
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-CD22-conjugated CdTe QDs co-loaded with doxorubicin and gambogic acid: a novel platform for lymphoma treatment†
Peipei Xu a,
Huaqin Zuoa,
Dangui Chenb,
Miaoxin Penga,
Ying Jianga,
Xu Liua,
Jian Ouyanga and
Bing Chen*b
a,
Huaqin Zuoa,
Dangui Chenb,
Miaoxin Penga,
Ying Jianga,
Xu Liua,
Jian Ouyanga and
Bing Chen*b
aDepartment of Hematology, Drum Tower Hospital, School of Medicine, Nanjing University, Nanjing, Jiangsu 210093, P. R. China
bDepartment of Hematology, Nanjing Drum Tower Hospital Clinical College of Nanjing Medical University, Nanjing 210036, P. R. China. E-mail: chenb211@163.com
First published on 5th July 2017
Abstract
Chemotherapy is the main treatment modality for lymphoma but may cause severe adverse effects. The number of patients who are refractory to conventional chemotherapeutic drugs continuously increases. We propose a novel drug delivery system comprising anti-CD22-conjugated cadmium–tellurium quantum dots (CdTe QDs) co-loaded with doxorubicin (DOX) and gambogic acid (GA; as a chemosensitizer). The system, designated as DOX/GA–CdTe–CD22, can be used for nanoparticle-based targeted combination chemotherapy. The system showed appropriate diameter, good dispersibility, high encapsulation efficiency, and high drug loading. The therapeutic and side effects were evaluated by in vitro and in vivo experiments. Results demonstrate that DOX/GA–CdTe–CD22 can precisely deliver drugs to tumor cells, thereby improving the antitumor activity of the drug and attenuating its toxicity against normal tissues. The enhanced efficacy could be due to increased apoptosis via the Bax/caspase-3/PARP pathway. This study suggests a novel and promising therapeutic option for lymphoma.
Introduction
Lymphoma, which originates from lymphoid tissues, is one of the most common cancers and is included in the top ten causes of cancer-related deaths.1 Lymphoma can be classified into Hodgkin's lymphomas and non-Hodgkin lymphomas (NHLs). Approximately 90% of lymphomas are NHLs, of which 85% originate from B cells.2 Currently, chemotherapy remains the main treatment modality for lymphoma. However, conventional chemotherapeutic agents can cause various adverse effects because of their non-specific biodistribution.3 Furthermore, the number of patients with lymphoma refractory to conventional chemotherapeutic drugs continuously increases.4 Both phenomena hamper the widespread clinical application of drugs. Therefore, scholars must focus on reducing the side effects of drugs on normal tissues and increasing the sensitivity of tumor cells to chemotherapeutic agents.Gambogic acid (GA) extracted from gamboge exhibit a wide spectrum of anticancer activity by targeting Bcl-2 family and NF-κB pathways.5,6 As a chemotherapy sensitizer, GA has gained increasing attention for improving the chemosensitivity and synergistically inducing apoptosis.7 GA can markedly sensitize refractory breast tumor cells to doxorubicin (DOX).8 The conjugation of GA and adriamycin exerts enhanced antitumor effects on lung cancer xenograft models through NF-κB and P-gp pathways.9
To overcome the limitations of the conventional chemotherapeutic treatments, nanoparticles, such as calcium phosphates, mesoporous silica, and carbon nanotubes, are leading the way toward increasing circulation times, controlling drug release, reducing drug toxicity, improving bioavailability, and targeting specific sites.10–13 Among these nanoparticles, quantum dots (QDs) are particularly ideal for drug delivery because their favorable features, including small size, versatile surface chemistry, and exquisite optical properties.14 Furthermore, cadmium–tellurium (CdTe) quantum dot (QD) nanoparticles have attracted considerable attention because of their unique physical and chemical properties.15,16 The biocompatibility of CdTe QDs can be improved through surface modification.17 As a drug delivery vehicle, CdTe QDs can release loaded drugs in a pH-triggered and controlled pattern; these particles can release drugs at low pH levels (pH < 7), which resemble the acidic microenvironment of solid tumors.18 Moreover, QDs with enhanced permeability and retention (EPR) effect can deliver drugs to tumor tissues.19 Nonetheless, the EPR effect only exhibits passive targeting and limited effects.20
Antibody-targeted chemotherapy (ATC) is an accepted therapeutic strategy, where a chemotherapeutic agent is coupled to monoclonal antibodies against a tumor-associated antigen.21 CD22, which is selectively expressed on B-cell lymphoma, has drawn much attention because mAbs bound to CD22 can be rapidly internalized into tumor cells, resulting in intracellular delivery of the drug.22
In this work, a novel drug delivery system (DOX/GA–CdTe–CD22) comprising anti-CD22-conjugated CdTe QDs co-loaded with DOX and GA is synthesized to develop targeted combination chemotherapy. This delivery system can precisely target lymphoma and facilitate preferential delivery of DOX and GA into tumor cells. The addition of GA as a chemosensitizer can enhance the antitumor effects of DOX and reduce the toxicity on normal tissues. The schematic of this process is shown in Fig. 1. The system was characterized, and its antitumor effects and systematic toxicity were evaluated by in vitro and in vivo experiments. The possible molecular mechanism of the system was further explored in this study.
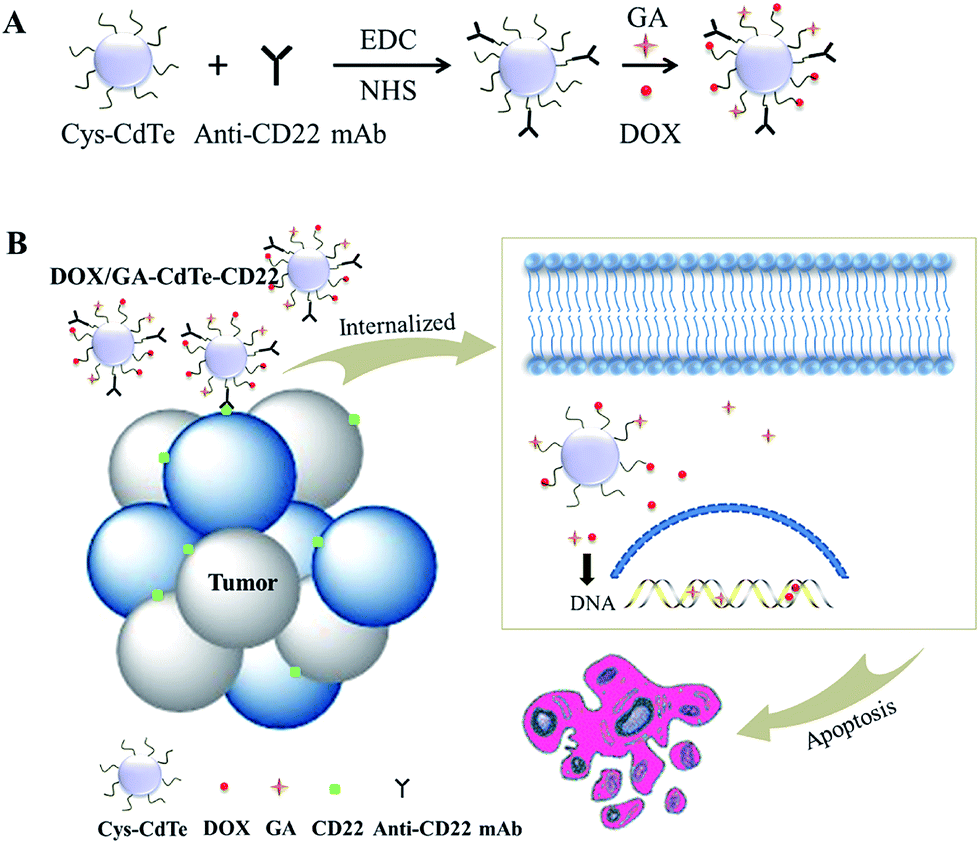 | ||
| Fig. 1 The schematic illustration of the structure and therapy strategy of DOX/GA–CdTe–CD22. (A) Schematic of DOX and GA binding to cys-CdTe QDs with the conjugation of anti-CD22 mAbs. (B) Therapy strategy of DOX/GA–CdTe–CD22. They can be internalized when actively targeting tumor cells through antigen–antibody binding. GA can sensitize the tumor cells to DOX and exert a synergistic effect to induce tumor cell apoptosis. | ||
Materials and methods
Materials
DOX and GA were purchased from Kanion Pharmaceutical Co., Ltd. (Jiangsu, China). EDC, sulfo-NHS and Pierce™ BCA Protein Assay Kit was purchased from Thermo Fisher Scientific (Massachusetts, USA). Roswell Park Memorial Institute medium (RPMI) 1640 was bought from Gibco Chemical Co. (Carlsbad, USA). Fetal bovine serum (FBS) was acquired from Wisent Inc. (Montreal, Canada). Dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich (St Louis, USA). 4,6-Diamidino-2-phenylindole (DAPI) staining solution was supplied by Beyotime Institute of Biotechnology (Jiangsu, China). The hematoxylin–eosin kit was provided by KeyGen Biotech Co., Ltd. (Jiangsu, China). Monoclonal antibodies for CD22, P-gp, Bax, caspase-3, cleaved PARP, and β-actin were purchased from Abcam, Plc. (Massachusetts, USA). All other reagents used in this study were analytically pure.A transmission electron microscope was obtained from Japan Electron Optics Laboratory, Ltd. (Tokyo, Japan). Fourier-transform infrared spectra were recorded using a Nicolet iS10 IR spectrometer from Thermo Fisher Scientific (MA, USA). Zetasizer NanoZS size analyzer was acquired from Malvern Instruments (Malvern, UK).
BALB/c-nude mice (equal numbers of males and females), aged 4–6 weeks and weighing 18–22 g, were purchased from the Shanghai National Center for Laboratory Animals (Shanghai, China). Mice were maintained in specific pathogen-free facilities and provided with food and water ad libitum. All experiments were approved by the Animal and Ethics Review Committee of the Affiliated Drum Tower Hospital of Nanjing University Medical School (Nanjing, China), and the experimental procedures were performed in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals.
Preparation of DOX/GA–CdTe–CD22
Cysteamine-modified CdTe QDs (cys-CdTe QDs) were synthesized as described elsewhere and used in our experiments.17 The anti-CD22 mAbs were coupled to CdTe QDs using EDC/NHS methods.23 5 × 10−8 mol of cys-CdTe QDs and 2 mg of anti-CD22 mAbs were mixed with 5 × 10−6 mol of sulfo-NHS and 5 × 10−5 mol of EDC in 1 ml of PBS buffer at pH 7.0 for 3 h under constant shaking of 100 rpm at room temperature. The anti-CD22-coupled CdTe QDs were purified after centrifugation at 10, 000 rpm for 20 min, and then resuspended in pH 7.4 PBS buffer. The CdTe QDs modified with mouse IgG1 used as isotype control was prepared with the similar method. The concentration of unconjugated antibodies was determined through bicinchoninic acid (BCA) assay and calculated by intrapolation with a standard curve to obtain the binding ratio of the antibodies to CdTe QDs.DOX and GA can efficiently be absorbed onto cys-CdTe QDs through electrostatic interaction. Briefly, 0.1 ml of DOX (1 mg ml−1) and 0.05 ml of mAbs-coupled cys-CdTe QDs (2.55 × 10−5 M) were mixed in 0.8 ml of deionized water under stirring for 6 h at room temperature in the dark. The mixture was then added with 0.05 ml of GA under stirring for another 16 h. DOX/GA–CdTe–CD22 was obtained after stepwise procedures involving centrifugation, discarding of the supernatant, washing, precipitation, and resuspension. Unbound DOX and GA in the supernatant were determined by high-performance liquid chromatography (HPLC).
Loading and encapsulation efficiencies were calculated using the following formulas: drug loading (DL) = (amount of drug in NPs/amount of NPs) × 100%; encapsulation efficiency (EE) = (amount of drug in NPs/amount of the total drug) × 100%. The morphological characteristics of the nanoparticles were observed under Transmission Electron Microscope (TEM). The characteristic bands were recorded by Fourier-transform infrared spectra. The hydrodynamic diameters of QDs were evaluated by dynamic light scattering (DLS). Meanwhile, protein bands of DOX/GA–CdTe, DOX/GA–CdTe–CD22, and anti-CD22 mAbs were stained with Coomassie Brilliant blue R250 following gel electrophoresis to determine whether the anti-CD22 mAbs were coupled to DOX/GA–CdTe QDs.
The DOX release behavior from DOX/GA–CdTe–CD22 (2.55 × 10−5 M of cys-CdTe QDs) were estimated by dynamic dialysis. In brief, 5 ml of DOX/GA–CdTe–CD22 was enclosed in dialysis bags (pH 7.4), which were then immersed in 95 ml PBS at pH 5.5 or 7.4 under constant shaking (100 rpm) at 37 °C. 0.2 ml of PBS was collected every 2 h and replaced with an equivalent volume of fresh PBS. The DOX concentration was quantified by HPLC.
Confocal fluorescence microscopy
Confocal microscopy was used to visually confirm drug loading and conjugation of anti-CD22 mAbs to QDs. Raji cells (a CD22 + cell line derived from B-cell lymphoma) and Jurkat cells (a CD22-cell line from T-cell leukemia), were treated with DOX, DOX/GA–CdTe, DOX/GA–CdTe–IgG1 (isotype control) or DOX/GA–CdTe–CD22 for 4 h. The cells were then centrifuged, washed, and resuspended in PBS. The cell suspension was dropped on a glass slide. After DAPI staining, confocal fluorescence images of the samples were taken with a confocal inverted microscope. The emission wavelengths of DAPI, DOX, and FITC were 454, 592, and 525 nm, respectively.Cell culture
Raji cells and Jurkat cells were obtained from Shanghai Institute of Cells (Shanghai, China). Raji cells were maintained in a RPMI 1640 medium supplemented with 10% fetal bovine serum at 37 °C with 5% CO2 in a humidified atmosphere. The cells undergo passage every 2–3 days.Cell viability assay
Raji cells and Jurkat cells were seeded in a 96-well plate with 1 × 104 cells per well and treated with PBS (as the control group), anti-CD22, DOX, DOX + GA, DOX–CdTe, DOX/GA–CdTe, and DOX/GA–CdTe–CD22. Each group has three duplicates. The concentration of DOX was 1.6 μg ml−1, and the concentration of GA was 0.4 μg ml−1. After incubation for 24, 48, or 72 h, each well was added with the CCK-8 solution (10 μl) and incubated for another 3 h. Growth inhibition rates (%) of Raji cells and Jurkat cells were calculated as (1 − ODtreatment/ODcontrol) × 100.Cell apoptosis study
Raji cells were seeded in a 24-well plate with 1.5 × 105 cells per well. After incubation with different treatments for 24 h, cells were collected and washed twice with cold PBS. The concentrations of DOX and GA were the same as mentioned above. The cells were then stained with 5 μl of Annexin V-FITC for 15 min and kept away from light. Apoptosis was quantitatively detected by flow cytometry (FCM). The experiments were repeated at least three times.Raji cells were processed as mentioned above to observe morphological changes associated with cell apoptosis. After staining with DAPI, the morphological changes of nuclei were observed with a fluorescence microscope.
Western blot assay
Raji cells were harvested after different treatments and subjected to western blot analysis. Proteins of Raji cells were extracted on ice using RIPA buffer (150 mM NaCl, 50 mM Tris–HCl pH 8, 0.5% sodium deoxycholate, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS)). Total proteins (25 μg) were size fractionated by SDS/PAGE and transferred to a PVDF membrane. After blocking with 5% BSA for 1 h, the following monoclonal antibodies were used for western blot analysis: P-gp, Bax, caspase-3, cleaved PARP, and β-actin (as internal control). The blots were detected by an enhanced chemiluminescence detection system (Amersham, UK).Nude mouse model of lymphoma xenografts
Tumor-bearing mouse models were established by subcutaneously injecting 200 μl of Raji cell suspension in PBS (2 × 107 cells per ml). Tumors were measured by a caliper. Tumor volume was calculated according to this formula: 1/2 × length × (width)2. The tumor-bearing model was established successfully when the tumor volume reached 80–150 mm3.In vivo tumor imaging
Tumor-bearing mice were intravenously injected with DOX, DOX/GA–CdTe, or DOX/GA–CdTe–CD22 (8 mg per kg per DOX). Anesthetized mice were imaged 24 h after injection by using an IVIS imaging system with the excitation wavelength of 485 nm and emission wavelength of 590 nm. The autofluorescence background was subtracted using Living Image Software (Xenogen).Tissue distribution of DOX in tumor-bearing mice
Tumor-bearing mice were intravenously injected with a single dose of DOX, DOX/GA–CdTe or DOX/GA–CdTe–CD22 (DOX 5 mg kg−1). At 4 h after injection, the mice were sacrificed and their main organs and tumor tissues were carefully isolated. The tissues were washed, lysed and homogenized. DOX in the tissue homogenate was extracted by acidified isopropanol. After centrifugation, the supernatant was analyzed in a spectrofluorometer with excitation and emission wavelengths of 485 and 590 nm, respectively.Determination of therapeutic efficacy
Tumor-bearing mice were randomly allocated to seven groups with three mice in each group: a (PBS as negative control), b (anti-CD22), c (DOX), d (DOX + GA), e (DOX–CdTe), f (DOX/GA–CdTe), and g (DOX/GA–CdTe–CD22). Each mouse received 200 μl of the treatment agent every 2 days via tail vein. The equivalent dosage of DOX for a single dose was 1 mg kg−1 each and concentration of GA was 0.25 mg kg−1 each. Tumor volume was measured 0, 2, 4, 6, 8, 10, and 12 days after the treatment. Relative tumor volume (RTV) was defined as RTV = V/V0, where V0 is the tumor volume before the treatment. The general condition of the mice, including eating, drinking, excretion, weight, and activity, were monitored after the treatment. On day 12, the serum concentrations of creatine kinase-MB (CK-MB), alanine transaminase (ALT), and blood urea nitrogen (BUN) were detected to evaluate myocardial injury, hepatic function, and renal function, respectively. Tumors and organs (heart, lung, liver, spleen, and kidney) were isolated from the sacrificed mice for further experiments.Histopathological examination
The major organs isolated from mice were fixed in 4% formaldehyde solution, embedded in paraffin, sectioned, and stained with hematoxylin–eosin. The resulting tissue sections were observed under an optical microscope.Immunofluorescence assay
Frozen sections of tumors were prepared and rinsed with PBS until room temperature was achieved. The sections were blocked for 1 h, added with diluted primary antibodies (P-gp, Bax, caspase-3, and cleaved PARP), and incubated overnight at 4 °C. FITC-Conjugated anti-mouse IgG was used as secondary antibody and incubated for 1 h at room temperature in the dark. After the nuclei were stained by DAPI, the cells were covered with coverslip slides with the anti-fade reagent. The covered sections were observed with a fluorescent microscope.Statistical analysis
Data are presented as means ± standard deviation and analyzed with SPSS software. One-way ANOVA followed by a Student–Newman–Keuls (SNK) test was used to calculate P values. Values with P < 0.05 were considered statistically significant.Results
Characterization of DOX/GA–CdTe–CD22
The TEM image indicated that DOX/GA–CdTe–CD22 exhibited a spherical morphology and dispersed well in water (Fig. 2A). The particle size distribution of DOX/GA–CdTe–CD22 was determined by DLS experiments. Fig. 2B shows that the average hydrodynamic diameter was 18.3 nm. The zeta potential of the cys-CdTe QDs, DOX/GA–CdTe, DOX/GA–CdTe–CD22 was −21.5 ± 3.1 mV, −10.4 ± 2.6 mV, and −10.6 ± 1.7 mV, respectively. The results of Coomassie Brilliant blue staining (Fig. 2C) indicated that anti-CD22 mAbs were successfully linked to CdTe QDs. Fourier-transform infrared spectroscopy was performed to verify successful loading of DOX and GA onto CdTe QDs. As shown in Fig. 2D, the spectra of DOX/GA–CdTe and DOX/GA–CdTe–CD22 showed characteristic bands of DOX at 1284 and 1716 cm−1 and those of GA at 1680 cm−1. This finding indicates the favorable loading of DOX and GA on CdTe QDs. Hence, DOX/GA–CdTe–CD22 was successfully synthesized.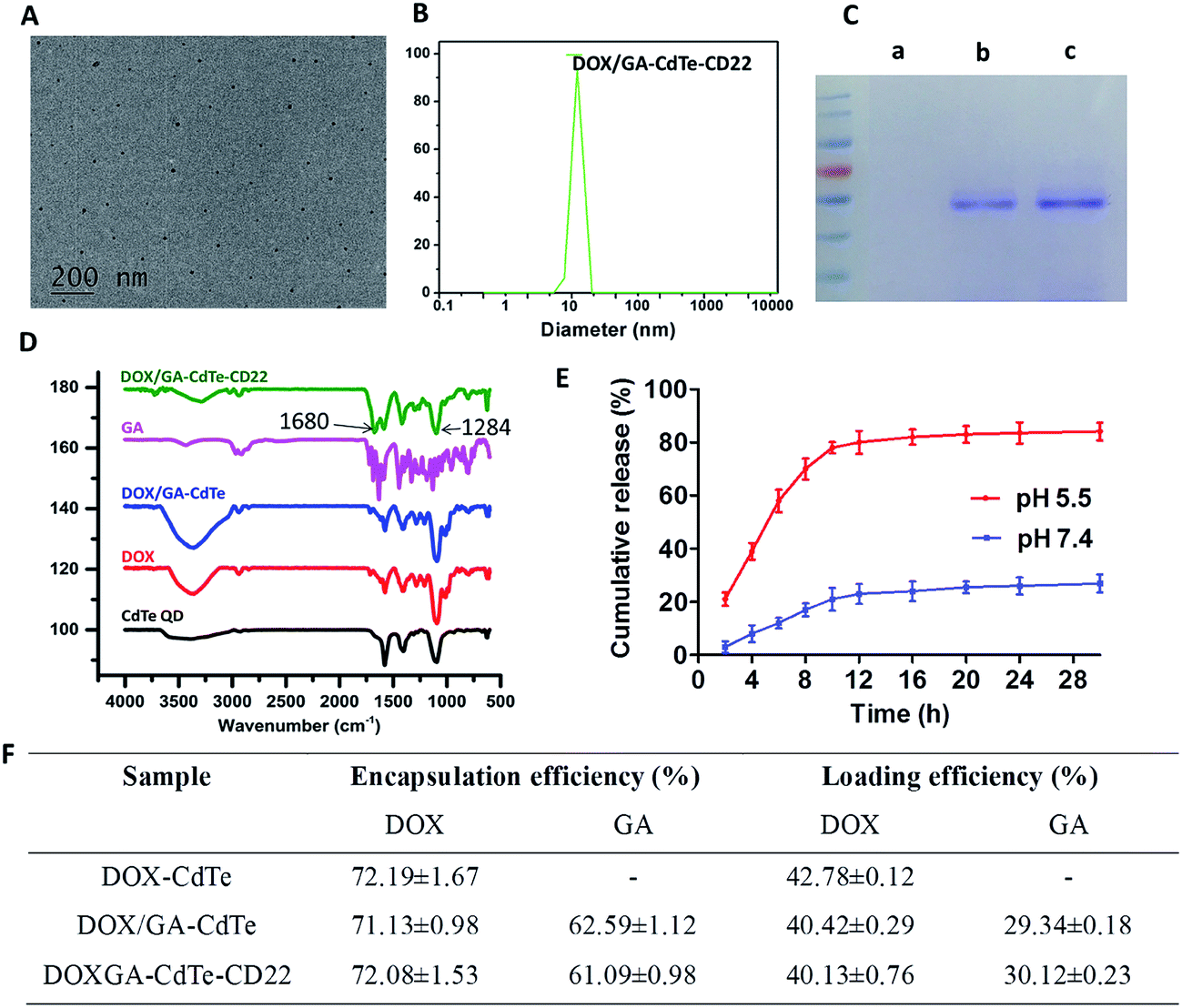 | ||
| Fig. 2 Characterization of DOX/GA–CdTe–CD22. (A) TEM image of DOX/GA–CdTe–CD22. (B) DLS analysis of DOX/GA–CdTe–CD22. (C) Protein bands of DOX/GA–CdTe (a), DOX/GA–CdTe–CD22 (b) and anti-CD22 mAbs (c) stained by Coomassie Brilliant blue R250. (D) Fourier transform infrared spectra of cys-CdTe QDs, DOX, GA, DOX/GA–CdTe and DOX/GA–CdTe–CD22. (E) DOX release behavior from DOX/GA–CdTe–CD22 at pH 5.5 and pH 7.4. (F) Encapsulation efficiency and drug loading of DOX–CdTe, DOX/GA–CdTe and DOX/GA–CdTe–CD22. | ||
The EE and DL of DOX and GA in DOX/GA–CdTe–CD22 were determined by HLPC. The EE and DL values were 72.08% ± 1.53% and 40.13% ± 0.76%, respectively, for DOX and 61.09% ± 0.98% and 30.12% ± 0.23%, respectively, for GA. According to the results of the BCA assay, approximately 70% of anti-CD22 mAbs were cross-linked to cys-CdTe QDs.
The cumulative release of DOX from DOX/GA–CdTe–CD22 is shown in Fig. 2E, which suggests a pH-triggered release behavior. The DOX release rate at pH 7.4 (the physiological environment) was relatively slow and sustained, while it was much faster at pH 5.5 (an acidic condition similar to tumor microenvironment). Therefore, a high concentration of DOX in tumor tissues could be obtained with a low DOX concentration in normal tissues.
The fluorescence images under a confocal microscope further confirmed that DOX and anti-CD22 mAbs were conjugated to the QDs (Fig. 3). Moreover, DOX markedly accumulated in Raji cells in the DOX/GA–CdTe–CD22 group compared with that in DOX alone, DOX/GA–CdTe and DOX/GA–CdTe–IgG1 group. Therefore, anti-CD22-conjugated CdTe QDs could enhance drug binding and intracellular delivery by actively targeting tumor cells.
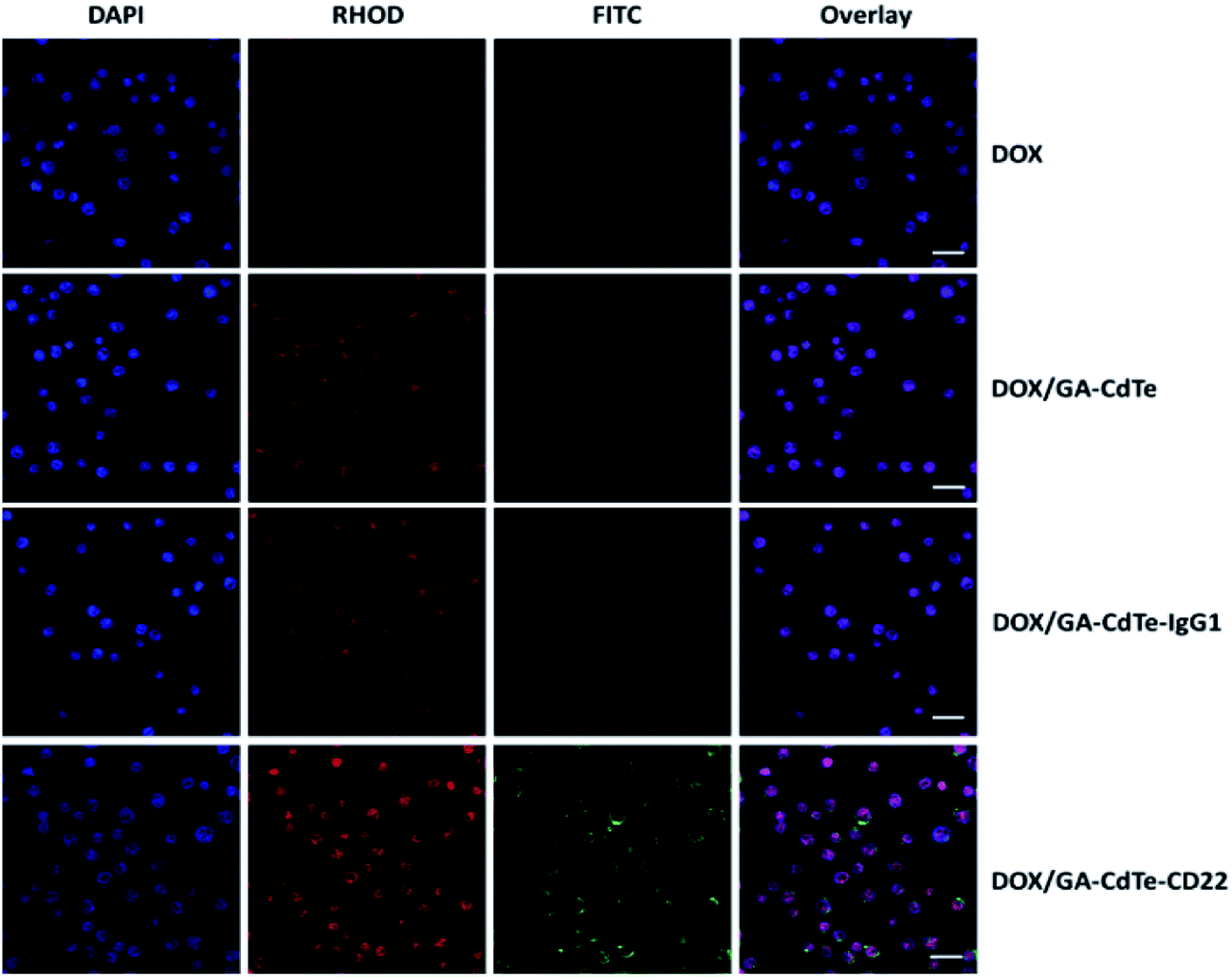 | ||
| Fig. 3 Fluorescence images of Raji cells respectively treated with DOX, DOX/GA–CdTe, DOX/GA–CdTe–IgG1 and DOX/GA–CdTe–CD22 after DAPI staining (scale bar: 20 μm, ×200). FITC-Conjugated anti-mouse IgG was used as the secondary antibodies for anti-CD22 mAbs. | ||
As a comparison, the fluorescence images under a confocal microscope of Jurkat cells (CD22−) showed there is no obvious differences between cell groups treated with DOX/GA–CdTe–CD22 and DOX/GA–CdTe (Fig. 4A, Fig. S1†), indicating the specificity of the binding of anti-CD22 loaded nanoparticles to CD22+ (Raji) cells. Additionally, the growth inhibition rate of Jurkat cells (Fig. 4B) treated with DOX/GA–CdTe–CD22 has no significant difference compared with DOX/GA–CdTe-treated cells at (P > 0.05). These results demonstrated that there is no significant difference between Jurkat cells (CD22−) treated with DOX/GA–CdTe–CD22 and those treated with DOX/GA–CdTe.
 | ||
| Fig. 4 (A) Fluorescence images of Jurkat cells respectively treated with DOX, DOX/GA–CdTe and DOX/GA–CdTe–CD22 after DAPI staining (scale bar: 20 μm, ×200). FITC-Conjugated anti-mouse IgG was used as the secondary antibodies for anti-CD22 mAbs. (B) Growth inhibition of Jurkat cells treated with PBS (control), anti-CD22, DOX, DOX + GA, DOX–CdTe, DOX/GA–CdTe, and DOX/GA–CdTe–CD22 at 24, 48 and 72 h (*P < 0.05 when compared to the control group at 24, 48 or 72 h; #P < 0.05). | ||
Cytotoxic effects of DOX/GA–CdTe–CD22 on Raji cells
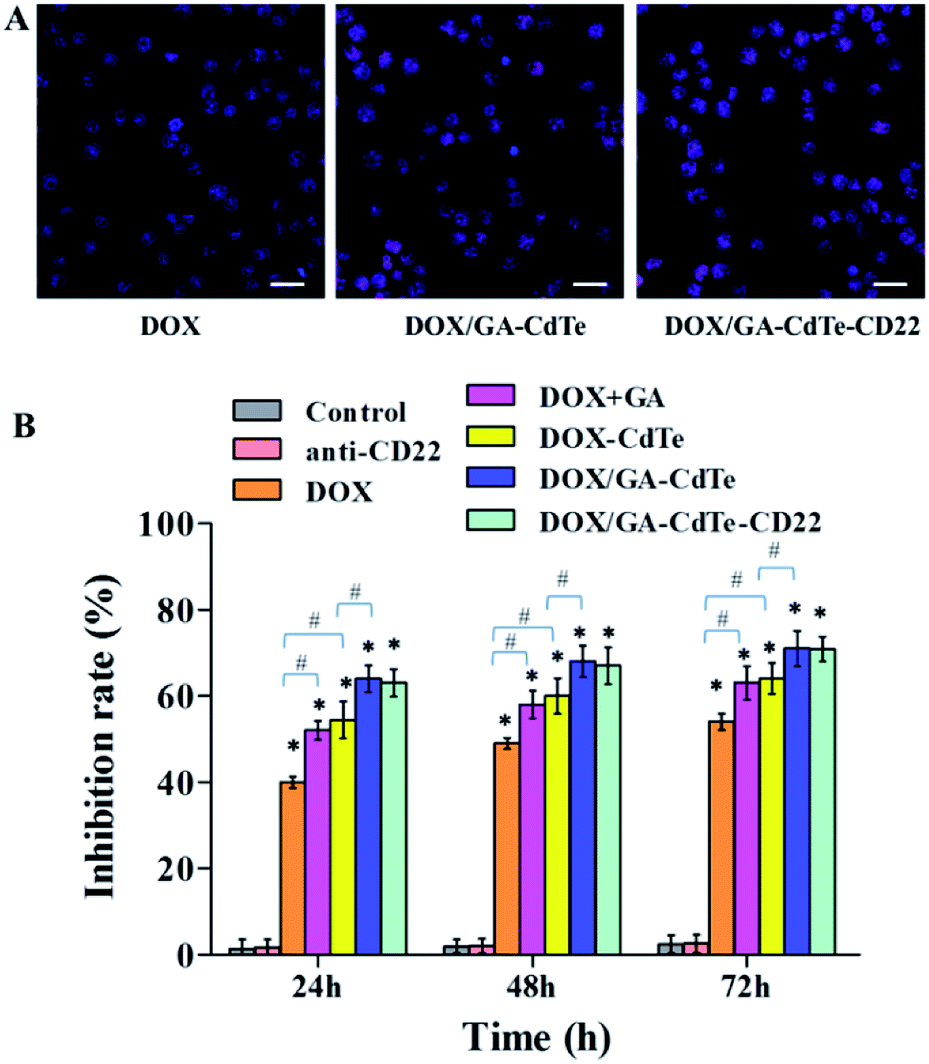
The cytotoxic effects of DOX/GA–CdTe–CD22 on Raji cells were evaluated using CCK-8 assay. The viability of Raji cells treated with DOX, DOX/GA–CdTe, or DOX/GA–CdTe–CD22 in different concentrations of DOX (0.2, 0.4, 0.8, 1.6 and 2.0 μg ml−1) was studied (Fig. 5A). The IC50 values for DOX, DOX/GA–CdTe, and DOX/GA–CdTe–CD22 were 1.77 μg ml−1, 1.13 μg ml−1, and 0.83 μg ml−1, respectively. As illustrated in Fig. 5B, the inhibition rates in Raji cells treated with DOX/GA–CdTe–CD22 were 79%, 85%, and 92% after incubation for 24, 48, and 72 h, respectively; these rates were significantly higher than those detected in the other groups (P < 0.05). High inhibition rates corresponded to high concentrations of DOX in Raji cells treated with DOX/GA–CdTe–CD22 (Fig. 3). Thus, CdTe QDs conjugated with anti-CD22 mAbs could enhance drug cytotoxicity by accumulating intracellular drugs through targeted delivery.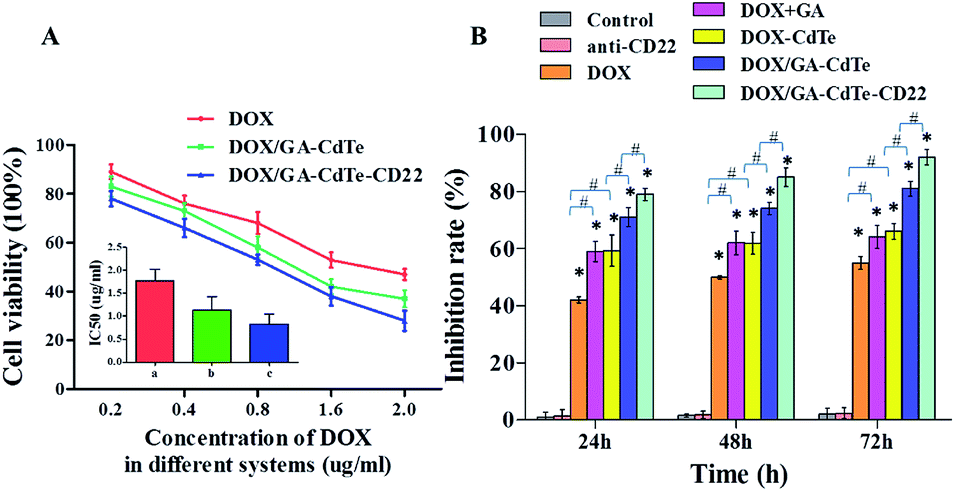 | ||
| Fig. 5 Cytotoxic effects of DOX/GA–CdTe–CD22 against Raji cells. (A) The cytotoxic effects of DOX, DOX/GA–CdTe and DOX/GA–CdTe–CD22 against Raji cells with different concentration of DOX; inset: the IC50 of DOX in different systems for Raji cells at 24 h. (a) DOX; (b) DOX/GA–CdTe; (c) DOX/GA–CdTe–CD22. (B) Growth inhibition of Raji cells treated with PBS (control), anti-CD22, DOX, DOX + GA, DOX–CdTe, DOX/GA–CdTe, and DOX/GA–CdTe–CD22 at 24, 48 and 72 h (*P < 0.05 when compared to the control group at 24, 48 or 72 h; #P < 0.05). | ||
Apoptosis of Raji cells
The apoptosis of Raji cells was evaluated by FCM (Fig. 6A). After different treatments, the corresponding total apoptosis rates were 3.0%, 3.1%, 18.9%, 35.3%, 36.8%, 49.6%, and 60.7% in the control, anti-CD22, DOX, DOX + GA, DOX–CdTe, DOX/GA–CdTe, and DOX/GA–CdTe–CD22 groups, respectively. Compared with the DOX group, apoptosis significantly increased in DOX + GA and DOX–CdTe groups (P < 0.05). Furthermore, the apoptosis rate significantly differed between Raji cells treated with DOX/GA–CdTe and DOX + GA. Particularly, the apoptosis rate in the DOX/GA–CdTe–CD22 group was the highest level among all groups. These results suggest that GA could enhance the antitumor activity of DOX by chemosensitization; moreover, the antitumor effects of DOX and GA co-loaded with anti-CD22-coupled CdTe QDs remarkably increased.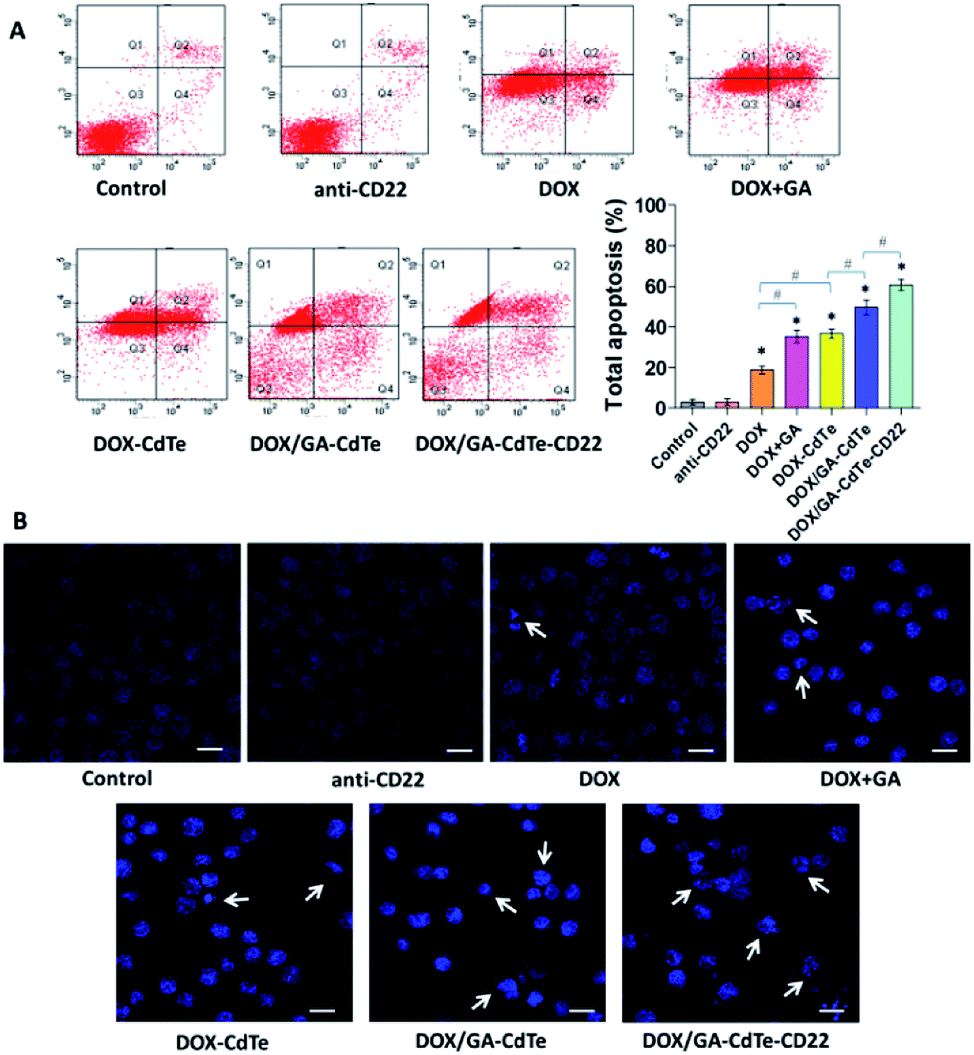 | ||
| Fig. 6 Apoptosis of Raji cells. (A) The apoptosis of Raji cells with different treatments was investigated using FCM; comparative apoptosis rates of Raji cells in different groups were showed in the bar graph (*P < 0.05 when compared with controls, #P < 0.05). (B) The fluorescence microscopy images of Raji cell nuclei stained by DAPI (scale bar: 10 μm, ×200). Apoptosis is indicated by “→”. | ||
The morphological changes associated with nuclear apoptosis in Raji cells were observed under a fluorescence microscope. The control and anti-CD22 groups showed homogeneously dispersed viable cell nuclei, demonstrating that no apoptosis occurred (Fig. 6B). In the groups of DOX, DOX + GA, DOX–CdTe, DOX/GA–CdTe, and DOX/GA–CdTe–CD22, the number of apoptotic cells increased; these cells exhibited the typical appearance of apoptosis, marked by chromatin condensation and nucleus segmentation. This finding is in accordance with the FCM results on apoptosis; hence, GA and antibody-conjugated CdTe QDs could improve the antitumor effects of DOX.
Western blot analysis
Western blot assay was performed to study the antitumor mechanism of DOX/GA–CdTe–CD22 (Fig. 7A). The apoptosis-associated protein expression levels of Bax, caspase-3, and cleaved PARP were gradually upregulated in Raji cells treated with DOX, DOX/GA–CdTe, and DOX/GA–CdTe–CD22 (Fig. 7B). By contrast, P-gp expression displayed a reverse trend. Compared with those in the DOX group, the expression levels of Bax, caspase-3, and cleaved PARP were higher in the groups of DOX + GA and DOX/GA–CdTe (P < 0.05), while the P-gp expression was lower. Moreover, these changes were particularly pronounced in the DOX/GA–CdTe–CD22 group (P < 0.05). These findings confirm that DOX/GA–CdTe–CD22 exhibited the highest antitumor activity among the others.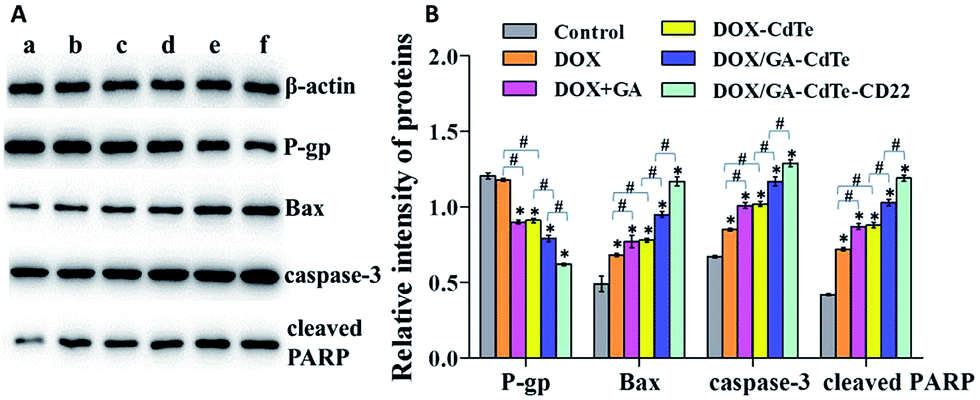 | ||
| Fig. 7 Expression of apoptosis-associated proteins. (a) Control; (b) DOX; (c) DOX + GA; (d) DOX–CdTe; (e) DOX/GA–CdTe; (f) DOX/GA–CdTe–CD22 (*P < 0.05 when compare with control, #P < 0.05). | ||
In vivo imaging
Mice were visualized with an in vivo imaging system 24 h after the treatment. Fig. 8A shows that the fluorescence signal intensity in the tumor tissues of DOX/GA–CdTe–CD22-treated mice was higher than that in DOX/GA–CdTe- or DOX-treated mice. These results reveal that CdTe QDs conjugated with anti-CD22 mAbs could increase the DOX accumulation in tumors; hence, these particles could be used to promote selectivity of drugs in tumor tissues.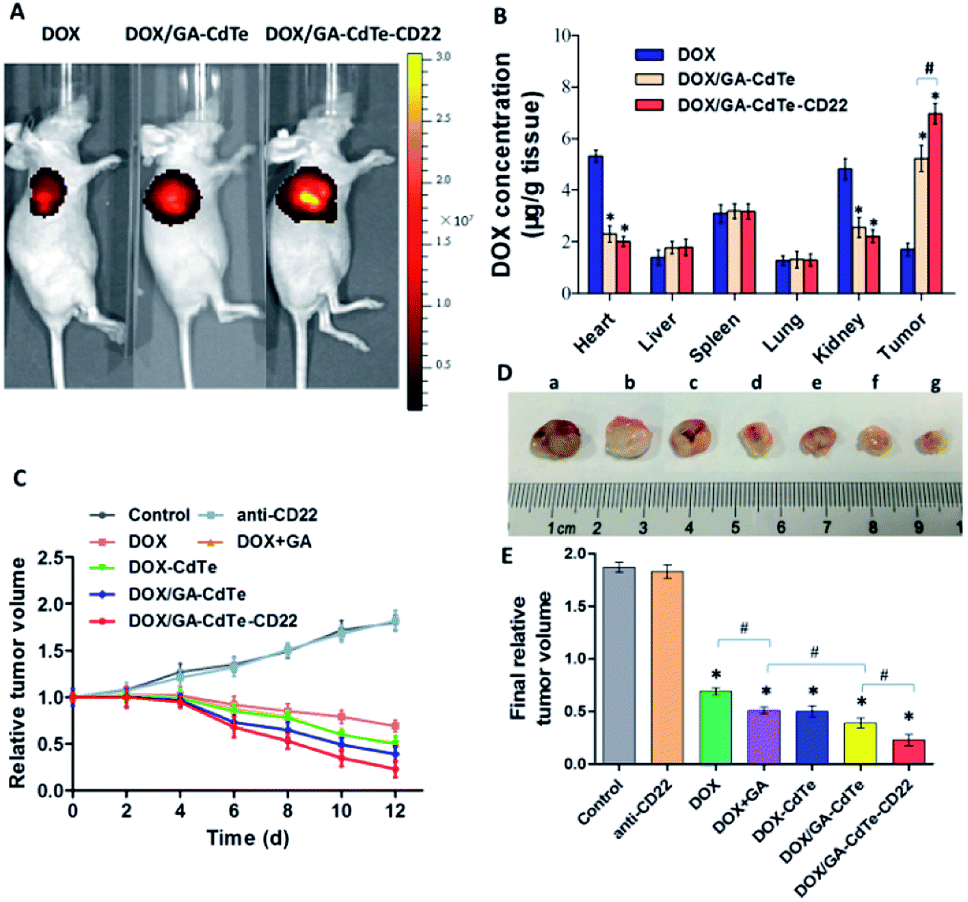 | ||
| Fig. 8 In vivo effects of DOX/GA–CdTe–CD22. (A) Fluorescence images of tumor-bearing mice after different treatments. (B) Tissue distribution of DOX at 4 h after injection with DOX, DOX/GA–CdTe or DOX/GA–CdTe–CD22. (C) Relative tumor volumes over time after administration of different treatments. (D and E) The final tumor volume of mice with different treatments. (a) Control; (b) anti-CD22; (c) DOX; (d) DOX + GA; (e) DOX–CdTe; (f) DOX/GA–CdTe; (g) DOX/GA–CdTe–CD22 (*P < 0.05 when compare with control, #P < 0.05). | ||
Tissue distribution of DOX
As shown in Fig. 8B, the mice treated with DOX had a high drug concentration in heart tissues with a low one in tumor tissues. On the contrary, the DOX concentration in heart tissues from mice treated with DOX/GA–CdTe or DOX/GA–CdTe–CD22 was significantly lower (P < 0.05). Furthermore, the tumor tissues of mice treated with DOX/GA–CdTe or DOX/GA–CdTe–CD22 had a higher DOX concentration than that with free DOX, especially the latter (P < 0.05). This finding reveals that DOX/GA–CdTe–CD22 can effectively accumulates DOX in tumor tissues and protect normal tissues from toxicity caused by high DOX concentration.Therapeutic effects in vivo
The tumor volume of mice in the control group increased continuously, whereas the tumor growth was inhibited over time after treatment in the other groups (Fig. 8C). As we can see from Fig. 8D and E, the differences in tumor response among various treatments in vivo were statistically significant (P < 0.05). The final volume of tumors from the DOX/GA–CdTe–CD22 group was significantly smaller than those from the DOX/GA–CdTe group, and the tumor volume in DOX/GA–CdTe group were significantly decreased compared with that in DOX + GA group. Additionally, the tumor size in the DOX–CdTe or DOX + GA groups was smaller than that in the DOX group. The smallest tumor size was observed in the DOX/GA–CdTe–CD22 group at the end of the experiments. Therefore, GA could sensitize tumor cells to DOX and antibody-conjugated CdTe QDs could enhance the anti-tumor activity of DOX, thereby suppressing tumor growth.Toxicity against normal tissues
No apparent changes in eating, behavior, physical features, or neurological conditions were observed in tumor-bearing mice injected with DOX, anti-CD22, DOX + GA, DOX–CdTe, DOX/GA–CdTe, or DOX/GA–CdTe–CD22. The CK-MB concentration significantly increased in mice treated with DOX and DOX + GA compared with that in the control group (P < 0.05). In the other groups, including DOX–CdTe, DOX/GA–CdTe, and DOX/GA–CdTe–CD22, none of these concentrations were significantly different from those in the control group (Fig. 9A). As shown in Fig. 9B, no apparent pathological changes were observed in the heart, lung, spleen, kidney, and liver of mice except those treated with DOX or DOX + GA. The heart tissues of mice treated with DOX or DOX + GA showed pathological changes. Therefore, DOX delivered by CdTe and anti-CD22 mAbs can protect the heart from exposure to cytotoxic drugs.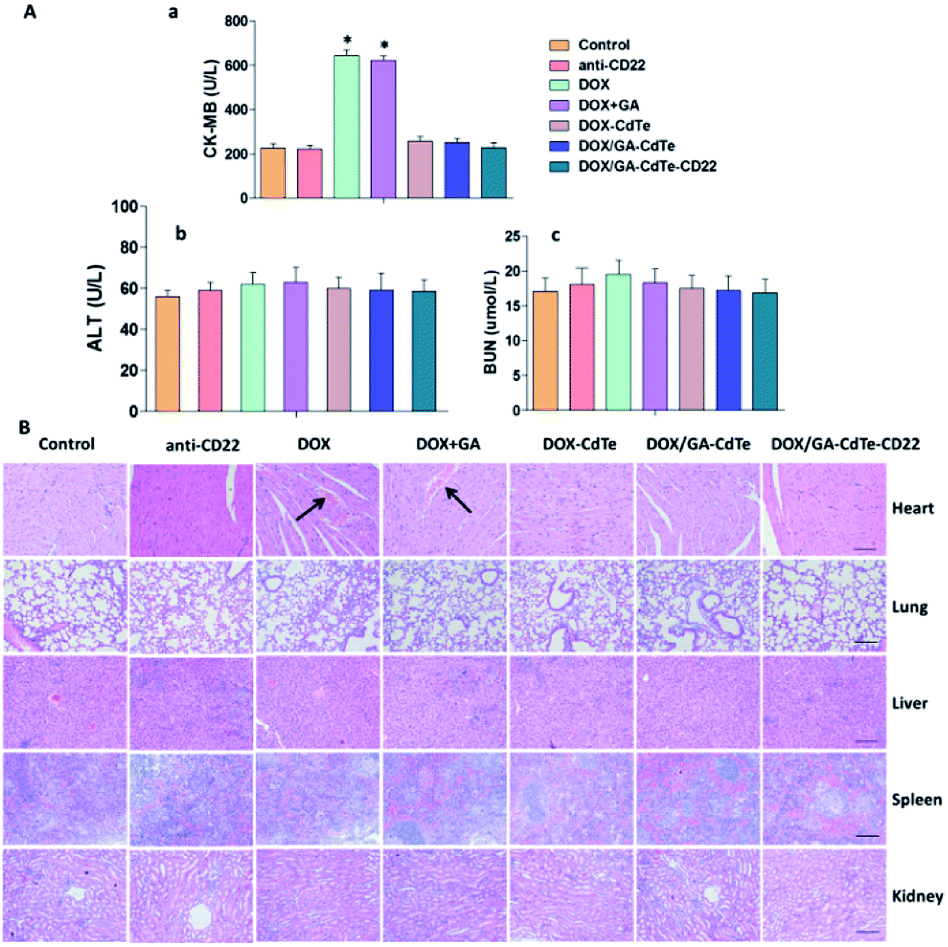 | ||
| Fig. 9 Toxicity on normal tissues in mice treated with different agents. (A) Serum concentration of ALT, BUN and CK-MB were determined at day 12 post-injection (*P < 0.05 when compared to control). (B) H&E-stained tissue sections from different groups (scale bar: 200 μm, ×100). Histopathological changes are indicated by “→”. | ||
Immunofluorescence staining
Immunofluorescence staining was conducted to evaluate the expression levels of P-gp, Bax, caspase-3, and cleaved PARP in tumor tissues. The results exhibited the same trends as the western blots of in vitro experiments (Fig. 10). The green fluorescence intensity of Bax, caspase-3, and cleaved PARP gradually increased in the tumor tissues of mice in the DOX, DOX + GA, DOX/GA–CdTe, and DOX/GA–CdTe–CD22 groups. The fluorescence intensity of P-gp sequentially decreased from the control/anti-CD22 to the DOX/GA–CdTe–CD22 group. These results demonstrate that genes of P-gp, Bcl-2, caspase-3, and cleaved PARP are associated with their antitumor activity.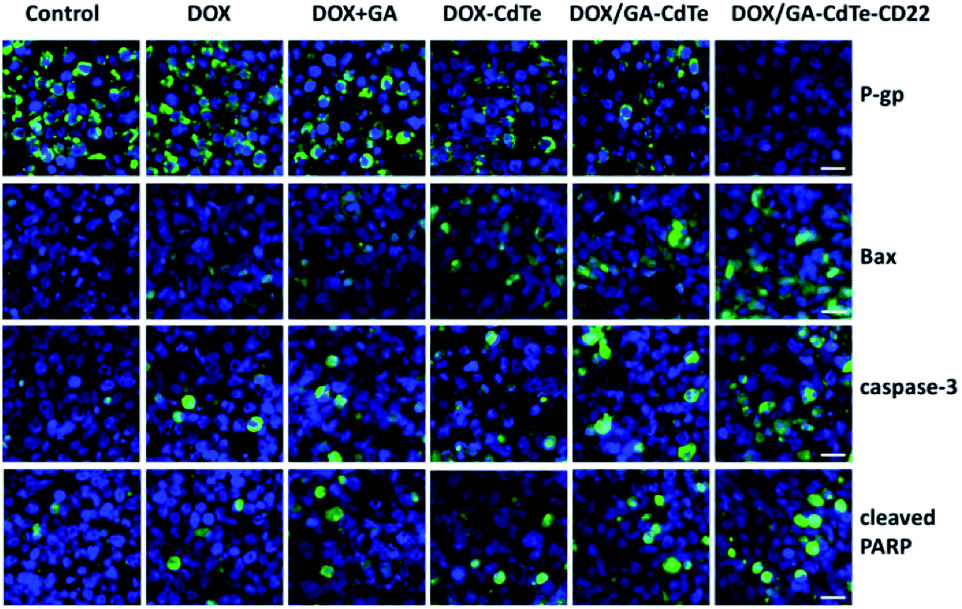 | ||
| Fig. 10 Immunofluorescence images of tumor sections from mice in different groups (scale bar: 10 μm, ×200). DAPI was used to stain cell nuclei and the secondary antibodies were cross-linked with FITC against primary antibodies for P-gp, Bax, caspase-3 and cleaved PARP. | ||
Discussion
The CHOP regimen, which comprises cyclophosphamide, DOX, vincristine, and prednisone, is the standard therapy for NHL.24 DOX plays an important part in treatment for lymphoma. DOX damages DNA to inhibit proliferation and induce apoptosis of tumor cells.25 However, the efficacy of DOX is dose dependent. High doses of DOX can achieve improved therapeutic effects but can also cause severe side effects, especially cardiotoxicity,26 thereby restricting the clinical use of this agent. Therefore, numerous patients with lymphoma had relapse after treatment, and some cells even became refractory to DOX because of chronic exposure to low doses. Thus, scholars must developed methods for reduction of adverse effects and enhancement of therapeutic effects.When combined with chemotherapeutic agents, GA exhibits synergistic antitumor effects and can sensitize tumor cells to drugs.27 The combination of DOX and GA could enhance cell proliferation inhibition and apoptosis induction, which are two main approaches through which drugs kill tumor cells (Fig. 5 and 6). Similarly, in vivo experiments showed that the tumor volume of mice injected with DOX and GA was smaller than that of mice treated with DOX alone. These results suggest that GA can sensitize tumor cells to DOX and enhance its antitumor activity. DOX combined with GA could exhibit the same efficacy at low doses, thus reducing side effects. The therapeutic effects of the combined treatment of DOX and GA can be improved by CdTe QDs. As a drug carrier, cysteamine-modified CdTe QDs can prolong the circulating period of loaded drugs with good biocompatibility and release drugs in a pH-triggered and controlled pattern.18 The drug concentration in tumor tissues can also be increased through EPR, thereby improving the effects of drugs.28 However, this accumulation is passive targeting with limited effects. We cross-linked anti-CD22 mAbs to cys-CdTe QDs to attain the active targeting of nanoparticle-based drug delivery. DOX/GA–CdTe–CD22 possessed good dispersibility and high EE and DL of the drugs. The particle diameter (18.3 nm) was optimal for in vivo delivery because sizes less than 10 nm could be rapidly cleared by the urinary system; meanwhile, the remaining particles with size more than 200 nm could be preferentially phagocytized by the reticuloendothelial system.29 cys-CdTe QDs conjugated with anti-CD22 mAbs could actively target lymphoma cells and precisely deliver co-loaded DOX and GA to tumor cells for targeted combination chemotherapy. In vitro (Fig. 3) and in vivo (Fig. 8A) experiments consistently show that DOX/GA–CdTe–CD22 targeted tumor cells and increased drug accumulation in tumor tissues. What should be noted is that our previous studies have indicated the high safety and low cytotoxicity of CdTe QDs at a low concentration.30,31 Furthermore, as another control group, the results (Fig. 5, 6, and 8) of anti-CD22 show that there was no significant difference between Raji cells or tumor-bearing mice treated with anti-CD22 and those with control, which confirmed that anti-CD22 have no therapeutic activity in vitro and in vivo.
Antibody-coupled CdTe QDs also could significantly attenuate the side effects of DOX by precisely delivering the drugs to tumor cells. DOX/GA–CdTe–CD22 caused no obvious functional and pathological changes in the major organs (Fig. 9). In particular, this drug delivery system ameliorated DOX-induced cardiotoxicity, as evident by the normal serum level of CK-MB and the lack of myocardial damage compared with those in the DOX group.
The expression of several proteins was detected by western blot and immunofluorescence staining analyses to elucidate the molecular mechanism of enhanced antitumor activity of DOX/GA–CdTe–CD22. P-Glycoprotein (P-gp) contributes to resistance of tumor cells to chemotherapeutic drugs via the efflux mechanism.32 P-gp expression significantly decreased in Raji cells and tumor tissues of mice treated with DOX + GA, DOX/GA–CdTe and DOX/GA–CdTe–CD22, thereby increasing the intracellular concentration of DOX and GA. Bax caspase-3 and cleaved PARP were highly expressed in the DOX/GA–CdTe–CD22 group. Bax, a pro-apoptotic member of the Bcl-2 family, can migrate from the cytosol to the mitochondrial membrane when stimulated by apoptosis signals;33 this phenomenon activates caspase-mediated apoptosis signaling.34 Caspase-3, an executioner in the caspase family, can cleave and inactivate poly ADP-ribose polymerase (PARP) when initiated;35 PARP is supposed to repair damaged DNA.36 Therefore, DNA interfered by DOX produced condensation and fragments without repair. Ultimately, proliferation was inhibited and apoptosis occurred in tumor cells. Hence, DOX/GA–CdTe–CD22 can increase cytotoxic effects on tumor cells by inducing apoptosis via the Bcl-2/caspase-mediated apoptotic pathway.
Conclusions
In summary, DOX/GA–CdTe–CD22 was successfully synthesized as a novel drug delivery system for combined chemotherapy by co-loading drugs with high encapsulation efficiency and drug loading. The system can precisely deliver drugs to tumor cells by active targeting, thereby enhancing the therapeutic effects and reducing the side effects of DOX. The system provides a novel platform for lymphoma treatment and can be expanded for other cancers.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20140100), the Technique Development Foundation of Nan Jing (Outstanding Youth Foundation, JQX15004), the Science and Technology Development Program of Nanjing, Medical Science and Technology Development Program of Nanjing (Ykk14069 &Ykk16099) and Peak of Six Talent in Jiangsu Province (2015-WSN-075).References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 10–29 CrossRef PubMed.
- K. R. Shankland, J. O. Armitage and B. W. Hancock, Lancet, 2012, 380, 848–857 CrossRef.
- A. J. Barsky, R. Saintfort, M. P. Rogers and J. F. Borus, JAMA, 2002, 287, 622–627 CrossRef PubMed.
- J. W. Friedberg, Hematology, 2011, 2011, 498–505 CrossRef PubMed.
- X. Wang and W. Chen, Anti-Cancer Agents Med. Chem., 2012, 12, 994–1000 CrossRef CAS PubMed.
- M. K. Pandey, B. Sung, K. S. Ahn, A. B. Kunnumakkara, M. M. Chaturvedi and B. B. Aggarwal, Blood, 2007, 110, 3517–3525 CrossRef CAS PubMed.
- P. Xu, R. Wang, J. Li, J. Ouyang and B. Chen, RSC Adv., 2015, 5, 61051–61059 RSC.
- S. Wang, L. Wang, M. Chen and Y. Wang, Chem.-Biol. Interact., 2015, 235, 76–84 CrossRef CAS PubMed.
- L. H. Wang, J. Y. Yang, S. N. Yang, Y. Li, G. F. Ping, Y. Hou, W. Cui, Z. Z. Wang, W. Xiao and C. F. Wu, Curr. Cancer Drug Targets, 2014, 14, 91–103 CrossRef CAS PubMed.
- W. A. Wani, S. Prashar, S. Shreaz and S. Gómez-Ruiz, Coord. Chem. Rev., 2016, 312, 67–98 CrossRef CAS.
- Y. Ellahioui, S. Prashar and S. Gómez-Ruiz, Curr. Med. Chem., 2017, 23, 4450–4467 CrossRef.
- S. K. Shukla, S. K. K. Shukla, P. P. Govender and N. G. Giri, RSC Adv., 2016, 6, 94325–94351 RSC.
- M. E. Godsey, S. Suryaprakash and K. W. Leong, RSC Adv., 2011, 3, 24794–24811 RSC.
- C. E. Probst, P. Zrazhevskiy, V. Bagalkot and X. Gao, Adv. Drug Delivery Rev., 2013, 65, 703–718 CrossRef CAS PubMed.
- A. M. Smith, H. Duan, A. M. Mohs and S. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS PubMed.
- S. Ghaderi, B. Ramesh and A. M. Seifalian, J. Drug Targeting, 2011, 19, 475–486 CrossRef CAS PubMed.
- S. Chang, B. Kang, X. Liu, Y. Dai and D. Chen, Acta Biochim. Biophys. Sin., 2012, 44, 241–248 CrossRef CAS PubMed.
- J. Li, C. Wu, P. Xu, L. Shi, B. Chen, M. Selke, H. Jiang and X. Wang, RSC Adv., 2013, 3, 6518–6525 RSC.
- S. Acharya and S. K. Sahoo, Adv. Drug Delivery Rev., 2010, 63, 170–183 CrossRef PubMed.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevickmuraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS PubMed.
- E. L. Sievers and M. Linenberger, Curr. Opin. Oncol., 2001, 13, 522–527 CrossRef CAS PubMed.
- J. F. Dijoseph, D. C. Armellino, E. R. Boghaert, K. Khandke, M. M. Dougher, L. Sridharan, A. Kunz, P. R. Hamann, B. Gorovits and C. Udata, Blood, 2004, 103, 1807–1814 CrossRef CAS PubMed.
- S. Wang, N. Mamedova, N. A. Kotov, W. Chen and J. Studer, Nano Lett., 2002, 2, 817–822 CrossRef CAS.
- R. I. Fisher and M. M. Oken, Cleveland Clinic Journal of Medicine, 1995, 62(1), S16–S142 Search PubMed.
- L. P. Swift, A. Rephaeli, A. Nudelman, D. R. Phillips and S. M. Cutts, Cancer Res., 2006, 66, 4863–4871 CrossRef CAS PubMed.
- T. L'Ecuyer, S. Sanjeev, R. Thomas, R. Novak, L. Das, W. Campbell and R. V. Heide, Heart Circ. Physiol., 2006, 291, H1273–H1280 CrossRef PubMed.
- Z. Zou, L. Xie, J. Wei, L. Yu, X. Qian, J. Chen, T. Wang and B. Liu, BMC Complementary Altern. Med., 2012, 12, 58 Search PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- V. I. Shubayev and S. Jin, Adv. Drug Delivery Rev., 2009, 61, 467–477 CrossRef CAS PubMed.
- Y. Zhou, R. Wang, B. Chen, D. Sun, Y. Hu and P. Xu, Int. J. Nanomed., 2016, 11, 5429–5442 CrossRef PubMed.
- H. Zuo, Z. Yi, D. Zhou, B. Chen and P. Xu, Nano, 2016, 12, 1750011 CrossRef.
- E. M. Leslie, R. G. Deeley and S. P. C. Cole, Toxicol. Appl. Pharmacol., 2005, 204, 216–237 CrossRef CAS PubMed.
- T. T. Renault, O. Teijido, B. Antonsson, L. M. Dejean and S. Manon, Int. J. Biochem. Cell Biol., 2013, 45, 64–67 CrossRef CAS PubMed.
- C. Weng, Y. Li, D. Xu, Y. Shi and H. Tang, J. Biol. Chem., 2005, 280, 10491–10500 CrossRef CAS PubMed.
- S. Lakhani, A. Masud, K. Kuida, G. A. Porter, C. J. Booth, W. Z. Mehal, I. Inayat and R. A. Flavell, Science, 2006, 311, 847–851 CrossRef CAS PubMed.
- J. Ame, C. Spenlehauer and G. De Murcia, BioEssays, 2004, 26, 882–893 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04056c |
| This journal is © The Royal Society of Chemistry 2017 |