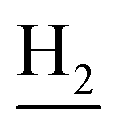 ). 13C NMR (125 MHz, DMSO-d6): δ 167.40, 158.11, 156.48, 151.24, 138.65, 135.01, 133.12, 131.55, 128.50, 125.63, 123.51, 122.25, 121.22, 119.85, 116.16, 111.15, 56.06, 48.86, 46.35, 28.61, 26.39, 24.24, 21.96, 20.85. HRMS (ESI): calcd for C25H27N3O2 [M + H]+ 402.2176, found 402.2169. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.63 min, 97.648%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.40, 158.11, 156.48, 151.24, 138.65, 135.01, 133.12, 131.55, 128.50, 125.63, 123.51, 122.25, 121.22, 119.85, 116.16, 111.15, 56.06, 48.86, 46.35, 28.61, 26.39, 24.24, 21.96, 20.85. HRMS (ESI): calcd for C25H27N3O2 [M + H]+ 402.2176, found 402.2169. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.63 min, 97.648%.
(E)-3-(3-Methoxyphenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (13). According to the procedure used to synthesize 8, 13 was synthesized from (E)-3-(3-methoxyphenyl)acrylic acid (0.15 g, 0.83 mmol) to give a white powder 0.11 g (33.03%). Mp 169–172 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.49–8.46 (m, 2H, ArH), 7.96 (br, 1H, CONH), 7.87 (t, J = 6.87 Hz, 1H, ArH), 7.78 (d, J = 7.50 Hz, 1H, ArH), 7.57 (t, J = 7.08 Hz, 1H, ArH), 7.40 (d, J = 15.81 Hz, 1H, COCH
C), 7.32 (t, J = 7.95 Hz, 1H, ArH), 7.13–7.10 (m, 2H, ArH and NH), 6.95 (dd, J1 = 7.95 Hz, J2 = 2.04 Hz, 1H, ArH), 6.57 (d, J = 15.81 Hz, 1H, COCCH), 4.02 (d, J = 5.65 Hz, 2H, 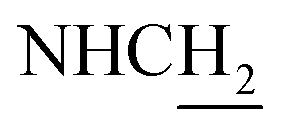 ), 3.77 (s, 3H, OCH3), 3.57 (d, J = 5.55 Hz, 2H,
), 3.77 (s, 3H, OCH3), 3.57 (d, J = 5.55 Hz, 2H, 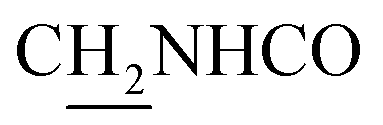 ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4–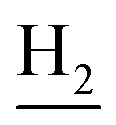 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–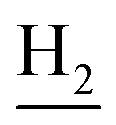 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–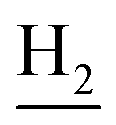 and C3–
and C3–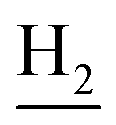 ). 13C NMR (125 MHz, CDCl3): δ 167.64, 159.86, 157.81, 151.14, 146.49, 141.27, 136.12, 129.83, 128.67, 127.50, 123.83, 122.92, 120.84, 120.39, 119.67, 115.72, 115.40, 113.16, 55.26, 49.87, 40.67, 33.37, 25.00, 22.93, 22.50. HRMS (ESI): calcd for C25H27N3O2 [M + H]+ 402.2176, found 402.2174. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.68 min, 98.401%.
). 13C NMR (125 MHz, CDCl3): δ 167.64, 159.86, 157.81, 151.14, 146.49, 141.27, 136.12, 129.83, 128.67, 127.50, 123.83, 122.92, 120.84, 120.39, 119.67, 115.72, 115.40, 113.16, 55.26, 49.87, 40.67, 33.37, 25.00, 22.93, 22.50. HRMS (ESI): calcd for C25H27N3O2 [M + H]+ 402.2176, found 402.2174. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.68 min, 98.401%.
(E)-3-(4-Methoxyphenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (14). According to the procedure used to synthesize 8, 14 was synthesized from (E)-3-(4-methoxyphenyl)acrylic acid (0.15 g, 0.83 mmol) to give a white powder 0.16 g (48.05%). Mp 180–182 °C. 1H NMR (300 MHz, DMSO-d6): δ 1H NMR (300 MHz, DMSO-d6): δ 8.47–8.44 (m, 2H, ArH), 7.93 (br, 1H, CONH), 7.72 (t, J = 7.01 Hz, 1H, ArH), 7.52 (d, J = 7.97 Hz, 2H, ArH), 7.46 (d, J = 15.93 Hz, 1H, COCH
C), 7.16–7.12 (m, 2H, ArH and NH), 6.99 (d, J = 7.98 Hz, 2H, ArH), 6.50 (d, J = 15.94 Hz, 1H, COCCH), 4.05 (d, J = 5.75 Hz, 2H, 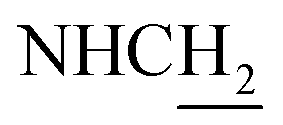 ), 3.83 (s, 3H, OCH3), 3.50 (d, J = 5.74 Hz, 2H,
), 3.83 (s, 3H, OCH3), 3.50 (d, J = 5.74 Hz, 2H, 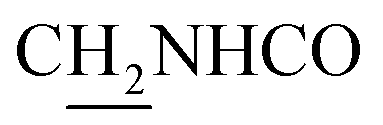 ), 2.89 (br, 2H, C4–
), 2.89 (br, 2H, C4–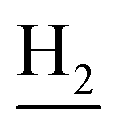 ), 2.72 (br, 2H, C1–
), 2.72 (br, 2H, C1–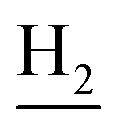 ), 1.86 (br, 4H, C2–
), 1.86 (br, 4H, C2–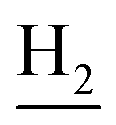 and C3–
and C3–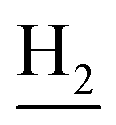 ). 13C NMR (125 MHz, DMSO-d6): δ 167.40, 158.11, 156.46, 151.26, 138.67, 135.01, 133.11, 131.55, 128.50, 125.62, 123.51, 122.24, 121.22, 119.87, 116.17, 112.16, 56.06, 48.86, 46.35, 28.62, 26.39, 24.24, 21.96, 20.86. HRMS (ESI): calcd for C25H27N3O2 [M + H]+ 402.2176, found 402.2182. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.44 min, 96.122%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.40, 158.11, 156.46, 151.26, 138.67, 135.01, 133.11, 131.55, 128.50, 125.62, 123.51, 122.24, 121.22, 119.87, 116.17, 112.16, 56.06, 48.86, 46.35, 28.62, 26.39, 24.24, 21.96, 20.86. HRMS (ESI): calcd for C25H27N3O2 [M + H]+ 402.2176, found 402.2182. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.44 min, 96.122%.
(E)-3-(2,3-Dimethoxyphenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (15). According to the procedure used to synthesize 8, 15 was synthesized from (E)-3-(2,3-dimethoxyphenyl)acrylic acid (0.17 g, 0.83 mmol) to give a white powder 0.2 g (55.87%). Mp 195–197 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.53–8.46 (m, 2H, ArH), 7.94 (br, 1H, CONH), 7.86 (t, J = 7.02 Hz, 1H, ArH), 7.78 (d, J = 7.71 Hz, 1H, ArH), 7.65–7.55 (m, 2H, COCH
C and ArH), 7.11–7.07 (m, 3H, NH and ArH), 6.59 (d, J = 15.96 Hz, 1H, COCCH), 4.05–3.98 (m, 2H, 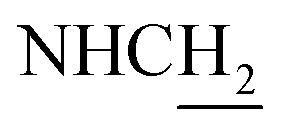 ), 3.81 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.56 (d, J = 5.37 Hz, 2H,
), 3.81 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.56 (d, J = 5.37 Hz, 2H, 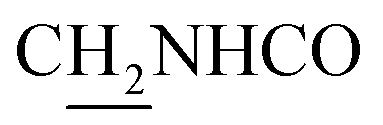 ), 2.87 (br, 2H, C4–
), 2.87 (br, 2H, C4–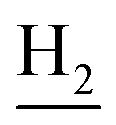 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–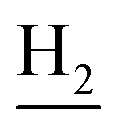 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–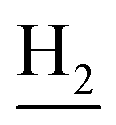 and C3–
and C3–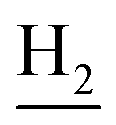 ). 13C NMR (125 MHz, DMSO-d6): δ 167.10, 156.69, 153.32, 147.97, 138.34, 134.39, 133.30, 128.70, 125.79, 125.61, 124.89, 123.09, 119.34, 116.03, 114.51, 111.96, 61.12, 60.23, 56.27, 48.73, 46.36, 28.43, 26.42, 24.23, 21.92, 21.01. HRMS (ESI): calcd for C26H29N3O3 [M + H]+ 432.2282, found 432.2278. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.21 min, 98.413%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.10, 156.69, 153.32, 147.97, 138.34, 134.39, 133.30, 128.70, 125.79, 125.61, 124.89, 123.09, 119.34, 116.03, 114.51, 111.96, 61.12, 60.23, 56.27, 48.73, 46.36, 28.43, 26.42, 24.23, 21.92, 21.01. HRMS (ESI): calcd for C26H29N3O3 [M + H]+ 432.2282, found 432.2278. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.21 min, 98.413%.
(E)-3-(2,5-Dimethoxyphenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (16). According to the procedure used to synthesize 8, 16 was synthesized from (E)-3-(2,5-dimethoxyphenyl)acrylic acid (0.17 g, 0.83 mmol) to give a white powder 0.14 g (39.11%). Mp 190–191 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.48–8.46 (m, 2H, ArH), 7.95 (br, 1H, CONH), 7.86 (t, J = 7.50 1H, ArH), 7.78 (d, J = 8.13 Hz, 1H, ArH), 7.65–7.55 (m, 2H, COCH
C and ArH), 7.04–6.93 (m, 3H, NH and ArH), 6.61 (d, J = 15.87 Hz, 1H, COCCH), 4.01 (d, J = 4.92 Hz, 2H, 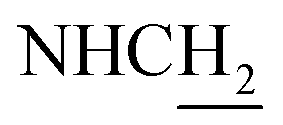 ), 3.79 (s, 3H, OCH3), 3.72 (s, 3H, OCH3), 3.56 (d, J = 5.22 Hz, 2H,
), 3.79 (s, 3H, OCH3), 3.72 (s, 3H, OCH3), 3.56 (d, J = 5.22 Hz, 2H, 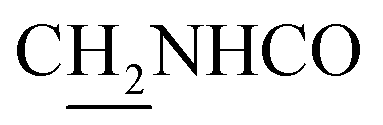 ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–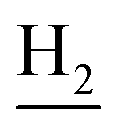 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–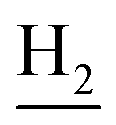 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–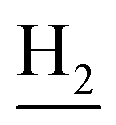 and C3–
and C3–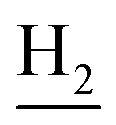 ). 13C NMR (125 MHz, DMSO-d6): δ 167.09, 156.69, 153.32, 147.96, 138.34, 134.39, 133.31, 128.70, 125.79, 125.61, 124.89, 123.09, 119.54, 119.14, 116.03, 114.51, 111.96, 61.12, 60.23, 56.27, 48.73, 46.36, 28.43, 26.42, 24.23, 21.92, 21.22, 20.80. HRMS (ESI): calcd for C26H29N3O3 [M + H]+ 432.2282, found 432.2277. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.63 min, 97.736%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.09, 156.69, 153.32, 147.96, 138.34, 134.39, 133.31, 128.70, 125.79, 125.61, 124.89, 123.09, 119.54, 119.14, 116.03, 114.51, 111.96, 61.12, 60.23, 56.27, 48.73, 46.36, 28.43, 26.42, 24.23, 21.92, 21.22, 20.80. HRMS (ESI): calcd for C26H29N3O3 [M + H]+ 432.2282, found 432.2277. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.63 min, 97.736%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(2,3,4-trimethoxyphenyl)acrylamide (17). According to the procedure used to synthesize 8, 17 was synthesized from (E)-3-(2,3,4-trimethoxyphenyl)acrylic acid (0.20 g, 0.83 mmol) to give a white powder 0.13 g (33.94%). Mp 206–208 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.50–8.47 (m, 2H, ArH), 8.00 (br, 1H, CONH), 7.88 (t, J = 7.46 Hz, 1H, ArH), 7.79 (d, J = 8.07 Hz, 1H, ArH), 7.62–7.51 (m, 2H, ArH and COCH
C), 7.28 (d, J = 8.82 Hz, 1H, ArH), 6.88 (d, J = 8.88 Hz, 1H, ArH), 6.53 (d, J = 15.84 Hz, 1H, COCCH), 4.03 (d, J = 5.25 Hz, 2H, 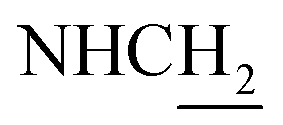 ), 3.83 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.57 (d, J = 5.82 Hz, 2H,
), 3.83 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.57 (d, J = 5.82 Hz, 2H, 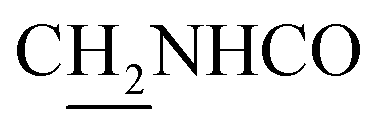 ), 2.96 (br, 2H, C4–
), 2.96 (br, 2H, C4–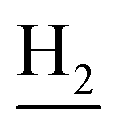 ), 2.69 (br, 2H, C1–
), 2.69 (br, 2H, C1–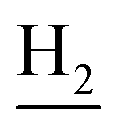 ), 1.84 (br, 4H, C2–
), 1.84 (br, 4H, C2–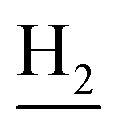 and C3–
and C3–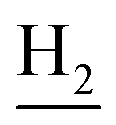 ). 13C NMR (125 MHz, DMSO-d6): δ 167.77, 158.45, 155.06, 152.73, 150.63, 147.46, 142.40, 134.25, 128.81, 128.31, 123.73, 123.48, 123.04, 121.74, 121.29, 120.56, 116.32, 108.89, 61.61, 60.87, 56.37, 48.79, 46.18, 34.05, 25.46, 23.25, 22.91. HRMS (ESI): calcd for C27H31N3O4 [M + H]+ 462.2387, found 462.2386. HPLC (80% methanol in water with 0.5% H3PO4): tR = 3.39 min, 96.333%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.77, 158.45, 155.06, 152.73, 150.63, 147.46, 142.40, 134.25, 128.81, 128.31, 123.73, 123.48, 123.04, 121.74, 121.29, 120.56, 116.32, 108.89, 61.61, 60.87, 56.37, 48.79, 46.18, 34.05, 25.46, 23.25, 22.91. HRMS (ESI): calcd for C27H31N3O4 [M + H]+ 462.2387, found 462.2386. HPLC (80% methanol in water with 0.5% H3PO4): tR = 3.39 min, 96.333%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (18). According to the procedure used to synthesize 8, 18 was synthesized from (E)-3-(3,4,5-trimethoxyphenyl)acrylic acid (0.20 g, 0.83 mmol) to give a white powder 0.17 g (44.39%). Mp 201–202 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.49–8.46 (m, 2H, ArH), 7.96–7.77 (m, 3H, CONH and ArH), 7.60–7.50 (m, 2H, COCH
C and ArH), 7.27 (d, J = 8.67 Hz, 1H, ArH), 6.86 (d, J = 8.79 Hz, 1H, NH) 6.51 (d, J = 15.75 Hz, 1H, COCCH), 4.02 (br, 2H, 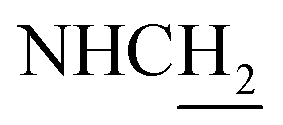 ), 3.82–3.54 (m, 11H, 3*OCH3 and
), 3.82–3.54 (m, 11H, 3*OCH3 and 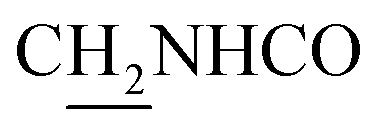 ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–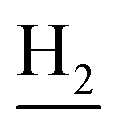 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–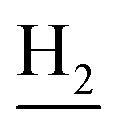 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–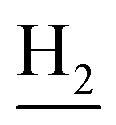 and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.43, 158.46, 153.57, 150.65, 150.63, 147.47, 139.61, 139.24, 130.97, 128.81, 128.32, 123.74, 123.48, 121.75, 120.55, 116.31, 105.48, 60.55, 56.34, 48.76, 34.05, 25.48, 23.25, 22.92. HRMS (ESI): calcd for C27H31N3O4 [M + H]+ 462.2387, found 462.2388. HPLC (70% methanol in water with 0.5% H3PO4): tR = 2.96 min, 98.112%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.43, 158.46, 153.57, 150.65, 150.63, 147.47, 139.61, 139.24, 130.97, 128.81, 128.32, 123.74, 123.48, 121.75, 120.55, 116.31, 105.48, 60.55, 56.34, 48.76, 34.05, 25.48, 23.25, 22.92. HRMS (ESI): calcd for C27H31N3O4 [M + H]+ 462.2387, found 462.2388. HPLC (70% methanol in water with 0.5% H3PO4): tR = 2.96 min, 98.112%.
(E)-3-(Benzo[d][1,3]dioxol-5-yl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (19)45. According to the procedure used to synthesize 8, 19 was synthesized from (E)-3-(benzo[d][1,3]dioxol-5-yl)acrylic acid (0.16 g, 0.83 mmol) to give a white powder 0.21 g (60.99%). Mp 197–199 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.48–8.42 (m, 2H, ArH), 7.95 (br, 1H, CONH), 7.80–7.77 (m, 2H, ArH), 7.56 (t, J = 7.32 Hz, 1H, ArH) 7.35 (d, J = 15.69 Hz, 1H, COCH
C), 7.14 (s, 1H, NH), 7.06 (d, J = 7.98 Hz, 1H, ArH), 6.94 (d, J = 7.98 Hz, 1H, ArH), 6.41 (d, J = 15.72 Hz, 1H, COCCH), 6.05 (s, 2H, OCH2O), 4.00 (d, J = 4.71 Hz, 2H,  ), 3.55 (d, J = 5.34 Hz, 2H,
), 3.55 (d, J = 5.34 Hz, 2H,  ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4– ), 2.69 (br, 2H, C1–
), 2.69 (br, 2H, C1– ), 1.82 (br, 4H, C2–
), 1.82 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 167.25, 156.50, 151.05, 149.12, 148.43, 139.75, 138.45, 133.19, 129.47, 125.76, 125.53, 123.96, 119.81, 119.64, 116.00, 111.89, 109.07, 106.70, 101.97, 48.87, 28.46, 24.21, 21.91, 20.80. HRMS (ESI): calcd for C25H25N3O3 [M + H]+ 416.1969, found 416.1964. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.44 min, 98.605%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.25, 156.50, 151.05, 149.12, 148.43, 139.75, 138.45, 133.19, 129.47, 125.76, 125.53, 123.96, 119.81, 119.64, 116.00, 111.89, 109.07, 106.70, 101.97, 48.87, 28.46, 24.21, 21.91, 20.80. HRMS (ESI): calcd for C25H25N3O3 [M + H]+ 416.1969, found 416.1964. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.44 min, 98.605%.
(E)-3-(2-Chlorophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (20). According to the procedure used to synthesize 8, 20 was synthesized from (E)-3-(2-chlorophenyl)acrylic acid (0.15 g, 0.83 mmol) to give a yellow powder 0.18 g (53.51%). Mp 201–203 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.60 (t, J = 5.70 Hz, 1H, ArH), 8.47 (d, J = 8.64 Hz, 1H, ArH) 7.89–7.84 (m, 2H, CONH and ArH), 7.80–7.66 (m, 3H, ArH and COCH
C), 7.60–7.51 (m, 2H, ArH), 7.43–7.38 (m, 2H, ArH and NH), 6.60 (d, J = 15.69 Hz, 1H, COCCH), 4.02 (d, J = 5.31 Hz, 2H,  ), 3.58 (d, J = 5.67 Hz, 2H,
), 3.58 (d, J = 5.67 Hz, 2H,  ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4– ), 2.67 (br, 2H, C1–
), 2.67 (br, 2H, C1– ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.36, 156.71, 151.06, 138.33, 135.12, 133.80, 133.32, 132.97, 131.60, 130.50, 128.32, 128.13, 125.77, 125.64, 125.01, 119.56, 116.05, 112.01, 48.51, 28.45, 24.24, 21.92, 20.81. HRMS (ESI): calcd for C24H24ClN3O [M + H]+ 406.1681, found 406.1675. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.69 min, 97.027%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.36, 156.71, 151.06, 138.33, 135.12, 133.80, 133.32, 132.97, 131.60, 130.50, 128.32, 128.13, 125.77, 125.64, 125.01, 119.56, 116.05, 112.01, 48.51, 28.45, 24.24, 21.92, 20.81. HRMS (ESI): calcd for C24H24ClN3O [M + H]+ 406.1681, found 406.1675. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.69 min, 97.027%.
(E)-3-(3-Chlorophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (21). According to the procedure used to synthesize 8, 21 was synthesized from (E)-3-(3-chlorophenyl)acrylic acid (0.15 g, 0.83 mmol) to give a yellow powder 0.17 g (50.53%). Mp 192–193 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.50–8.45 (m, 2H, ArH) 7.94–7.78 (m, 2H, CONH and ArH), 7.78 (d, J = 8.07 Hz, 1H, ArH) 7.62–7.51 (m, 3H, ArH and COCH
C), 7.44–7.39 (m, 3H, ArH and NH), 6.63 (d, J = 15.81 Hz, 1H, COCCH), 4.02 (d, J = 4.80 Hz, 2H,  ), 3.57 (d, J = 5.31 Hz, 2H,
), 3.57 (d, J = 5.31 Hz, 2H,  ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4– ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1– ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.59, 156.65, 150.99, 138.32, 137.43, 134.17, 133.34, 131.29, 129.78, 127.77, 126.56, 125.80, 125.61, 123.51, 119.52, 115.96, 111.91, 48.61, 28.39, 24.20, 21.89, 20.78. HRMS (ESI): calcd for C24H24ClN3O [M + H]+ 406.1681, found 406.1671. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.15 min, 95.756%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.59, 156.65, 150.99, 138.32, 137.43, 134.17, 133.34, 131.29, 129.78, 127.77, 126.56, 125.80, 125.61, 123.51, 119.52, 115.96, 111.91, 48.61, 28.39, 24.20, 21.89, 20.78. HRMS (ESI): calcd for C24H24ClN3O [M + H]+ 406.1681, found 406.1671. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.15 min, 95.756%.
(E)-3-(4-Chlorophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (22). According to the procedure used to synthesize 8, 22 was synthesized from (E)-3-(4-chlorophenyl)acrylic acid (0.15 g, 0.83 mmol) to give a yellow powder 0.2 g (59.45%). Mp 200–202 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.52–8.45 (m, 2H, ArH), 7.95 (br, 1H, CONH), 7.89–7.76 (m, 2H, ArH and COCH
C), 7.57 (br, 2H, ArH and NH), 7.48–7.40 (m, 3H, ArH), 6.57 (d, J = 15.81 Hz, 1H, COCCH), 4.02 (d, J = 5.07 Hz, 2H,  ), 3.57 (d, J = 5.37 Hz, 2H,
), 3.57 (d, J = 5.37 Hz, 2H,  ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4– ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1– ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.72, 156.63, 150.96, 138.53, 134.59, 134.08, 133.31, 129.80, 129.50, 125.80, 125.60, 122.61, 119.59, 116.00, 111.94, 48.65, 28.43, 24.21, 21.90, 20.79. HRMS (ESI): calcd for C24H24ClN3O [M + H]+ 406.1681, found 406.1673. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.23 min, 95.101%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.72, 156.63, 150.96, 138.53, 134.59, 134.08, 133.31, 129.80, 129.50, 125.80, 125.60, 122.61, 119.59, 116.00, 111.94, 48.65, 28.43, 24.21, 21.90, 20.79. HRMS (ESI): calcd for C24H24ClN3O [M + H]+ 406.1681, found 406.1673. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.23 min, 95.101%.
(E)-3-(4-Fluorophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (23). According to the procedure used to synthesize 8, 23 was synthesized from (E)-3-(4-fluorophenyl)acrylic acid (0.14 g, 0.83 mmol) to give a white powder 0.14 g (43.37%). Mp 198–199 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.50–8.45 (m, 2H, ArH), 7.96 (br, 1H, CONH), 7.89–7.76 (m, 2H, ArH), 7.64–7.55 (m, 3H, ArH and NH), 7.43 (d, J = 15.78 Hz, 1H, COCH
C), 7.24 (t, J = 8.64 Hz, 2H, ArH), 6.52 (d, J = 15.81 Hz, 1H, COCCH), 4.02 (d, J = 4.80 Hz, 2H,  ), 3.57 (d, J = 5.07 Hz, 2H,
), 3.57 (d, J = 5.07 Hz, 2H,  ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4– ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1– ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2– and C3–
and C3–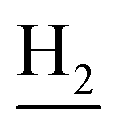 ). 13C NMR (125 MHz, DMSO-d6): δ 166.90, 164.09, 162.45, 156.65, 150.96, 138.71, 138.30, 133.32, 131.74, 130.28, 125.69, 121.68, 119.51, 116.51, 116.36, 115.95, 111.88, 48.74, 46.22, 28.38, 24.19, 21.89, 20.77. HRMS (ESI): calcd for C24H24FN3O [M + H]+ 390.1976, found 390.1968. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.30 min, 95.458%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.90, 164.09, 162.45, 156.65, 150.96, 138.71, 138.30, 133.32, 131.74, 130.28, 125.69, 121.68, 119.51, 116.51, 116.36, 115.95, 111.88, 48.74, 46.22, 28.38, 24.19, 21.89, 20.77. HRMS (ESI): calcd for C24H24FN3O [M + H]+ 390.1976, found 390.1968. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.30 min, 95.458%.
(E)-3-(4-Bromophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (24). According to the procedure used to synthesize 8, 24 was synthesized from (E)-3-(4-bromophenyl)acrylic acid (0.19 g, 0.83 mmol) to give a brown powder 0.19 g (50.91%). Mp 203–205 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.53 (br, 1H, ArH), 8.41 (d, J = 8.61 Hz, 1H, ArH), 7.79 (br, 2H, CONH and ArH), 7.62 (d, J = 8.31 Hz, 2H, ArH), 7.52 (d, J = 8.25 Hz, 4H, ArH and NH), 7.41 (d, J = 15.59 Hz, 1H, COCH
C), 6.60 (d, J = 15.81 Hz, 1H, COCCH), 3.93 (d, J = 5.70 Hz, 2H, 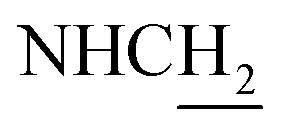 ), 3.55 (d, J = 5.34 Hz, 2H,
), 3.55 (d, J = 5.34 Hz, 2H, 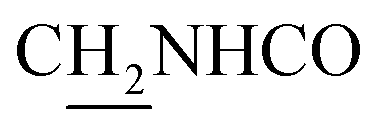 ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–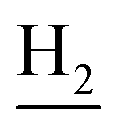 ), 2.69 (br, 2H, C1–
), 2.69 (br, 2H, C1–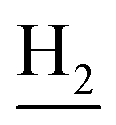 ), 1.82 (br, 4H, C2–
), 1.82 (br, 4H, C2–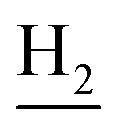 and C3–
and C3–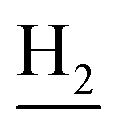 ). 13C NMR (125 MHz, DMSO-d6): δ 166.05, 158.46, 156.65, 150.59, 147.47, 138.16, 134.62, 132.33, 129.93, 128.84, 128.31, 123.75, 123.47, 123.27, 123.13, 120.60, 116.41, 116.36, 48.63, 46.20, 34.05, 25.48, 23.26, 22.93. HRMS (ESI): calcd for C24H24BrN3O [M + H]+ 450.1176, found 450.1181. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.79 min, 95.458%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.05, 158.46, 156.65, 150.59, 147.47, 138.16, 134.62, 132.33, 129.93, 128.84, 128.31, 123.75, 123.47, 123.27, 123.13, 120.60, 116.41, 116.36, 48.63, 46.20, 34.05, 25.48, 23.26, 22.93. HRMS (ESI): calcd for C24H24BrN3O [M + H]+ 450.1176, found 450.1181. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.79 min, 95.458%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(2-(trifluoromethyl)phenyl)acrylamide (25). According to the procedure used to synthesize 8, 25 was synthesized from (E)-3-(2-(trifluoromethyl)phenyl)acrylic acid (0.18 g, 0.83 mmol) to give a white powder 0.22 g (60.41%). Mp 183–184 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.62 (t, J = 5.43 Hz, 1H, ArH), 8.46 (d, J = 8.7 Hz, 1H, ArH) 7.89–7.84 (m, 2H, CONH and ArH), 7.80–7.73 (m, 4H, ArH and COCH
C), 7.70–7.68 (m, 1H, ArH), 7.62–7.55 (m, 2H, ArH and NH), 6.62 (d, J = 15.60 Hz, 1H, COCCH), 4.03 (d, J = 5.40 Hz, 2H, 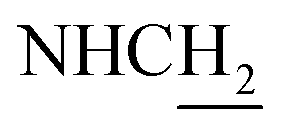 ), 3.59 (d, J = 5.76 Hz, 2H,
), 3.59 (d, J = 5.76 Hz, 2H, 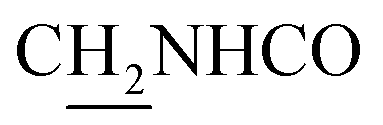 ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4–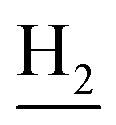 ), 2.67 (br, 2H, C1–
), 2.67 (br, 2H, C1–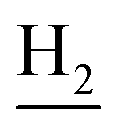 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–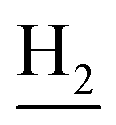 and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 165.95, 156.56, 138.43, 134.64, 133.61, 133.26, 130.23, 128.30, 127.40, 127.20, 126.64, 126.41, 125.74, 125.59, 123.73, 119.66, 48.40, 46.33, 28.47, 26.41, 26.38, 24.23, 21.88, 20.78. HRMS (ESI): calcd for C25H24F3N3O [M + H]+ 440.1944, found 440.1936. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.72 min, 97.899%.
). 13C NMR (125 MHz, DMSO-d6): δ 165.95, 156.56, 138.43, 134.64, 133.61, 133.26, 130.23, 128.30, 127.40, 127.20, 126.64, 126.41, 125.74, 125.59, 123.73, 119.66, 48.40, 46.33, 28.47, 26.41, 26.38, 24.23, 21.88, 20.78. HRMS (ESI): calcd for C25H24F3N3O [M + H]+ 440.1944, found 440.1936. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.72 min, 97.899%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(4-(trifluoromethyl)phenyl)acrylamide (26). According to the procedure used to synthesize 8, 26 was synthesized from (E)-3-(4-(trifluoromethyl)phenyl)acrylic acid (0.18 g, 0.83 mmol) to give a white powder 0.18 g (49.42%). Mp 182–184 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.62 (br, 1H, ArH), 8.47 (d, J = 8.61 Hz, 1H, ArH), 7.92–7.84 (m, 2H, CONH and ArH), 7.77 (br, 4H, ArH and NH), 7.58 (t, J = 7.44 Hz, 1H, ArH), 7.50 (d, J = 15.84 Hz, 1H, COCH
C), 6.62 (d, J = 15.60 Hz, 1H, COCCH), 4.03 (d, J = 5.40 Hz, 2H,  ), 3.59 (d, J = 5.76 Hz, 2H,
), 3.59 (d, J = 5.76 Hz, 2H,  ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4– ), 2.67 (br, 2H, C1–
), 2.67 (br, 2H, C1– ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.40, 156.60, 151.16, 139.21, 133.28, 138.38, 138.16, 133.28, 128.72, 126.34, 126.31, 125.76, 125.60, 125.45, 124.65, 123.65, 119.61, 116.02, 111.96, 48.50, 28.44, 24.24, 21.90, 20.80. HRMS (ESI): calcd for C25H24F3N3O [M + H]+ 440.1944, found 440.1948. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.36 min, 98.618%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.40, 156.60, 151.16, 139.21, 133.28, 138.38, 138.16, 133.28, 128.72, 126.34, 126.31, 125.76, 125.60, 125.45, 124.65, 123.65, 119.61, 116.02, 111.96, 48.50, 28.44, 24.24, 21.90, 20.80. HRMS (ESI): calcd for C25H24F3N3O [M + H]+ 440.1944, found 440.1948. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.36 min, 98.618%.
(E)-3-(2-Nitrophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (27). According to the procedure used to synthesize 8, 27 was synthesized from (E)-3-(2-nitrophenyl)acrylic acid (0.16 g, 0.83 mmol) to give a yellow powder 0.23 g (66.64%). Mp 216–218 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.63 (br, 1H, ArH), 8.45 (d, J = 8.55 Hz, 1H, ArH), 8.25 (d, J = 8.70 Hz, 2H, ArH), 7.87–7.76 (m, 4H, CONH and ArH and NH), 7.59–7.51 (m, 2H, ArH and COCH
C), 6.65 (d, J = 15.84 Hz, 1H, COCCH), 4.00 (d, J = 4.62 Hz, 2H,  ), 3.57 (d, J = 5.64 Hz, 2H,
), 3.57 (d, J = 5.64 Hz, 2H,  ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–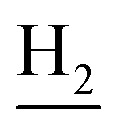 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–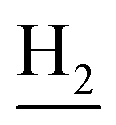 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–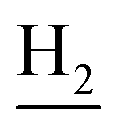 and C3–
and C3–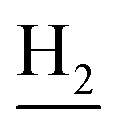 ). 13C NMR (125 MHz, DMSO-d6): δ 166.01, 156.51, 151.16, 148.77, 138.53, 135.02, 134.38, 133.23, 130.90, 130.30, 129.21, 126.44, 125.75, 125.59, 125.16, 119.76, 116.08, 112.07, 48.48, 28.53, 24.21, 21.91, 20.82. HRMS (ESI): calcd for C24H24N4O3 [M + H]+ 417.1921, found 417.192. HPLC (70% methanol in water with 0.5% H3PO4): tR = 2.97 min, 99.720%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.01, 156.51, 151.16, 148.77, 138.53, 135.02, 134.38, 133.23, 130.90, 130.30, 129.21, 126.44, 125.75, 125.59, 125.16, 119.76, 116.08, 112.07, 48.48, 28.53, 24.21, 21.91, 20.82. HRMS (ESI): calcd for C24H24N4O3 [M + H]+ 417.1921, found 417.192. HPLC (70% methanol in water with 0.5% H3PO4): tR = 2.97 min, 99.720%.
(E)-3-(3-Nitrophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (28). According to the procedure used to synthesize 8, 28 was synthesized from (E)-3-(3-nitrophenyl)acrylic acid (0.16 g, 0.83 mmol) to give a yellow powder 0.22 g (63.74%). Mp 210–213 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.57 (t, J = 5.55 Hz, 1H, ArH), 8.47 (d, J = 8.76 Hz, 1H, ArH), 8.37 (s, 1H, CONH), 8.21 (d, J = 6.87 Hz, 1H, ArH), 8.01 (d, J = 7.86 Hz, 1H, ArH) 7.94 (br, 1H, NH), 7.87 (t, J = 7.59 Hz, 1H, ArH), 7.77 (d, J = 8.43 Hz, 1H, ArH), 7.70 (t, J = 8.00 Hz, 1H, ArH), 7.60–7.53 (m, 2H, ArH and COCH
C), 6.75 (d, J = 15.81 Hz, 1H, COCCH), 4.03 (d, J = 5.31 Hz, 2H, 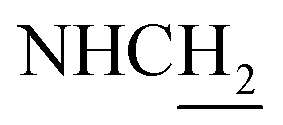 ), 3.59 (d, J = 5.58 Hz, 2H,
), 3.59 (d, J = 5.58 Hz, 2H, 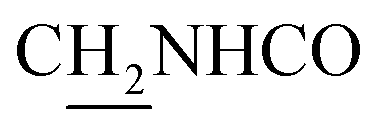 ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–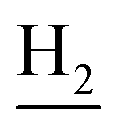 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–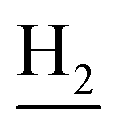 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.25, 156.54, 151.16, 148.76, 137.46, 137.01, 134.38, 133.22, 131.02, 125.76, 125.59, 124.80, 124.40, 122.16, 119.59, 116.03, 111.94, 48.40, 28.41, 24.24, 21.92, 20.78. HRMS (ESI): calcd for C24H24N4O3 [M + H]+ 417.1921, found 417.1921. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.19 min, 98.916%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.25, 156.54, 151.16, 148.76, 137.46, 137.01, 134.38, 133.22, 131.02, 125.76, 125.59, 124.80, 124.40, 122.16, 119.59, 116.03, 111.94, 48.40, 28.41, 24.24, 21.92, 20.78. HRMS (ESI): calcd for C24H24N4O3 [M + H]+ 417.1921, found 417.1921. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.19 min, 98.916%.
(E)-3-(4-Nitrophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (29). According to the procedure used to synthesize 8, 29 was synthesized from (E)-3-(4-nitrophenyl)acrylic acid (0.16 g, 0.83 mmol) to give a yellow powder 0.24 g (69.54%). Mp 215–217 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.64 (br, 1H, ArH), 8.46 (d, J = 8.73 Hz, 1H, ArH), 8.04 (d, J = 7.92 Hz, 1H, ArH), 7.86 (t, J = 7.25 Hz, 2H, CONH and ArH), 7.79–7.74 (m, 3H, ArH and COCH
C), 7.68–7.55 (m, 3H, ArH and NH), 6.55 (d, J = 15.54 Hz, 1H, COCCH), 4.02 (d, J = 5.43 Hz, 2H, 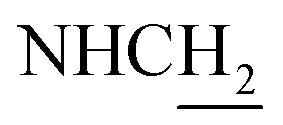 ), 3.59 (d, J = 5.58 Hz, 2H,
), 3.59 (d, J = 5.58 Hz, 2H, 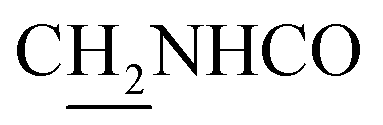 ), 2.96 (br, 2H, C4–
), 2.96 (br, 2H, C4–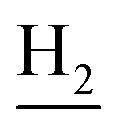 ), 2.67 (br, 2H, C1–
), 2.67 (br, 2H, C1–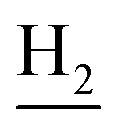 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–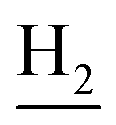 and C3–
and C3–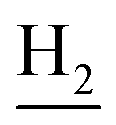 ). 13C NMR (125 MHz, DMSO-d6): δ 166.14, 156.43, 151.24, 148.06, 141.68, 138.55, 137.43, 133.16, 129.13, 126.08, 125.68, 125.55, 124.59, 123.22, 119.80, 116.09, 112.05, 48.43, 28.56, 24.26, 21.92, 20.84. HRMS (ESI): calcd for C24H24N4O3 [M + H]+ 417.1921, found 417.1914. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.27 min, 97.468%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.14, 156.43, 151.24, 148.06, 141.68, 138.55, 137.43, 133.16, 129.13, 126.08, 125.68, 125.55, 124.59, 123.22, 119.80, 116.09, 112.05, 48.43, 28.56, 24.26, 21.92, 20.84. HRMS (ESI): calcd for C24H24N4O3 [M + H]+ 417.1921, found 417.1914. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.27 min, 97.468%.
(E)-3-(4-Chloro-3-nitrophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (30). According to the procedure used to synthesize 8, 30 was synthesized from (E)-3-(4-chloro-3-nitrophenyl)acrylic acid (0.19 g, 0.83 mmol) to give a white powder 0.19 g (50.84%). Mp 208–210 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.56 (br, 1H, ArH), 8.47 (d, J = 8.55 Hz, 1H, ArH), 8.30–8.26 (m, 1H, ArH), 7.92–7.77 (m, 5H, CONH and ArH and NH), 7.58 (t, J = 7.52 Hz, 1H, ArH), 7.48 (d, J = 15.87 Hz, 1H, COCH
C), 6.71 (d, J = 15.84 Hz, 1H, COCCH), 4.02 (d, J = 5.52 Hz, 2H, 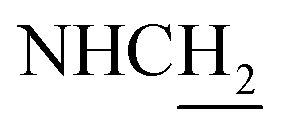 ), 3.58 (d, J = 5.64 Hz, 2H,
), 3.58 (d, J = 5.64 Hz, 2H, 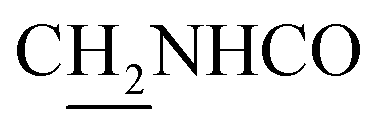 ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4–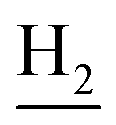 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–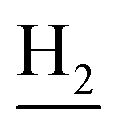 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.16, 156.61, 151.00, 148.44, 138.28, 136.52, 135.84, 133.31, 132.79, 132.64, 125.76, 125.60, 125.25, 124.67, 119.52, 115.94, 111.91, 48.47, 46.34, 28.39, 24.21, 21.88, 20.78. HRMS (ESI): calcd for C24H24ClN4O3 [M + H]+ 451.1532, found 451.1528. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.23 min, 98.389%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.16, 156.61, 151.00, 148.44, 138.28, 136.52, 135.84, 133.31, 132.79, 132.64, 125.76, 125.60, 125.25, 124.67, 119.52, 115.94, 111.91, 48.47, 46.34, 28.39, 24.21, 21.88, 20.78. HRMS (ESI): calcd for C24H24ClN4O3 [M + H]+ 451.1532, found 451.1528. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.23 min, 98.389%.
(E)-N-(2-((1,2,3,4-Tetrahydroacridin-9-yl)amino)ethyl)-3-(4-(trifluoromethoxy)phenyl)acrylamide (31). According to the procedure used to synthesize 8, 31 was synthesized from (E)-3-(4-(trifluoromethoxy)phenyl)acrylic acid (0.19 g, 0.83 mmol) to give a white powder 0.13 g (34.44%). Mp 188–189 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.54–8.48 (m, 2H, ArH), 7.94–7.79 (m, 3H, CONH and ArH), 7.56–7.38 (m, 5H, ArH and NH and COCH
C), 6.70–6.62 (m, 1H, COCCH), 4.02 (br, 2H,  ), 3.58 (br, 2H,
), 3.58 (br, 2H,  ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4– ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1– ), 1.82 (br, 4H, C2–
), 1.82 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.51, 156.62, 151.02, 149.28, 138.33, 138.18, 137.65, 133.32, 131.47, 127.10, 125.78, 125.60, 123.85, 122.37, 121.38, 120.28, 119.68, 119.55, 115.97, 111.92, 48.59, 28.41, 24.20, 21.89, 20.78. HRMS (ESI): calcd for C25H24FN3O2 [M + H]+ 456.1893, found 456.1886. HPLC (70% methanol in water with 0.5% H3PO4): tR = 6.05 min, 98.933%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.51, 156.62, 151.02, 149.28, 138.33, 138.18, 137.65, 133.32, 131.47, 127.10, 125.78, 125.60, 123.85, 122.37, 121.38, 120.28, 119.68, 119.55, 115.97, 111.92, 48.59, 28.41, 24.20, 21.89, 20.78. HRMS (ESI): calcd for C25H24FN3O2 [M + H]+ 456.1893, found 456.1886. HPLC (70% methanol in water with 0.5% H3PO4): tR = 6.05 min, 98.933%.
(E)-Methyl-(4-(3-oxo-3-((2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)amino)prop-1-en-1-yl)phenyl)-carbonate (32). According to the procedure used to synthesize 8, 32 was synthesized from (E)-3-(4-((methoxycarbonyl)oxy)phenyl)acrylic acid (0.18 g, 0.83 mmol) to give a white powder 0.2 g (54.17%). Mp 201–203 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.56–8.46 (m, 2H, ArH), 7.95 (br, 1H, CONH), 7.87 (t, J = 7.47 Hz, 1H, ArH), 7.78 (d, J = 8.16 Hz, 1H, ArH), 7.63–7.58 (m, 3H, ArH and NH), 7.45 (d, J = 15.78 Hz, 1H, COCH
C), 7.28 (d, J = 8.37 Hz, 1H, ArH), 6.55 (d, J = 15.78 Hz, 1H, COCCH), 4.02 (d, J = 4.74 Hz, 2H,  ), 3.83 (s, 3H, OCH3), 3.58 (br, 2H,
), 3.83 (s, 3H, OCH3), 3.58 (br, 2H,  ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4–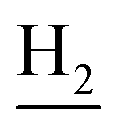 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–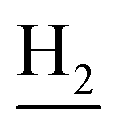 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–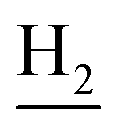 and C3–
and C3–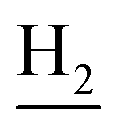 ). 13C NMR (125 MHz, DMSO-d6): δ 166.82, 156.64, 153.85, 151.98, 138.79, 133.31, 133.14, 129.36, 125.80, 125.59, 122.36, 122.13, 119.53, 115.97, 111.91, 56.05, 54.06, 48.69, 42.31, 36.93, 28.41, 24.20, 21.89, 20.79, 18.55, 17.19, 12.98. HRMS (ESI): calcd for C26H27N3O4 [M + H]+ 446.2074, found 446.2077. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.06 min, 95.785%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.82, 156.64, 153.85, 151.98, 138.79, 133.31, 133.14, 129.36, 125.80, 125.59, 122.36, 122.13, 119.53, 115.97, 111.91, 56.05, 54.06, 48.69, 42.31, 36.93, 28.41, 24.20, 21.89, 20.79, 18.55, 17.19, 12.98. HRMS (ESI): calcd for C26H27N3O4 [M + H]+ 446.2074, found 446.2077. HPLC (70% methanol in water with 0.5% H3PO4): tR = 3.06 min, 95.785%.
(E)-3-(2-(Benzyloxy)phenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (33). According to the procedure used to synthesize 8, 33 was synthesized from 7–33 (0.21 g, 0.83 mmol) to give a white powder 0.19 g (48.00%). Mp 116–118 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.48 (br, 2H, ArH), 7.95 (br, 1H, CONH), 7.88–7.75 (m, 3H, ArH), 7.57–7.51 (m, 2H, ArH), 7.46–7.32 (m, 6H, ArH and NH and COCH
C), 7.14 (d, J = 8.25 Hz, 1H, ArH), 6.98 (t, J = 7.41 Hz, 1H, ArH), 6.58 (d, J = 15.87 Hz, 1H, COCCH), 5.21 (s, 2H, OCH2), 4.02 (d, J = 4.92 Hz, 2H, 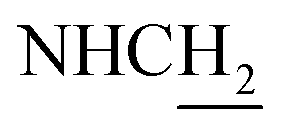 ), 3.56 (d, J = 5.22 Hz, 2H,
), 3.56 (d, J = 5.22 Hz, 2H, 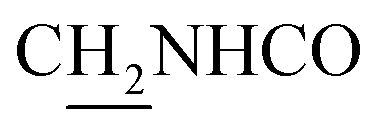 ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–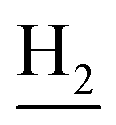 ), 2.66 (br, 2H, C1–
), 2.66 (br, 2H, C1–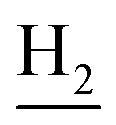 ), 1.80 (br, 4H, C2–
), 1.80 (br, 4H, C2–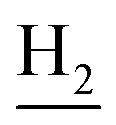 and C3–
and C3–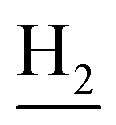 ). 13C NMR (125 MHz, DMSO-d6): δ 167.28, 156.92, 156.61, 150.94, 138.33, 137.35, 134.71, 133.29, 131.53, 129.00, 128.41, 128.00, 127.88, 125.80, 125.55, 123.86, 121.93, 121.48, 119.52, 115.94, 113.64, 111.88, 70.01, 48.91, 28.39, 24.17, 21.87, 20.77. HRMS (ESI): calcd for C31H31N3O2 [M + H]+ 478.2489, found 478.2476. HPLC (70% methanol in water with 0.5% H3PO4): tR = 7.96 min, 95.046%.
). 13C NMR (125 MHz, DMSO-d6): δ 167.28, 156.92, 156.61, 150.94, 138.33, 137.35, 134.71, 133.29, 131.53, 129.00, 128.41, 128.00, 127.88, 125.80, 125.55, 123.86, 121.93, 121.48, 119.52, 115.94, 113.64, 111.88, 70.01, 48.91, 28.39, 24.17, 21.87, 20.77. HRMS (ESI): calcd for C31H31N3O2 [M + H]+ 478.2489, found 478.2476. HPLC (70% methanol in water with 0.5% H3PO4): tR = 7.96 min, 95.046%.
(E)-3-(3-(Benzyloxy)phenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (34). According to the procedure used to synthesize 8, 34 was synthesized from 7–34 (0.21 g, 0.83 mmol) to give a white powder 0.18 g (45.48%). Mp 112–115 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.49 (br, 2H, ArH), 7.97 (br, 1H, CONH), 7.89 (t, J = 7.59 Hz, 1H, ArH), 7.79 (d, J = 7.95 Hz, 1H, ArH), 7.58 (t, J = 7.05 Hz, 1H, ArH), 7.46–7.32 (m, 7H, ArH and COCH
C), 7.20 (br, 1H, NH), 7.14 (d, J = 7.53 Hz, 1H, ArH), 7.03 (d, J = 6.57 Hz, 1H, ArH), 6.57 (d, J = 15.87 Hz, 1H, COCCH), 5.13 (s, 2H, OCH2Ar), 4.02 (d, J = 6.81 Hz, 2H, 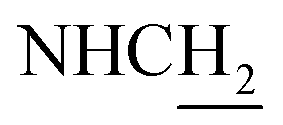 ), 3.57 (d, J = 5.25 Hz, 2H,
), 3.57 (d, J = 5.25 Hz, 2H, 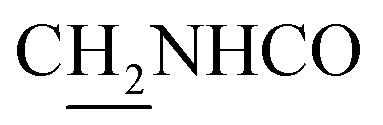 ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4–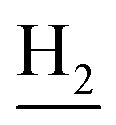 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–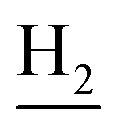 ), 1.83 (br, 4H, C2–
), 1.83 (br, 4H, C2–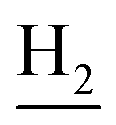 and C3–
and C3–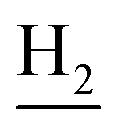 ). 13C NMR (125 MHz, DMSO-d6): δ 166.74, 159.13, 155.58, 152.06, 139.50, 137.37, 136.66, 132.36, 130.51, 128.93, 128.36, 128.24, 125.42, 125.27, 122.46, 120.87, 120.53, 116.61, 114.23, 112.43, 69.67, 48.43, 29.18, 24.51, 22.13, 21.05. HRMS (ESI): calcd for C31H31N3O2 [M + H]+ 478.2489, found 478.2483. HPLC (70% methanol in water with 0.5% H3PO4): tR = 8.69 min, 96.547%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.74, 159.13, 155.58, 152.06, 139.50, 137.37, 136.66, 132.36, 130.51, 128.93, 128.36, 128.24, 125.42, 125.27, 122.46, 120.87, 120.53, 116.61, 114.23, 112.43, 69.67, 48.43, 29.18, 24.51, 22.13, 21.05. HRMS (ESI): calcd for C31H31N3O2 [M + H]+ 478.2489, found 478.2483. HPLC (70% methanol in water with 0.5% H3PO4): tR = 8.69 min, 96.547%.
(E)-3-(4-(Benzyloxy)phenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (35). According to the procedure used to synthesize 8, 35 was synthesized from 7–35 (0.21 g, 0.83 mmol) to give a white powder 0.21 g (53.06%). Mp 120–122 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.49 (br, 2H, ArH), 7.99 (br, 1H, CONH), 7.86 (t, J = 7.58 Hz, 1H, ArH), 7.78 (d, J = 7.74 Hz, 1H, ArH), 7.59–7.32 (m, 9H, ArH and COCH
C and NH), 7.04 (d, J = 8.71 Hz, 2H, ArH), 6.43 (d, J = 15.78 Hz, 1H, COCCH), 5.13 (s, 2H, OCH2Ar), 4.01 (d, J = 5.01 Hz, 2H, 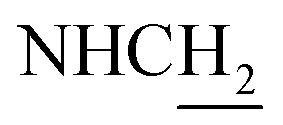 ), 3.55 (d, J = 5.37 Hz, 2H,
), 3.55 (d, J = 5.37 Hz, 2H, 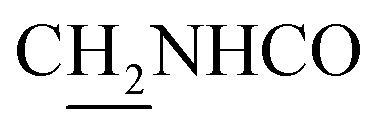 ), 2.94 (br, 2H, C4–
), 2.94 (br, 2H, C4–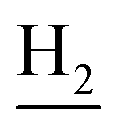 ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1–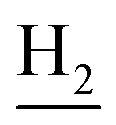 ), 1.82 (br, 4H, C2–
), 1.82 (br, 4H, C2– and C3–
and C3– ). 13C NMR (75 MHz, CDCl3): δ 167.39, 159.64, 157.91, 150.29, 146.63, 140.48, 135.98, 128.91, 128.15, 127.92, 127.64, 127.11, 126.96, 123.24, 122.28, 119.47, 117.61, 115.52, 114.64, 69.55, 49.43, 40.15, 33.29, 24.58, 22.51, 22.15. HRMS (ESI): calcd for C31H31N3O2 [M + H]+ 478.2489, found 478.2488. HPLC (70% methanol in water with 0.5% H3PO4): tR = 8.40 min, 96.871%.
). 13C NMR (75 MHz, CDCl3): δ 167.39, 159.64, 157.91, 150.29, 146.63, 140.48, 135.98, 128.91, 128.15, 127.92, 127.64, 127.11, 126.96, 123.24, 122.28, 119.47, 117.61, 115.52, 114.64, 69.55, 49.43, 40.15, 33.29, 24.58, 22.51, 22.15. HRMS (ESI): calcd for C31H31N3O2 [M + H]+ 478.2489, found 478.2488. HPLC (70% methanol in water with 0.5% H3PO4): tR = 8.40 min, 96.871%.
(E)-3-(4-(Benzyloxy)-3-methoxyphenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (36). According to the procedure used to synthesize 8, 36 was synthesized from 7–36 (0.24 g, 0.83 mmol) to give a white powder 0.25 g (59.43%). Mp 109–111 °C. 1H NMR (300 MHz, CDCl3): δ 7.99–7.90 (m, 2H, ArH), 7.59 (d, J = 15.57 Hz, 1H, COCH
C), 7.50–7.29 (m, 7H, ArH), 7.04–7.01 (m, 2H, ArH and NH), 6.86 (d, J = 8.04 Hz, 1H, ArH), 6.61 (br, 1H, CONH), 6.35 (d, J = 15.60 Hz, 1H, COCCH), 5.18 (s, 2H, OCH2Ar), 3.90 (s, 3H, OCH3), 3.76–3.71 (m, 4H, 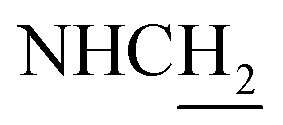 and
and 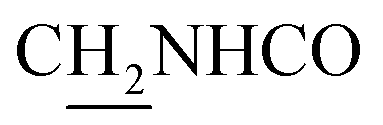 ), 3.02 (br, 2H, C4–
), 3.02 (br, 2H, C4– ), 2.66 (br, 2H, C1–
), 2.66 (br, 2H, C1– ), 1.85 (br, 4H, C2–
), 1.85 (br, 4H, C2– and C3–
and C3– ). 13C NMR (75 MHz, CDCl3): δ 167.50, 156.41, 151.86, 149.32, 149.18, 140.60, 136.09, 128.42, 128.11, 127.61, 127.52, 126.77, 126.16, 123.31, 122.55, 121.24, 118.61, 118.04, 114.53, 113.05, 110.04, 70.37, 55.46, 49.52, 39.99, 31.02, 24.36, 22.31, 21.77. HRMS (ESI): calcd for C32H34N3O3 [M + H]+ 508.2595, found 508.2591. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.97 min, 96.738%.
). 13C NMR (75 MHz, CDCl3): δ 167.50, 156.41, 151.86, 149.32, 149.18, 140.60, 136.09, 128.42, 128.11, 127.61, 127.52, 126.77, 126.16, 123.31, 122.55, 121.24, 118.61, 118.04, 114.53, 113.05, 110.04, 70.37, 55.46, 49.52, 39.99, 31.02, 24.36, 22.31, 21.77. HRMS (ESI): calcd for C32H34N3O3 [M + H]+ 508.2595, found 508.2591. HPLC (70% methanol in water with 0.5% H3PO4): tR = 5.97 min, 96.738%.
(E)-3-(2-Aminophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (37). 27 (0.2 g, 0.48 mmol) and stannous chloride dehydrate (0.54 g, 2.40 mmol) were dissolved in 10 mL EtOH, the mixture was stirred at 80 °C for 6 h. Then the reaction cooled to room temperature and acidized by AcOH. The reaction was filtered and the concentrated the filtrate. Then the filtrate was purified by column chromatography (CH2Cl2/MeOH = 40
:1, v/v, with 0.1% triethylamine) to give an orange powder 0.07 g (37.63%). Mp 205–206 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.81 (br, 1H, ArH), 8.55 (d, J = 7.71 Hz, 1H, ArH), 8.05 (br, 2H, CONH and ArH), 7.83 (br, 1H, ArH), 7.69 (d, J = 15.54 Hz, 1H, COCCH), 7.55 (br, 1H, ArH), 7.26 (d, J = 7.50 Hz, 1H, ArH), 7.02 (br, 1H, NH), 6.68 (d, J = 7.71 Hz, 1H, ArH), 6.53 (br, 1H, ArH), 6.40 (d, J = 15.57 Hz, 1H, COCCH), 5.42 (br, 2H, NH2), 4.02 (br, 2H, 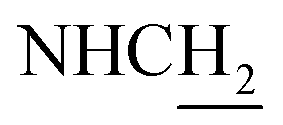 ), 3.54 (br, 2H,
), 3.54 (br, 2H, 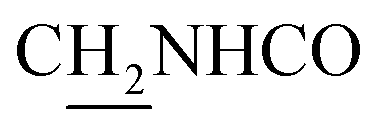 ), 2.73 (br, 2H, C1–
), 2.73 (br, 2H, C1– ), 1.82 (br, 4H, C2–
), 1.82 (br, 4H, C2– and C3–
and C3–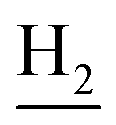 ). 13C NMR (125 MHz, DMSO-d6): δ 166.11, 156.23, 151.50, 148.09, 141.72, 137.47, 132.98, 130.80, 129.15, 126.14, 125.61, 125.49, 124.62, 123.39, 123.24, 116.30, 112.25, 48.41, 28.78, 24.32, 21.98, 20.92, 20.80. HRMS (ESI): calcd for C24H26N4O2 [M + H]+ 387.2179, found 387.2186. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.42 min, 96.101%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.11, 156.23, 151.50, 148.09, 141.72, 137.47, 132.98, 130.80, 129.15, 126.14, 125.61, 125.49, 124.62, 123.39, 123.24, 116.30, 112.25, 48.41, 28.78, 24.32, 21.98, 20.92, 20.80. HRMS (ESI): calcd for C24H26N4O2 [M + H]+ 387.2179, found 387.2186. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.42 min, 96.101%.
(E)-3-(3-Aminophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (38). According to the procedure used to synthesize 37, 38 was synthesized from 28 (0.2 g, 0.48 mmol) to give an orange powder 0.06 g (32.26%). Mp 207–209 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.78 (br, 1H, ArH), 8.58 (d, J = 7.60 Hz, 1H, ArH), 8.10 (br, 2H, CONH and ArH), 7.78 (br, 1H, ArH), 7.70 (d, J = 15.90 Hz, 1H, COCH
C), 7.59 (br, 1H, ArH), 7.22 (d, J = 7.66 Hz, 1H, ArH), 7.03 (br, 1H, NH), 6.66 (d, J = 7.80 Hz, 1H, ArH), 6.49 (br, 1H, ArH), 6.38 (d, J = 15.88 Hz, 1H, COCCH), 5.37 (br, 2H, NH2), 4.00 (br, 2H, 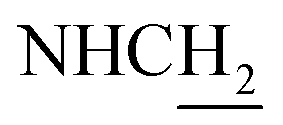 ), 3.59 (br, 2H,
), 3.59 (br, 2H, 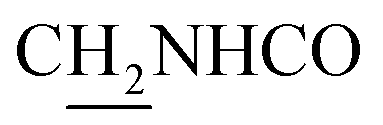 ), 2.70 (br, 2H, C1–
), 2.70 (br, 2H, C1–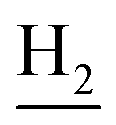 ), 1.81 (br, 4H, C2–
), 1.81 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): 167.68, 162.35, 148.16, 140.69, 135.83, 132.91, 130.78, 128.33, 127.04, 125.76, 125.49, 124.39, 122.36, 122.19, 116.62, 111.75, 48.39, 28.39, 24.44, 21.98, 21.82, 20.74, 20.56. HRMS (ESI): calcd for C24H26N4O2 [M + H]+ 387.2179, found 387.2183. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.69 min, 98.131%.
). 13C NMR (125 MHz, DMSO-d6): 167.68, 162.35, 148.16, 140.69, 135.83, 132.91, 130.78, 128.33, 127.04, 125.76, 125.49, 124.39, 122.36, 122.19, 116.62, 111.75, 48.39, 28.39, 24.44, 21.98, 21.82, 20.74, 20.56. HRMS (ESI): calcd for C24H26N4O2 [M + H]+ 387.2179, found 387.2183. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.69 min, 98.131%.
(E)-3-(4-Aminophenyl)-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)acrylamide (39). According to the procedure used to synthesize 37, 39 was synthesized from 29 (0.2 g, 0.48 mmol) to give an orange powder 0.07 g (37.63%). Mp 208–209 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.46 (d, J = 8.61 Hz, 1H, ArH), 8.38 (br, 1H, ArH), 7.87–7.79 (m, 4H, CONH and ArH and NH), 7.54 (t, J = 6.68 Hz, 1H, ArH), 7.30–7.20 (m, 3H, COCH
C and ArH), 6.54 (d, J = 8.28 Hz, 2H, ArH), 6.24 (d, J = 15.66 Hz, 1H, COCCH), 5.61 (br, 2H, NH2), 3.97 (d, J = 4.32 Hz, 2H,  ), 3.52 (d, J = 5.16 Hz, 2H,
), 3.52 (d, J = 5.16 Hz, 2H,  ), 2.95 (br, 2H, C4–
), 2.95 (br, 2H, C4– ), 2.68 (br, 2H, C1–
), 2.68 (br, 2H, C1– ), 1.81 (br, 4H, C2–
), 1.81 (br, 4H, C2– and C3–
and C3– ). 13C NMR (125 MHz, DMSO-d6): δ 166.12, 156.27, 148.09, 145.91, 141.72, 137.48, 133.03, 130.80, 129.15, 126.13, 125.64, 125.51, 124.62, 123.25, 120.10, 116.27, 112.21, 48.41, 28.74, 24.32, 21.97, 20.91, 20.79. HRMS (ESI): calcd for C24H26N4O2 [M + H]+ 387.2179, found 387.2175. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.33 min, 97.356%.
). 13C NMR (125 MHz, DMSO-d6): δ 166.12, 156.27, 148.09, 145.91, 141.72, 137.48, 133.03, 130.80, 129.15, 126.13, 125.64, 125.51, 124.62, 123.25, 120.10, 116.27, 112.21, 48.41, 28.74, 24.32, 21.97, 20.91, 20.79. HRMS (ESI): calcd for C24H26N4O2 [M + H]+ 387.2179, found 387.2175. HPLC (70% methanol in water with 0.5% H3PO4): tR = 4.33 min, 97.356%.
In vitro inhibitory evaluations on AChE and BuChE
The investigation of the inhibitory effects of the test compounds was performed followed the method of Ellman et al., using a Shimadzu 160 spectrophotometer. AChE (EC 3.1.1.7, Type VI-S, from Electric Eel, C3389; from human, C1682) and BuChE (EC 3.1.1.8, from equine serum, C0663; from human, B4186), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, D218200), acetylthiocholine iodide (ATC, A5751), and butyrylthiocholine iodide (BTC, B3253) were purchased from Sigma-Aldrich (St. Louis, MO, USA). AChE/BuChE stock solution was diluted before use to give 2.5 units per mL (for eeAChE, eqBuChE and huAChE) or 0.5 units per mL for huBuChE. ATC/BTC iodide solution (0.075 M) was prepared in deionized water. DTNB solution (0.01 M) was prepared in water containing 0.15% (w/v) sodium bicarbonate. For buffer preparation, potassium dihydrogen phosphate (1.36 g, 10 mmol) was dissolved in 100 mL of water. The pH of the solution was adjusted to 8.0 ± 0.1 by KOH. Stock solutions of the test compounds were dissolved in ethanol to give a final concentration of 10−4 M when diluted to the final volume of 3.32 mL. For each compound, a dilution series of at least five different concentrations (normally 10−5 to 10−9 M) were prepared.For measurement, a cuvette containing 3.0 mL of phosphate buffer, 100 μL of AChE or BuChE, 100 μL of DTNB and 100 μL of the test compound solution were added. After the addition of 20 μL of ATC or BTC, the reaction was initiated and solution was mixed immediately. Two minutes (eeAChE and eqBuChE) or fifteen minutes (huAChE and huBuChE) after substrate addition, the absorption was determined at 25 °C (eeAChE and eqBuChE) or 37 °C (huAChE and huBuChE) at 412 nm. For the reference value, 100 μL of water replaced the test compound solution. For determining the blank value, additionally 100 μL of water replaced the enzyme solution. The measurement for each concentration was performed in triplicate. The inhibition curve was fitted by plotting percentage enzyme activity (100% for the reference) versus logarithm of test compound concentration. The IC50 values were calculated by GraphPad Prism 5 and the data were shown in mean ± SEM.
Kinetic studies of AChE inhibition
Kinetic studies were performed in the same manner to the determination of ChEs inhibition, while the substrate (ATC/BTC) was used in concentrations of 25, 50, 90, 150, 226, and 452 μM. The concentrations of test compounds were set to 0, 20, 60, 100, 200 nM for 36. The enzymatic reaction was extended to 4 min (eeAChE and eqBuChE) or 20 min (huAChE and huBuChE) before the determination of the absorption. Vmax and Km values of the Michaelis–Menten kinetics were calculated by nonlinear regression from substrate–velocity curves using GraphPad Prism 5. Linear regression was used for fitting the Lineweaver–Burk plots.Molecular modeling studies
The docking study was performed by CDOCKER module implemented in Discovery Studio (version 3.0, BIOVIA, USA). CDOCKER is a grid-based molecular docking method that employs CHARMm.46 Random ligand conformations are generated from the initial ligand structure through high temperature molecular dynamics, followed by random rotations. The random conformations are refined by grid-based simulated annealing and a final grid-based or full forcefield minimization. The solutions are then clustered according to position and conformation and ranked by energy.The co-crystal structure of huAChE bound with donepezil was selected for molecular docking. The structure was download from Protein Data Bank (PDB, ID: 4EY7). It was prepared by “Prepare Protein” module in DS for further docking. Missed sidechains were added and the water molecules were removed, then it was protonated at pH 7.4. For the test compounds docked into huAChE, they were first sketched in DS, and then prepared by using “Prepare Ligands” module to protonate at pH 7.4. The resulted molecules were minimized by “Minimize Ligands” module. The “Smart Minimizer” algorithm was used to perform the minimization, with max steps set to 2000, RMS Gradient set to 0.01. Other parameters were set as default.47
A sphere (in 10 Å radius) around donepezil was determined as the binding site, including both CAS and PAS of huAChE. For the simulated annealing, heating steps and cooling steps was set to 2000 and 5000, respectively, while heating and cooling temperature was set to 700, and 310, respectively. Other parameters were kept as default. After docking, ten top-ranked conformations were retained for analysis. Binding patterns of the docked molecules were described by DS visualizer.48
Inhibition of self-induced Aβ1–42 aggregation
Inhibition of self-induced Aβ1–42 aggregation was measured using a Thioflavin T (ThT)-(T3516, Sigma-Aldrich, St. Louis, MO, USA) binding assay as previously described.49 Aliquots of 2.0 μL of Aβ1–42 (AS-64129-05 Anaspec Inc.), lyophilized from 2 mg mL−1 HFIP (1,1,1,3,3,3-hexafluoro-2-propanol, 52517, Sigma-Aldrich, St. Louis, MO, USA) and dissolved in DMSO, were incubated for 24 h at room temperature in 0.215 M sodium phosphate buffer (pH 8.0) at a final concentration of 500 μM. Test compounds were dissolved in DMSO and then diluted by buffer to a final concentration of 20 μM. After incubation, the samples were diluted to a final volume of 150 μL with 50 mM glycine–NaOH buffer (pH 8.5) containing 5 mM thioflavin T. Fluorescence signal was determined (excitation wavelength 450 nm, emission wavelength 485 nm) on a SpectraMax Paradigm Multimode Reader (Molecular Device, USA).The inhibitory rate of Aβ1–42 aggregation was calculated according to the following equation: (1-IFi/IFc) × 100%. Here, IFi and IFc were the fluorescence intensities obtained for absorbance in the presence and absence of inhibitors, respectively, after subtracting the background fluorescence of the 5 mM thioflavin T solution. Each measurement was measured in triplicate. The inhibitory rate of the test compound was shown in mean ± SD.
Cyto-protection and cell toxicity against PC-12 neuroblastoma cells
Cytotoxicity was determined by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. The PC-12 cell line was purchased from Cell Culture Center at the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences. MTT was purchased from Sigma (M2128, St. Louis, MO). It was dissolved in phosphate buffered saline (PBS) to a stock concentration of 5 mg mL−1 and stored at −20 °C. PC-12 cells were plated in 96-well plates, raised to a population of 1 × 104 cells per well, and incubated overnight. After cells were treated with density gradient of test compounds or DMSO for 24 h at 37 °C or treated with 500 μM H2O2 for another 12 h, 20.0 μL of MTT solution was added into each well of the plate and incubated for 4 h. Then the solution was removed and 150.0 μL of DMSO was added into each well to dissolve the MTT-formazan crystals. DMSO was used as a negative control. The absorbance values (OD value) were read at 570 nm by Elx800 Absorbance Microplate Reader (BioTek, Vermont, USA). The inhibitory rate for each concentration of the test compound was calculated by the equation as follows:| IR = [1 − (ODtest − ODblank)/(ODcontrol − ODblank)] × 100%. |
A statement identifying the institutional and/or licensing committee experimental approval
All animal studies were performed in compliance with the Chinese Academy of Medical Sciences and the Animal Ethics Committees of the Institute of Materia Medica. The animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with the approval of the Center for New Drug Evaluation and Research of China Pharmaceutical University (Nanjing, China).Behavioral studies
Behavioral studies were performed by using adult male ICR mice (8–10 weeks old, weight 20–25 g), which were purchased from the Yangzhou University Medical Center (Yangzhou, China). Scopolamine hydrobromide was supplied by Aladdin Reagents (H1507073, Shanghai, China). Tacrine was synthesized in our lab with >95% purity as determined by HPLC.The mice were separated into five groups as follows: (i) vehicle as blank control, (ii) scopolamine as model group, (iii) tacrine plus scopolamine as positive control, (iv) compound 36 plus scopolamine as test group, and (v) compound 35 plus scopolamine as test group. Tacrine, 36 and 35 (20 μmol kg−1 body weight) were orally administered to mice in groups (iii), (iv), and (v), respectively, 30 min before the ip administration of scopolamine (1 mg kg−1) or saline for 10 consecutive days.
Cognitive function was evaluated by the Morris water maze analysis-management system (Panlab SMART 3.0, America), according to the method previously described.37 The maze was placed in a lit room with visual cues at 25 °C. An escape platform (10 cm diameter) was located in the center of one quadrant of the circular pool (120 cm diameter, 60 cm height) with a depth of 40 cm water. The behavioral study of each mouse included 5 days of learning and memory training and a probe trial on day 6. The animal starting positions faced to the pool wall, and were pseudorandomized for each trial. For the cognitive evaluation, each mouse was individually evaluated on both visible-platform (days 1–2) and hidden-platform (days 3–5) versions of the water maze. All mice received nonspatial pretraining during the first two training days, which prepared them for the subsequent spatial learning test. During the two days, mice were trained to find the platform that was labeled by a small flag (5 cm tall). The hidden-platform version was used to determine the retention of memory to find the platform. During the hidden-platform training trials, the escape platform was placed 1 cm below the surface of the water. On each day, the animal was subjected to two trials, each of which lasted for 90 s. The time for the mouse to find the platform (a successful escape) was recorded. If a mouse failed to reach the platform within 90 s, the test was terminated and the animal was gently navigated to the platform by hand. Whether a mouse was successful or failed to reach the platform within 90 s, it was kept on the platform for 30 s. On the last day (day 6), the platform was removed from its location and the animals were given a probe trial in which they had 90 s to search for the platform. The time taken to reach the missing platform and the number of times the animals crossed the platform location were recorded.
Data for the time of escape latency, the trajectory traveled, and the number of platform location crossings were recorded by Panlab SMART 3.0 and processed by Graphpad Prism 5.
Hepatotoxicity studies
Hepatotoxicity was evaluated according to the method previously described32 by using adult male ICR mice (8–10 weeks old, weighing 20–25 g) obtained from the Yangzhou University Medicine Centre (Yangzhou, China). Tacrine and the test compounds were dissolved in a sodium carboxymethyl cellulose (CMC-Na) solution (0.5 g CMC-Na in 100 mL distilled water). Concentration of 3 mg/100 g body wt of tacrine, corresponding to 151.5 μmol kg−1 body wt, was administered intragastrically (ig). Equimolar dose of test compounds to that of tacrine was administered ig. 8, 22, and 36 h after the administration, heparinized serum was collected from the retrobulbar plexus and subjected to hepatotoxicity evaluation. The activity of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), two indicators of liver damage, was determined using corresponding assay kit (EF551 and EF550 for ALT, EH027 and EF548 for AST, Wako, Japan). The data were processed by Biochemical Analyzer (HITACHI 7020, Japan).1 h after the collection of retrobulbar blood, mice were sacrificed and livers were harvested for morphological studies by using immunohistochemical method. Two 3 mm sections of each liver extending from the hilus to the margin of the left lateral lobe were isolated by Ultra-Thin Semiautomatic Microtome (Leica RM2245, Germany) and immediately placed in 10% buffered formaldehyde, fixed for two days, and embedded together in one paraffin block by using Paraffin Embedding Station (Leica EG1150H, Germany). Subsequently, 5 μm sections were prepared from these paraffin blocks. They were deparaffinated and stained with hematoxylin and eosin or by means of the periodic acid-Schiff procedure for glycogen.
Author contributions
C. Y. wrote the result and discussion of main manuscript text. S. P. wrote the introduction part. L. Z. wrote the methods and materials of the manuscript. Z. J., G. K., L. Q., H. Y. and L. X. were responsible for the synthesis of the inhibitor. P. Q. was responsible for the characterization of the compounds. B. Y. and W. L. were responsible for the biological determination. T. X. revised the manuscript. All authors reviewed the manuscript.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
We gratefully thank the support from the grants 81402851 and 81573281 of National Natural Science Foundation of China, BK20140957 of Natural Science Foundation of Jiangsu Province. We also thank the support from Fundamental Research Funds for the Central Universities (2015ZD009), Jiangsu Qing Lan Project, Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP-PPZY2015A070) and Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- V. Tumiatti, A. Minarini, M. L. Bolognesi, A. Milelli, M. Rosini and C. Melchiorre, Curr. Med. Chem., 2010, 17, 1825–1838 CrossRef CAS PubMed.
- H. Tang, H. T. Zhao, S. M. Zhong, Z. Y. Wang, Z. F. Chen and H. Liang, Bioorg. Med. Chem. Lett., 2012, 22, 2257–2261 CrossRef CAS PubMed.
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741–766 CAS.
- J. Hardy, Neuron, 2006, 52, 3–13 CrossRef CAS PubMed.
- P. T. Francis, A. M. Palmer, M. Snape and G. K. Wilcock, J. Neurol., Neurosurg. Psychiatry, 1999, 66, 137–147 CrossRef CAS.
- E. Karran, M. Mercken and B. De Strooper, Nat. Rev. Drug Discovery, 2011, 10, 698–712 CrossRef CAS PubMed.
- C. Ballatore, V. M. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2007, 8, 663–672 CrossRef CAS PubMed.
- K. M. Webber, A. K. Raina, M. W. Marlatt, X. Zhu, M. I. Prat, L. Morelli, G. Casadesus, G. Perry and M. A. Smith, Mech. Ageing Dev., 2005, 126, 1019–1025 CrossRef CAS PubMed.
- L. Tapia-Arancibia, E. Aliaga, M. Silhol and S. Arancibia, Brain Res. Rev., 2008, 59, 201–220 CrossRef CAS PubMed.
- B. Su, X. Wang, A. Nunomura, P. I. Moreira, H. G. Lee, G. Perry, M. A. Smith and X. Zhu, Curr. Alzheimer Res., 2008, 5, 525–532 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- K. Jomova and M. Valko, Toxicology, 2011, 283, 65–87 CrossRef CAS PubMed.
- L. Devi and H. K. Anandatheerthavarada, Biochim. Biophys. Acta, 2010, 1802, 11–19 CrossRef CAS PubMed.
- U. Holzgrabe, P. Kapkova, V. Alptuzun, J. Scheiber and E. Kugelmann, Expert Opin. Ther. Targets, 2007, 11, 161–179 CrossRef CAS PubMed.
- J. L. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker and I. Silman, Science, 1991, 253, 872–879 CrossRef CAS PubMed.
- Y. Bourne, P. Taylor, Z. Radic and P. Marchot, EMBO J., 2003, 22, 1–12 CrossRef CAS PubMed.
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary, J. Love, M. C. Franklin and J. J. Height, J. Med. Chem., 2012, 55, 10282–10286 CrossRef CAS PubMed.
- M. Bartolini, C. Bertucci, V. Cavrini and V. Andrisano, Biochem. Pharmacol., 2003, 65, 407–416 CrossRef CAS PubMed.
- N. C. Inestrosa, J. P. Sagal and M. Colombres, Subcell. Biochem., 2005, 38, 299–317 CAS.
- S. Darvesh, D. A. Hopkins and C. Geula, Nat. Rev. Neurosci., 2003, 4, 131–138 CrossRef CAS PubMed.
- C. Chianella, D. Gragnaniello, P. Maisano Delser, M. F. Visentini, E. Sette, M. R. Tola, G. Barbujani and S. Fuselli, Eur. J. Clin. Pharmacol., 2011, 67, 1147–1157 CrossRef CAS PubMed.
- A. V. Terry Jr and J. J. Buccafusco, J. Pharmacol. Exp. Ther., 2003, 306, 821–827 CrossRef CAS PubMed.
- M. Bajda, N. Guzior, M. Ignasik and B. Malawska, Curr. Med. Chem., 2011, 18, 4949–4975 CrossRef CAS PubMed.
- S. Leone, A. Ottani and A. Bertolini, Curr. Top. Med. Chem., 2007, 7, 265–275 CrossRef CAS PubMed.
- A. Minarini, A. Milelli, E. Simoni, M. Rosini, M. L. Bolognesi, C. Marchetti and V. Tumiatti, Curr. Top. Med. Chem., 2013, 13, 1771–1786 CrossRef CAS PubMed.
- R. A. de Aquino, L. V. Modolo, R. B. Alves and A. de Fatima, Curr. Drug Targets, 2013, 14, 378–397 CAS.
- M. Benchekroun, M. Bartolini, J. Egea, A. Romero, E. Soriano, M. Pudlo, V. Luzet, V. Andrisano, M. L. Jimeno, M. G. Lopez, S. Wehle, T. Gharbi, B. Refouvelet, L. de Andres, C. Herrera-Arozamena, B. Monti, M. L. Bolognesi, M. I. Rodriguez-Franco, M. Decker, J. Marco-Contelles and L. Ismaili, ChemMedChem, 2015, 10, 523–539 CrossRef CAS PubMed.
- M. A. Ceschi, J. S. da Costa, J. P. Lopes, V. S. Camara, L. F. Campo, A. C. Borges, C. A. Goncalves, D. F. de Souza, E. L. Konrath, A. L. Karl, I. A. Guedes and L. E. Dardenne, Eur. J. Med. Chem., 2016, 121, 758–772 CrossRef CAS PubMed.
- M. Digiacomo, Z. Chen, S. Wang, A. Lapucci, M. Macchia, X. Yang, J. Chu, Y. Han, R. Pi and S. Rapposelli, Bioorg. Med. Chem. Lett., 2015, 25, 807–810 CrossRef CAS PubMed.
- R. S. Keri, C. Quintanova, S. M. Marques, A. R. Esteves, S. M. Cardoso and M. A. Santos, Bioorg. Med. Chem., 2013, 21, 4559–4569 CrossRef CAS PubMed.
- J. S. Lan, S. S. Xie, S. Y. Li, L. F. Pan, X. B. Wang and L. Y. Kong, Bioorg. Med. Chem., 2014, 22, 6089–6104 CrossRef CAS PubMed.
- S. Y. Li, N. Jiang, S. S. Xie, K. D. Wang, X. B. Wang and L. Y. Kong, Org. Biomol. Chem., 2014, 12, 801–814 CAS.
- S. Liao, H. Deng, S. Huang, J. Yang, S. Wang, B. Yin, T. Zheng, D. Zhang, J. Liu, G. Gao, J. Ma and Z. Deng, Bioorg. Med. Chem. Lett., 2015, 25, 1541–1545 CrossRef CAS PubMed.
- J. Misik, J. Korabecny, E. Nepovimova, A. Kracmarova and J. Kassa, Neurosci. Lett., 2016, 612, 261–268 CrossRef CAS PubMed.
- E. Nepovimova, J. Korabecny, R. Dolezal, K. Babkova, A. Ondrejicek, D. Jun, V. Sepsova, A. Horova, M. Hrabinova, O. Soukup, N. Bukum, P. Jost, L. Muckova, J. Kassa, D. Malinak, M. Andrs and K. Kuca, J. Med. Chem., 2015, 58, 8985–9003 CrossRef CAS PubMed.
- B. N. Saglik, S. Ilgin and Y. Ozkay, Eur. J. Med. Chem., 2016, 124, 1026–1040 CrossRef CAS PubMed.
- X. Zha, D. Lamba, L. Zhang, Y. Lou, C. Xu, D. Kang, L. Chen, Y. Xu, L. Zhang, A. De Simone, S. Samez, A. Pesaresi, J. Stojan, M. G. Lopez, J. Egea, V. Andrisano and M. Bartolini, J. Med. Chem., 2016, 59, 114–131 CrossRef CAS PubMed.
- C. Zhang, Q. Y. Du, L. D. Chen, W. H. Wu, S. Y. Liao, L. H. Yu and X. T. Liang, Eur. J. Med. Chem., 2016, 116, 200–209 CrossRef CAS PubMed.
- B. Sameem, M. Saeedi, M. Mahdavi and A. Shafiee, Eur. J. Med. Chem., 2017, 128, 332–345 CrossRef CAS PubMed.
- L. Fang, B. Kraus, J. Lehmann, J. Heilmann, Y. Zhang and M. Decker, Bioorg. Med. Chem. Lett., 2008, 18, 2905–2909 CrossRef CAS PubMed.
- Y. Chen, J. Sun, L. Fang, M. Liu, S. Peng, H. Liao, J. Lehmann and Y. Zhang, J. Med. Chem., 2012, 55, 4309–4321 CrossRef CAS PubMed.
- P. R. Carlier, Y. F. Han, E. S. Chow, C. P. Li, H. Wang, T. X. Lieu, H. S. Wong and Y. P. Pang, Bioorg. Med. Chem., 1999, 7, 351–357 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres Jr and R. M. Feather-Stone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- S. L. Cockroft, C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Am. Chem. Soc., 2005, 127, 8594–8595 CrossRef CAS PubMed.
- C. Quintanova, R. S. Keri, S. M. Marques, M. G. Fernandes, S. M. Cardoso, M. L. Serralheiro and M. A. Santos, MedChemComm, 2015, 6, 1969–1977 RSC.
- G. Wu, D. H. Robertson, C. L. Brooks 3rd and M. Vieth, J. Comput. Chem., 2003, 24, 1549–1562 CrossRef CAS PubMed.
- Y. Chen, X. Xu, T. Fu, W. Li, Z. Liu and H. Sun, RSC Adv., 2015, 5, 90288–90294 RSC.
- M. Bartolini, C. Bertucci, M. L. Bolognesi, A. Cavalli, C. Melchiorre and V. Andrisano, ChemBioChem, 2007, 8, 2152–2161 CrossRef CAS PubMed.
- L. L. Xu, X. Zhang, Z. Y. Jiang and Q. D. You, Bioorg. Med. Chem., 2016, 24, 3540–3547 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04385f |
| This journal is © The Royal Society of Chemistry 2017 |