
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of integrated microstructure on the performance of LiNi0.8Co0.15Al0.05O2 as a cathodic material for lithium ion batteries†
Yongjie Chenab,
Ping Lib,
Sijia Zhaoa,
Yan Zhuangab,
Shiyong Zhaoc,
Qun Zhou*a and
Junwei Zheng *ab
*ab
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China. E-mail: zhq@suda.edu.cn
bCollege of Physics, Optoelectronics and Energy, Soochow University, Suzhou 215006, P. R. China. E-mail: jwzheng@suda.edu.cn
cZhangjiagang Guotai Huarong New Chemical Material Co., Ltd, Zhangjiagang, P. R. China
First published on 5th June 2017
Abstract
Microstructures of active materials may definitively determine the performance of lithium ion batteries. Herein, we develop a facile approach to synthesize porous LiNi0.8Co0.15Al0.05O2 (NCA) with uniform Al distribution by a two-step solid reaction with assistance of spray drying. Relative to the randomly aggregated counterpart, the NCA microspheres with an integrated framework and porous structure result in not only a profitable accessibility of the electrolyte, but also a favorable interfacial behavior. The porous NCA spheres exhibit a superior electrochemical performance with a discharge capacity of 202.1 mA h g−1 at 0.1C and 151 mA h g−1 at 2C, and capacity retention of 74.5% after 500 cycles at 2C. These are ascribed to the integrated network accumulating the stress generated during cycling to maintain the structural stability of the spheres. As a result, less solid electrolyte interphase (SEI) film is formed at the interface of the resulting electrode, consequently leading to a lower resistance of charge transfer, and better rate capability and cycling performance, compared to those of the electrode with the aggregated counterpart. Thereby, a purposeful engineering of the microstructures of the NCA materials would be important to achieve an optimal electrochemical performance of the electrode material.
1. Introduction
Nickel-rich layered LiNiO2-based materials have drawn great attention as the cathode materials for lithium ion batteries, in particular to meet the requirements as the power sources for hybrid electric vehicles (HEV) and electric vehicles (EV), due to their high capacity, low cost and toxicity.1,2 Among those layered materials, LiNi0.8Co0.15Al0.05O2 (NCA) has been successfully used in lithium ion batteries in commercialized EVs. So far, various methods have been developed for the synthesis of NCA, including sol–gel synthesis,3 the PVA precursor method,4 hydrothermal ion-exchange,5 spray pyrolysis,6 solution combustion,7 and co-precipitation.8–10 Co-precipitation is the common method widely employed for the production of NCA, benefiting from the advantages such as homogenous elemental distribution in the materials and well-controlled spherical morphology.11–13 However, the complicated conditions for manufacturing such as precise controlling of pH, addition of chelating agent,14 stringent requirement of aluminum source and handling of massive wastes still constrain the large-scale production. In this regard, an efficient approach for the synthesis of NCA is desirable to meet the increasing market of the NCA materials for the application in HEV and EVs.On the other hand, fast fading in the capacity and power upon storage and cycling is still the major problem for the application of the NCA-based lithium ion batteries.15,16 Formation of the poor conductive NiO-like byproducts and Li2CO3 on the surface of the NCA primary particles,17 cation mixing caused by the migration of Ni2+ and Co2+ into Li+ slab,18 dissolution of the transitional metals by the attachment of HF generated from decomposition of the electrolyte,19 and degradation of the morphologies of the NCA particles have been suggested to be the main factors responsible for the capacity and power fading.20 Several strategies have been proposed to overcome those drawbacks. Surface coating is the common method employed to modify the surface of the NCA particles in order to prevent the particles from direct contact with the electrolytes and to suppress the side reactions at the interface of the electrodes. A great improvement in the cycling performance and capacity retention has been achieved by surface coating of various materials, such as MnO2,21 LiMnPO4,22 ZnO,23 Li2TiO3,24 Ni3(PO4)2,25 AlF3,26 etc. The capacity and power fading could also be restrained by Al gradient doping27 and fluorine surface doping.28 Nevertheless, a deep insight into the mechanism associated with the fading of the capacity and power of the NCA materials would be beneficial to the design of the materials. Recently, the microstructure evolution during electrochemical processes has been recognized and suggested to play a key role in the deterioration of the NCA materials. In particular, the micro-cracks could be developed at the triple junctions and grain boundaries of the cathode active materials during the first delithiation process, leading to the infiltration of electrolyte into the interior of the aggregated particle along the micro-crack network, and consequently the formation of a thick solid electrolyte interphase (SEI) film and NiO-phase.29–31 Consequently, the loss of innerparticle connectivity and increased polarization contribute remarkably to the degradation of performance and capacity fading. In this regard, a purposeful engineering of the microstructure of the NCA particles might be one of the efficient ways to achieve good performance of the NCA materials.
Herein, we report a facile approach to synthesize the microsized NCA spheres with a rigid porous structure by a two-step solid-state process with assistance of spray drying. It is demonstrated that the formation of the SEI film on the active materials are strongly related to the microstructure of the materials. Relative to the randomly aggregated NCA particles, the spherical NCA particles with a rigid porous structure may not only provide good access of the electrolyte to electrode surfaces, but also accommodate the cyclic strains generated in the lithiation/delithiation process, leading to a better stability of the microstructure and less SEI film formed on the NCA material. Consequently, a better contact among the primary NCA particles and conductivity would bring about a superior electrochemical performance of the NCA materials.
2. Experimental
2.1. Material synthesis
Stoichiometric amount of Ni(OH)2, Co3O4, Al2O3, and Li2CO3 were mixed by ball milling in an alcohol media, followed by being dried at 90 °C to obtain fine gray powder. Then, the dried powder was calcinated under flowing oxygen at 700 °C for 8 h. The obtained precursor was ball milled again in deionized water to form slurry. A certain amount of starch as the binder was added. The slurry was dried by spray drying under the condition of the inlet temperature of 220 °C, the outlet temperature of 90 °C and the compressed air pressure of 0.2 MPa to produce the spherical precursor. Finally, the spherical precursor was annealed at 720 °C under oxygen for 28 h in muffle furnace to obtain LiNi0.8Co0.15Al0.05O2 (SD-NCA). For comparison, the common-dried counterpart (CD-NCA) was prepared by direct annealing of the ball-milled precursor slurry under the same conditions.2.2. Structural characterization
The crystal structures of the samples were examined by X-ray diffraction (XRD) using X Pert-ProMPd (Holand) D/max-A X-ray diffractometer with Cu Kα radiation (λ = 0.1514178 nm), operated at 40 kV and 40 mA. The morphologies of the samples were obtained on a scanning electron microscopy (SEM) (Hitachi S4700). The N2 adsorption/desorption isotherms were measured on a Quadrasorb 2MP surface area & porosity analyzer (Micromeritics Instrument Corp.) at 77 K. The analysis of the particle size and size distribution was carried out on a Malvern mastersizer 3000 particle size analyzer.2.3. Electrochemical characterization
Electrochemical behaviors during charge/discharge cycles were examined using a two-electrode test cell with lithium foil as the negative electrode. A positive electrode was made by coating a paste of LiNi0.8Co0.15Al0.05O2 active material, acetylene black, and polyvinylidene fluoride (PVDF) binder (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 wt%) on an aluminum-foil collector. The positive electrodes were dried at 120 °C for 12 h in a vacuum oven. Battery assembly of CR2032 coin cells was carried out in an argon filled glove box with an electrolyte of 1 M LiPF6 in a 50:50 v/v mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) solution. The coin cells were cycled between 2.8 and 4.3 V (vs. Li+/Li) on a LAND battery testing system and the current density of 1C is set as 200 mA g−1. Cyclic voltammetry measurements were carried out on an electrochemical workstation (CHI660D, CH Instruments) between 2.5 and 4.5 V at a scan rate of 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) measurements were performed via PARSTAT 2273 electrochemical workstation system (Princeton Applied Research, AMETEK, America) over frequency range of 100 kHz–10 mHz with amplitude of 10 mV.
:10:10 wt%) on an aluminum-foil collector. The positive electrodes were dried at 120 °C for 12 h in a vacuum oven. Battery assembly of CR2032 coin cells was carried out in an argon filled glove box with an electrolyte of 1 M LiPF6 in a 50:50 v/v mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) solution. The coin cells were cycled between 2.8 and 4.3 V (vs. Li+/Li) on a LAND battery testing system and the current density of 1C is set as 200 mA g−1. Cyclic voltammetry measurements were carried out on an electrochemical workstation (CHI660D, CH Instruments) between 2.5 and 4.5 V at a scan rate of 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) measurements were performed via PARSTAT 2273 electrochemical workstation system (Princeton Applied Research, AMETEK, America) over frequency range of 100 kHz–10 mHz with amplitude of 10 mV.
3. Results and discussion
The key strategy of the two-step solid-state reaction approach for the synthesis of the NCA spheres is to generate the NCA fine crystals in the precursor, which would then combine with each other through a simultaneous crystal growth eventually to form an integrated framework. The mixture of Ni, Co, Al, and Li salts were annealed at a NCA forming temperature (700 °C) to form a precursor in the first step. Under the conditions, the fine NCA crystals might be formed inside the precursor. Then, the precursor was treated by ball milling again, to ensuring a uniform Al distribution in the powder sample. In contrast to the traditional spray-drying method, in which the mixed solution of the Co, Ni, and Al salts were directly dried by spray-drying to produce the solid spheres, in this case, a spray-drying treatment was combined in the second step with the treated precursor dispersed in water to generate the spherical secondary particles. The spherical precursor was further annealed at high temperature, so that the NCA crystals formed in precursor would simultaneously grow to produce the NCA primary particles, leading to the formation of the spherical SD-NCA secondary particles with an integrated framework. A comparison of the typical XRD patterns of the precursor, SD-NCA, and CD-NCA are shown in Fig. 1. Evidence of the formation of the fine NCA crystals in the precursor comes from the appearance of the characteristic bands at 18.8°, 37.8°, 43.9°, 64.3°, and 77.1° in Fig. 1(a). The appearance of those bands has been ascribed to the formation of mixed crystals Li1−xNi1+xO2, i.e., of NiO and LiNiO2.32 The rest minor reflections may be from the undecomposed Li2CO3.33 No obvious bands corresponding to Co and Al species was detected for the treated precursor, likely implying that the components was well dispersed in the treated precursor. That is, the NCA crystals were exactly formed and dispersed in the precursor in the first step of the synthesis process. For CD-NCA and SD-NCA, all the reflections can be indexed to the layered α-NaFeO2 structure with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.34 The similarity in the XRD patterns implies that CD-NCA and SD-NCA possess similar crystal structure and lattice constants. Moreover, the distinct peak splitting of (006)/(102) and (018)/(110) pairs, as an indicative of the well-developed layered structure, can be clearly observed for both CD-NCA and SD-NCA. Additionally, the excellent hexagonal ordering in the crystal structure is revealed by the low R factor, defined by intensity ratio of I(006)/(I(101) + I(102)), which is calculated to be ca. 0.095 and 0.089 for CD-NCA and SD-NCA, respectively. Furthermore, the integrated intensity ratio of I(003)/I(104), which is sensitive to the cation mixing in the lattice, is 1.349 and 1.516 for CD-NCA and SD-NCA, respectively. The values are much higher than the typical value of 1.2 for the undesirable cation mixing samples.34 The dominating Ni 2p3/2 peak located at 856.0 eV (Fig. S1, ESI†) indicates that Ni mainly existed in the Ni3+ state, as the combination of one Li+ ion is accompanied by a simultaneous change of valence from Ni2+ to Ni3+.35 The large difference in the ionic radii of Ni3+ and Li+ (rNi3+ = 0.56 Å, rLi+ = 0.74 Å) would lead to less chance for the occurrence of the cation mixing.19
m space group.34 The similarity in the XRD patterns implies that CD-NCA and SD-NCA possess similar crystal structure and lattice constants. Moreover, the distinct peak splitting of (006)/(102) and (018)/(110) pairs, as an indicative of the well-developed layered structure, can be clearly observed for both CD-NCA and SD-NCA. Additionally, the excellent hexagonal ordering in the crystal structure is revealed by the low R factor, defined by intensity ratio of I(006)/(I(101) + I(102)), which is calculated to be ca. 0.095 and 0.089 for CD-NCA and SD-NCA, respectively. Furthermore, the integrated intensity ratio of I(003)/I(104), which is sensitive to the cation mixing in the lattice, is 1.349 and 1.516 for CD-NCA and SD-NCA, respectively. The values are much higher than the typical value of 1.2 for the undesirable cation mixing samples.34 The dominating Ni 2p3/2 peak located at 856.0 eV (Fig. S1, ESI†) indicates that Ni mainly existed in the Ni3+ state, as the combination of one Li+ ion is accompanied by a simultaneous change of valence from Ni2+ to Ni3+.35 The large difference in the ionic radii of Ni3+ and Li+ (rNi3+ = 0.56 Å, rLi+ = 0.74 Å) would lead to less chance for the occurrence of the cation mixing.19
 | ||
| Fig. 1 XRD patterns of (a) SD-NCA precursor, (b) SD-NCA and (c) CD-NCA. | ||
The morphologies of the CD-NCA and SD-NCA samples were determined by SEM images, the results are shown in Fig. 2(a) and (b). The CD-NCA particles showed a wide size distribution due to irregular aggregation of the primary particles with average particle size of 410 nm to form the large bulks (Fig. 2(a)). On the other hand, as seen in Fig. 2(b), SD-NCA exists as microsized spherical particles composed of uniform primary particles. The mean size of the microspheres was ca. 12.6 μm in diameter (Fig. S2(a), ESI†). A careful comparison of the SEM images for the spheres before and after annealing at high temperature in Fig. 2(c)–(f) reveals that upon annealing, an increase in the size of the primary particles from around ca. 50 nm to 400 nm, which is comparable to the size of the primary particles in the CD-NCA aggregates. This strongly suggests that the annealing treatment resulted in not only the formation of the well-crystallized NCA structure, but also a growth of the primary particles. As we demonstrated previously, being properly annealed at high temperature, the primary particles of the metal oxides could be partially fused and solidly connected through the mutual molten growth mechanism to generate the rigid porous crystal structure.36 In this case, it is also reasonable that the primary particles in SD-NCA microspheres are interconnected with each other to form a rigid porous framework due to the simultaneous growth of the NCA crystals in the integrated NCA primary particles of the spherical precursor. The nanosized pores formed among the neighboring SD-NCA primary particles can be clearly observed in Fig. 2(d), which uniformly distributed all over the surface of the spheres. The interconnected opened nanosized channels and continuous network in the microspheres of the metal oxides may provide the favorable paths for the penetration of the electrolyte and charge transportation through the active material to facilitate the battery performance. The porous structures of the SD-NCA spheres are further verified by N2 adsorption–desorption isotherms. A typical type IV isotherm with type H3 hysteresis loop according to BDDT classification was observed (Fig. S2(b), ESI†). The specific surface area of SD-NCA was estimated to be 2.051 m2 g−1, using the Brunauer–Emmett–Teller (BET) method. The pore size distribution derived from desorption data indicates that the mean diameter of the pores in SD-NCA was ca. 52.9 nm with a broad size distribution. The EDS-mapping shown in Fig. 2 further confirms that Co, Ni, and Al are homogeneously distributed in the SD-NCA particles.
 | ||
| Fig. 2 SEM images of (a) CD-NCA, (b) SD-NCA, (c) spherical precursor, and (d) SD-NCA sphere. Magnified SEM graphs of (e) precursor and (f) SD-NCA. EDX mapping of (g) Ni, (h) Co and (i) Al distribution on the surface of SD-NCA. | ||
The electrochemical performance of CD-NCA and SD-NCA was firstly investigated by cyclic voltammetry (CV) in the coin-type cells with metallic lithium foil as the reference electrode, the results are shown in Fig. 3(a) and (b). In general, LiNiO2-based materials may experience phase transfer among four different phases (one monoclinic phase, M and three hexagonal phases, H1, H2, and H3) during the charge–discharge cycle.37,38 As a result, the CV curve would exhibit three pairs of peaks, corresponding to the coexistence of H1 and M, M and H2, and H2 and H3, respectively. As expected, for SD-NCA in the first cycle, three pairs of redox peaks can be clearly observed with the cathodic/anodic peaks at 3.84 V/3.70 V for H1 → M, 3.99 V/3.96 V for M → H2, and 4.19 V/4.17 V for H2 → H3, respectively. In the second cycle, the cathodic peak for H1 → M shifted largely to 3.69 V, which slightly shifted to 3.67 V in the third cycle. The shift of this cathodic peak has been ascribed to an inner structural adjustment, owing to the phase transfer processes and the formation of the SEI film during the first cycle.14 No major change in the CV curves was observed in the subsequent cycles, indicating the good stability of SD-NCA during the lithiation and delithiation process. It can also be confirmed with the SEM images of SD-NCA after the first and the third CV cycle, no obvious structure changes were observed (Fig. S3, ESI†). The CV behaviors of CD-NCA are quite different from those of SD-NCA. The cathodic peaks for H1 → M and M → H2 were merged together as a broad band maximized at 3.96 V in the first cycle. However, the three pairs of the redox peaks became distinguishable in the second cycle, with the cathodic/anodic peaks located at 3.86 V/3.74 V for H1 → M, 4.05 V/4.00 V for M → H2, and 4.24 V/4.18 V for H2 → H3, respectively. A large shift in the redox peaks for H1 → M from 3.86 V/3.74 V to 3.73 V/3.67 V was still observed in the third cycle, along with the slight potential shifts for the M → H2 and H2 → H3 peaks. The phenomena could not be solely attributed to the alteration of the inner structure induced by the phase transfer. As reported in the literature, in most cases, the inner structure alteration would be completed during the first cycle. Because the crystallinity and size of the primary particles in the CD-NCA spheres are comparable to those of the SD-NCA particles, as revealed by the XRD patterns in Fig. 1 and SEM images in Fig. 2, the difference in the CV behaviors of CD-NCA and SD-NCA has to be related to the difference in the formation of the SEI films on the two materials. In particular, the rigid porous structure of SD-NCA would have profound impact on the formation of the SEI film, which is confirmed by the electrochemical impedance spectroscopic characterization discussed later.
 | ||
| Fig. 3 Cyclic voltammograms of (a) SD-NCA and (b) CD-NCA electrodes. | ||
The major difference in the electrochemical behaviors of CD-NCA and SD-NCA was further revealed by the charge–discharge cycling measurements. The typical initial charge–discharge curves of CD-NCA and SD-NCA are shown in Fig. 4(a). In the voltage range of 2.8–4.3 V at 0.1C (1C = 200 mA g−1), SD-NCA delivered an initial charge and discharge specific capacities of 240.6 and 205.1 mA h g−1, respectively, with an efficiency of 85.2%, whereas the CD-NCA exhibited the corresponding capacities of 218.7 and 168.7 mA h g−1 with an efficiency of 77.1%. Thus, SD-NCA with the porous structure exhibited a much better initial capacity and efficiency than CD-NCA. The rate performance of the two samples is further compared in Fig. 4(b). SD-NCA delivered discharge capacities of 179.8 mA h g−1 at 0.5C, and 150.9 mA h g−1 at 2C. For CD-NCA, the discharge capacities were only 135.9 and 92.5 mA h g−1 at 0.5C and 2C, respectively. Evidently, the porous structure of the SD-NCA spheres can also improve the rate capability, probably due to the better accessibility of electrolyte in the porous structure.
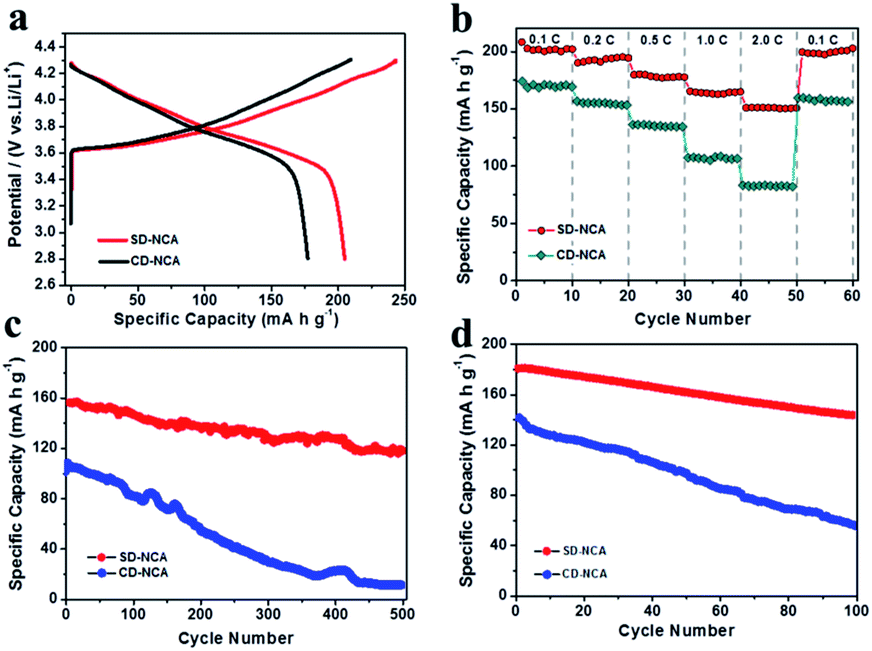 | ||
| Fig. 4 (a) Initial charge–discharge curves at 0.1C (1C = 200 mA g−1), (b) rate performance, and cycling performance (c) at 2C, room temperature and (d) at 1C, 55 °C for CD-NCA and SD-NCA. | ||
The cycleability of the two samples are examined at 2C at room temperature, the result is shown in Fig. 4(c). The representative data of the capacity and capacity retention are listed in Table 1. Besides having much high discharge capacity, SD-NCA showed almost no change in the capacity during the first 10 cycles. However, the discharge capacity for CD-NCA slightly decreased from 107.6 mA h g−1 to 105.2 mA h g−1 with capacity retention of 97.8%. The difference in the capacity retention became noticeable upon 100 cycles. SD-NCA can deliver a discharge capacity of 146.1 mA h g−1 with a capacity retention as high as 93.3%. In contrast, for CD-NCA, only a discharge capacity of 82.5 mA h g−1 can be obtained, and the corresponding capacity retention was ca. 76.7%. Prolonging cycling resulted in a decrease in the discharge capacity for both SD-NCA and CD-NCA. Nevertheless, the capacity retention for the SD-NCA was still about 81.2% and 75.4% after 300 and 500 cycles, respectively. The values are much higher than those for CD-NCA (29.1% for 300 cycles, 11.2% for 500 cycles). Similarly, as shown in Fig. 4(d), the SD-NCA exhibited much better capacity retention as the cells was cycled at 1C rate at elevated temperature (55 °C). The capacity retention was 79.5% for SD-NCA, and 39.2% for CD-NCA after 100 cycles. The high discharge capacities at elevated temperature relative to those at room temperature are due to the improved ion conductivity.
| Cycle number | CD-NCA | SD-NCA | ||
|---|---|---|---|---|
| Capacity (mA h g−1) | Retention (%) | Capacity (mA h g−1) | Retention (%) | |
| 1 | 107.6 | 100 | 156.6 | 100 |
| 10 | 105.2 | 97.8 | 156.1 | 99.7 |
| 50 | 98.4 | 91.4 | 151.7 | 96.9 |
| 100 | 82.5 | 76.7 | 146.1 | 93.3 |
| 200 | 57.9 | 53.8 | 136.7 | 87.3 |
| 300 | 31.3 | 29.1 | 127.1 | 81.2 |
| 500 | 12.0 | 11.2 | 118.0 | 75.4 |
In order to gain further insight into the electrochemical behaviors of CD-NCA and SD-NCA, the EIS measurements were carried out for the cells with SD-NCA and CD-NCA as the working electrodes. Fig. 5 depicts the EIS spectra obtained after different cycles for CD-NCA and SD-NCA at frequencies ranging from 10 mHz to 100 kHz. All the spectra consist of two separated semicircles and a sloping line. The first semicircle at high frequency is mainly caused by the formation of the passive film or SEI film on the surface of the electrode materials due to the interaction of the electrode materials with the electrolytes.39 The magnitude of this semicircle has been ascribed to the resistance of lithium ion migration through the SEI film, and mainly determined by the thickness of the SEI film. In the medium-low frequency region, the diameter of the semicircle is a measure of the charge transfer resistance, related to the contact between the particles and/or between the electrode and the electrolyte. The sloping line is associated with the semi-infinite diffusion of the lithium ions in the bulk of the active electrode materials.40 From the magnified EIS spectra inserted in Fig. 5, it can be clearly seen that both of the semi-circles, which are closely related to the resistance of the SEI film and charge transfer, varied with the cycle number. To gain a better view, the EIS spectra were simulated by using the simulation circuit illustrated in Fig. 5, the data of the resistance of the SEI film (RSEI) and the resistance of the charge transfer (RCT) at different cycling stage are obtained and plotted against the cycle number, the results are shown in Fig. 6(a) and (b). After the first cycle, the values of the RSEI are 8.14 Ω for the SD-NCA and 25.16 Ω for CD-NCA, respectively. For both of the CD-NCA and SD-NCA, the RSEI decreases during the following few cycles, probably because of the activation of the active materials of the electrodes. After 30 cycles, the RSEI increases to 37.27 Ω for the SD-NCA, and then only slight variation in the RSEI was observed even upon 500 cycles. However, the RSEI for the CD-NCA continuously increases from 51.76 Ω at the 10th cycle to 69.57 Ω at 300th cycle, and eventually became almost stable. As aforementioned, the value of the RSEI is strongly associated with the thickness of the SEI film. A relatively large RSEI and variation in the RSEI during cycling for CD-NCA suggest that a thicker SEI film was formed on the surface of the CD-NCA particles during the first few cycles, and it may grow much fast in the following cycles. On the other hand, the SEI film on SD-NCA was mainly formed in the early cycles, and then grew very slowly in the following cycles. The RCT curves in Fig. 6(b) indicate that for both of CD-NCA and SD-NCA, the RCT persistently increases from the 10th cycle to the 200th cycle, and then became steady. Nevertheless, for SD-NCA, the absolute value of RCT is only 3.56 Ω at the 10th cycle, and slightly increases to 14.05 Ω at 200th cycle, while the corresponding RCT values for the CD-NCA are 18.74 Ω and 41.03 Ω, respectively.
 | ||
| Fig. 5 Nyquist plots of (a) CD-NCA; (b) SD-NCA after various cycles at 1C. Inset: magnified Nyquist plots in the high-frequency range and equivalent circuit for simulation. | ||
 | ||
| Fig. 6 Plots of (a) RSEI and (b) RCT as function of cycle number. | ||
Considering the variation behaviors of RSEI in Fig. 6(a), which is associated with the formation of the SEI films for SD-NCA and CD-NCA, we believe that the different growth behaviors of the SEI film on the active materials could lead to the changes in the microstructure and the electrical contact between the particles, and those, in turn, would influence the cycling behaviors of SD-NCA and CD-NCA. Watanabe et al.41 demonstrated that after the cycle test in the ΔDOD of 0–100%, many micro-cracks would be generated in the inter-surface between the primary particles which are physically constrained by surrounding primary particles through aggregation, and subject to the cyclic strains during repeated charge–discharges. The electrolyte would infiltrate into the NCA particles through the micro-cracks. As a result, new SEI films are surely formed on the primary particle surface of NCA along the micro-cracks, and the ionic/electronic conductivity of the NCA particle would become worse remarkably. In this case, most of the CD-NCA particles existed in the aggregated state, as revealed by the SEM images in Fig. 2(a). Similarly, the micro-cracks could also be continuously generated during cycling, leading to the formation of new SEI films. Consequently, the newly formed SEI films would result in lack of contact between primary particles. As a result, both of RSEI and RCT largely increase with the cycle number, which may be the main factors responsible for the significant capacity fading and impedance increase of CD-NCA. In this regard, the primary particles of SD-NCA exist in an integrated framework due to the crystal growth of the fine NCA crystals. To a certain degree, the microsized spheres of SD-NCA could have less chance to generate the micro-cracks, due to that the integrated network of SD-NCA may accumulate the cyclic strains during cycling. This is verified by the fact that the spherical structure of SD-NCA was well maintained on the electrode after 100 cycles (Fig. S4, ESI†). That is, the electrical contact among the primary particles in the microsized spheres of SD-NCA is preserved, leading to less SEI film formed on the particles, and hence a superior cycling performance of the material.
4. Conclusion
In summary, the microsized spherical NCA cathode material for lithium-ion batteries was successfully synthesized by a solid-state process in combination with a spray drying method. The growth of the primary particles leads to the formation of the interconnected nanopores with opened channels, which may provide the favorable paths for penetration of the electrolyte. The porous spherical particles exhibited a superior electrochemical performance with the discharge capacity of 202.1 mA h g−1 at 0.1C and 151 mA h g−1 at 2C, capacity retention of 74.5% after 500 cycles at 2C. The variation in the RSEI and RCT during cycling suggests that the integrated porous structure of the NCA spheres may prevent the formation of the thick SEI films, and hence lead to a low resistance for the charge transfer at the surface of the active material. Those might be the main factors to determine the cycling performance of the materials. Thereby, the microstructures of the NCA materials could have a great impact on the electrochemical performance of the materials.Acknowledgements
We gratefully acknowledge financial supports from the Nature Science Foundation of China (21336005), Ministry of Science and Technology of China (2014EG111224).References
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS.
- W. Liu, P. Oh, X. Liu, M. J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef CAS PubMed.
- C. J. Han, J. H. Yoon, W. I. Cho and H. Jang, J. Power Sources, 2004, 136, 132–138 CrossRef CAS.
- P.-H. Duvigneaud and T. Segato, J. Eur. Ceram. Soc., 2004, 24, 1375–1380 CrossRef CAS.
- W. Chen, Y. Li, D. Yang, X. Feng, X. Guan and L. Mi, Electrochim. Acta, 2016, 190, 932–938 CrossRef CAS.
- S. Ju, H. Jang and Y. Kang, Electrochim. Acta, 2007, 52, 7286–7292 CrossRef CAS.
- S. Majumder, S. Nieto and R. Katiyar, J. Power Sources, 2006, 154, 262–267 CrossRef CAS.
- G. Hu, W. Liu, Z. Peng, K. Du and Y. Cao, J. Power Sources, 2012, 198, 258–263 CrossRef CAS.
- M. Guilmard, C. Pouillerie, L. Croguennec and C. Delmas, Solid State Ionics, 2003, 160, 39–50 CrossRef CAS.
- Y. Kim and D. Kim, ACS Appl. Mater. Interfaces, 2012, 4, 586–589 CAS.
- K. K. Cheralathan, N. Y. Kang, H. S. Park, Y. J. Lee, W. C. Choi, Y. S. Ko and Y.-K. Park, J. Power Sources, 2010, 195, 1486–1494 CrossRef CAS.
- Y. Kim, ACS Appl. Mater. Interfaces, 2012, 4, 2329–2333 CAS.
- H. Xie, G. Hu, K. Du, Z. Peng and Y. Cao, J. Alloys Compd., 2016, 666, 84–87 CrossRef CAS.
- H. Xie, K. Du, G. Hu, J. Duan, Z. Peng, Z. Zhang and Y. Cao, J. Mater. Chem. A, 2015, 3, 20236–20243 CAS.
- K. Karin, M. Julia, M. Michael, J. Peter, N. Peter, S. Stefan, L. Verena and E. Helmut, ACS Appl. Mater. Interfaces, 2015, 7, 19589–19600 Search PubMed.
- K. Du, H. Xie, G. Hu, Z. Peng, Y. Cao and F. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 17713–17720 CAS.
- K. X. Wang, X. H. Li and J. S. Chen, Adv. Mater., 2015, 27, 527–545 CrossRef CAS PubMed.
- F. Fu, Q. Wang, Y.-P. Deng, C.-H. Shen, X.-X. Peng, L. Huang and S.-G. Sun, J. Mater. Chem. A, 2015, 3, 5197–5203 CAS.
- H. Liu, Y. Yang and J. Zhang, J. Power Sources, 2006, 162, 644–650 CrossRef CAS.
- H. Visbal, S. Fujiki, Y. Aihara, T. Watanabe, Y. Park and S. Doo, J. Power Sources, 2014, 269, 396–402 CrossRef CAS.
- B. Huang, X. Li, Z. Wang, H. Guo, L. Shen and J. Wang, J. Power Sources, 2014, 252, 200–207 CrossRef CAS.
- J. Duan, C. Wu, Y. Cao, K. Du, Z. Peng and G. Hu, Electrochim. Acta, 2016, 221, 14–22 CrossRef CAS.
- Y.-Q. Lai, M. Xu, Z.-A. Zhang, C.-H. Gao, P. Wang and Z.-Y. Yu, J. Power Sources, 2016, 309, 20–26 CrossRef CAS.
- N. Wu, H. Wu, H. Liu and Y. Zhang, J. Alloys Compd., 2016, 665, 48–56 CrossRef CAS.
- D.-J. Lee, B. Scrosati and Y.-K. Sun, J. Power Sources, 2011, 196, 7742–7746 CrossRef CAS.
- S.-H. Lee, C. S. Yoon, K. Amine and Y.-K. Sun, J. Power Sources, 2013, 234, 201–207 CrossRef CAS.
- J. Duan, G. Hu, Y. Cao, C. Tan, C. Wu, K. Du and Z. Peng, J. Power Sources, 2016, 326, 322–330 CrossRef CAS.
- X. Li, Z. Xie, W. Liu, W. Ge, H. Wang and M. Qu, Electrochim. Acta, 2015, 174, 1122–1130 CrossRef CAS.
- S. Zheng, R. Huang, Y. Makimura, Y. Ukyo, C. A. Fisher, T. Hirayama and Y. Ikuhara, J. Electrochem. Soc., 2011, 158, A357–A362 CrossRef CAS.
- S. Muto, Y. Sasano, K. Tatsumi, T. Sasaki, K. Horibuchi, Y. Takeuchi and Y. Ukyo, J. Electrochem. Soc., 2009, 156, A371–A377 CrossRef CAS.
- D. Mori, H. Kobayashi, M. Shikano, H. Nitani, H. Kageyama, S. Koike, H. Sakaebe and K. Tatsumi, J. Power Sources, 2009, 189, 676–680 CrossRef CAS.
- H. Wulff, M. Mohan Rao and F. Scholz, Chem. Mater., 2003, 15, 988–993 CrossRef CAS.
- R. Amin, D. B. Ravnsbæk and Y.-M. Chiang, J. Electrochem. Soc., 2015, 162, A1163–A1169 CrossRef CAS.
- T. Ohzuku, A. Ueda and M. Nagayama, J. Electrochem. Soc., 1993, 140, 1862–1870 CrossRef CAS.
- J. Duan, G. Hu, Y. Cao, C. Tan, C. Wu, K. Du and Z. Peng, J. Power Sources, 2016, 326, 322–330 CrossRef CAS.
- Z. Jia, Q. Zhou, X. Li, Y. Fu, H. Ming and J. Zheng, Electrochim. Acta, 2015, 156, 216–222 CrossRef CAS.
- X.-J. Zhu, H.-X. Liu, X.-Y. Gan, M.-H. Cao, J. Zhou, W. Chen, Q. Xu and S.-X. Ouyang, J. Electroceram., 2006, 17, 645–649 CrossRef CAS.
- H. Y. Tran, G. Greco, C. Täubert, M. Wohlfahrt-Mehrens, W. Haselrieder and A. Kwade, J. Power Sources, 2012, 210, 276–285 CrossRef CAS.
- C. F. Chen and P. P. Mukherjee, Phys. Chem. Chem. Phys., 2015, 17, 9812 RSC.
- J. Li, L. E. Downie, L. Ma, W. Qiu and J. Dahn, J. Electrochem. Soc., 2015, 162, A1401–A1408 CrossRef CAS.
- S. Watanabe, M. Kinoshita, T. Hosokawa, K. Morigaki and K. Nakura, J. Power Sources, 2014, 258, 210–217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04206j |
| This journal is © The Royal Society of Chemistry 2017 |