
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct synthesis and characterization of mixed-valent Li0.5−δCoPO4, a Li-deficient derivative of the Cmcm polymorph of LiCoPO4†
Jennifer Ludwiga,
Carlos Alarcón-Suescaa,
Stephan Geprägsb,
Dennis Nordlundc,
Marca M. Doeffd,
Inés Puente Orenchef and
Tom Nilges *a
*a
aTechnical University of Munich, Department of Chemistry, Synthesis and Characterization of Innovative Materials, Lichtenbergstr. 4, 85747 Garching, Germany. E-mail: tom.nilges@lrz.tum.de
bWalther Meissner Institute, Bavarian Academy of Sciences and Humanities, Walther-Meissner-Str. 8, 85747 Garching, Germany
cStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, 2575 Sand Hill Rd, Menlo Park, CA 94025, USA
dLawrence Berkeley National Laboratory, Energy Storage and Distributed Resources Division, 1 Cyclotron Rd, Berkeley, CA 94720, USA
eInstituto de Ciencia de Materiales de Aragón, Pedro Cerbuna 12, 50009 Zaragoza, Spain
fInstitut Laue-Langevin, 71 Avenue des Martyrs, B.P. 156, 38042 Grenoble Cedex 9, France
First published on 26th May 2017
Abstract
While the majority of research activities on LiCoPO4 is focussed on the thermodynamically stable olivine-type Pnma polymorph, the metastable Pna21 and Cmcm modifications have recently attracted considerable attention due to their interesting material properties. In this study, we present the first Li-deficient structural derivative of the Cmcm modification with the nominal composition Li0.5−δCoPO4. As opposed to the substoichiometric olivine (Pnma) phases LixCoPO4 (x = 0; 2/3), which are exclusively accessible by electrochemical or chemical Li extraction techniques, this is also the first time that a direct soft-chemical synthesis route towards a LixCoPO4-type material is accomplished. X-ray and neutron diffraction studies indicate that Cmcm-type Li0.5−δCoPO4 shows vacancies on both the Li and Co sites, whereas X-ray absorption spectra demonstrate that the structure features heterovalent Co ions (+2/+3) to compensate for the Li deficit. Magnetic measurements reveal a long-range antiferromagnetic order below 10.5 K. A thorough investigation of the thermal stability using thermogravimetric analysis, differential scanning calorimetry, and temperature-dependent in situ X-ray powder diffraction demonstrates that Li0.5−δCoPO4 is metastable and exhibits a complex, multi-step thermal decomposition mechanism. In the first step at 394 °C, it decomposes to α-Co2P2O7 (P21/c) and LiCoPO4 (Cmcm) upon O2 release. The LiCoPO4 (Cmcm) intermediate is then irreversibly transformed to olivine-type LiCoPO4 (Pnma) at 686 °C. The material properties of Li0.5−δCoPO4 are further compared to the fully lithiated, isostructural LiCoPO4 (Cmcm) phase, for which an improved structure solution as well as Co L2,3-edge X-ray absorption spectra are reported for the first time.
Introduction
In the last two decades, thermodynamically stable, olivine-type (space group: Pnma) LiCoPO4 polymorphs have been extensively studied as a high-voltage cathode material for lithium-ion batteries (operating voltage: ∼4.8 V vs. Li/Li+; theoretical capacity: 167 mA h g−1).1–4 The three-dimensional network structure features [CoO6] octahedra, [PO4] tetrahedra, and Li+ ions in octahedral voids.5 The majority of research activities have been focused on optimizing the electrochemical performance of the material.4,6,7 However, despite intensive efforts, the nature of the intermediate phase LixCoPO4, which occurs upon the two-step Li insertion–extraction reaction of LiCoPO4,8 is still under investigation and debated in the literature. Earlier investigations suggested compositions of Li0.7CoPO4 (ref. 8) and Li0.6CoPO4,9 respectively, whereas a later report10 stated a LixCoPO4 (x = 0.20–0.45) composition. Recently, the lithiation state of the intermediate was determined to be Li2/3CoPO4 by two independent studies.11,12 Since the completely delithiated phase CoPO4 is unstable and undergoes amorphization when exposed to air or moisture,8,9 the application of inert gas atmospheres and/or in situ techniques is crucial. According to Bramnik et al.,13 both lithium-poor, Co3+-containing phases are intrinsically instable and exhibit a low thermal stability. Charged LiCoPO4 electrodes were shown to decompose rapidly at temperatures below 200 °C, leading to gas evolution and the crystallization of LiCoPO4 (only in the case of LixCoPO4) and Co2P2O7. In contrast, Theil et al.14 claimed that LixCoPO4 is thermally stable up to 550 °C and that the thermal instability of charged LiCoPO4 electrodes can be solely ascribed to the instability of the CoPO4 phase. To the best of our knowledge, these Li-deficient phases are exclusively accessible by electrochemical or chemical Li extraction from LiCoPO4 (Pnma),9,13 and a direct synthesis route has not been reported to date. Interestingly, in contrast to previous reports,15,16 we recently demonstrated17 that also the fully lithiated olivine-type LiCoPO4 does not exhibit unlimited thermal stability since it transforms to the (at room temperature) metastable Pna21-LiCoPO4 phase around 900 °C.The less common, metastable LiCoPO4 modifications, which crystallize in the space groups Pna21 (ref. 16, 18 and 19) and Cmcm, (ref. 16, 20 and 21) have recently attracted attention because of their interesting material properties and potential applicability as cathode materials for Li-ion batteries. The Pna21 modification exhibits a network of [PO4] and [CoO4] tetrahedra and Li+ ions on tetrahedral sites.18 To date, the polymorph has only been accessible by microwave-assisted synthesis techniques.16,18,19 Pna21-type LiCoPO4 shows the highest redox potential of ∼5.0 V vs. Li/Li+ compared to the other two LiCoPO4 polymorphs.16,18 A single redox peak was observed upon cycling, indicating that the compound is delithiated in one step. However, the electrochemical performance was found to be poor (maximum capacity: 33 mA h g−1).16,18,19 Magnetic measurements indicated a paramagnetic Curie–Weiss-like behavior at high temperatures, and a long-range antiferromagnetic order below TN = 11 K.18 Recently, a structure redetermination suggested that the material is non-stoichiometric and shows Li–Co anti-site defects, which provide an explanation for this poor performance.19 A thorough investigation of the thermal stability revealed that LiCoPO4 (Pna21) converts to the olivine LiCoPO4 (Pnma) modification at 527 °C.19 Interestingly, the Pna21 structure re-emerges as a stable high-temperature phase above 800 °C.19
The LiCoPO4 (Cmcm) polymorph was first reported by Amador et al.20 using a high-pressure, high-temperature synthesis route (6 GPa, 900 °C). Alternative pathways using low-temperature procedures such as microwave-assisted solvothermal16 and polyol21 synthesis have been demonstrated recently. The structure is built from [CoO6] and [PO4] units, with Li+ ions occupying tetrahedral sites. LiCoPO4 (Cmcm) shows a single redox peak at ∼4.3 V vs. Li/Li+ upon cycling,16 which corresponds to the lowest redox potential of all the LiCoPO4 polymorphs. A discharge capacity of only 6 mA h g−1 has been reported, which was associated with the poor conductivity of the material.16 The magnetic characterization suggested a long-range antiferromagnetic order below TN = 11 K at low fields (10 kOe) and the presence of a metamagnetic transition.21 Investigations on the thermal stability showed that the structure is metastable and transforms to olivine-type LiCoPO4 (Pnma) at 575 °C, which then transforms to the Pna21 modification at 675 °C. The thermodynamically stable Pnma-LiCoPO4 phase was obtained after cooling.21
Based on our previous work on the three LiCoPO4 polymorphs,17,19,21 we herein present the first Li-deficient structural derivative of the Cmcm modification with the nominal composition Li0.5−δCoPO4. To the best of our knowledge, this is the first time that a sub-stoichiometric LixCoPO4 phase has been synthesized directly (bottom-up) by a soft-chemical polyol approach as opposed to electrochemical or electrochemical Li extraction (top-down) techniques described in the literature.9,13 The structure, morphology, oxidation state as well as electrochemical and magnetic properties of the novel Cmcm-type phase Li0.5−δCoPO4 are investigated. Moreover, the thermal properties are studied using thermogravimetry, differential scanning calorimetry, and temperature-dependent in situ X-ray powder diffraction. The results are discussed in context of the ‘fully lithiated’ LiCoPO4 (Cmcm) phase, for which an improved structure solution (revealing a sub-stoichiometry reflected by the revised empirical formula Li1−γCoPO4) as well as X-ray absorption spectra are presented for the first time.
Experimental
Synthesis of Li0.5−δCoPO4 (Cmcm) and Li1−γCoPO4 (Cmcm)
Cmcm-type LiCoPO4 samples with varied Li contents were obtained from a polyol process as described in our previous work21 using LiCH3COO (ChemPur, 99+%), (Co(CH3COO)2·4H2O, Merck, 99.99%), and H3PO4 (Merck, 85 wt%) as precursors, and tetraethylene glycol (TTEG, Merck, ≥99.0%) as solvent. For the synthesis of Li1−γCoPO4 (Cmcm), a Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co:P molar ratio of 3:1:10 was used, whereas Li0.5−δCoPO4 (Cmcm) was obtained from a modified process using a ratio of 1:1:10. First, H3PO4 was added dropwise to a solution of cobalt acetate in 125 mL TTEG. Then, a second solution containing lithium acetate in 75 mL TTEG was added. The resulting mixture was refluxed at 185 °C for 14 h in a round-bottom flask. After cooling, the precipitate was recovered by centrifugation (1500 rpm, 20 min, three times) and washed with ethanol (VWR AnalaR NORMAPUR, 99.95%). The light pink powder (cf. graphical abstract) was collected by filtration, washed with acetone (99%), and dried in air at 100 °C for 14 h. Note that in contrast to the delithiated Pnma structures LixCoPO4 and in particular CoPO4, which are sensitive to air and moisture,8,9 the Cmcm-derivative Li0.5−δCoPO4 is stable under air for at least several months.
:Co:P molar ratio of 3:1:10 was used, whereas Li0.5−δCoPO4 (Cmcm) was obtained from a modified process using a ratio of 1:1:10. First, H3PO4 was added dropwise to a solution of cobalt acetate in 125 mL TTEG. Then, a second solution containing lithium acetate in 75 mL TTEG was added. The resulting mixture was refluxed at 185 °C for 14 h in a round-bottom flask. After cooling, the precipitate was recovered by centrifugation (1500 rpm, 20 min, three times) and washed with ethanol (VWR AnalaR NORMAPUR, 99.95%). The light pink powder (cf. graphical abstract) was collected by filtration, washed with acetone (99%), and dried in air at 100 °C for 14 h. Note that in contrast to the delithiated Pnma structures LixCoPO4 and in particular CoPO4, which are sensitive to air and moisture,8,9 the Cmcm-derivative Li0.5−δCoPO4 is stable under air for at least several months.
X-ray powder diffraction (PXRD) and Rietveld refinement details
Room-temperature PXRD data of the ground powders sealed in borosilicate glass capillaries (0.5 mm, Hilgenberg) were collected on a Stoe STADI P diffractometer (Mo Kα1 radiation, λ = 0.70930 Å; Ge(111) monochromator; Dectris MYTHEN DCS 1K silicon solid-state detector) in a 2θ range of 3–60° (PSD step: 0.015°; time per step: 30 s, three ranges, total measurement time: 12 h). The diffraction patterns were calibrated using an external silicon standard. The Jana2006 software22 was used for the structure refinement by the Rietveld method, using the recently reported structure solution of LiCoPO4 (Cmcm; ICSD no. 432186)21 as a starting model. Details on the Rietveld refinement strategy and parameters used can be found in our previous work.21Neutron powder diffraction (NPD) experiments
Neutron powder diffraction data were collected using the diffractometer D2B at the Institut Laue-Langevin (ILL, Grenoble, France), working at a calibrated wavelength of 1.5942 Å in a 2θ range of 5–160°. The data were recorded at 296 K with a collection time of 4 h per pattern. The data were analyzed by the Rietveld method with the FULLPROF program.23 The line shape of the diffraction peaks was generated by a pseudo-Voigt function. The instrumental contribution to the peak broadening was determined using an instrument resolution function built from the refinement of a Na2Ca3Al2F14 standard, while the wavelength was refined using a Si standard.Elemental analysis
Analysis of the Li, Co, and P contents was carried out by atomic absorption spectroscopy (AAS, Varian AA280FS sequential device) and photometry (Shimadzu UV-160 photometer). A Hekatech Euro EA CHNSO combustion analyzer was used to determine the C, H, N, and S amounts.Soft X-ray absorption spectroscopy (soft XAS)
Co L2,3-edge soft XAS spectra were collected at beamline 8–2 of Stanford Synchrotron Radiation Lightsource (SSRL), operating the spherical grating monochromator (SGM, ruling: 1100 mm−1) with 40 × 40 μm slits (resolution: ∼0.3 eV), as described in our previous work.17 The XAS spectra presented in this report are derived from the total electron yield (TEY), measured via the drain current (probing depth: 2–5 nm). We also recorded Auger electron yield (AEY) and total fluorescence yield (FY) spectra via a cylindrical mirror analyzer and a silicon diode (AXUV100). These modes, which probe ∼2 nm and 50–100 nm deep, respectively, ensured that the best quality TEY spectra was bulk representative and that the contribution from surface contamination was small. All spectra were normalized to the incoming flux and the energy scale was calibrated to match that of ref. 24, followed by a background subtraction to a line, and a final area normalization for comparison. In order to produce the most stable and reproducible fits, the fitting was limited to the region just around the L3 edge (774–784 eV), in which the normalization was further restricted through another line subtraction and area normalization.Fourier-transform infrared (FTIR) spectroscopy
IR spectra were collected on a Varian 670 FTIR spectrometer equipped with a PIKE GladiATR diamond ATR stage. The measurements were performed using 132 scans in a wavenumber range of 400–4000 cm−1.Magnetic measurements
DC magnetization measurements were performed using a Quantum Design MPMS XL7 SQUID magnetometer in the temperature range from 2 K to 300 K with a magnetic field of up to 7 T. The powder samples were placed inside gelatin capsules. The temperature-dependent magnetic moments were recorded under an applied magnetic field of 1 kOe after cooling the samples under a magnetic field of 7 T (field-cooled, FC) and 0 T (zero field-cooled, ZFC). Magnetic hysteresis measurements were carried out with magnetic field strengths between −7 T and 7 T at 2 K, 11 K, and 300 K, respectively.Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)
The stability of the material (specimen weight: ∼10 mg) upon heating was assessed using a simultaneous Mettler Toledo TGA/DSC 1 STAR device, which did not allow to monitor the cooling cycle due to the setup. The measurement was run in a temperature range of 30–900 °C (heating rate: 10 °C min−1) in an argon stream (10 mL min−1), and additionally synthetic air (10 mL min−1) in order to ensure comparability of the data with the in situ PXRD experiment.Temperature-dependent in situ X-ray powder diffraction (PXRD)
Temperature-controlled PXRD was performed in situ under air using a PANalytical X'Pert Pro diffractometer (Bragg–Brentano geometry; Cu Kα radiation; XCelerator detector) equipped with an Anton Parr HTK-1200 hot stage and a TCU 1000N temperature controller. The material was placed inside a corundum flat plate sample holder and heated up to 900 °C in steps of 100 °C (heating rate: 5 °C min−1), with each temperature being held for 5 min before collecting the data. The scans were recorded in a 2θ range of 15–70° (step: 0.022°; time per step: 209.5 s; total experiment time: 145.5 h).Results and discussion
Rietveld refinement of X-ray and neutron powder diffraction data
Fig. 1a shows the Rietveld fit of the X-ray powder diffraction pattern of the as-prepared title compound Li0.5−δCoPO4. The pattern of a reproduced sample of Cmcm-type LiCoPO4 (Fig. 1b) is in good agreement with our previous work.21 In both cases, all the reflections can be indexed in the orthorhombic space group Cmcm and no additional reflections are observed, indicating that both materials are phase pure. While the pattern of Li0.5−δCoPO4 appears to be roughly similar to the one of LiCoPO4, suggesting that the crystal structure of the Li-deficient compound is strongly correlated with the one of the fully lithiated material, some shifts in the peak positions can be recognized. The most significant feature of the Li0.5−δCoPO4 pattern is the narrowing of the (200) and (112) reflections at 15.0° and 15.9° 2θ. Furthermore, a completely different peak pattern can be observed in the 2θ region of 25.5–27.5° (for a detailed view, please refer to Fig. S1, ESI†).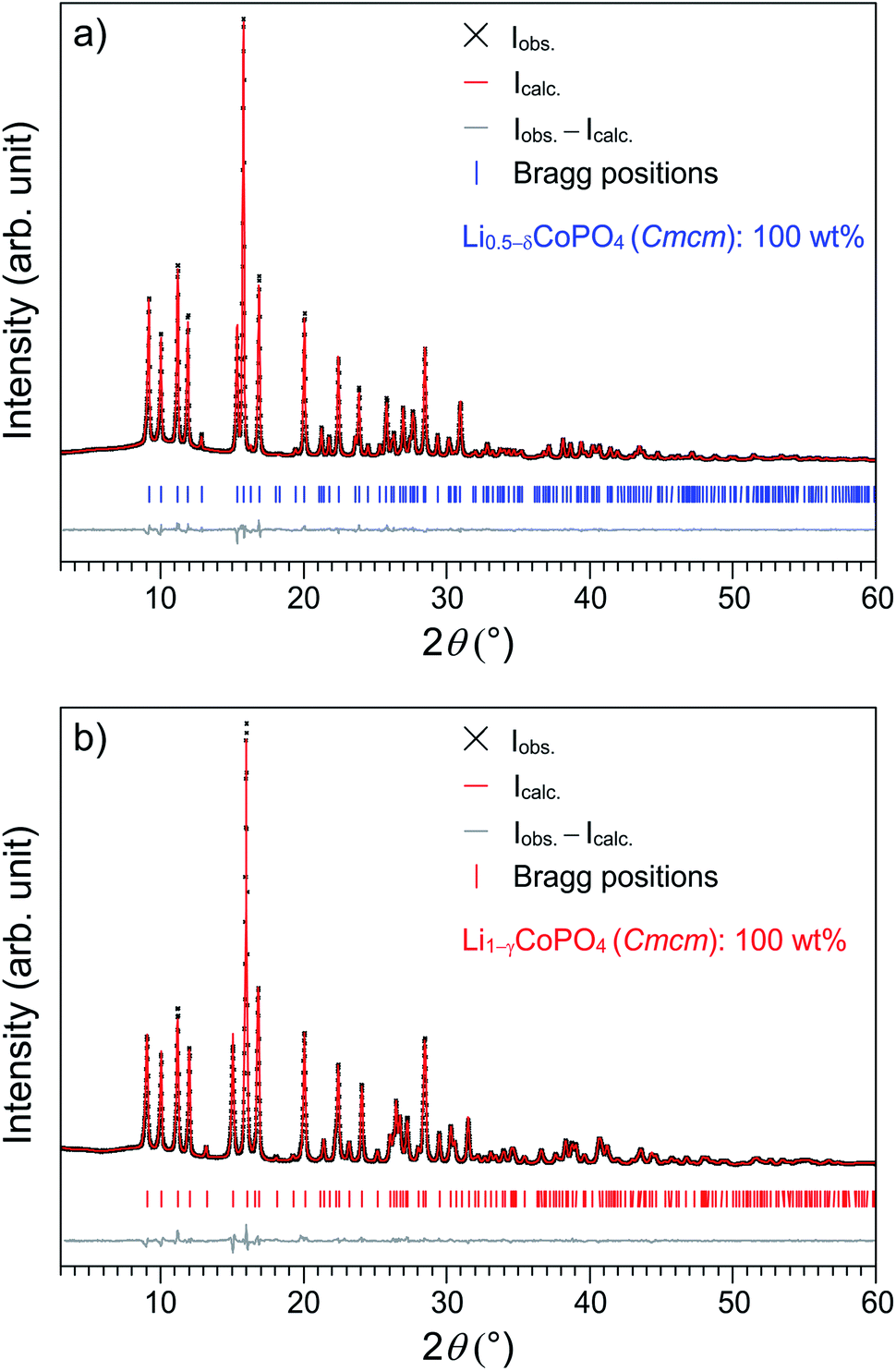 | ||
| Fig. 1 Rietveld fits of the X-ray powder diffraction data (transmission geometry, Mo Kα1 radiation) of (a) Li0.5−δCoPO4 (Cmcm; refined composition: Li0.39(2)Co0.96(1)PO4, δ = 0.11(2)), and (b) Li1−γCoPO4 (Cmcm, refined composition: Li0.94(2)Co0.96(1)PO4, γ = 0.06(2)). | ||
To gain further insights into the structural differences causing the peak shifts, a Rietveld refinement was performed, using the previously published structure solution of Cmcm-type LiCoPO4 (ICSD no. 143186)21 as a starting model. Since the elemental analysis indicated an approximate 50% deficit in Li for Li0.5−δCoPO4 compared to Cmcm-LiCoPO4 (cf. Table 2) within standard deviations, the structures were at first refined with fixed Li site occupancy factors of 50% and 100%, respectively, resulting in good reliability factors (Table S1, ESI†). Taking into account that the empirical formulas derived from elemental analysis indicated a deficit in both Li and Co for the two materials (empirical formulas: Li0.45(5)Co0.93(3)P1.00(2)O4 and Li0.93(5)Co0.91(3)P1.00(2)O4; cf. Table 2), we tentatively refined the occupancy factors of the Li and Co sites after having applied an absorption correction.25 In both cases, the free refinement resulted in statistically significant values for the occupancies (39(2)% Li and 96.4(5)% Co for Li0.5−δCoPO4; δ = 0.11(2) and 94(2)% Li and 95.5(5)% Co for LiCoPO4, cf. Table S2, ESI†), indicating that both structures feature vacancies in the cationic substructures and are non-stoichiometric. In both cases, the reliability factors were significantly improved over the previous structure models with fixed occupancies (cf. Tables 1 and S1, ESI†). To simplify the sum formulas of both compounds while still reflecting the off-stoichiometry from the idealized formulas Li0.5CoPO4 and LiCoPO4 (within three standard deviations), the compounds are referred to as Li0.5−δCoPO4 for the Li-deficient phase, and Li1−γCoPO4 (with γ = 0.06(2)) for Cmcm-type LiCoPO4 in this work. It is worth noting that on basis of these refinements, there was no indication for the occurrence of anti-site defects, which are profound for materials synthesized at low temperatures (as observed e.g. in Pna21-type LiCoPO4).19 Furthermore, in contrast to Li0.94(2)Co0.96(1)PO4, the composition of the Li-deficient Cmcm derivative Li0.39(2)Co0.96(1)PO4 would not be charge-balanced assuming that Co is only present in the oxidation state +2. We therefore assume that the deficit in positive electric charge caused by the lower Li+ content is compensated by Co3+ in the framework, which was confirmed by X-ray absorption spectroscopic studies discussed later.
| a The estimated standard deviations were calculated by the Berar's procedure and are indicated in parentheses. | ||
|---|---|---|
| Sample | (a) Li0.5−δCoPO4 | (b) Li1−γCoPO4 |
| Empirical formula | Li0.39(2)Co0.96(1)PO4 | Li0.94(2)Co0.96(1)PO4 |
| Mr (g mol−1) | 154.3 | 158.1 |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group (no.) | Cmcm (63) | Cmcm (63) |
| Z | 4 | 4 |
| a (Å) | 5.3385(2) | 5.4432(3) |
| b (Å) | 8.1763(3) | 8.1695(4) |
| c (Å) | 6.3716(2) | 6.2128(3) |
| V (Å3) | 278.116(19) | 276.28(2) |
| F(000) | 297 | 302 |
| ρ (calcd) (g cm−3) | 3.684(1) | 3.800(1) |
| Rp | 0.0257 | 0.0196 |
| Rwp | 0.0327 | 0.0255 |
| Rexp | 0.0267 | 0.0252 |
| RF | 0.0143 | 0.0106 |
| RB | 0.0254 | 0.0184 |
| χ2 | 1.23 | 1.01 |
| Data/restraints/parameter | 3800/0/59 | 3835/0/57 |
The refined cell parameters (Table 1) indicate a significant contraction along the a axis and an expansion along the c axis for Li0.5−δCoPO4 compared to Li1−γCoPO4, while b is not significantly changed, hence providing an explanation for the peak shifts observed in the PXRD patterns. Furthermore, the respective cell volumes (V = 278.116(19) Å3 vs. 276.28(2) Å3, corresponding to an increase of 0.7%) reveal that the Li0.5−δCoPO4 structure is less dense, which is consistent with the decrease in crystal densities. This is surprising since for the delithiated phases LixCoPO4 and CoPO4 derived from olivine-type LiCoPO4 (Pnma), a significant decrease in cell volume of up to ∼7% (CoPO4) was observed due to the smaller ionic radius of Co3+ compared to Co2+.8,9,11 The slight increase in cell volume might be explained by the fact that our Li-poor Cmcm material was produced from a kinetically controlled synthesis as opposed to Pnma-type CoPO4, which was obtained by electrochemical Li extraction. As a result, the Li ions and voids are likely to be statistically distributed within the Cmcm structure. This would also be in line with investigations on olivine-type LiFePO4, which revealed that materials synthesized at low temperature are prone to disorder, resulting in larger cell volumes than expected.26 However, a thorough investigation of the Li+/vacancy distributions in the structures is beyond this work.
In order to further verify the Li contents and the structure model, we performed additional neutron powder diffraction (NPD) studies of both samples. Rietveld refinements of the NPD data were carried out using the structure parameters obtained from the PXRD experiment as a starting model. Then, the atomic coordinates, thermal parameters as well as occupation factors of Li and Co were refined (see Fig. S2 (ESI†) for the Rietveld fits). The refined lattice parameters (Table S4, ESI†) are in good agreement with the X-ray data. The refinement of the site occupancy factors revealed 37(4)% Li and 98(2)% Co for Li0.5−δCoPO4 (δ = 0.13(4)), and 90(3)% Li and 95(6)% Co for Li1−γCoPO4 (γ = 0.10(3)) (Table S5, ESI†). The compositions and structural parameters of Li0.37(4)Co0.98(1)PO4 and Li0.90(2)Co0.95(6)PO4 are very close to the ones derived from the X-ray experiment and hence confirm the structure model.
Crystal structure
The structures of both Cmcm-type LiCoPO4 materials feature rows of edge-sharing, distorted [CoO6] octahedra which are running along the [001] direction as illustrated in Fig. 2a.20,27 The [CoO6]∞ rows are cross-linked along [100] by alternating pairs of edge-sharing, asymmetric [PO4] and [LiO4] units (note that the tetrahedrally coordinated Li ions are shown in Fig. 2 instead of [LiO4] tetrahedra for clarity). As a result, layers of the composition ([CoO6][LiO4][PO4])∞ are formed in the ac plane (Fig. 2b). In these layers, every [CoO6] octahedron shares two opposite O2–O2 edges with neighboring [CoO6] units, and two apical O2 atoms with two different [PO4] and [LiO4] tetrahedra, which connect the [CoO6]∞ strands (for details see Fig. S3, ESI†). The layers are in turn stacked in the sequence AB (indicated by black and grey boxes in Fig. 2a and c) along [010] with a displacement of a/2 via the apical O1 atoms of the [CoO6] units so that a three-dimensional framework is formed. It has to be emphasized that despite the fact that Li channels seem to occur along [100] (Fig. 2a), the Li–Li distances are very large (d = 5.3385(4) Å and 5.4432(5) Å, cf. Table S3b and d, ESI†), so that these sites are considered isolated. Hence, Li migration will require a very high activation energy along this path and the Li mobility is extremely low in both structures.20,27 The lack of suitable Li migration pathways is confirmed by electrochemical measurements (see Section 5 and Fig. S4, ESI†) that revealed a very poor performance for both Cmcm materials (discharge capacities of < 3 mA h g−1), and is also in line with a previous report.16 Hence, due to the intrinsically low Li-ion conductivity, the Li0.5−δCoPO4 and Li1−γCoPO4 phases cannot be considered suitable for battery applications.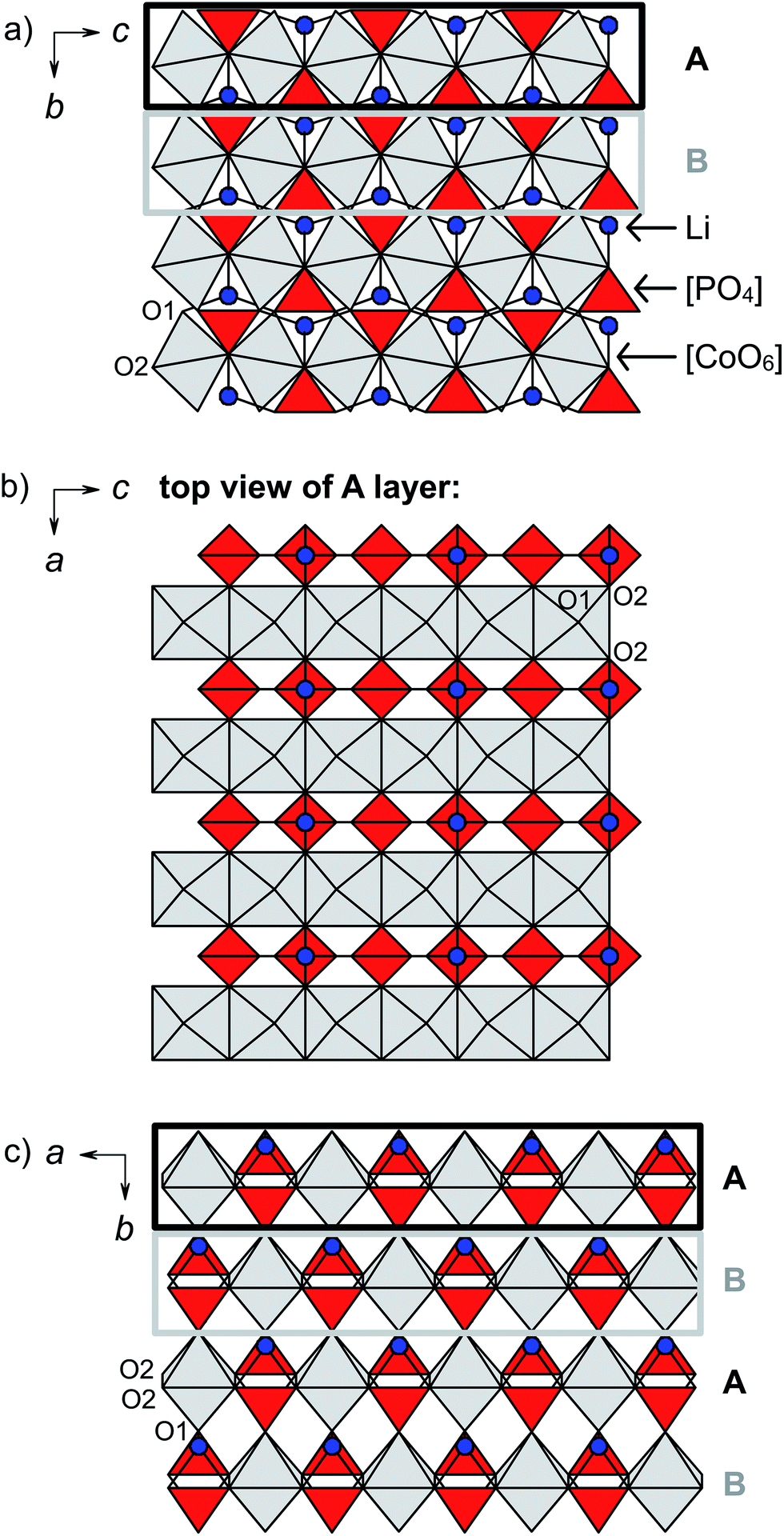 | ||
| Fig. 2 Polyhedral illustration of the crystal structures of Cmcm-type Li0.5−δCoPO4 and Li1−γCoPO4 viewed along (a) [100], (b) [010] (showing one A layer as indicated by the black boxes in a, c), and (c) [001]. [CoO6] octahedra are displayed in grey, [PO4] tetrahedra in red, and tetrahedrally coordinated Li ions in blue (for clarity, no Td representation was used for the [LiO4] units). The structure is built from ([CoO6][LiO4][PO4])∞ layers in the ac plane (b), which are stacked along b in the sequence AB (c). The layers consist of rows of edge-sharing [CoO6] octahedra (a), which are connected by alternating pairs of [LiO4] and [PO4] units. The occupancies on the Li and Co sites are 39(2)% Li and 96.5(5)% Co for Li0.5−δCoPO4, and 94(2)% Li and 95.5(5)% Co for Li1−γCoPO4. | ||
The structural differences between Li1−γCoPO4 and its Li-deficient analogue Li0.5−δCoPO4 can be derived from the cell parameters (Table 1) and the interatomic distances (Table S3b and d, ESI†). In general, the framework of Li0.5−δCoPO4 is contracted by ∼0.10 Å in the a dimension and expanded by ∼0.16 Å and ∼0.01 Å along c and b, respectively, as also indicated by the distances between the Co centers in and between the layers. The average Co–O (2.122 Å in Li0.5−δCoPO4 vs. 2.123 Å in LiCoPO4) and P–O (1.539 Å vs. 1.538 Å) distances in the [CoO6] and [PO4] units remain virtually unchanged, which is surprising since one would expect a decrease in the Co–O distances due to the occurrence of a definite amount of the smaller Co3+ ion compared to Co2+ in the structure. The individual bond lengths, however, reveal that both the [CoO6] and [PO4] units show a higher degree of distortion. This is reflected by the fact that the Co–O2 and P–O2 distances (in the ([CoO6][LiO4][PO4])∞ layers) are shortened, whereas the Co–O1 and P–O1 bonds (connecting the layers along b) are expanded, resulting in an increase of the distance between the A–B layers (cf. Co–P distances). On the other hand, the mean Li–O distances are increased by ∼0.4 Å, leading to an expansion of the “channels” along [100]. This is consistent with the increase of the Li–Li distances by ∼0.10 Å. However, it has to be noted that we cannot provide any information about the ordering of the Li+, Co2+, and Co3+ ions or the vacancies in the structure. Based on considerations on the charge distribution, it is likely that the vacancies in the Li0.5−δCoPO4 framework are located next to the Co3+ centers, which would be consistent with DFT (density functional theory) studies12 on the Li+/vacancy distribution in olivine-type Li2/3CoPO4.
Elemental analysis
The results of the elemental analysis of Li0.5−δCoPO4 (Cmcm) are compared to the values of Cmcm-type LiCoPO4 from our recent report21 in Table 2. The CHNS analyses are similar and show small amounts of hydrogen and carbon in both materials, which arise from residual tetraethylene glycol (TTEG) solvent or the decomposition products of TTEG and the acetate precursors. Whereas the Co and P contents (in wt%) are comparable within three standard deviations, it is evident that the novel compound contains about half the amount of Li (2.0(2) wt%) compared to the LiCoPO4 (Cmcm) material (4.1(2) wt%). This is confirmed by the respective Li:Co:P molar ratios of 0.45(5):0.93(3):1.00(2) and 0.93(5):0.91(5):1.00(2). EDS measurements further substantiate the Co:P ratios found by elemental analyses. (Images of the obtained crystal morphologies and semi-quantitative EDS analyses are provided in Fig. S5, ESI†.) The empirical sum formulas derived from elemental analysis, Li0.45(5)Co0.93(3)P1.00(2)O4 and Li0.93(5)Co0.91(3)P1.00(2)O4, indicate a deficit both in Li and Co for both compounds and are also consistent with the results of the Rietveld refinements (cf. Table 1) within standard deviations. Based on the results of the elemental analysis, δ in this case is 0.05(5) in Li0.5−δCoPO4 (whereas γ is 0.07(5) in Li1−γCoPO4), which is slightly smaller than derived from X-ray and neutron diffraction. Due to the kinetically controlled synthesis procedure, however, it is possible that the composition will slightly vary from batch to batch. Summarizing all efforts from diffraction experiments and elemental analysis to determine the Li and Co contents of the present phases, the notations Li0.5−δCoPO4 and Li1−γCoPO4 seem therefore to best reflect the off-stoichiometries found by the different techniques within three standard deviations.
| a The molar composition is calculated from the experimental values and normalized to the P content (standard deviations in parentheses).b The N and S contents were below the detection limit in both samples (= 0). | ||
|---|---|---|
| Sample | (a) Li0.5−δCoPO4 | (b) Li1−γCoPO4 |
| C (wt%) | 0.4(3) | 0.8(3) |
| H (wt%) | 0.5(3) | 0.4(3) |
| Li (wt%) | 2.0(2) | 4.1(2) |
| Co (wt%) | 35(1) | 34(1) |
| P (wt%) | 20.0(3) | 19.6(3) |
| n(Li):n(P) |
0.45(5) | 0.93(5) |
| n(Co):n(P) |
0.93(3) | 0.91(3) |
| Empirical formula | Li0.45(5)Co0.93(3)P1.00(2)O4 | Li0.93(5)Co0.91(3)P1.00(2)O4 |
As shown in our previous work21 on Cmcm-type LiCoPO4, the synthesis method strongly affects the phase composition. Whereas it was observed that the composition (and also the morphology) can be slightly varied by changing the synthesis technique (solvothermal vs. polyol), the molar ratio of the Li, Co, and P precursors represents another, more effective approach towards compositional tuning. For the synthesis of Li1−γCoPO4, the Li:Co:P molar ratio of the starting materials used was 3:1:10, whereas Li0.5−δCoPO4 was obtained from a Li:Co:P ratio of 1:1:10 (cf. experimental part). As a result, different amounts of Li are incorporated in the crystal structures of the products, although the molar amounts of the precursors and contents found in the obtained materials are not correlated linearly. Adjusting the molar ratio of the precursors might therefore provide a synthetic strategy towards other lithium cobalt or transition metal phosphate materials with modified Li contents. Given the fact that the delithiated Pnma phases Li2/3CoPO4 and CoPO4 are only accessible by chemical or electrochemical Li extraction from the fully lithiated olivine-type LiCoPO4 (Pnma) material9,13 and also very instable, this bottom-up approach might also pave the way towards the direct and simple soft-chemical preparation of these Li-deficient intermediates. In that matter, the influence of the synthesis route on the chemical composition will have to be thoroughly examined in further experiments.
X-ray absorption spectroscopy
L2,3-edge X-ray absorption spectroscopy is highly sensitive to the oxidation state as well as the spin state and chemical environment of transition metals.28,29 We have therefore applied Co L2,3-edge XAS to investigate the chemical state (valency and symmetry) of the cobalt ions in Li0.5−δCoPO4 (Cmcm) and Li1−γCoPO4 (Cmcm). Fig. 3 displays the normalized TEY spectra over the L3-edge region from this study along with reference spectra (adapted from (ref. 24 and 30)) for octahedral (Oh) high-spin (HS) Co2+ as well as both low-spin (LS) and high-spin (HS) Co3+. Li1−γCoPO4 exhibits all characteristic features of octahedrally (Oh) coordinated Co2+, including the low-energy peak at 776.4 eV that is uniquely ascribed to Oh Co2+.24,29 Furthermore, overall sharp features can be observed, which indicate a low degree of covalency and energetic alignment with an average octahedral crystal field strength in the order of 1 eV.29 In fact, a broadening of the CoO spectrum results in a near identical spectrum to Li1−γCoPO4, indicating high purity Co2+, with local disorder being the main difference.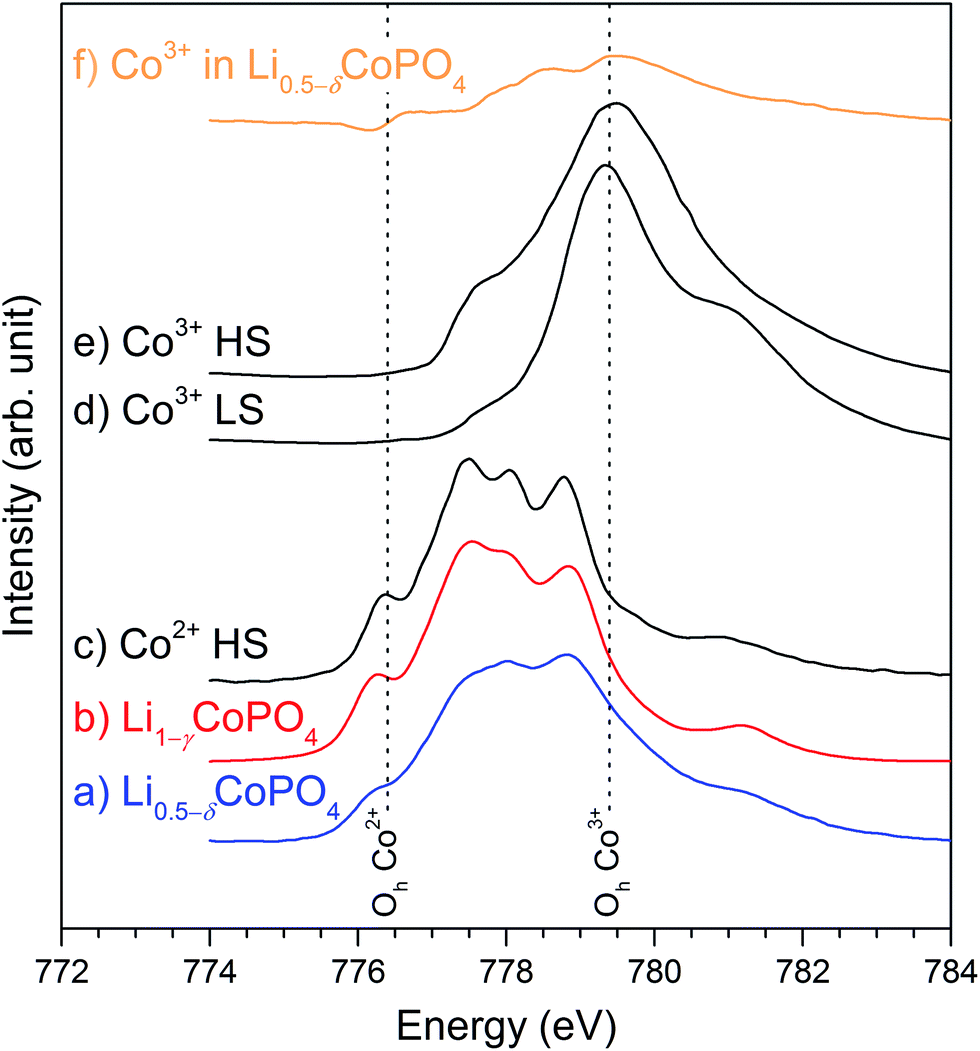 | ||
| Fig. 3 Normalized Co L3-edge XAS spectra in the TEY mode for (a) Li0.5−δCoPO4 (Cmcm, blue), (b) Li1−γCoPO4 (Cmcm, red) along with reference spectra for (c) Oh high-spin Co2+ in CoO (adapted from ref. 24, black), and (d) low- and (e) high-spin Co3+ in EuCoO3 and Sr2CoO3Cl (from ref. 30, both black). (f) shows the difference spectrum resulting from a subtraction of 71% Co2+ from Li0.5−δCoPO4 (orange, Li1−γCoPO4 subtraction), representing the trivalent Co3+ ion in the compound. The Co3+ association and the lower energy shoulder spectral weight are apparent (see text). All datasets have been aligned to match the common energy scale of ref. 24. The vertical dashed lines indicate the energies corresponding to Oh Co2+ (776.4 eV) and Oh Co3+ (779.4 eV). | ||
Charge balance arguments suggest that Li0.5−δCoPO4 (Cmcm) bears cobalt ions in nominal oxidation states of both +2 and +3. Fitting of the spectrum using principal Co2+ and Co3+ components results in relative contributions of approximately (71 ± 3)% Co2+ and (29 ± 3)% Co3+ (as opposed to (97 ± 3)% Co2+ and (3 ± 3)% Co3+ for Li1−γCoPO4), where the symmetry and spin state of the trivalent Co has some, but not dominating effects on the distribution. These values are in line with the Co2+ and Co3+ contents expected on basis of the nominal composition Li0.5−δCoPO4 (∼50% Co2+ and ∼50% Co3+), but where the Co3+ contribution derived from XAS is lower. The discrepancy can partly be explained by a small but noticeable reduction at the surface, in line with the fact that Co3+ is significantly less stable than Co2+. Moreover, the material was produced using a TTEG solvent, which also acts as a weak reducing agent31,32 and hence, might reduce the Co3+ concentration on the particle surface (cf. probing depth of TEY: 2−5 nm). We note that the more bulk sensitive spectra (FY, not shown) indicated higher spectral weight towards higher energies (and thus more Co3+ in the bulk), consistent with this hypothesis, but the spectra are not of high enough quality to be analyzed or discussed further.
In order to learn more about the symmetry and spin of the Co3+ sites, we have subtracted the fitted Co2+ contribution from the Li0.5−δCoPO4 spectrum (Fig. 3f). We note that while the main intensity difference is centered around the energy associated with the main peak of Co3+ (779.4 V), there is significant intensity on the low-energy side of this peak that is not accounted for by the LS Co3+. Comparison with Co3+ ref. 30 of different spin indicates that the trivalent Co ions are primarily high-spin, which can be rationalized based on the tetragonal distortions in the [CoO6] octahedra (cf. Table S3, ESI†) and the analogous HS Co3+ L-edge spectral assignment upon axial elongations and equatorial contractions in various perovskites30,33,34. The XAS thus indicates that the Li vacancies are indeed inducing distortions near the Co3+ sites that favor the HS Co3+ state. The HS Co3+ state is also consistent with a larger average Co–O distance (i.e., larger ionic radius) than what would be expected from the (smaller) LS Co3+ that only occupies the t2g orbitals and is associated with a stronger, more covalent Co–O interaction (cf. Table S3, ESI†). The presence of HS Co3+ is further consistent with the large magnetic moment (see later).
Infrared spectroscopy
Fig. 4 compares the infrared spectra of Li0.5−δCoPO4 (Cmcm) and Li1−γCoPO4 (Cmcm; data reproduced from ref. 21) in the range of 400–1700 cm−1. The omitted region from 1700 cm−1 to 4000 cm−1 is presented in Fig. S6 (ESI†) and does not show any absorption bands of water or other impurities. As expected, both spectra are dominated by the four fundamental vibrations of the pseudo-tetrahedral phosphate groups. Two regions can be distinguished, with the symmetric (ν1) and asymmetric (ν3) stretching vibrations of P–O being observed at higher frequencies, as well as the symmetric (ν2) and asymmetric (ν4) bending vibrations of the O–P–O group at lower frequencies. According to previous reports,16,21 the absorption peaks of Cmcm-type Li1−γCoPO4 (Fig. 4b) can be assigned as follows: the ν1 and ν3 stretching modes are observed at 1070 cm−1, 1016 cm−1, and 939 cm−1, whereas the ν2 and ν4 deformation vibrations appear in the region of 455–602 cm−1. Despite the structural similarity of the compounds, the IR spectrum of Li0.5−δCoPO4 (Cmcm, Fig. 4a) shows some distinct differences, in particular a shift of the band frequencies to slightly lower energies and band broadening, and the appearance of additional weak bands at 1512 cm−1 and 1138 cm−1.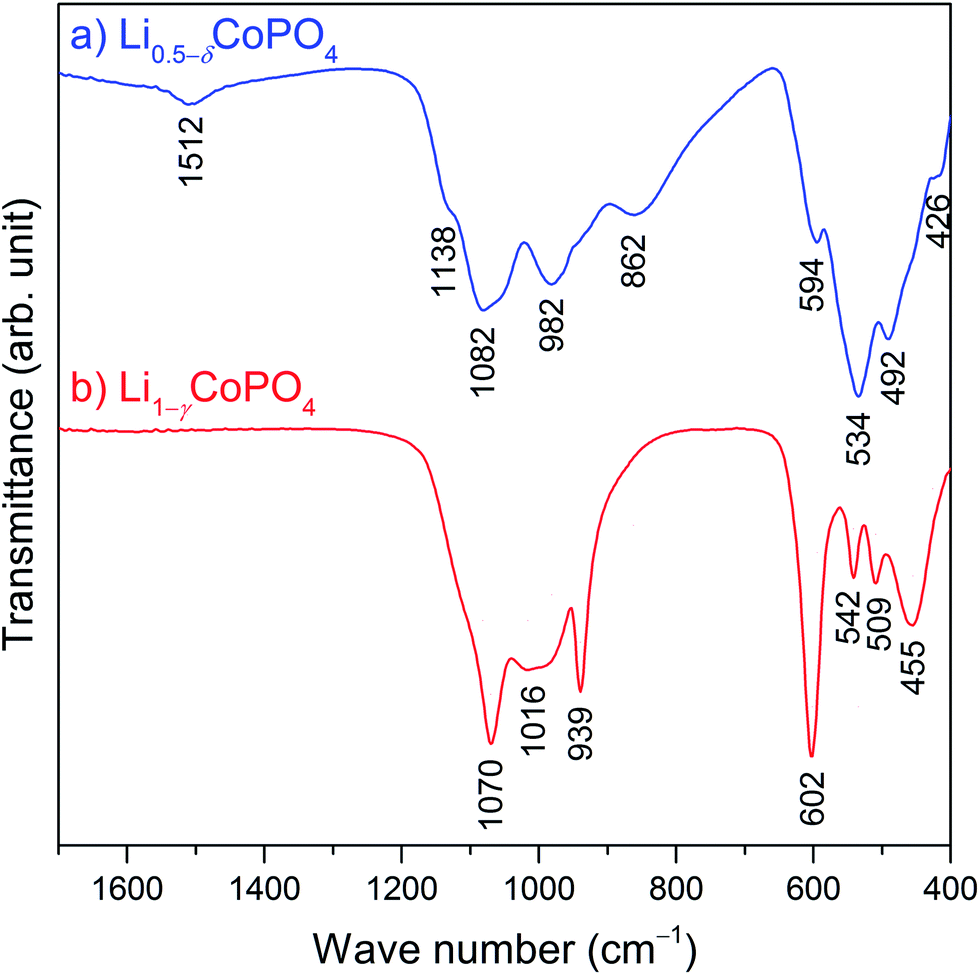 | ||
| Fig. 4 Comparison of the FTIR spectra of (a) Li0.5−δCoPO4 (Cmcm, blue), and (b) Li1−γCoPO4 (Cmcm, red, data reproduced from ref. 21). The omitted region of 1700–4000 cm−1 (cf. Fig. S6, ESI†) does not show any absorption bands of water or other impurities. | ||
The changes observed for the Li-deficient phase Li0.5−δCoPO4 are in line with reports35,36 on olivine-type LixFePO4 (0 ≤ x ≤ 1) which demonstrated that the absorption modes of the [PO4]3− groups are extremely sensitive to the delithiation of LiFePO4 and the associated oxidation of Fe2+ to Fe3+. As discussed, the average P–O distances in Li1−γCoPO4 (Cmcm) remain virtually unchanged when less Li is incorporated in the structure (cf. Table S3, ESI†). Hence, the energies of the absorption modes are not changed drastically, indicating similar local structures of the [PO4]3− units. This is in good agreement with a report by Popović and co-workers,37 which suggested a linear correlation between the P–O bond lengths and stretching frequencies. The observed band splitting, on the other hand, is correlated with interactions between ions, in this case between the [PO4]3− units and the adjacent Li+ and Co2+/3+ cations, i.e. the stronger the interaction, the larger the factor group splitting effects.36 In fact, the P–Li and P–Co distances are reduced in Li0.5−δCoPO4 compared to Li1−γCoPO4 (Table S3, ESI†), which leads to stronger interactions. Furthermore, it was shown38,39 that the factor group splitting of the ν3 modes increases with the second ionization potential of the transition metal due to the formation of strong bonds with the oxygen atoms of the [PO4] units, which causes a redistribution of electron density in the P–O bonds. This is reflected in the observation that the P–O1 bonds are expanded, and the P–O2 bonds shortened by ∼0.02 Å each in Li0.5−δCoPO4 (cf. Table S3, ESI†). Due to the significantly higher ionization potential of Co3+ compared to Co2+, the larger factor group splitting in the IR spectrum of Li0.5−δCoPO4 is therefore the result of the mixed valence state of the Co ions in the structure (Co2+/Co3+) as opposed to Li1−γCoPO4 which contains Co2+ only. However, a thorough analysis of the spectra, including the assignment of the additional absorption band at 1512 cm−1, would require a complete structural model, including the ordering of the Li+, Co2+, and Co3+ ions in the framework as well as DFT calculations, which is beyond the scope of this work. It is likely that the occurrence of additional modes is the result of a lower local symmetry in Li0.5−δCoPO4 due to a higher defect concentration.
Magnetic properties
Fig. 5 shows the magnetic susceptibility as function of temperature measured at a magnetic field of 0.1 T in field-cooled (FC) condition as well as the magnetic hysteresis recorded at a temperature of 2 K of Li0.5−δCoPO4 in comparison with Li1−γCoPO4 (both Cmcm). The temperature dependence of the magnetic susceptibilities indicates a long-range antiferromagnetic to paramagnetic transition at TN = 10.5 K for Li0.5−δCoPO4, and TN = 12 K for Li1−γCoPO4, respectively. The low transition temperature TN of 12 K of the Li1−γCoPO4 (Cmcm) phase is in agreement with our previous study21 on Cmcm-type LiCoPO4 and is comparable to the non-olivine, metastable LiCoPO4 (Pna21) phase (TN = 11 K),18 but much lower than the well investigated olivine-type LiCoPO4 (Pnma, TN = 21.6 K).40,41 This finding demonstrates the close relation between structural and magnetic properties. However, the further reduction of the observed transition temperature TN of the Li-deficient Li0.5−δCoPO4 (Cmcm) compound might be related to the vacancies on the Co site, weakening the Co–O–Co super exchange interaction. Above the transition temperature, the magnetic susceptibility of both compounds follow the Curie–Weiss law (see Fig. S7, ESI†), χ = χ0 + (NAμeff2)/(3kb(T − θC)), where χ0 is a temperature-independent contribution, μeff the effective magnetic moment, NA the Avogadro number, θC the Weiss temperature, and kb the Boltzmann constant. Fitting the high-temperature magnetic susceptibility with the Curie–Weiss law yields a higher Weiss temperature of −21.8 K of Li0.5−δCoPO4 compared to the stoichiometric Li1−γCoPO4 (−28.2 K; cf. inset of Fig. S7, ESI†), which reflects the difference in transition temperatures TN. Furthermore, similar effective magnetic moments of μeff = (5.20 ± 0.02) μB for Li0.5−δCoPO4 and (5.08 ± 0.02) μB for Li1−γCoPO4 were found. Both values exceed the spin-only value of high-spin Co2+ and Co3+ (3.9 μB and 4.8 μB, respectively), demonstrating a non-negligible orbital contribution.42 Taking into account the finite angular momentum, effective magnetic moments of 5.2 μB and 5.5 μB for high-spin Co2+ and Co3+ in the Oh symmetry (5t2g and 4t1g ground state) are expected in the weak spin–orbit coupling limit. Assuming the presence of only Co2+ in the Li1−γCoPO4 compound, the observed values are slightly smaller than the expected ones. However, the larger effective magnetic moment μeff of Li0.5−δCoPO4 clearly supports the findings of a mixed valence state of Co in the Li-deficient compound as discussed in the previous sections.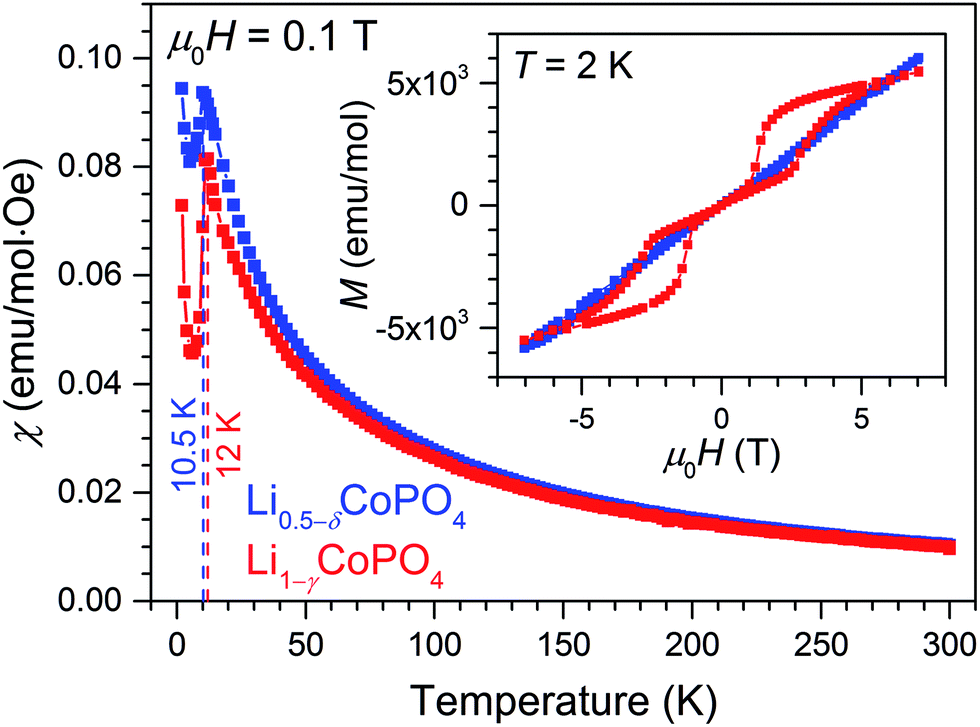 | ||
| Fig. 5 Magnetic susceptibility as function of temperature of Li0.5−δCoPO4 (Cmcm, blue data points) and Li1−γCoPO4 (Cmcm, red) measured at a magnetic field of 0.1 T in field-cooled condition. The vertical dashed lines mark the respective transition temperatures of both phases (10.5 K and 12 K). The inset shows the corresponding magnetic hysteresis curves recorded at T = 2 K. | ||
The inset of Fig. 5 reveals a more distinct difference of the magnetic properties of Li1−γCoPO4 and the Li-deficient Li0.5−δCoPO4 compounds. LiCoPO4 exhibits a magnetic double-hysteresis loop at 2 K, demonstrating an antiferromagnetic ground state at 0 T (for the hysteresis curves at 11 K and 300 K see Fig. S8, ESI†). Furthermore, the double-hysteresis loop indicates a spin-flip transition at a critical field of around ±3 T, which is much lower than for Pnma-type LiCoPO4.43 Again, this can be attributed to the different structural properties. In contrast, an almost linear dependence of the magnetization as a function of the applied field is observed for Li0.5−δCoPO4 below TN. No hysteresis with a finite remanence caused by a weak ferromagnetic phase due to the mixed-valence state of Co ions were found.40 However, the difference of the magnetic susceptibility recorded under field-cooled (FC) and zero field-cooled (ZFC) conditions suggests the formation of magnetic domains below T ≈ 5 K. Below this temperature, the finite amount of Co3+ ions as well as the observed defects on the Co sites might cause competing magnetic interactions resulting in a complex antiferromagnetic state as, for instance, described by Jensen and co-workers.44
Thermal stability
The thermal stability of Li0.5−δCoPO4 (Cmcm) was investigated using TGA/DSC (Fig. 6) and temperature-dependent in situ X-ray powder diffraction, both performed under air (Fig. 7). The thermal behavior of LiCoPO4 (Cmcm) has been discussed in detail in ref. 21. In the TGA curve (Fig. 6a), an overall mass loss of ∼4.8 wt% is observed between 30 °C and 900 °C, which proceeds in several steps. The mass loss of ∼1.3 wt% up to 360 °C is probably correlated with the decomposition of residues of the TTEG solvent and the acetate precursors (cf. CHNS analysis, Table 2). The DSC curve shows two pronounced, broad endothermic peaks at 394 °C and 686 °C, respectively, with the first signal being accompanied by a weight loss step of ∼2.5 wt% and the second one of ∼0.1 wt%. The corresponding X-ray powder diffraction pattern (Fig. 6b, refinement details see Tables S7–S11†) of the dark violet powder (cf. graphical abstract) obtained after the TGA/DSC measurement reveals that a mixture of 44.9(6) wt% olivine-type LiCoPO4 (Pnma, ICSD no. 431999)45 and 55.1(6) wt% of the low-temperature modification of cobalt pyrophosphate, α-Co2P2O7 (P21/c, ICSD no. 280959)46 was formed. Note that data obtained from a similar experiment using an argon atmosphere are in good agreement (Fig. S9 and S10, Tables S12–S16, ESI†).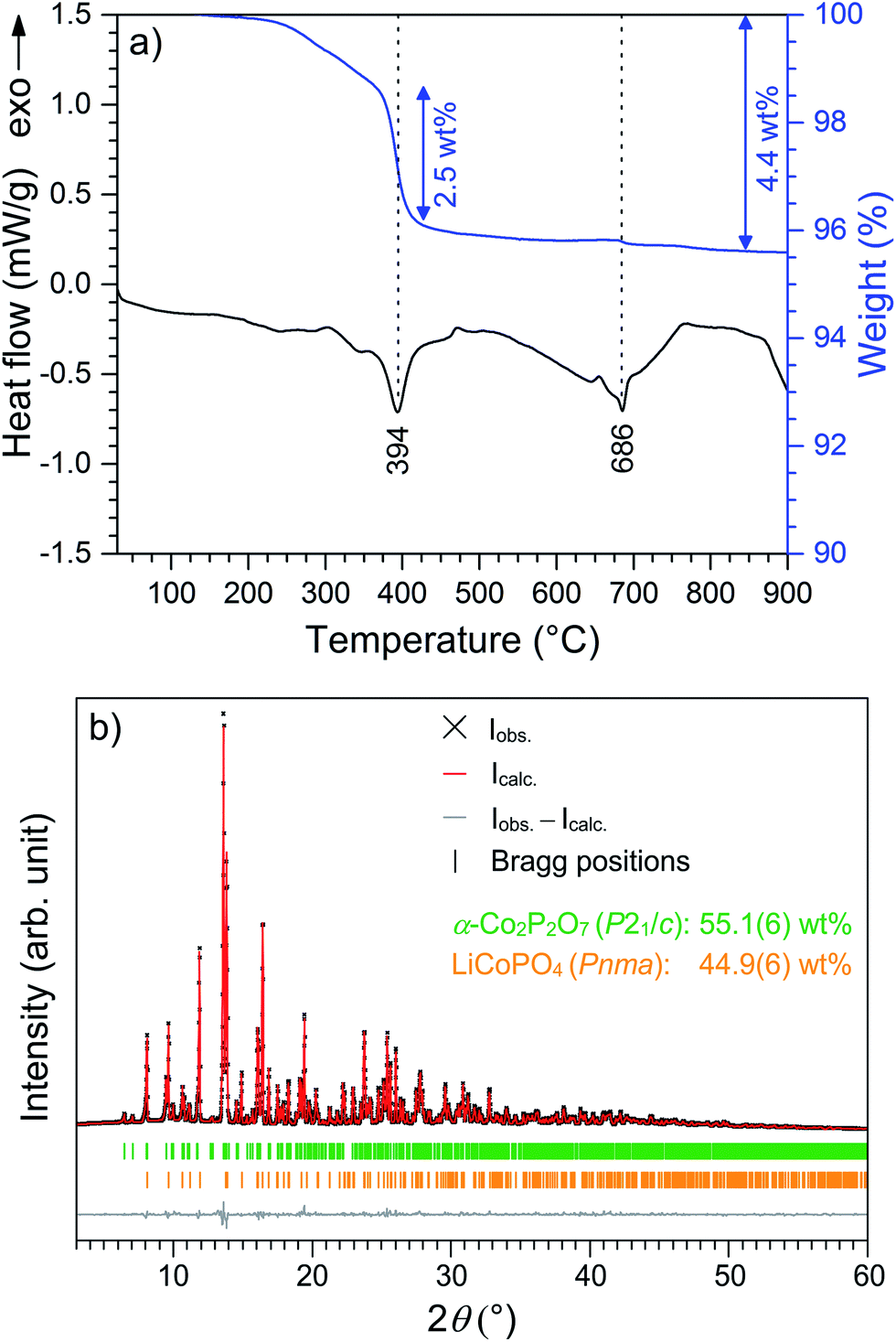 | ||
| Fig. 6 (a) DSC (black) and TGA (blue) curves of Li0.5−δCoPO4 (Cmcm) measured in a temperature range of 30–900 °C (heating rate: 10 °C min−1, atmosphere: synthetic air). Two endothermic DSC signals are observed at 394 °C (accompanied by a TGA weight loss step of ∼2.5 wt%) and 686 °C, respectively. (b) Rietveld fit of the X-ray powder diffraction data (transmission geometry, Mo Kα1 radiation) of the dark violet post TGA/DSC-material demonstrating that mixture of olivine-type LiCoPO4 (Pnma) and α-Co2P2O7 (P21/c) was formed upon heating. | ||
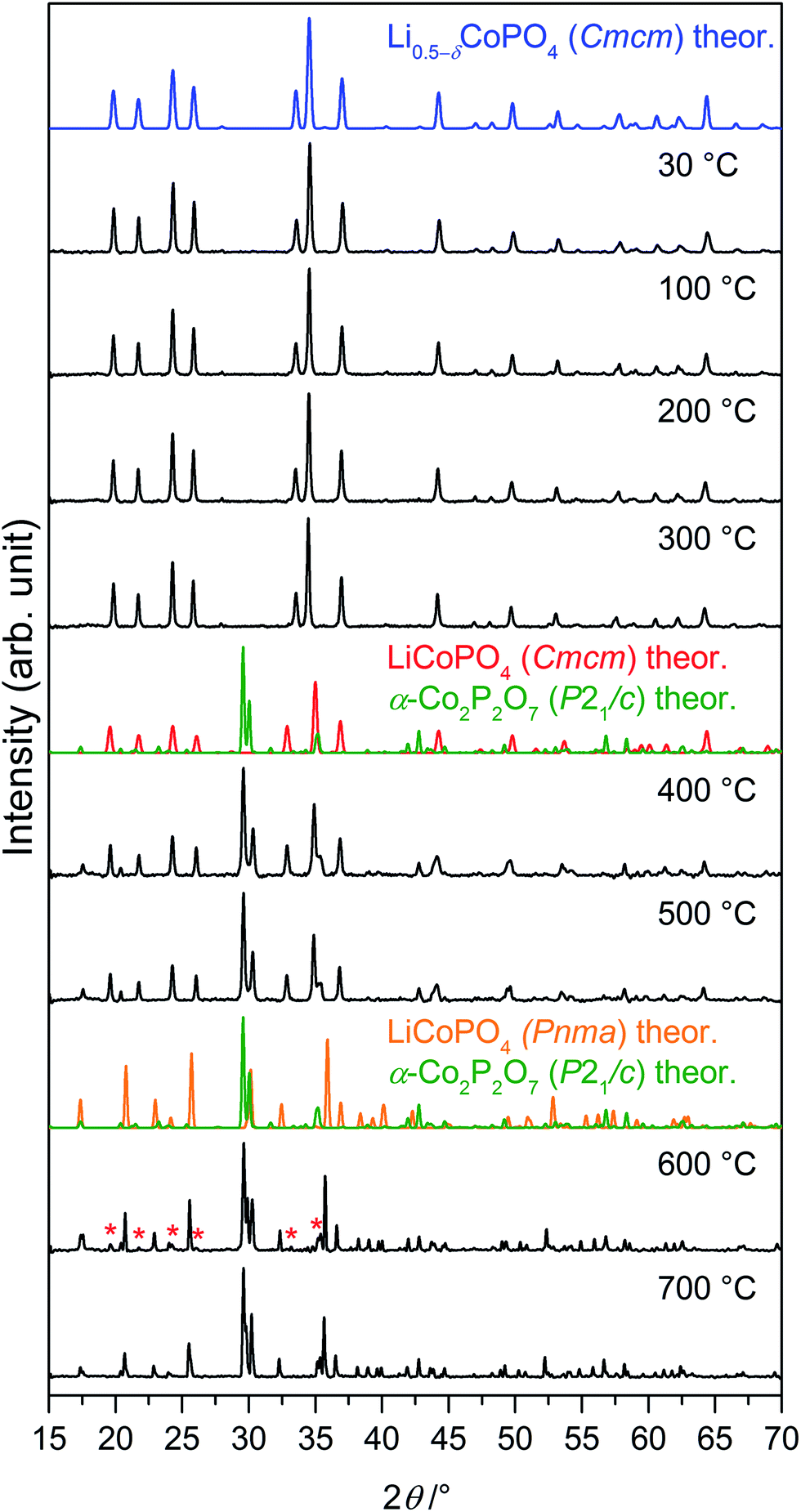 | ||
Fig. 7 In situ X-ray powder diffraction patterns (Bragg–Brentano geometry, Cu Kα radiation) of Li0.5−δCoPO4 (Cmcm) measured between 30 °C and 700 °C under air (heating rate: 5 °C min−1; for patterns at 800 °C, 900 °C, and after cooling see Fig. S12, ESI†). The phase undergoes several transitions upon heating. The theoretical patterns of the involved phases, which were calculated from room temperature data, are displayed in color. Between 300 °C and 400 °C, Li0.5−δCoPO4 (Cmcm, this work, blue) decomposes to LiCoPO4 (Cmcm, ICSD no. 432186,21 red) and α-Co2P2O7 (P21/c, ICSD no. 280959,46 green). Above 500 °C, an irreversible transition of LiCoPO4 (Cmcm, blue) to olivine-type LiCoPO4 (Pnma, ICSD no. 431999,45 orange) occurs. The reflections marked with red asterisks ( ) in the pattern at 600 °C can be assigned to residues of LiCoPO4 (Cmcm). ) in the pattern at 600 °C can be assigned to residues of LiCoPO4 (Cmcm). | ||
In order to understand the signals observed in the TGA/DSC study and to identify possible intermediates of the heating process, we performed a temperature-controlled in situ PXRD experiment between room temperature (30 °C) and 900 °C with a step size of 100 °C (Fig. 7). The Rietveld fits of the individual PXRD patterns at each temperature up to 700 °C can be found in Fig. S11 (ESI†). The refined phase fractions and crystallographic details (atomic coordinates, thermal displacements parameters, bond lengths), reflecting the structural changes, are presented in Tables S17–S25.† (Note that the patterns at T ≥ 800 °C, which are shown in Fig. S12 (ESI†), were of insufficient quality for a refinement because of the occurrence of strong reflections from the corundum sample holder.) Up to 300 °C, no change of the diffraction patterns occurs, indicating that the Li0.5−δCoPO4 (Cmcm) phase is thermally stable up to that temperature. Between 300 °C and 400 °C, a mixture of the ‘fully lithiated’ Li1−γCoPO4 (Cmcm) structure (referred to as LiCoPO4 with γ = 0 in Fig. 7 since ICSD no. 432186 (ref. 21) was used for the theoretical pattern) and α-Co2P2O7 (P21/c) is observed. Hence, the endothermic DSC signal at 395 °C can be explained by the decomposition of Li0.5−δCoPO4 (Cmcm) to these phases (the simultaneous mass loss will be explained later in the text). Between 500 °C and 600 °C, the Cmcm-type Li1−γCoPO4 intermediate starts to convert to the thermodynamically more stable olivine Pnma structure, whereas the peaks of α-Co2P2O7 remain unaltered. The fact that the low-temperature α-modification of Co2P2O7 (P21/c) does not transform to the high-temperature β-Co2P2O7 (A2/m)47 phase, which would be expected at ∼480 °C (ref. 48) for the pure material, suggests that the transformation is either kinetically hindered or very slow. Hence, the DSC signal at 688 °C corresponds to the transformation of the Cmcm to the Pnma LiCoPO4 phase. The lower transition temperature found in the in situ PXRD study compared to the DSC data might be related to slightly different atmospheres (air vs. synthetic air) and heating rates (10 °C min−1 vs. 5 °C min−1) being used. The phase transformation is not completed until 700 °C because traces of Li1−γCoPO4 (Cmcm) can be identified in the diffraction pattern at 600 °C. Compared to the thermal stability of the pure, lithiated LiCoPO4 (Cmcm) material (transformation to single-phase LiCoPO4 (Pnma) at 575 °C),21 the phase transition temperature is significantly increased. No further phase changes are observed in the PXRD patterns up to 700 °C, yet the diffraction peaks are shifted to lower angles, indicating bigger lattice dimensions due to thermal expansion. Unfortunately, it cannot be derived from the patterns at T ≥ 800 °C (see Fig. S12, ESI†) whether the Pna21-type LiCoPO4 structure reappears as a high-temperature phase as reported for all three, fully lithiated LiCoPO4 polymorphs (Pnma,17 Pna21,19 and Cmcm21). The pattern of the cooled sample (25 °C; Fig. S12, ESI†) is consistent with the ex situ PXRD experiment (Fig. 6b) and shows reflections of α-Co2P2O7 and Pnma-type LiCoPO4, indicating that both phase transitions are irreversible.
The results of the thorough investigation of the thermal stability of the Li-deficient compound Li0.5−δCoPO4 demonstrate that the phase exhibits a complex behavior upon heating which involves several phase transitions. Based on the combined approach using TGA, DSC (Fig. 6), and in situ PXRD studies (Fig. 7), a decomposition mechanism can be proposed according to Scheme 1. Note that since it is not possible to determine the composition of the decomposition products and intermediates (which are likely to be deficient in Li and Co as well), the mechanism is presented on basis of the nominal composition Li0.5−δCoPO4 with δ = 0. In the first step (eqn (I)), LiCoPO4 (Cmcm) and Co2P2O7 (P21/c; both with Co oxidation state +2) are formed from four equivalents of the Co2+/Co3+ mixed-valent starting material Li0.5−δCoPO4 (Cmcm). This step is based on a redox reaction, in which the two Co3+ equivalents from the four Li0.5−δCoPO4 (δ = 0) units are reduced by O2− ions (eqn (Ia)), which are released upon the pyrophosphate formation (i.e., coupling of isolated [PO4] tetrahedra to [P2O7] units via shared corners). The O2− ions are in turn oxidized to form elemental oxygen (eqn (Ib)) that is released. The O2 release from the phosphate groups corresponds to an approximate mass loss of ∼2.5 wt% which in full agreement with the TGA mass loss step observed at 394 °C (cf. Fig. 6a). The estimated mass fractions of Li1−γCoPO4 (Cmcm, with γ = 0) and Co2P2O7 after the oxygen release are ∼52.4 wt% and ∼47.6 wt%, respectively, and are very close to the refined values obtained from our Rietveld analysis (cf. Fig. 6, and Table S17, ESI†). The slight deviation of our refined values from the calculated ones can be explained by the fact that the nominal composition was used for this hypothesis and that the refinement of phase fractions on basis of the reflection intensities depends on the particle size and crystallinity. In the second step at 686 °C (eqn (II)), which was not accompanied by a significant mass loss in the TGA (Fig. 6a), LiCoPO4 (Cmcm) is transformed to the thermodynamically more stable olivine-type LiCoPO4 (Pnma) as observed in the PXRD study (Fig. 6b).
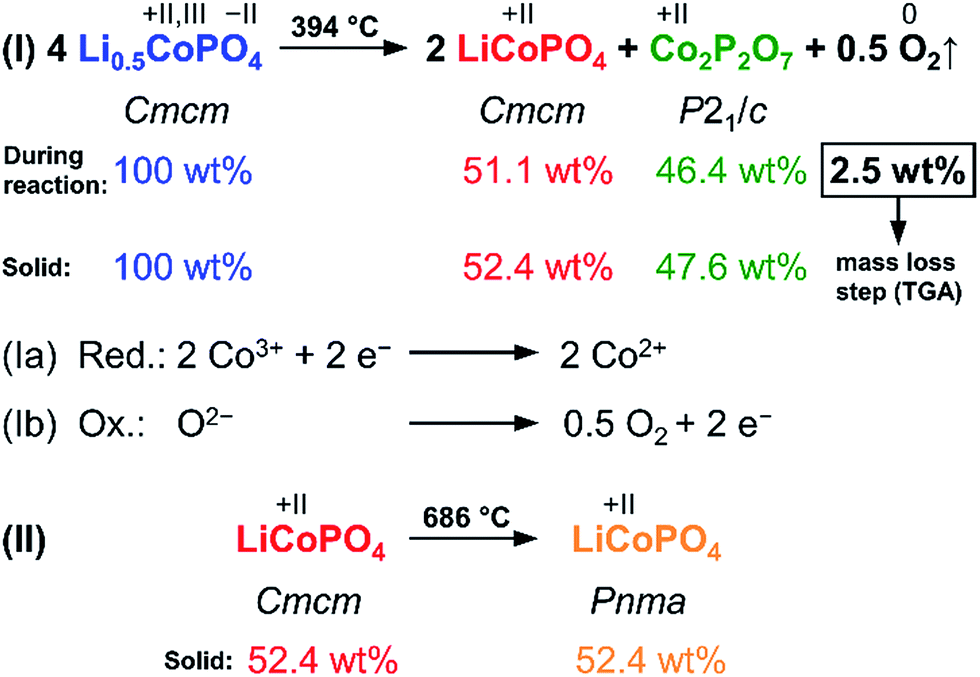 | ||
| Scheme 1 Proposed two-step mechanism of the thermal decomposition of Li0.5−δCoPO4 (Cmcm, with δ = 0) and theoretically expected weight fractions (in wt%) of the involved phases during the reaction as well as in the solid remainder after O2 release. It can be inferred that the TGA weight loss (cf. Fig. 7) at 394 °C is due to the release of oxygen as a result of a redox process. The color code of the phases is related to Fig. 6 and 7. | ||
Interestingly, the decomposition leads to the crystallization of same phases (Pnma-type LiCoPO4 and Co2P2O7) under oxygen evolution as reported for the olivine-like Li-poor phase LixCoPO4.13 However, there are some significant mechanistic differences between Li0.5−δCoPO4 (Cmcm) and LixCoPO4 (Pnma), which might be due to the fact that our studies are based on the pristine material whereas the studies on LixCoPO4 were based on charged LiCoPO4 electrodes. First of all, as reported by Bramnik et al.,13 the decomposition of LixCoPO4 cathodes occurs at much lower temperatures (<200 °C) suggesting that the Li-poor, Cmcm-type Li0.5−δCoPO4 phase (decomposition at 394 °C) is significantly more stable. (Note that compared to the results of Theil et al.,14 on the other hand, Cmcm-type Li0.5−δCoPO4 seems to be less stable than LixCoPO4, which was found to be thermally stable at least up to 550 °C. This discrepancy might be explained by different particle sizes and carbon contents.). Furthermore, in contrast to our material, the Co2P2O7 crystallization was found to be not proceeding simultaneously, but at higher temperature than the LixCoPO4 (Pnma) decomposition, which was related to a possibly amorphous intermediate.13 In addition, the decomposition process was suggested to be promoted by carbon present in samples, which reacts with the released oxygen to form CO2 gas.13 The crucial influence/destabilizing effect of carbon on the thermal stability was also confirmed for charged LiCoPO4 electrodes containing CoPO4.14 Based on this work, however, there is no indication that carbon affects the decomposition of the pristine Li0.5−δCoPO4 (Cmcm) material since the carbon content of our material is not significant (0.4(3) wt%, cf. Table 2). To clarify whether this is also the case for the pristine, carbon-free LixCoPO4 (x = 0, 0.7; space group Pnma) olivine phases (as opposed to the studies13,14 based on carbon-containing charged LiCoPO4 electrodes), our direct synthetic approach might provide a pathway to get a deeper understanding of their intrinsic thermal stabilities as well.
Conclusions
In this study, a polyol synthesis pathway towards the first Li-deficient structural derivative of the Cmcm polymorph of LiCoPO4 with the nominal composition Li0.5−δCoPO4 and its material properties were presented. To the best of our knowledge, this is also the first time that a sub-stoichiometric LixCoPO4 phase was synthesized using a soft-chemical bottom-up approach as opposed to common chemical and electrochemical Li extraction techniques starting from LiCoPO4-type materials.Neutron and X-ray powder diffraction experiments as well as elemental analysis suggested that Li0.5−δCoPO4 (Cmcm) is non-stoichiometric and deficient both in Li and Co, which generates vacancies on both cation sub-lattices in the crystal structure. The occurrence of vacancies was also observed in the course of a structure redetermination of the ‘fully lithiated’ Cmcm phase, resulting in the revised formula Li1−γCoPO4. Co L2,3-edge X-ray absorption spectroscopy indicated that, unlike Li1−γCoPO4 which exclusively contains octahedrally coordinated Co2+ ions, the Li-deficient structure bears both Co2+ and Co3+ ions to compensate for the Li deficit. Due to the reduced Li content and amount of electrochemically active Co2+ ions, the material exhibited a poor electrochemical performance. The thermal stability of Li0.5−δCoPO4 has been studied thoroughly using thermogravimetry, differential scanning calorimetry, and temperature-dependent in situ X-ray powder diffraction experiments. Li0.5−δCoPO4 (Cmcm) is metastable and shows a complex, two-step decomposition mechanism upon heating. At 394 °C, it decomposes to α-Co2P2O7 (P21/c) and Li1−γCoPO4 (Cmcm) in an endothermic reaction upon which oxygen is released as a result of a redox reaction. The Li1−γCoPO4 (Cmcm) phase then irreversibly converts to the thermodynamically more stable LiCoPO4 (Pnma) olivine structure at 686 °C.
To conclude, the present work paves the way towards the direct and simple soft-chemical preparation and investigation of Li-deficient structures derived from lithium transition-metal phosphates. Our methodology provides fundamental insights into the material properties, and hence to study Li-deficient intermediates that are probably involved in the charge/discharge steps of LiCoPO4-type cathodes. It further helps to understand the complex structure chemistry of this class of cathode materials for Li-ion batteries. In that matter, future studies should focus on compositional tuning (e.g. by modifying the amounts of the precursors in the synthesis) in order to identify other partially lithiated structural derivatives.
Author contributions
J. L. performed the material characterization using in situ and ex situ PXRD, SEM/EDS, IR spectroscopy, and electrochemical measurements, and analyzed the data (under supervision of T. N. and M. M. D.). C. A.-S. performed the synthesis. S. G. and D. N. performed and analyzed magnetic and XAS measurements, respectively. I. P. O. and C. A.-S. conducted neutron diffraction experiments. J. L. wrote the manuscript. All authors have given approval to the final version of the manuscript.Funding sources
This work has been funded by the Fonds der Chemischen Industrie and the TUM Graduate School. The use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, and Office of Basic Energy Sciences (contract no. DE-AC02-76SF00515). We further thank the Institut Laue-Langevin (ILL) for beam time allocation, and the D2B team for technical support (proposal no. 5-31-2531).Abbreviations
| AAS | Atomic absorption spectroscopy |
| AEY | Auger electron yield |
| DFT | Density functional theory |
| DSC | Differential scanning calorimetry |
| EDS | Energy-dispersive X-ray spectroscopy |
| FC | Field-cooled |
| FTIR | Fourier-transform infrared |
| FY | Fluorescence yield |
| HS | High-spin |
| ICSD | Inorganic Crystal Structure Database |
| LS | Low-spin |
| NPD | Neutron powder diffraction |
| PXRD | Powder X-ray diffraction |
| SEM | Scanning electron microscope |
| SQUID | Superconducting quantum interference device |
| TEY | Total electron yield |
| TGA | Thermogravimetric analysis |
| TTEG | Tetraethylene glycol |
| XAS | X-ray absorption spectroscopy |
| ZFC | Zero field-cooled |
Acknowledgements
The authors thank U. Ammari for elemental analysis, and D. Haering for TGA/DSC measurements. J. L. and C. A.-S. are grateful to the TUM Graduate School, the Fonds der Chemischen Industrie, DAAD and Colciencias for financial support of their Ph.D. work.References
- K. Amine, H. Yasuda and M. Yamachi, Electrochem. Solid-State Lett., 2000, 3, 178–179 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- K. Zaghib, A. Guerfi, P. Hovington, A. Vijh, M. Trudeau, A. Mauger, J. B. Goodenough and C. M. Julien, J. Power Sources, 2013, 232, 357–369 CrossRef CAS.
- A. Yamada, M. Hosoya, S.-C. Chung, Y. Kudo, K. Hinokuma, K.-Y. Liu and Y. Nishi, J. Power Sources, 2003, 119–121, 232–238 CrossRef CAS.
- S. Okada, S. Sawa, M. Egashira, J. Yamaki, M. Tabuchi, H. Kageyama, T. Konishi and A. Yoshino, J. Power Sources, 2001, 97–98, 430–432 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- N. N. Bramnik, K. Nikolowski, C. Baehtz, K. G. Bramnik and H. Ehrenberg, Chem. Mater., 2007, 19, 908–915 CrossRef CAS.
- H. Ehrenberg, N. N. Bramnik, A. Senyshyn and H. Fuess, Solid State Sci., 2009, 11, 18–23 CrossRef CAS.
- H. Ju, J. Wu and Y. Xu, Int. J. Energy Environ. Eng., 2013, 4(22), 26 Search PubMed.
- M. Kaus, I. Issac, R. Heinzmann, S. Doyle, S. Mangold, H. Hahn, V. S. K. Chakravadhanula, C. Kuebel, H. Ehrenberg and S. Indris, J. Phys. Chem. C, 2014, 118, 17279–17290 CAS.
- F. C. Strobridge, R. J. Clement, M. Leskes, D. S. Middlemiss, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, P. J. Chupas and C. P. Grey, Chem. Mater., 2014, 26, 6193–6205 CrossRef CAS PubMed.
- N. N. Bramnik, K. Nikolowski, D. M. Trots and H. Ehrenberg, Electrochem. Solid-State Lett., 2008, 11, A89–A93 CrossRef CAS.
- S. Theil, M. Fleischhammer, P. Axmann and M. Wohlfahrt-Mehrens, J. Power Sources, 2013, 222, 72–78 CrossRef CAS.
- X. Huang, J. Ma, P. Wu, Y. Hu, J. Dai, Z. Zhu, H. Chen and H. Wang, Mater. Lett., 2005, 59, 578–582 CrossRef CAS.
- K. J. Kreder, G. Assat and A. Manthiram, Chem. Mater., 2015, 27, 5543–5549 CrossRef CAS.
- J. Ludwig, C. Marino, D. Haering, C. Stinner, D. Nordlund, M. M. Doeff, H. A. Gasteiger and T. Nilges, RSC Adv., 2016, 6, 82984–82994 RSC.
- C. Jaehne, C. Neef, C. Koo, H.-P. Meyer and R. Klingeler, J. Mater. Chem. A, 2013, 1, 2856–2862 CAS.
- J. Ludwig, D. Nordlund, M. M. Doeff and T. Nilges, J. Solid State Chem., 2017, 248, 9–17 CrossRef CAS.
- U. Amador, J. M. Gallardo-Amores, G. Heymann, H. Huppertz, E. Moran and M. E. Arroyo-de Dompablo, Solid State Sci., 2009, 11, 343–348 CrossRef CAS.
- C. Alarcón-Suesca, J. Ludwig, V. Hlukhyy, C. Stinner and T. Nilges, Inorganics, 2016, 4, 35 CrossRef.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr.–Cryst. Mater., 2014, 229, 345–352 CAS.
- J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef CAS.
- A. M. Hibberd, H. Q. Doan, E. N. Glass, F. M. F. de Groot, C. L. Hill and T. Cuk, J. Phys. Chem. C, 2015, 119, 4173–4179 CAS.
- D. T. Cromer and D. A. Liberman, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1981, 37, 267–268 CrossRef.
- J. Chen and M. S. Whittingham, Electrochem. Commun., 2006, 8, 855–858 CrossRef CAS.
- O. Garcia-Moreno, M. Alvarez-Vega, F. Garcia-Alvarado, J. Garcia-Jaca, J. M. Gallardo-Amores, M. L. Sanjuan and U. Amador, Chem. Mater., 2001, 13, 1570–1576 CrossRef CAS.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter, 1990, 42, 5459–5468 CrossRef CAS.
- F. M. F. de Groot, M. Abbate, J. van Elp, G. A. Sawatzky, Y. J. Ma, C. T. Chen and F. Sette, J. Phys.: Condens. Matter, 1993, 5, 2277–2288 CrossRef CAS.
- Z. Hu, H. Wu, M. W. Haverkort, H. H. Hsieh, H. J. Lin, T. Lorenz, J. Baier, A. Reichl, I. Bonn, C. Felser, A. Tanaka, C. T. Chen and L. H. Tjeng, Phys. Rev. Lett., 2004, 92, 207402 CrossRef CAS PubMed.
- D.-H. Kim and J. Kim, Electrochem. Solid-State Lett., 2006, 9, A439–A442 CrossRef CAS.
- M. K. Devaraju and I. Honma, Adv. Energy Mater., 2012, 2, 284–297 CrossRef CAS.
- M. Abbate, J. C. Fuggle, A. Fujimori, L. H. Tjeng, C. T. Chen, R. Potze, G. A. Sawatzky, H. Eisaki and S. Uchida, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 16124–16130 CrossRef CAS.
- S. Y. Istomin, O. A. Tyablikov, S. M. Kazakov, E. V. Antipov, A. I. Kurbakov, A. A. Tsirlin, N. Hollmann, Y. Y. Chin, H. J. Lin, C. T. Chen, A. Tanaka, L. H. Tjeng and Z. Hu, Dalton Trans., 2015, 44, 10708–10713 RSC.
- C. M. Burba and R. Frech, J. Electrochem. Soc., 2004, 151, A1032–A1038 CrossRef CAS.
- C. M. Burba and R. Frech, Spectrochim. Acta, Part A, 2006, 65, 44–50 CrossRef PubMed.
- L. Popović, D. De Waal and J. C. A. Boeyens, J. Raman Spectrosc., 2005, 36, 2–11 CrossRef.
- M. T. Paques-Ledent and P. Tarte, Spectrochim. Acta, Part A, 1974, 30, 673–689 CrossRef.
- A. Ait Salah, P. Jozwiak, J. Garbarczyk, K. Benkhouja, K. Zaghib, F. Gendron and C. M. Julien, J. Power Sources, 2005, 140, 370–375 CrossRef.
- N. F. Kharchenko, Y. N. Kharchenko, R. Szymczak, M. Baran and H. Schmid, Low Temp. Phys., 2001, 27, 895–898 CrossRef CAS.
- J. P. Rivera, Ferroelectrics, 1994, 161, 147–164 CrossRef CAS.
- S. H. Baek, R. Klingeler, C. Neef, C. Koo, B. Buechner and H. J. Grafe, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 134424 CrossRef.
- N. F. Kharchenko, V. M. Khrustalev and V. N. Savitskii, Low Temp. Phys., 2010, 36, 558–564 CrossRef CAS.
- M. H. Jensen and P. Bak, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 6853–6868 CrossRef.
- J. Ludwig, C. Marino, D. Haering, C. Stinner, H. A. Gasteiger and T. Nilges, J. Power Sources, 2017, 342, 214–223 CrossRef CAS.
- B. El Bali and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, i32–i33 CAS.
- A. El Belghitti, A. Boukhari and E. M. Holt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 482–484 CrossRef.
- M. Trojan and D. Brandova, Sb. Ved. Pr. - Vys. Sk. Chemickotechnol. Pardubice, 1985, 47, 33–42 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Comparison of the PXRD patterns of Li0.5−δCoPO4 and Li1−γCoPO4, (2) Rietveld refinement details from PXRD data, (3) Rietveld refinement details from neutron powder diffraction data, (4) additional illustrations of the crystal structure, (5) electrochemical characterization, (6) SEM and EDS analysis, (7) full IR spectra, (8) additional magnetic data, (9) Rietveld refinement details of the PXRD pattern of the sample obtained from the TGA/DSC measurement of Li0.5−δCoPO4 under air, (10) thermal stability under Ar (TGA/DSC, Rietveld refinement), (11) Rietveld fits and crystallographic data of the in situ PXRD patterns (30–700 °C; air), (12) additional in situ PXRD patterns (800 °C, 900 °C, and 25 °C; air). The cif files containing the crystallographic data of Li0.5−δCoPO4 and Li1−γCoPO4 can be obtained from FIZ Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: +49 7247 808 666; e-mail: crysdata@fiz-karlsruhe.de), on quoting the CSD deposition numbers 432850 and 432851. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04043a |
| This journal is © The Royal Society of Chemistry 2017 |