DOI:
10.1039/C7RA04133K
(Paper)
RSC Adv., 2017,
7, 29713-29721
Conformation dynamics of the intrinsically disordered protein c-Myb with the ff99IDPs force field†
Received
12th April 2017
, Accepted 30th May 2017
First published on 7th June 2017
Abstract
The intrinsically disordered protein c-Myb plays a critical role in cellular proliferation and differentiation. Loss of c-myb function results in embryonic lethality due to failure of fetal hepatic hematopoiesis. The conformation dynamics of the intrinsically disordered c-Myb are still unknown. Here, molecular dynamics (MD) simulations with the intrinsically disordered protein force field ff99IDPs were used to study the conformation dynamics. In comparison with ff99SBildn, ff99IDPs can reproduce more diverse disordered conformers of c-Myb. The predicted secondary chemical shift under ff99IDPs is more close to that of experiment data than that under ff99SBildn. Therefore, ff99IDPs can sample native molten globule, native pre-molten globule and native coil conformers for c-Myb. These results are consistent with those of other intrinsically disordered proteins. Kinetic analysis of MD simulations shows that c-Myb folds via a two-state process and indicates that c-Myb folds in the order of tertiary folding and helical folding. The folding nucleus of KEL plays an essential role in stabilizing the folding state with dynamic correlation networks. The influences of solvent models for TIP3P, TIP4P-EW and TIP5P were also investigated and it was found that TIP3P and ff99IDPs are the best combination to research the conformer sampling of c-Myb. These results reveal the conformation dynamics of c-Myb and confirm that the ff99IDPs force field can be used to research the relationship between structure and function of other intrinsically disordered proteins.
Introduction
The MYB family of transcriptional regulators, including A, B and C Myb proteins, plays a critical role in cellular proliferation and differentiation. Furthermore, the c-myb protein (c-Myb) is significantly expressed in different hemopoietic lineages1–4 of both normal and tumoral immature cells.
c-myb5 was originally identified as an oncogene carried by the chicken leukemia viruses avian myeloblastosis virus and E26,6–8 and is a significant conserved transcription factor.9 The previous work reports that high levels of c-myb are expressed in immature hematopoietic cells,10 while its expression is down-regulated during differentiation.11 This indicates that the c-myb gene plays a significant role in cellular proliferation of immature hematopoietic cells.
Three functional domains are found for c-Myb: an N-terminal highly conserved DNA binding domain, a central transcriptional activation domain, and a C-terminal negative regulation domain.12 c-Myb is one of intrinsically disordered proteins (IDPs),13 since it does not fold spontaneously into well-organized globular structures in the absence of stabilizing interactions. The conformation dynamics of intrinsically disordered c-Myb is still unknown, therefore we used molecular dynamics (MD) simulations to study the conformation dynamics. Dynamic conformation plays a key role in the function of intrinsically disordered proteins.14–18
However, the accuracy of the force field remains an issue in applications of MD simulation, while most traditional widely used force fields can't well reproduce intrinsically disordered proteins, especially on backbone dihedrals.19 In order to better reproduce the disordered conformation, a set of AMBER force fields ff99IDPs20,21 and others22 were used, which were developed specially for IDPs with grid-based energy correction maps (CMAP) term23,24 to correct the dihedral of backbone for eight disordered-promotion residues of alanine, arginine, glycine, glutamine, serine, glutamic acid, lysine, and proline. By adding CMAP energy term, the Φ/Ψ distributions of the eight disorder-promoting residues have been improved with respect to the benchmark data of IDPs, while the root mean squared percentage deviation is less than 0.15% between the benchmark and the simulation.20
In this study, we compared the performances of ff99IDPs and ff99SBildn to reproduce intrinsically disordered c-Myb. Based on the results, ff99IDPs was used to reveal the conformation dynamics of c-Myb.
Materials and methods
Molecular dynamics simulation
The atomic coordinates of the c-Myb were extracted from the ternary complex of PDB database (PDB code: 2AGH).25 AMBER12 was used to perform efficient simulations with periodic boundary conditions.26 Hydrogen atoms were added using the LEaP module of AMBER12. Counter-ions were used to maintain system neutrality. C-Myb was solvated in a truncated octahedron box of TIP3P, TIP4P-EW, and TIP5P solvent models with a buffer of 10 Å, respectively.27–29 The pairwise interactions (van der Waals and direct Coulomb) were computed with a cutoff distance of 8 Å. Particle Mesh Ewald (PME) was employed to treat long-range electrostatic interactions in AMBER12.30 The ff99SBildn and ff99IDPs force fields were used for the intramolecular interactions, respectively. The Langevin thermostat was used in the preparation runs with a friction constant of 1 ps−1 and the Berendsen thermostat was used in the production runs.31 All MD simulations were accelerated with the CUDA version of PMEMD in GPU cores of NVIDIA® Tesla K20.
To relieve any further structural clash in the solvated systems, initial minimization was performed with 2000-step conjugate gradient minimization. After minimization, a 400 ps' heating up in the NVT ensemble and a 200 ps' equilibration in the NPT ensemble at 298 K were performed before MD simulation. To compare the performances between ff99IDPs and ff99SBildn for the sampling of c-Myb, multiple independent trajectories with the same initial structure but different velocities under each force field were simulated. Total 5.8 μs trajectories were collected at 298 K. The simulation conditions are gathered in Table 1.
Table 1 Simulation condition for c-Myb
| Force field |
Solvent |
Trajectories |
Time (ns) |
| ff99SBildn |
TIP3P |
10 |
100 |
| TIP4P-EW |
5 |
| TIP5P |
5 |
| ff99IDPs |
TIP3P |
10 |
190 |
| TIP4P-EW |
5 |
| TIP5P |
5 |
Data analysis
The PTRAJ module in AMBER12 and Amber Tools 13 were used to calculate root-mean-square deviations (RMSDs) and root-mean-square fluctuations (RMSFs) for c-Myb.32 Kclust program in the MMTSB toolset were used to analysis the structural cluster on the basis of phi/psi dihedral.33 Native contact was assigned using in-house tools.34–36 In order to illustrate the convergence for sampling the disordered c-Myb, cluster numbers within 5 ns period along all the trajectories were plotted. Dictionary of Secondary Structure of Proteins (DSSP) algorithm was applied to assign the secondary structures of c-Myb. Experimental Cα chemical shift data were offered by Arai et al.37 SPARTA version 1.01 was used to calculate the secondary chemical shift data for the simulated structures.38 Radius of gyration (RG) and RMSD were both separated into eight bins. All of the structural visualizations were generated using PyMOL 1.7.
Dynamics correlation network
Every amino acid was defined as one node for dynamics correlation network. The fluctuation correlation between any pair of nodes i and j was calculated with eqn (1).| |
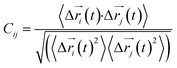 | (1) |
where 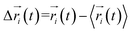 ,
, 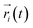 is the position of node i at time t, and 〈·〉 represents a time averaging. These elements were conveniently organized as a covariance matrix for simulated system. This general method has been successfully applied in several systems.39–42 In the current study, the covariance matrix for each system was constructed using snapshots (every 2 ps) of all simulated trajectories. Besides nodes, “edge” that transfers allosteric information from one node to another is defined between any two nodes without covalent bond and the distance between two heavy atoms from two nodes are closer than 4.5 Å over 75% sampling time. The strength of the edge between nodes i and j is defined as the absolute value of the inter-node correlation (Cij). The number of connected edges at each node is defined as the degree of the node. Correlation-weighted degree, which is the summation of strengths of all edges connected to a given node, indicates the importance of the node. After the network construction, Cytoscape3.1.1 was used to calculate the network topological parameters.43 The shortest path between any two nodes in the network was identified with the Floyd–Warshall algorithm.44
is the position of node i at time t, and 〈·〉 represents a time averaging. These elements were conveniently organized as a covariance matrix for simulated system. This general method has been successfully applied in several systems.39–42 In the current study, the covariance matrix for each system was constructed using snapshots (every 2 ps) of all simulated trajectories. Besides nodes, “edge” that transfers allosteric information from one node to another is defined between any two nodes without covalent bond and the distance between two heavy atoms from two nodes are closer than 4.5 Å over 75% sampling time. The strength of the edge between nodes i and j is defined as the absolute value of the inter-node correlation (Cij). The number of connected edges at each node is defined as the degree of the node. Correlation-weighted degree, which is the summation of strengths of all edges connected to a given node, indicates the importance of the node. After the network construction, Cytoscape3.1.1 was used to calculate the network topological parameters.43 The shortest path between any two nodes in the network was identified with the Floyd–Warshall algorithm.44
Results and discussion
Comparison of force fields
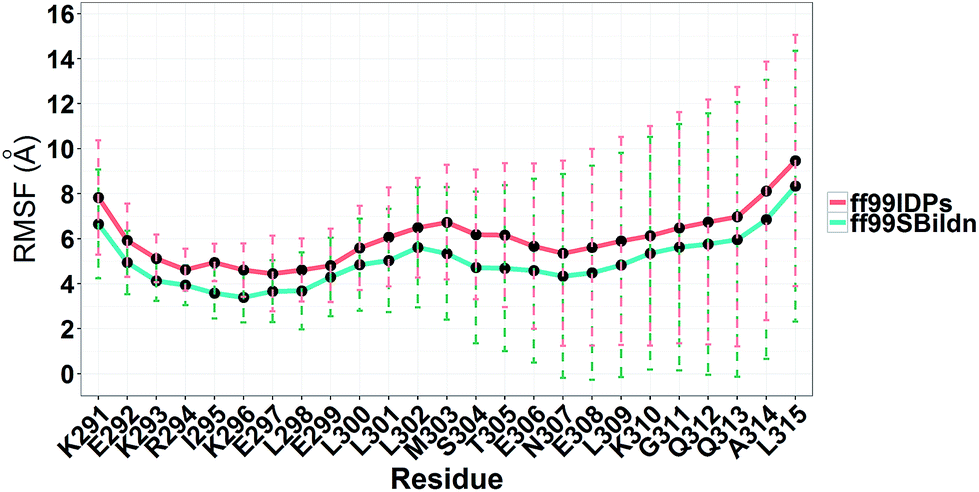
MD simulations were performed to model the conformation of c-Myb. The RMSD, Cα fluctuation (RMSF), structural clustering, secondary Cα chemical shift, helicity content, and other structural indices was calculated under ff99IDPs and ff99SBildn, respectively. For each force field, the RMSD and Cα fluctuation (RSMF) were calculated to ensure that both force field were able to sample rational conformations. The RMSDs relative to initial structure are shown in Fig. 1. This figure indicates that that 100 ns simulations are sufficient for the sampling conformer for c-Myb at room temperature under ff99SBildn and 190 ns under ff99IDPs. The Cα fluctuation (RMSF)s under ff99SBildn and ff99IDPs are shown in Fig. 2. The results suggest that the fluctuation of ff99IDPs is higher than that of ff99SBildn.
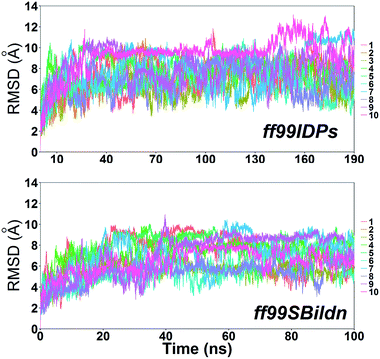 |
| | Fig. 1 RMSD under ff99SBildn and ff99IDPs force fields. | |
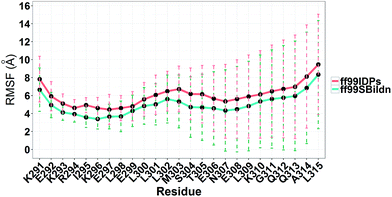 |
| | Fig. 2 RMSF under ff99SBildn and ff99IDPs force fields. | |
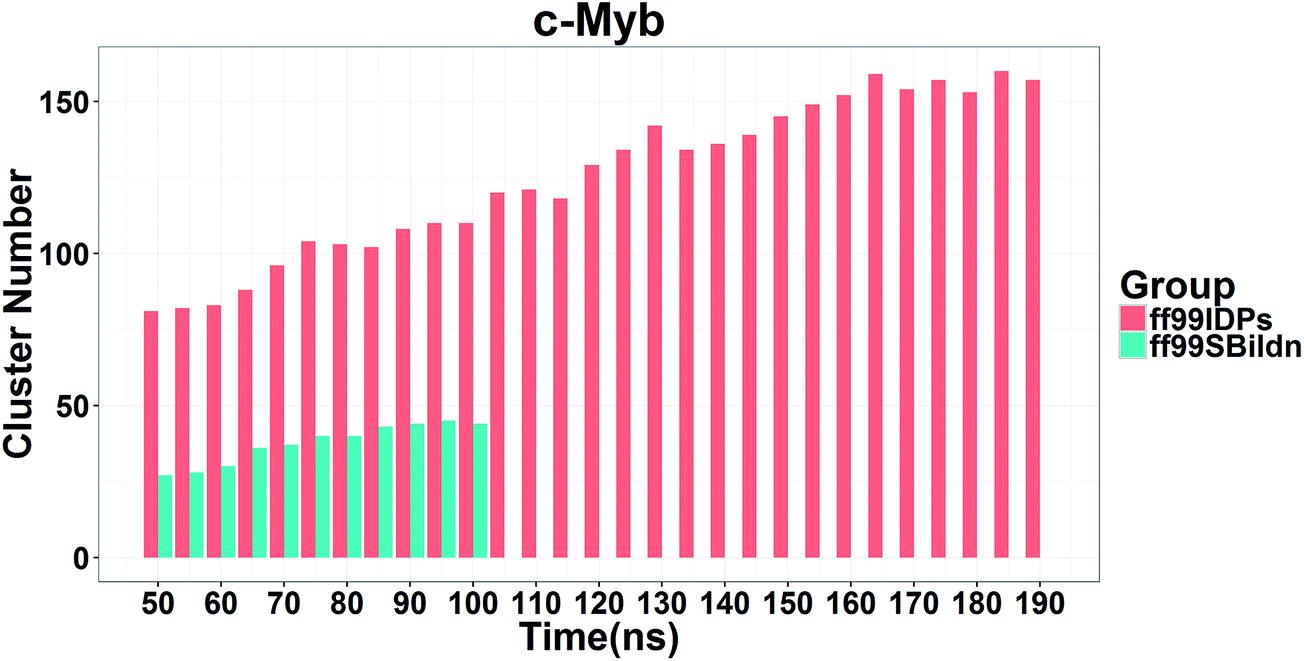
In order to ensure the convergence of the conformation sampling, the numbers of cluster that occupy 70% of the conformations over accumulated simulation time under both force fields are shown in Fig. 3. The conformer cluster suggests that the numbers of clusters do not change significantly at the end of the simulations. This indicates that the chosen simulation lengths are appropriate for sampling c-Myb.
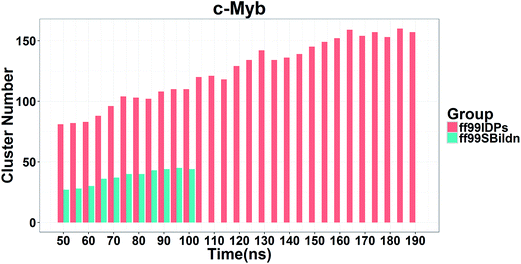 |
| | Fig. 3 Numbers of clusters that can occupy 70% of the conformations within accumulated 5 ns period under both force fields. | |
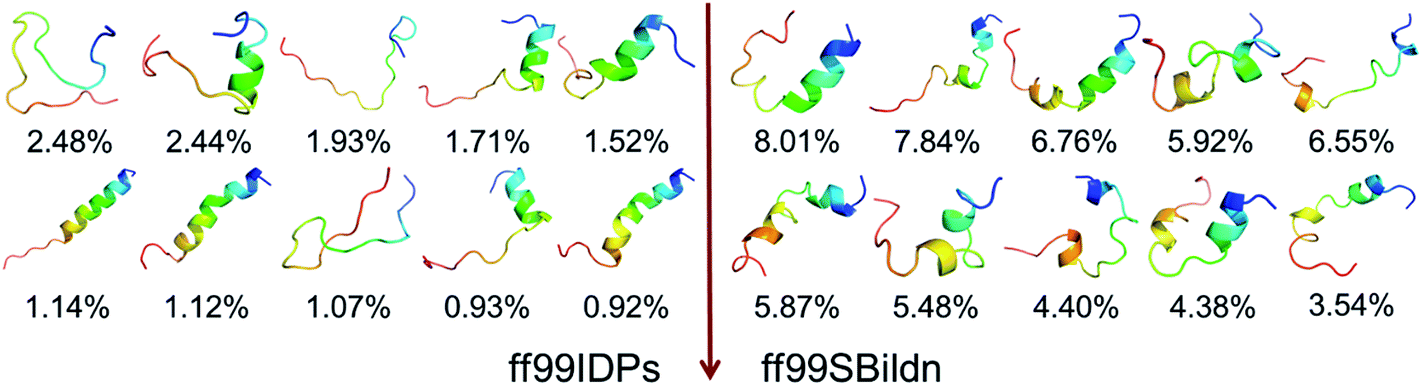
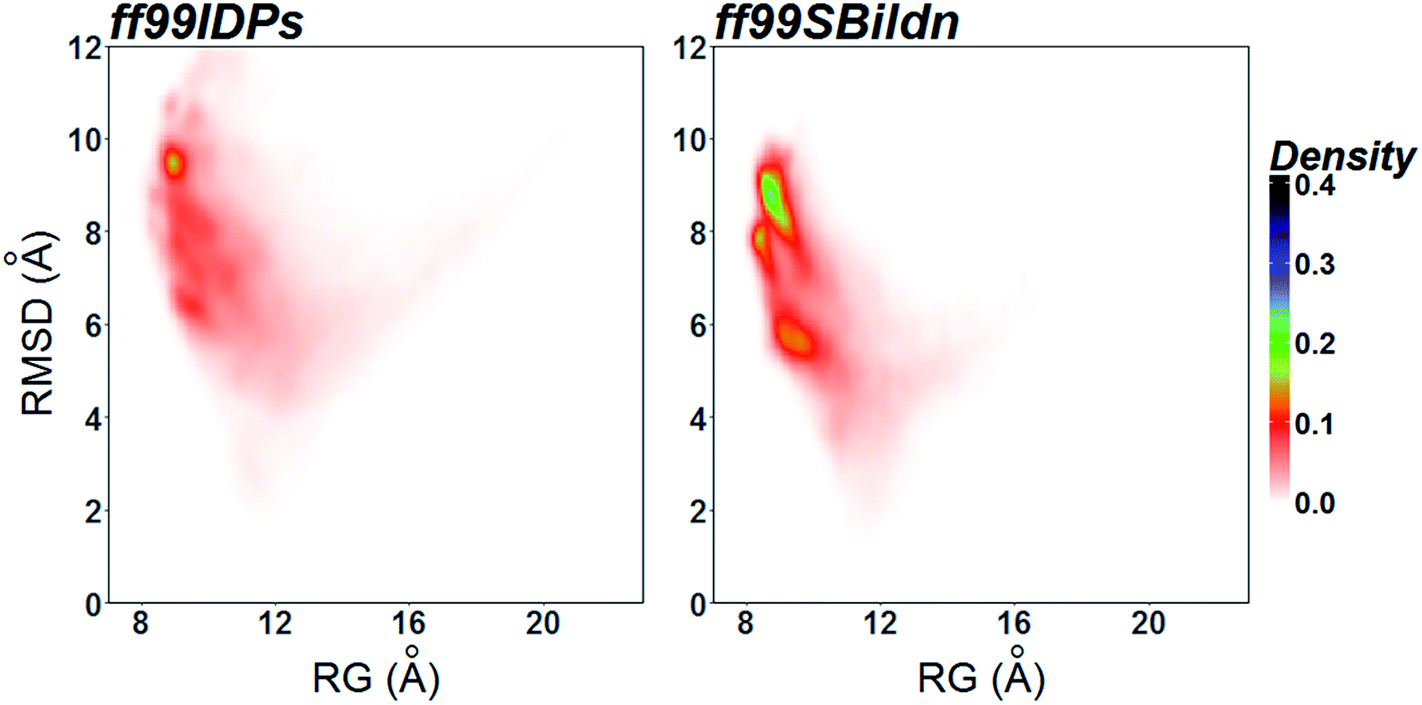
The clustering, secondary Cα chemical shift, helicity of c-Myb under ff99IDPs and ff99SBildn are shown in Fig. 4–6. Apparent differences were found in the structural clustering. The top 10 clusters under ff99SBildn occupy 58.75% of the total conformations, of which the top 14 clusters account for 70%. However, under ff99IDPs, the top 10 clusters occupy only 15.26% of the total conformations, while the top 195 clusters account for 70%. It was apparent that the clusters under ff99IDPs have a higher ratio of population for disordered structures than those under ff99SBildn. Most conformers show disordered state and include few secondary structural elements especially in C-terminal domain. The PMF free energy landscapes in respect to RMSD and RG show that the distribution of conformers from ff99SBildn is located at RMSDs from 2 to 10 Å and RGs from 8 to ∼15 Å, while that from ff99IDPs is located at RMSDs from 2 to 12 Å and RGs from 8 to ∼20 Å, indicating that ff99IDPs samples more flexible conformers than ff99SBildn, which is consistent with the clustering result. In order to compare with the experimental data, secondary Cα chemical shifts were calculates from the clustered representative structures that occupy no less than 70% of the conformations and their occupancies. The full-length RMSD between the data under ff99SBildn and the experimental data was 1.123 ppm, while it is 0.573 ppm between the data under ff99IDPs and the experimental data (shown in Fig. 6A). This shows that the performance of ff99IDPs is much better than that of ff99SBildn to simulate c-Myb. The main difference between simulation under ff99IDPs and experiment is focused on the N-terminal domain by the motif of structure promoting residues of KRI. Force field parameters based on the CMAP correction of these residues indeed improve the sampling of c-Myb. ff99IDPs force field can sample more flexible conformers than ff99SBildn. Furthermore, the helical content under ff99IDPs is also lower than that under ff99SBildn and consistent with the results of chemical shift (shown in Fig. 6B). The result is consistent with the property of IDPs, therefore, the trajectories under ff99IDPs were chosen to study the conformation dynamics for intrinsically disordered c-Myb.
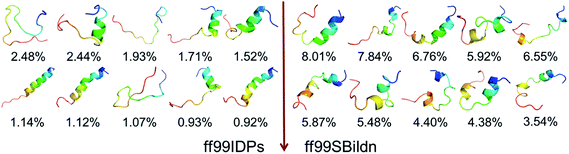 |
| | Fig. 4 Representative conformers of the top 10 clusters and their occupations. | |
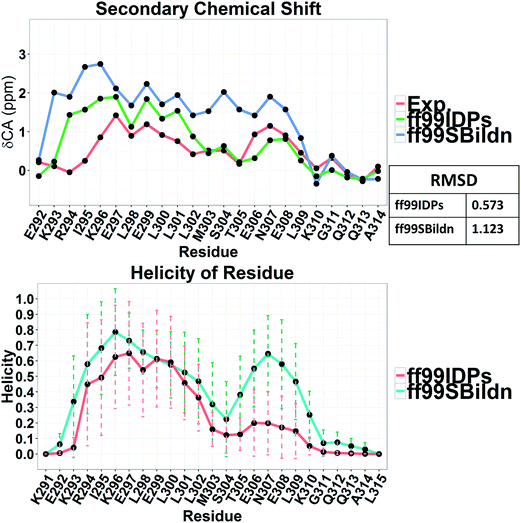 |
| | Fig. 5 RMSD between experimental and prediction data for secondary chemical shift and helicity under force fields of ff99SBildn and ff99IDPs. | |
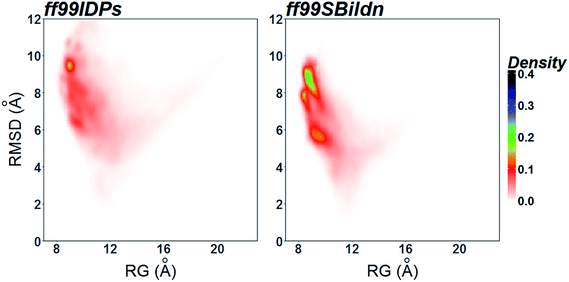 |
| | Fig. 6 PMF free energy landscapes on the 2D space of radius of gyration (RG) and root-mean-square deviation (RMSD). | |
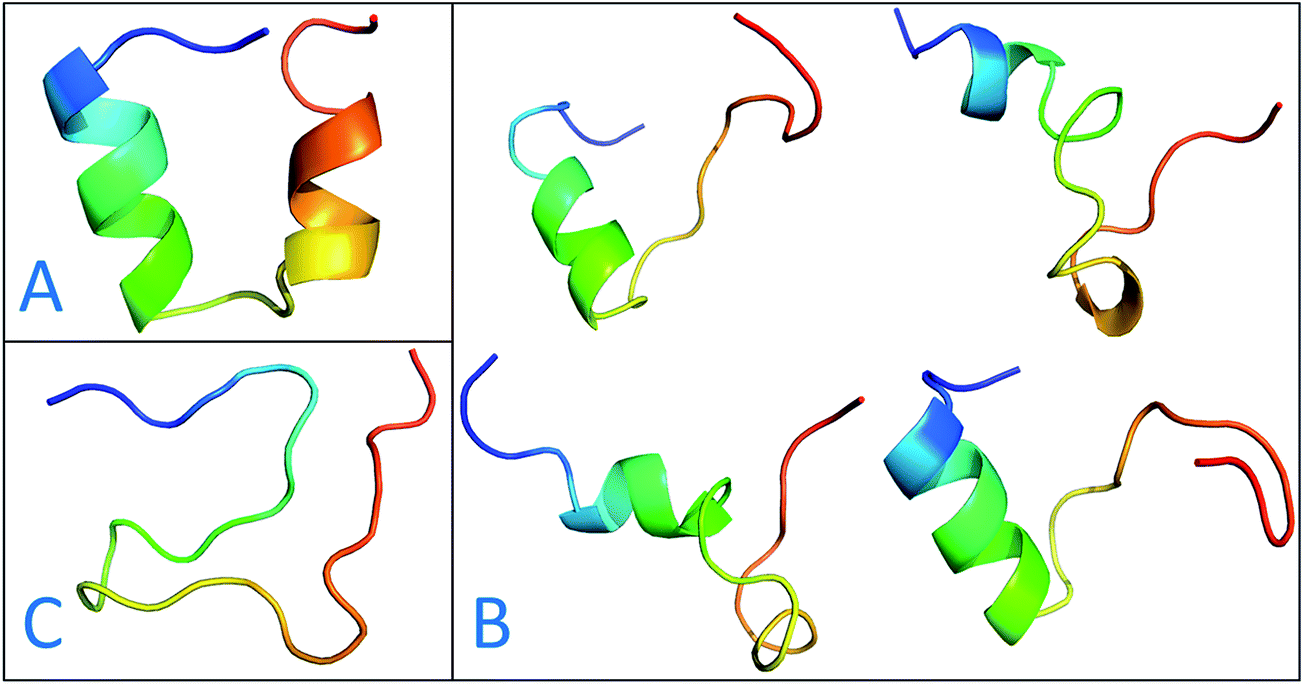
The previous work reports that IDPs were grouped into three structurally different subclasses, native molten globules, native pre-molten globules and native coils.45 According to the content of secondary structure for the conformers of c-Myb, these structure subclasses are shown in Fig. 7. The native molten globule states include high properties of helical structure, pre-molten globule with partly helical conformer and native coil corresponding to extended disordered c-Myb.
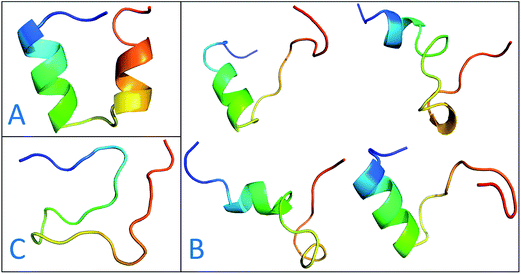 |
| | Fig. 7 Representative structures of intrinsically disordered proteins from ff99IDPs. (A) Native molten globules. (B) Native pre-molten globules. (C) Native coils. | |
Conformation dynamics of c-Myb
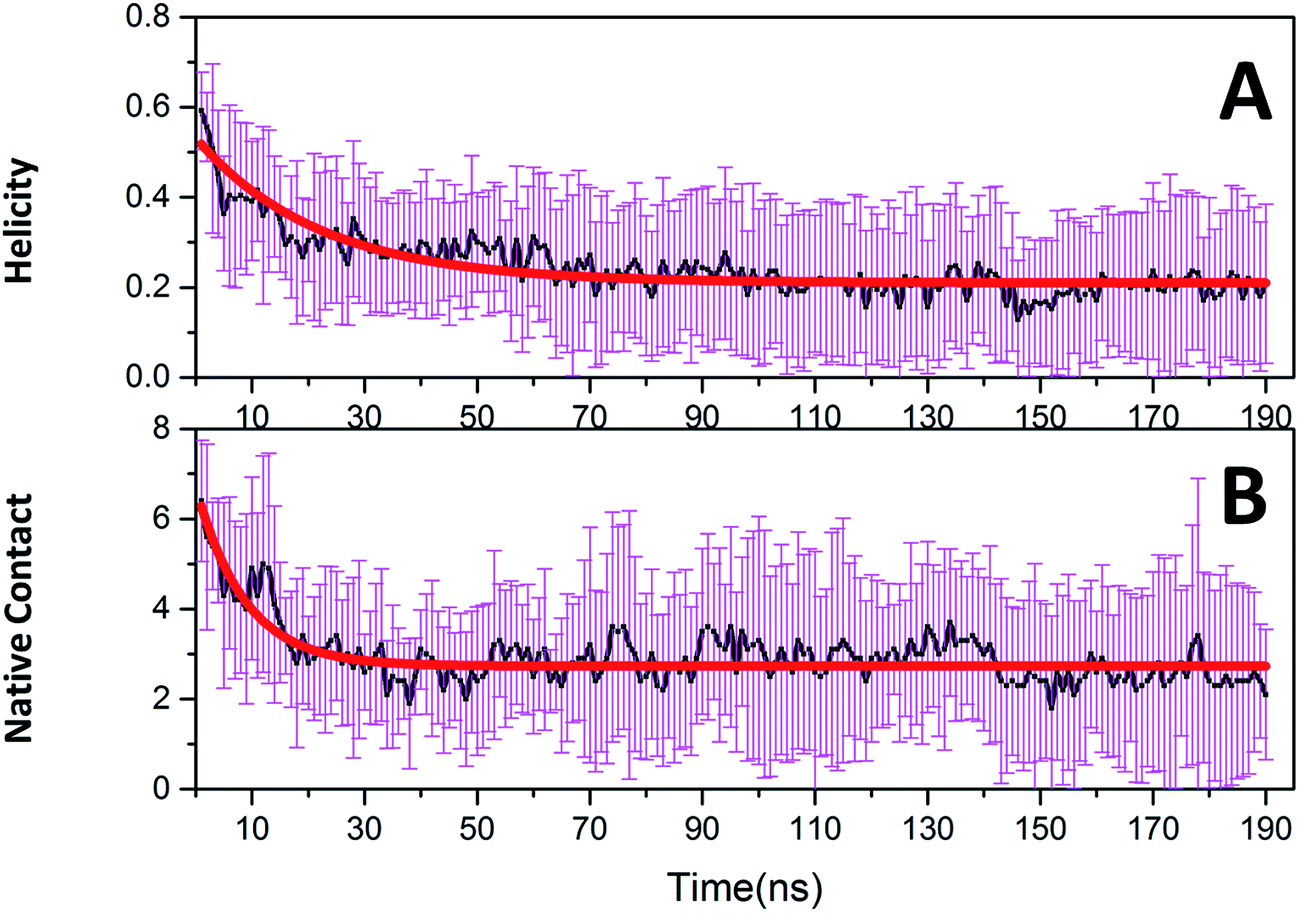
Native tertiary contacts (Qf) and native helical content (Qh) were used to monitor the tertiary and secondary folding kinetics. Time evolutions of Qf and Qh for c-Myb are shown in Fig. 8. Apparently, the tertiary and secondary folding kinetics can be represented well by single exponential function (Table 2). This indicates that the tertiary and secondary folding process obeys first order kinetics at room temperature. Our kinetics analysis shows that the folding half-time of tertiary structure is 8.694 ± 0.783 ns, and the unfolding half-time of helical structure is 21.975 ± 1.546 ns. This indicates that the tertiary unfolding is much faster than the helical structure, that is, the secondary structure unfolding of c-Myb depends on the tertiary unfolding. That is, the unfolding of the tertiary structure happened before the issue of the secondary structure.
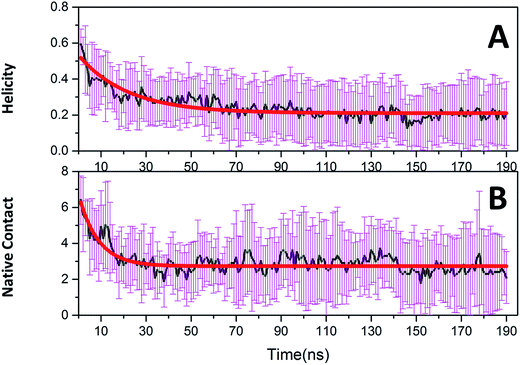 |
| | Fig. 8 Folding kinetics of c-Myb for native contact and helicity content under ff99IDPs. (A) Native contact; (B) helicity content. | |
Table 2 Kinetics parameters for c-Myb
| y = A × exp(−x/τ) + B |
| |
Native contact |
Helicity |
| A |
3.994 ± 0.209 |
0.323 ± 0.012 |
| B |
2.725 ± 0.032 |
0.210 ± 0.004 |
| τ |
8.694 ± 0.783 |
21.975 ± 1.546 |
| R2 |
0.760 |
0.831 |
In order to further reveal the folding kinetics, the dynamic correlation network analysis was used to illustrate the residue fluctuation correlation. To construct the correlation network, the covariance matrices were first calculated. Then fluctuation correlation networks were built. The topology parameters of network for three stages according to the time scale of folding kinetics are listed in Table 3.
Table 3 Topology parameters for correlation network
| |
0–8.7 ns |
8.7–21.9 ns |
21.9–190 ns |
| Clustering coefficient |
0.213 |
0.157 |
0 |
| Network centralization |
0.132 |
0.134 |
0.056 |
| Average number of neighbors |
4.348 |
3.304 |
1.76 |
| Network density |
0.198 |
0.150 |
0.073 |
| Network heterogeneity |
0.453 |
0.159 |
0.332 |
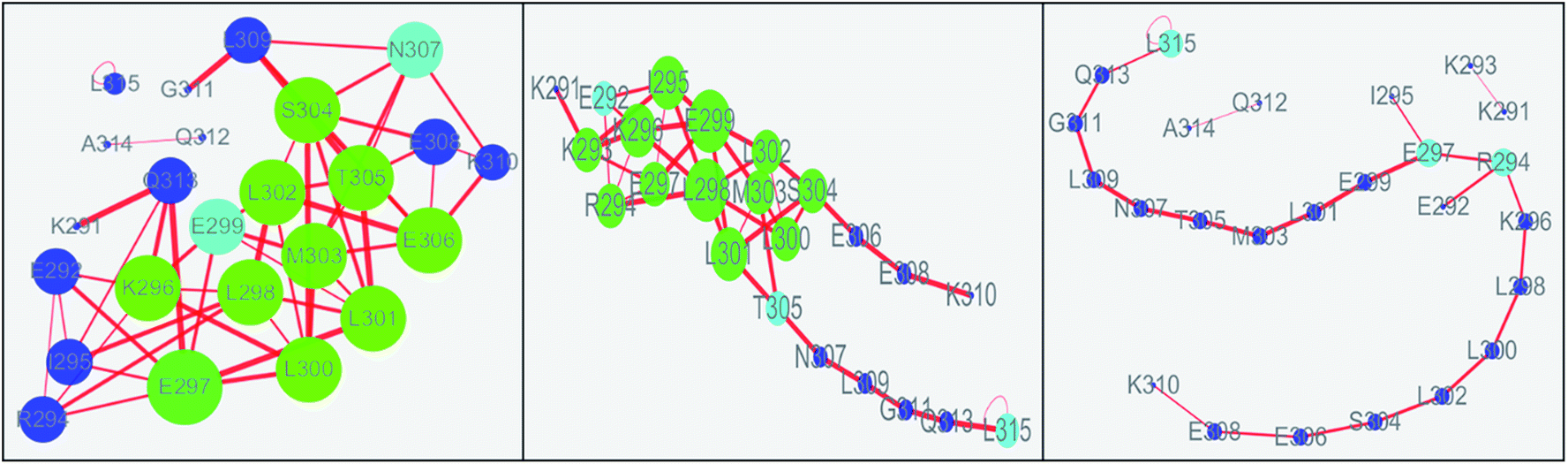
The values of topology parameters for network of the first stage are the highest among these four stages. This suggests that the characters of network for the last stages are significant different from those of other stages. The dynamics correlation networks for three stages are shown in Fig. 9. It shows that the number of nodes with weighted degree higher than 10 (more than 10 edges) is 17 in the first stage network and more than that of the other stages. Q313, E292, I295, K294, E297, L300, L298, K296, E299, M303, L302, S304, T305, E306, N307, E308, K310, with higher degree were marked in the first stage and most of them were located on the N-terminal and middle regions. At the second stage, there are just 14 nodes with high weighted degree. The changed nodes are mostly focused on the middle region. For the last stage, the network is separated into three parts. This suggests that the folding of c-Myb almost finished and C-terminal and most middle regions changed into disordered. However, there are two nodes with high weighted degree remaining. These high weighted nodes might be as folding nucleus to stabilize the structure of c-Myb.
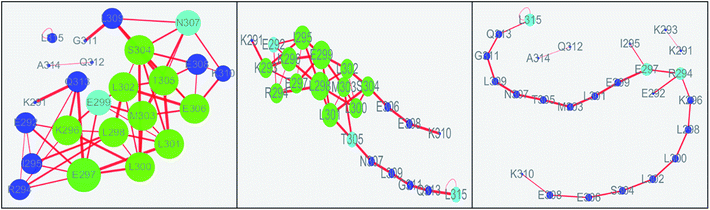 |
| | Fig. 9 Dynamics correlation networks of c-Myb under ff99IDPs for three different stages. | |
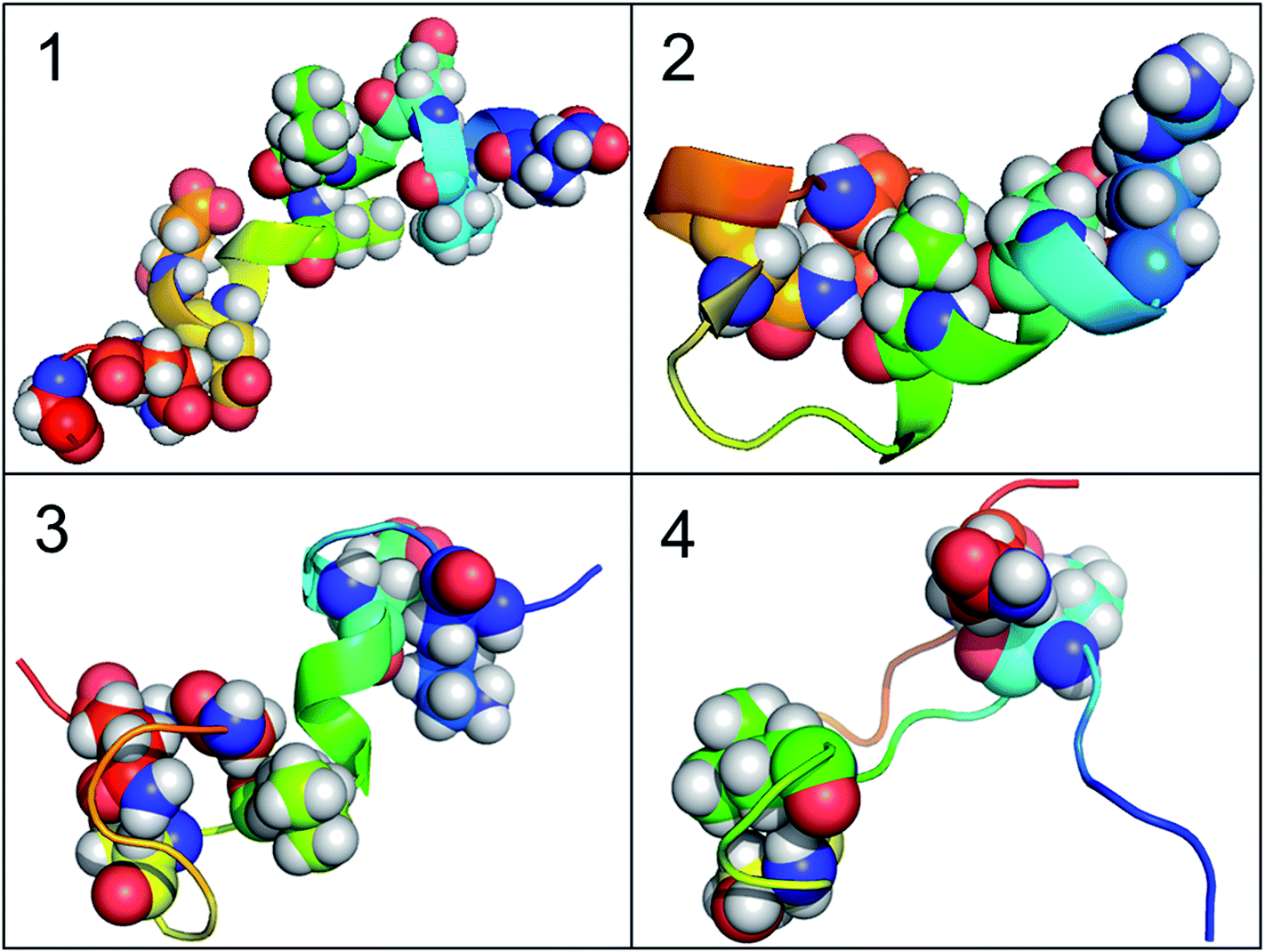
According to the results of correlation network analysis, the folding pathway corresponding to the representative structure of c-Myb is shown in Fig. 10. From the folding pathway, we can find a significant folding site, which located between K296 and L298 residues for KEL. These structures are corresponding to the initial state which includes enough native contacts, tertiary folding state which discards part of native contacts, secondary folding state with part helical structure, and folding state with folding nucleus.
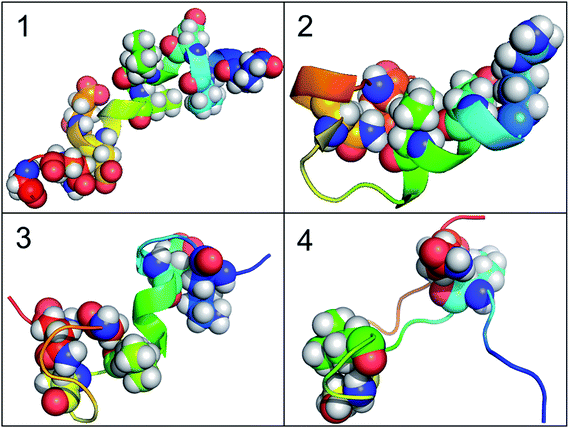 |
| | Fig. 10 The folding pathway of c-Myb. (1) Folded state. (2) Tertiary folding. (3) Helical folding (4). | |
Effects of solvent models in simulations of c-Myb
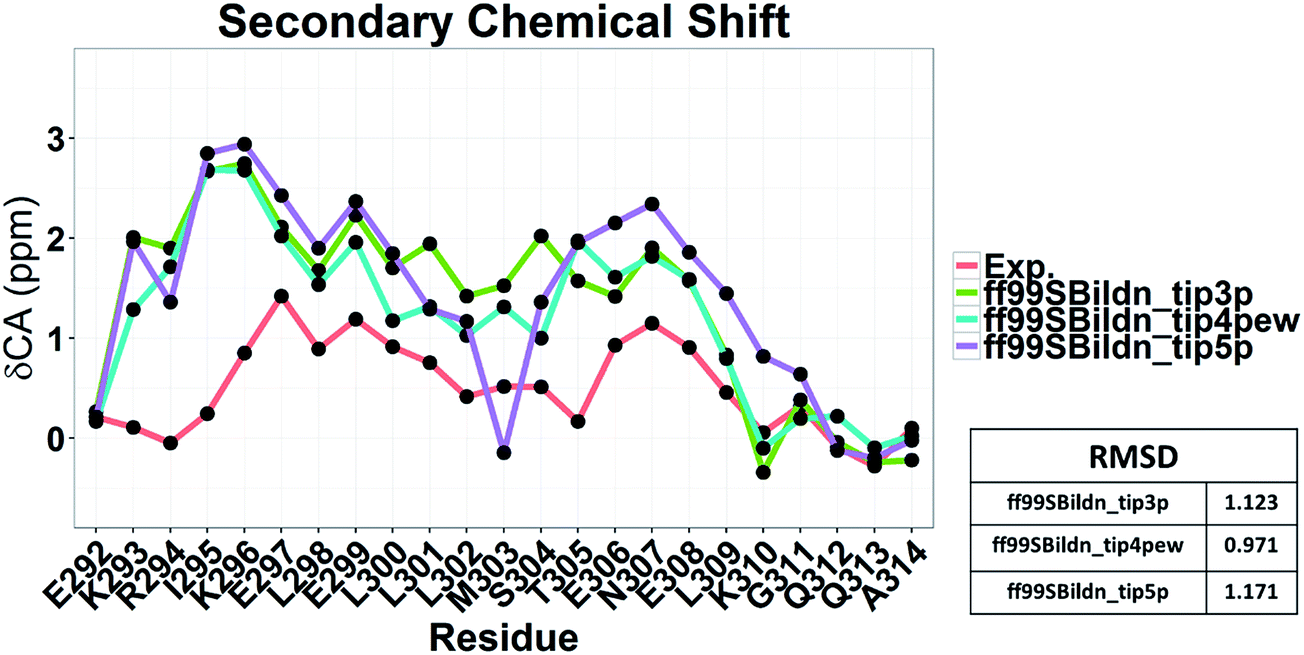
To illustrate the influence of solvent models with generic protein force fields in simulation of c-Myb, we tested TIP3P, TIP4P-EW, and TIP5P models with ff99SBildn. The results are shown in Fig. 11. These data show that the differences due to the use of different solvent models do exist, though the effect is small in the final agreement between simulation and experiment. Therefore, force field correction is necessary for improved structural sampling of c-Myb.
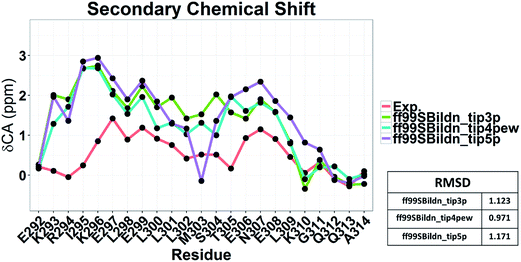 |
| | Fig. 11 Secondary chemical shift predictions from different solvent models with ff99SBildn. | |
Nevertheless, the small effects of solvent models may be found by the biases in ff99SBildn force fields. We thus hypothesized that the solvent model might still play a larger role in sampling c-Myb with ff99IDPs. To verify this hypothesis, MD simulations with TIP3P, TIP4P-EW, and TIP5P solvent models were conducted in ff99IDPs, respectively. The comparison with NMR experiment is shown in Fig. 12. The analysis suggests that the ff99IDPs with the modern solvent models of TIP3P, TIP4P-EW and TIP5P do improve the accuracy of c-Myb conformer and better than ff99SBildn. In general, the result of predicted secondary chemical shift with ff99IDPs/TIP3P was similar to that with ff99IDPs/TIP4P-EW. The hydrogen bond between water and c-Myb for different solvent models is shown in ESI Fig. S1.† In general, the number of hydrogen bond for TIP5P was significant different from that for TIP3P or TIP4P-EW. This is consistent with the difference of chemical shift. In summary, these detailed comparison between simulation and experiment for c-Myb further supports the accuracy of ff99IDPs with the TIP3P or TIP4P-EW solvent in modeling IDPs structures.
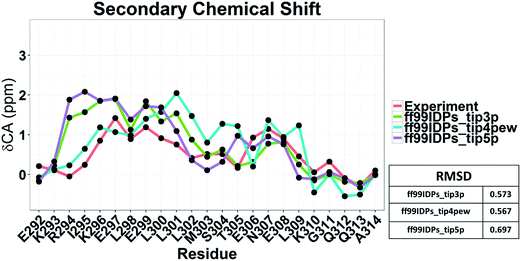 |
| | Fig. 12 Secondary chemical shift prediction from different solvent models with ff99IDPs. | |
Conclusion
Multiple trajectories molecular dynamics (MD) simulations with ff99IDPs and ff99SBildn force fields was used to study the conformation dynamics of c-Myb. Comparison with ff99SBildn, ff99IDPs can reproduce more diverse disordered conformers of c-Myb. The predicted secondary chemical shift under ff99IDPs is more approach to that of experiment than under ff99SBildn. Therefore, ff99IDPs can sample native molten globule, native pre-molten globule and native coil conformers. These results are consistent with other IDPs. Kinetic analysis of MD simulations shows that c-Myb folds via a two-state process and indicates that c-Myb folds in the order of tertiary folding and folding of helix. Folding nucleus of KEL plays essential role in stabilizing the folding state with dynamics correlation networks. The influences of solvent models for TIP3P, TIP4P-EW and TIP5P were also investigated and found that ff99IDPs and TIP3P or TIP4P-EW is the best combination to research the conformer sampling of c-Myb. These results reveal the conformation dynamics of c-Myb and confirm that ff99IDPs force field can be used to research the relationship between structure and function for IDPs.
Conflict of interest
The authors declare that there is no conflict of interest.
Acknowledgements
This work was supported by Center for HPC at Shanghai Jiao Tong University, the National High-tech R&D Program of China (863 Program) (2014AA021502), the National Natural Science Foundation of China (31620103901), Medical Engineering Cross Fund of Shanghai Jiao Tong University (YG2014MS47 and YG2015MS56), and National Institutes of Health/NIGMS (GM093040 & GM079383).
References
- G. L. Shen-Ong, The myb oncogene, Biochim. Biophys. Acta, 1990, 1032, 39–52 CAS.
- B. Luscher and R. N. Eisenman, New light on Myc and Myb. Part II. Myb, Genes Dev., 1990, 4, 2235–2241 CrossRef CAS PubMed.
- M. Introna, M. Luchetti, M. Castellano, M. Arsura and J. Golay, The myb oncogene family of transcription factors: potent regulators of hematopoietic cell proliferation and differentiation, Semin. Cancer Biol., 1994, 5, 113–124 CAS.
- M. A. Thompson and R. G. Ramsay, Myb: an old oncoprotein with new roles, BioEssays, 1995, 17, 341–350 CrossRef CAS PubMed.
- K. T. Greig, S. Carotta and S. L. Nutt, Critical roles for c-Myb in hematopoietic progenitor cells, Semin. Immunol., 2008, 20, 247–256 CrossRef CAS PubMed.
- M. Roussel, S. Saule, C. Lagrou, C. Rommens, H. Beug, T. Graf and D. Stehelin, Three new types of viral oncogene of cellular origin specific for haematopoietic cell transformation, Nature, 1979, 281, 452–455 CrossRef CAS PubMed.
- L. M. Souza, J. N. Strommer, R. L. Hillyard, M. C. Komaromy and M. A. Baluda, Cellular sequences are present in the presumptive avian myeloblastosis virus genome, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 5177–5181 CrossRef CAS.
- D. G. Bergmann, L. M. Souza and M. A. Baluda, Vertebrate DNAs contain nucleotide sequences related to the transforming gene of avian myeloblastosis virus, J. Virol., 1981, 40, 450–455 CAS.
- C. J. Davidson, R. Tirouvanziam, L. A. Herzenberg and J. S. Lipsick, Functional evolution of the vertebrate Myb gene family: B-Myb, but neither A-Myb nor c-Myb, complements Drosophila Myb in hemocytes, Genetics, 2005, 169, 215–229 CrossRef CAS PubMed.
- M. B. Kastan, D. J. Slamon and C. I. Civin, Expression of protooncogene c-myb in normal human hematopoietic cells, Blood, 1989, 73, 1444–1451 CAS.
- C. E. Burns, J. L. Galloway, A. C. Smith, M. D. Keefe, T. J. Cashman, E. J. Paik, E. A. Mayhall, A. H. Amsterdam and L. I. Zon, A genetic screen in zebrafish defines a hierarchical network of pathways required for hematopoietic stem cell emergence, Blood, 2009, 113, 5776–5782 CrossRef CAS PubMed.
- H. Sakura, C. Kanei-Ishii, T. Nagase, H. Nakagoshi, T. J. Gonda and S. Ishii, Delineation of three functional domains of the transcriptional activator encoded by the c-myb protooncogene, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 5758–5762 CrossRef CAS.
- T. Zor, B. M. Mayr, H. J. Dyson, M. R. Montminy and P. E. Wright, Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators, J. Biol. Chem., 2002, 277, 42241–42248 CrossRef CAS PubMed.
- X. Chu, F. Liu, B. A. Maxwell, Y. Wang, Z. Suo, H. Wang, W. Han and J. Wang, Dynamic conformational change regulates the protein-DNA recognition: an investigation on binding of a Y-family polymerase to its target DNA, PLoS Comput. Biol., 2014, 10, e1003804 Search PubMed.
- X. Chu, Y. Wang, L. Gan, Y. Bai, W. Han, E. Wang and J. Wang, Importance of electrostatic interactions in the association of intrinsically disordered histone chaperone Chz1 and histone H2A.Z-H2B, PLoS Comput. Biol., 2012, 8, e1002608 CAS.
- J. Wang, Y. Wang, X. Chu, S. J. Hagen, W. Han and E. Wang, Multi-scaled explorations of binding-induced folding of intrinsically disordered protein inhibitor IA3 to its target enzyme, PLoS Comput. Biol., 2011, 7, e1001118 CAS.
- F. Liu, X. Chu, H. P. Lu and J. Wang, Molecular mechanism of multispecific recognition of Calmodulin through conformational changes, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3927–E3934 CrossRef CAS PubMed.
- X. Chu, L. Gan, E. Wang and J. Wang, Quantifying the topography of the intrinsic energy landscape of flexible biomolecular recognition, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2342–E2351 CrossRef CAS PubMed.
- D. Song, W. Wang, W. Ye, D. Ji, R. Luo and H. F. Chen, ff14IDPs force field improving the conformation sampling of intrinsically disordered proteins, Chem. Biol. Drug Des., 2017, 89, 5–15 CAS.
- W. Wang, W. Ye, C. Jiang, R. Luo and H. F. Chen, New force field on modeling intrinsically disordered proteins, Chem. Biol. Drug Des., 2014, 84, 253–269 CAS.
- W. Ye, D. Ji, W. Wang, R. Luo and H. F. Chen, Test and Evaluation of ff99IDPs Force Field for Intrinsically Disordered Proteins, J. Chem. Inf. Model., 2015, 55, 1021–1029 CrossRef CAS PubMed.
- D. Song, R. Luo and H. F. Chen, The IDP-Specific Force Field ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins, J. Chem. Inf. Model., 2017, 57, 1166–1178 CrossRef CAS PubMed.
- A. D. MacKerell Jr, M. Feig and C. L. Brooks 3rd, Improved treatment of the protein backbone in empirical force fields, J. Am. Chem. Soc., 2004, 126, 698–699 CrossRef PubMed.
- A. D. Mackerell Jr, M. Feig and C. L. Brooks 3rd, Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef PubMed.
- R. N. De Guzman, N. K. Goto, H. J. Dyson and P. E. Wright, Structural basis for cooperative transcription factor binding to the CBP coactivator, J. Mol. Biol., 2006, 355, 1005–10013 CrossRef CAS PubMed.
- D. A. Case, et al., AMBER 12, in University of California, San Francisco, 2012 Search PubMed.
- H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura and T. Head-Gordon, Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew, J. Chem. Phys., 2004, 120, 9665–9678 CrossRef CAS PubMed.
- M. W. Mahoney, A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions, J. Chem. Phys., 2000, 112, 8910–8922 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N˙
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(N) method for Ewald sums in large systems., J. Chem. Phys., 1993, 98, 4 CrossRef.
log(N) method for Ewald sums in large systems., J. Chem. Phys., 1993, 98, 4 CrossRef. - R. Baron and N. A. Vellore, LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12509–12514 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham 3rd, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- M. Feig, J. Karanicolas and C. L. Brooks 3rd, MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology, J. Mol. Graphics Modell., 2004, 22, 377–395 CrossRef CAS PubMed.
- H. F. Chen and R. Luo, Binding induced folding in p53-MDM2 complex, J. Am. Chem. Soc., 2007, 129, 2930–2937 CrossRef CAS PubMed.
- H. F. Chen, Mechanism of Coupled Folding and Binding in the siRNA-PAZ Complex, J. Chem. Theory Comput., 2008, 4, 1360–1368 CrossRef CAS PubMed.
- F. Qin, Y. Chen, M. Wu, Y. Li, J. Zhang and H. F. Chen, Induced fit or conformational selection for RNA/U1A folding, RNA, 2010, 16, 1053–1061 CrossRef CAS PubMed.
- M. Arai, K. Sugase, H. J. Dyson and P. E. Wright, Conformational propensities of intrinsically disordered proteins influence the mechanism
of binding and folding, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9614–9619 CrossRef CAS PubMed.
- Y. Shen and A. Bax, Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology, J. Biomol. NMR, 2007, 38, 289–302 CrossRef CAS PubMed.
- W. Wang, C. Jiang, J. Zhang, W. Ye, R. Luo and H. F. Chen, Dynamics Correlation Network for Allosteric Switching of PreQ1 Riboswitch, Sci. Rep., 2016, 6, 31005 CrossRef CAS PubMed.
- L. Xu, W. Ye, C. Jiang, J. Yang, J. Zhang, Y. Feng, R. Luo and H. F. Chen, Recognition mechanism between Lac repressor and DNA with correlation network analysis, J. Phys. Chem. B, 2015, 119, 2844–2856 CrossRef CAS PubMed.
- J. Yang, H. Liu, X. Liu, C. Gu, R. Luo and H. F. Chen, Synergistic Allosteric Mechanism of Fructose-1,6-bisphosphate and Serine for Pyruvate Kinase M2 via Dynamics Fluctuation Network Analysis, J. Chem. Inf. Model., 2016, 56, 1184–1192 CrossRef CAS PubMed.
- J. Zhang, H. Luo, H. Liu, W. Ye, R. Luo and H. F. Chen, Synergistic Modification Induced Specific Recognition between Histone and TRIM24 via Fluctuation Correlation Network Analysis, Sci. Rep., 2016, 6, 24587 CrossRef CAS PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- R. W. Floyd, Algorithm 97: shortest path, Commun. ACM, 1962, 5 Search PubMed.
- V. N. Uversky, Intrinsically disordered proteins and their environment: effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding, Protein J., 2009, 28, 305–325 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04133k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ac
*ac
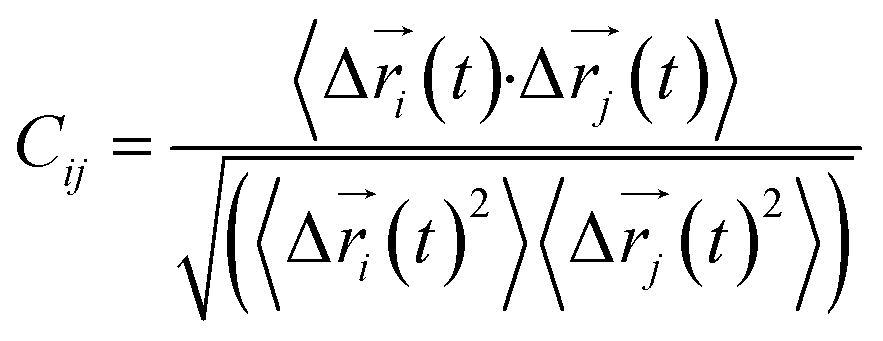
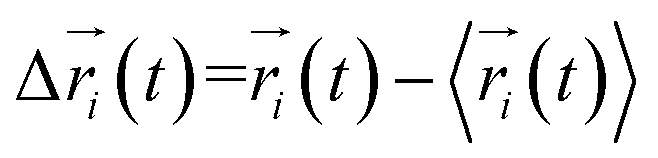 ,
, 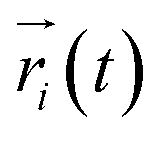 is the position of node i at time t, and 〈·〉 represents a time averaging. These elements were conveniently organized as a covariance matrix for simulated system. This general method has been successfully applied in several systems.39–42 In the current study, the covariance matrix for each system was constructed using snapshots (every 2 ps) of all simulated trajectories. Besides nodes, “edge” that transfers allosteric information from one node to another is defined between any two nodes without covalent bond and the distance between two heavy atoms from two nodes are closer than 4.5 Å over 75% sampling time. The strength of the edge between nodes i and j is defined as the absolute value of the inter-node correlation (Cij). The number of connected edges at each node is defined as the degree of the node. Correlation-weighted degree, which is the summation of strengths of all edges connected to a given node, indicates the importance of the node. After the network construction, Cytoscape3.1.1 was used to calculate the network topological parameters.43 The shortest path between any two nodes in the network was identified with the Floyd–Warshall algorithm.44
is the position of node i at time t, and 〈·〉 represents a time averaging. These elements were conveniently organized as a covariance matrix for simulated system. This general method has been successfully applied in several systems.39–42 In the current study, the covariance matrix for each system was constructed using snapshots (every 2 ps) of all simulated trajectories. Besides nodes, “edge” that transfers allosteric information from one node to another is defined between any two nodes without covalent bond and the distance between two heavy atoms from two nodes are closer than 4.5 Å over 75% sampling time. The strength of the edge between nodes i and j is defined as the absolute value of the inter-node correlation (Cij). The number of connected edges at each node is defined as the degree of the node. Correlation-weighted degree, which is the summation of strengths of all edges connected to a given node, indicates the importance of the node. After the network construction, Cytoscape3.1.1 was used to calculate the network topological parameters.43 The shortest path between any two nodes in the network was identified with the Floyd–Warshall algorithm.44