
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of (4E,6Z,10Z)-hexadeca-4,6,10-trien-1-ol and (4E,6E,10Z)-hexadeca-4,6,10-trien-1-ol, the pheromone components of cocoa pod borer moth Conopomorpha cramerella
Fei Huangb,
Yushun Zhanga,
Yun Yaoa,
Wanqiu Yang*c and
Yunhai Tao *a
*a
aSchool of Chemical Science and Technology, Yunnan University, Kunming 650091, PR China. E-mail: cloudsea2000@sina.com; Fax: +86-871-67429991; Tel: +86-871-65031119
bKunming Biohome Technology Co. Ltd., Kunming 650501, PR China
cDepartment of Chemical Science and Technology, Kunming University, Kunming 650214, PR China
First published on 17th July 2017
Abstract
A concise and efficient synthesis of the pheromone components of the cocoa pod borer moth, namely (4E,6Z,10Z)-hexadeca-4,6,10-trien-1-ol and (4E,6E,10Z)-hexadeca-4,6,10-trien-1-ol, starting from commercially available materials, was reported. The overall yield was 30.4% and 27.4%, respectively. The stereoselective formation of (E,Z)- or (E,E)-conjugated double bond relied on Sonogashira coupling with (E)-5-bromopent-4-en-1-ol prepared from (E)-5-bromopent-4-enal and the stereoselective hydrogenation of the enyne, while the 10Z-double bond was formed by Wittig reaction from 4-hydroxybutanal and n-hexyltriphenylphosphonium bromide.
Introduction
(4E,6Z,10Z)-Hexadeca-4,6,10-trien-1-ol (1a), (4E,6E,10Z)-hexadeca-4,6,10-trien-1-ol (2a), and their acetates (1b and 2b) are the pheromone components of cocoa pod borer moth (Conopomorpha cramerella, Lepidoptera: Gracillariidae) which is the most important pest damaging cocoa plantations in Southeast Asia.1 The life cycle of the insect starts once the egg is laid on the pod surface of the cocoa plant (this cycle lasts for 2–7 days). Then, the larval stage is characterized by the formation of tunnels at the pod surface that reaches the sclerotic layer of the husk to reach food, causing scarification and sticking of the beans. Damage to the funicles of the pods results in beans that are deformed and under-sized, significantly reducing the quality and thus the value of the processed beans. Insect feeding in the pods also causes a change of the color of the beans into yellow or uneven and premature ripening, which do not match with the ripeness standards for harvesting.2 Consequently, the production losses can exceed 50% of the crop.1In 1986 Beevor et al. isolated and identified the pheromone components of the insect, and synthesized those compounds using a long and non-stereospecific route.1 In 1992 Yen et al. reported a shorter synthesis for the pheromone components, in eight steps and with an overall yield of 32%.3 Later, Pereira and Cabezas reported a new method for the synthesis of 1,5-diynes using the reaction of 1,3-dilithiopropyne with propargyl chlorides, and applied this new method to prepare 1a with an overall yield of 51%.4 Yet, the preparation of 1,5-diynes was complicated, and this compound could not be used as the starting material of isomer 2a. The general utilization of insecticides is limited by their high cost, environmentally unfriendliness and the development of insecticide-resistance. Considerable efforts have been made for the development and evaluation of new integrated pest management techniques, such as the usage of pheromone compounds (Fig. 1).
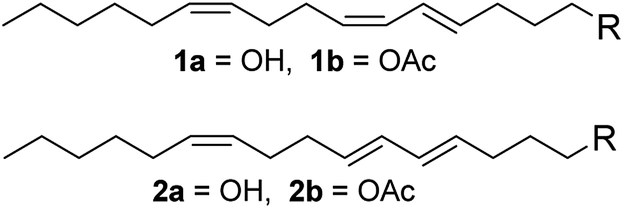 | ||
| Fig. 1 Structure of the pheromone components of the cocoa pod borer moth Conopomorpha cramerella. | ||
Previously, we reported the efficient syntheses of (5Z,7E)-dodecadien-1-ol, (7Z,11Z,13E)-hexadecatrienal, (8E,10Z)-tetradecadienal, and (9Z,11E)-hexadecadienal from (4Z,6E)-7-bromohepta-4,6-dienal (3a),5 and developed an useful compound, namely (E)-5-bromopent-4-enal (4a).5c As a part of an ongoing investigation, we reported herein a concise and stereoselective synthesis of 1a and 2a from 4a, which successfully overcame the major limitations of the previous strategies published.
Results and discussion
Compound 4a was prepared by the tandem addition reaction of acrolein with acetylene using Pd(OAc)2 as catalyst. Previously, Lu et al. reported the preparation of 3a.6 By repeating the reported strategy, we found that the method was efficient only in small scale. In the large-scale preparations, 4a was produced in considerable amount as a by-product. Based on this result, we optimized the experimental conditions to afford uniquely 4a (Scheme 1).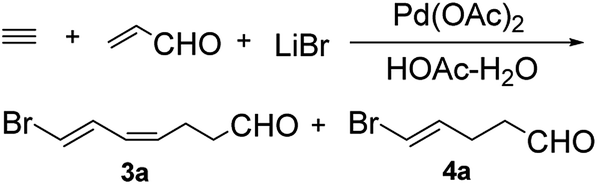 | ||
| Scheme 1 Pd(II)-catalyzed coupling reaction of lithium bromide, acetylene, and acrolein. | ||
We studied the effect of different reaction conditions on the ratio of 3a and 4a. The reaction time, reactant concentration and temperature were tested (Table 1). A high concentration of acrolein (a decrease of the quantity of HOAc and H2O) did not increase the total conversion rate, but it resulted in a better selectivity of the reaction (Table 1, entries 1–3). Increasing the temperature resulted in a better selectivity of the reaction, but it decreased the total conversion rate (Table 1, entries 3–5), because acrolein and the two products were not stable at high temperature. When the reaction time was prolonged, significant improvements were observed in terms of conversion rate and selectivity (Table 1, entries 4, 6, and 7). Lower acetylene pressure allowed a better selectivity, but it did not affect strongly the total conversion rate (Table 1, entries 7–9). These results showed the same trend in both large-scale and small-scale preparation. Previously, Lu et al. explained the formation of 3a (Scheme 2, path a).6 First, transhalopalladation of acetylene lead to the (E)-vinylpalladium intermediate 5, which was inserted by a second molecule of acetylene to form (E,Z)-dienylpalladium 6. After the insertion of acrolein, (2-oxoalkyl)palladium intermediate 7 underwent protonolysis (path a) to afford 3a. We suggest an explanation of the formation of 4a based on the path a (Scheme 2, path b). (E)-Vinylpalladium intermediate 5 could react with acrolein to give (2-oxoalkyl)palladium intermediate 8, which underwent protonolysis to afford 4a. The crucial factors that decided which path to go were the acetylene pressure and the acrolein concentration. At a fixed acetylene pressure, a high acrolein concentration obviously increased the selectivity of the reaction.
| Entry | HOAc/H2O (mol mol−1) | Reaction time (h) | Temp. (°C) | Acetylene pressure (atm) | Yield (%) | |
|---|---|---|---|---|---|---|
| 3a | 4a | |||||
| a Pd(OAc)2 (1 mmol), LiBr (1 mol), and acrolein (1.2 mol). | ||||||
| 1 | 4.5/3 | 24 | 5 | 1 | 16 | 15 |
| 2 | 3/2 | 24 | 5 | 1 | 18 | 21 |
| 3 | 1.5/1 | 24 | 5 | 1 | 3.2 | 35 |
| 4 | 1.5/1 | 24 | 25 | 1 | 1.0 | 38 |
| 5 | 1.5/1 | 24 | 45 | 1 | 0.8 | 31 |
| 6 | 1.5/1 | 48 | 25 | 1 | 1.2 | 59 |
| 7 | 1.5/1 | 72 | 25 | 1 | 1.3 | 73 |
| 8 | 1.5/1 | 72 | 25 | 0.8 | 0.5 | 70 |
| 9 | 1.5/1 | 72 | 25 | 1.2 | 3.3 | 73 |
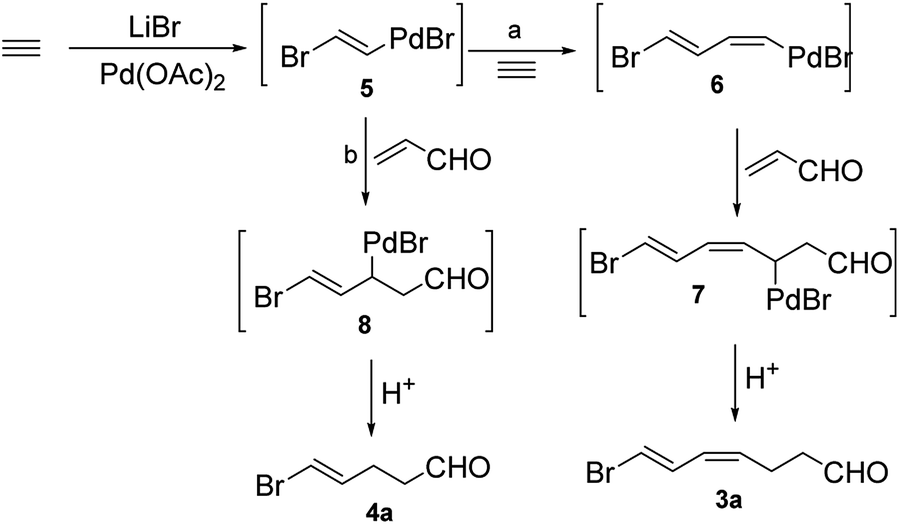 | ||
| Scheme 2 Possible mechanisms for Pd(II)-catalyzed coupling reaction of halide, acetylene, and acrolein. | ||
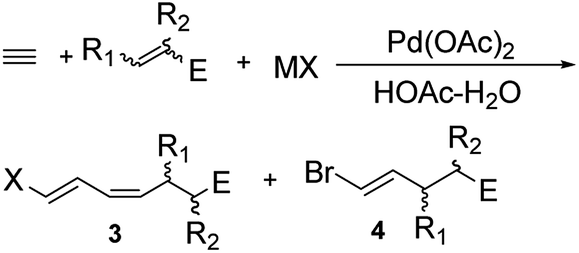
We studied further the effect of different counter ions and reactants on the products distribution (Table 2). Br− and Cl− ions allowed satisfying total conversion rate and selectivity (Table 2, entries 1–3). In contrast, I− ion resulted in poor total conversion rate and selectivity (Table 2, entry 4). The results indicated that a difference in the electron-deficiency of the alkenes could have a striking effect on the products distribution. The total conversion rate and the selectivity of the reaction were in agreement with the electrophilicity of the alkenes (enal or enone > acrolein dimethyl acetal > allyl bromide or allyl alcohol) (Table 2, entries 5–10). However, crotonaldehyde resulted in poor total conversion rate and selectivity, which probably resulted from the steric hindrance of the CH3 group. The reaction with acrolein dimethyl acetal afforded a mixture, because acrolein dimethyl acetal and the products 3g and 4g were partially hydrolyzed by acetic acid. The reaction with allyl bromide gave 3h and 4h after 12 h reaction, as did the corresponding reaction with allyl alcohol resulting in 3j and 4j, but these chemicals were completely converted into 3i and 4i after 24 h reaction. These results could be explained by the fact that both Br and OH groups were good leaving groups, supporting a β-heteroatom elimination.7
|
||||||
|---|---|---|---|---|---|---|
| Entry | MX | R1 | R2 | E | Product (yield, %) | |
| a Pd(OAc)2 (1 mmol), halides (1 mol), and electron-deficient alkenes (1.2 mol), HOAc (1.5 mol), H2O (1 mol), acetylene pressure (0.8 atm), 25 °C, 72 h. | ||||||
| 1 | n-Bu4NBr | H | H | CHO | 3a (2.6) | 4a (71) |
| 2 | n-Bu4NCl | H | H | CHO | 3b (2.8) | 4b (73) |
| 3 | LiCl | H | H | CHO | 3b (0.9) | 4b (69) |
| 4 | LiI | H | H | CHO | 3c (2.2) | 4c (25) |
| 5 | LiBr | H | H | COCH3 | 3d (2.3) | 4d (56) |
| 6 | LiBr | H | H | COC2H5 | 3e (2.4) | 4e (65) |
| 7 | LiBr | H | CH3 | CHO | 3f (1.1) | 4f (68) |
| 8 | LiBr | H | H | CH(OMe)2 | 3a (7.6) | 4a (21) |
| 3g (6.8) | 4g (19) | |||||
| 9 | LiBr | H | H | CH2Br | 3h (traces) | 4h (traces) |
| 3i (12) | 4i (9.5) | |||||
 |
 |
|||||
| 10 | LiBr | H | H | CH2OH | 3j (traces) | 4j (traces) |
| 3i (15) | 4i (12) | |||||
| 11 | LiBr | CH3 | H | CHO | 3k (8.3) | 4k (14) |
Both pheromone components of the cocoa pod borer moth were synthesized by an efficient and stereoselective route (Schemes 3 and 4). Preparation of the key intermediate 13 involved the following five steps. Hydration of 2,3-dihydrofuran in wet acid according to our improved method based on the literature8 to afforded 4-hydroxybutanal (9b) in 84% yield. Construction of the 10Z-double bond relied on Wittig reaction. To ensure the cis selectivity of the Wittig olefination, potassium bis(trimethylsilyl) amide was selected as the base. The ylide was prepared from n-hexyltriphenylphosphonium bromide by treatment with potassium bis(trimethylsilyl) amide in dry THF at −78 °C. Subsequently, the ylide reacted with aldehyde 9b to afford enol 10 in 78% yield and with 98% isomeric purity (GC).9 Enal 11 was prepared by oxidation of enol 10 using pyridinium chlorochromate in dichloromethane in 86% yield.9b The aldehydic carbonyl group was converted into the carbon–carbon triple bond via the standard Corey–Fuchs protocol. Enal 11 reacted with carbon tetrabromide and triphenylphosphine in dichloromethane to afford dibromoolefin 12 in 87% yield.10 Subsequently, dibromoolefin 12 was converted cleanly and rapidly into enyne 13 in 91% yield by treatment with 2 equivalents of n-butyllithium followed by aqueous work-up.10
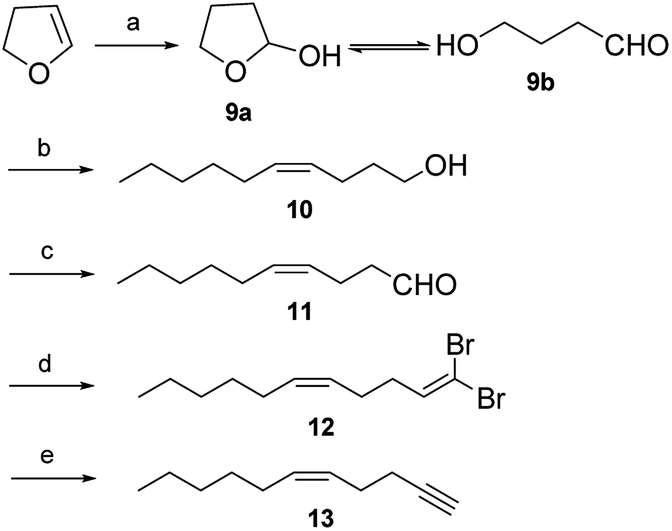 | ||
| Scheme 3 Synthetic route of key intermediate 13. Reagents and conditions: (a) HCl/H2O, rt, 5 h; (b) n-hexyltriphenylphosphonium bromide, KN[Si(Me)3]2, THF, −78 °C, 12 h; (c) PCC/Celite, CH2Cl2, rt, 5 h; (d) PPh3, CBr4, CH2Cl2, 0 °C, rt, 12 h; (e) n-BuLi, THF, −50 °C, 2 h. | ||
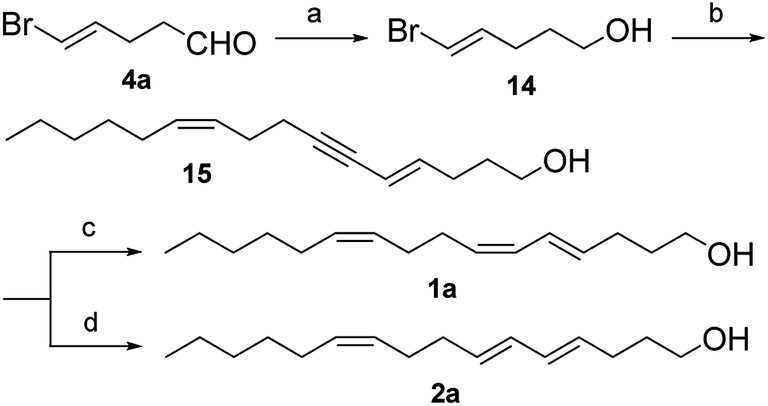 | ||
| Scheme 4 Synthetic route of the target compounds. Reagents and conditions: (a) NaBH4, CH3OH, 0 °C, 3 h; (b) alkyne 7, Pd(PPh3)4, CuI, Et2NH, rt, 12 h; (c) Zn, 1,2-dibromoethane, LiBr, CuBr, EtOH, reflux, 4 h; (d) LiAlH4, diglyme, reflux, 24 h. | ||
On the other hand, reduction of enal 4a to enol 14 was achieved in 86% yield by treatment with an excess of sodium borohydride in methanol, without affecting the double bonds. The Sonogashira coupling of enol 14 with enyne 13 using Pd(PPh3)4 as catalyst in dry diethylamine at room temperature gave enynol 15 in 83% yield.11 The Sonogashira coupling maintained the excellent isomeric purity owing to the moderate conditions used. Enynol 15 was hydrogenated using Zn to form trienol 1a in 82% yield and with a high isomeric purity (>98%, GC).12 The reduction of enynol 15 with LiAlH4 provided the corresponding trienol 2a in 74% yield and with a high isomeric purity (>98%, GC).13
Conclusions
In conclusion, preparation of (E)-5-bromopent-4-enal (4a) was optimized. (4E,6Z,10Z)-Hexadeca-4,6,10-trien-1-ol (1a) and (4E,6E,10Z)-hexadeca-4,6,10-trien-1-ol (2a) were prepared stereoselectively in 8 steps starting from commercially available materials with an overall yield of 30.4% and 27.4%, respectively. The stereoselective formation of (E,Z)- or (E,E)-conjugated double bond relied on the Sonogashira coupling with (E)-5-bromopent-4-en-1-ol (14) prepared from 4a and stereoselective hydrogenation of (Z)-undec-5-en-1-yne (13), while the 10Z-double bond was formed by the Wittig reaction of 4-hydroxybutanal and n-hexyltriphenylphosphonium bromide. (E)-5-Bromopent-4-enal (4a) may be used in biologically active natural products including insect pheromones.Experimental
General
All reagents used in reactions were obtained commercially in China. All anhydrous solvents were dried and purified by standard techniques just before use. All nonaqueous reactions were performed under an inert atmosphere (nitrogen) with rigid exclusion of moisture from reagents, and all reaction vessels were oven dried. 1H NMR spectra were recorded on Bruker-Ascend 400 spectrometer at 400 MHz. Chemical shifts are reported in parts per million (ppm) relative to TMS. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet), coupling constants, and integration. 13C NMR were recorded on Bruker-Ascend 400 spectrometer at 100 MHz. Chemical shifts are reported in parts per million measured relative to TMS. In preparation of (E)-5-bromopent-4-enal (4a), some reaction conditions may be different from those cited in the text (small scale) since they have been chosen for large scale reactions.(E)-5-Bromopent-4-enal (4a)
Acetylene (passed through concentrated sulfuric acid) was tardily passed through a mixture of Pd(OAc)2 (1122 mg, 5 mmol), LiBr (261 g, 3 mol), and acrolein (168 g, 3 mol) in HOAc (500 mL) and H2O (100 mL) at room temperature for 72 h. The reaction mixture was poured into a saturated aqueous solution of Na2CO3 and then extracted with CH2Cl2 (500 mL × 3). The extract was washed with water, dried over Na2SO4, and then concentrated by evaporation. The residue was purified through a silica gel column (eluent: 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether/ethyl acetate) to afford enal 4a as a colorless oil (337 g, 69%, isomeric purity > 99%, GC). 1H NMR (300 MHz, CDCl3) (δ, ppm): 9.76 (s, 1H, CHO), 6.15–6.12 (m, 2H, CH
:1 petroleum ether/ethyl acetate) to afford enal 4a as a colorless oil (337 g, 69%, isomeric purity > 99%, GC). 1H NMR (300 MHz, CDCl3) (δ, ppm): 9.76 (s, 1H, CHO), 6.15–6.12 (m, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 2.55–2.50 (m, 2H, CH2), 2.44 (m, 2H, CH2); 13C NMR (75 MHz, CDCl3) (δ, ppm): 202.1, 131.5, 130.8, 42.9, 21.3; EI-MS (m/z): 164/162 (M+), 121/119, 108/106, 95/93, 83, 65, 55.
CH), 2.55–2.50 (m, 2H, CH2), 2.44 (m, 2H, CH2); 13C NMR (75 MHz, CDCl3) (δ, ppm): 202.1, 131.5, 130.8, 42.9, 21.3; EI-MS (m/z): 164/162 (M+), 121/119, 108/106, 95/93, 83, 65, 55.
4-Hydroxybutanal (9b)
After adding concentrated hydrochloric acid (2.0 g, 20 mmol) and H2O (10.8 g, 0.6 mol) to CH2Cl2 (600 mL), 2,3-dihydrofuran (28 g, 0.4 mol) was added to the mixture. The resulting mixture was stirred for 5 h at room temperature, neutralized with Na2CO3, dried over Na2SO4, and then concentrated to afford 4-hydroxybutanal (9b) as a colorless oil (29.6 g, 84%). The product was directly used for the Wittig reaction without further purification. Tetrahydrofuran-2-ol (9a): 1H NMR (CDCl3, 400 MHz) (δ, ppm): 5.55–5.50 (m, 1H, CHOH), 3.95–3.81 (m, 2H, OCH2), 2.09–2.01 (m, 1H, CH2), 2.0–1.8 (m, 3H, CH2CH2); 13C NMR (CDCl3, 100 MHz) (δ, ppm): 100.3/98.6, 67.6/67.4, 33.5/32.6, 23.8. 4-Hydroxybutanal (9b): 1H NMR (CDCl3, 400 MHz) (δ, ppm): 9.75 (s, 1H, CHO), 3.70–3.64 (m, 2H, CH2OH), 2.51–2.48 (m, 2H, CH2CHO), 2.0–1.8 (m, 2H, CH2); 13C NMR (CDCl3, 100 MHz) (δ, ppm): 203.0, 66.4, 41.4, 23.0; EI-MS (m/z): 87.0 (M+ − 1), 71.1.(Z)-Dec-4-en-1-ol (10)
Potassium bis(trimethylsiyl)amide (300 mL, 0.3 mol, 1 M in THF) was added dropwise over 30 min to a suspension of n-hexylphosphonium bromide (128 g, 0.3 mol) in dry THF (600 mL) at 0 °C under nitrogen. The resulting orange solution was further stirred for 1 h at 0 °C and then cooled to −78 °C. A solution of aldehyde 9b (17.6 g, 0.2 mol) in dry THF (200 mL) was added dropwise to the above mixture over 1 h, maintaining the temperature below −70 °C. The resulting yellow solution was allowed to slowly warm up to room temperature over a period of 10 h. Then the reaction mixture was quenched with a saturated solution of NH4Cl. After the complete evaporation of THF, the reaction mixture was extracted with EtOAc (300 mL × 3). The extract was washed with H2O and brine, dried over Na2SO4, and then concentrated. The residue was purified through a silica gel column (eluent: 10:1 petroleum ether/ethyl acetate) to afford enol 10 as a colorless oil (24.3 g, 78%). 1H NMR (400 MHz, CDCl3) (δ, ppm): 5.45–5.37 (m, 2H, Z–CHCH), 3.67 (t, J = 6.42 Hz, 2H, CH2OH), 2.14 (m, 2H, CHCH2), 2.06 (m, 2H, CHCH2), 1.65 (m, 2H, CH2), 1.40–1.29 (m, 6H, –CH2–), 0.91 (t, 3H, J = 7.5 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 131.2, 129.2, 62.9, 33.0, 32.2, 30.1, 27.6, 24.0, 23.1, 14.5.
(Z)-Dec-4-enal (11)
Enol 10 (15.6 g, 0.1 mol) was stirred at room temperature for 5 h with a slurry of pyridinium chlorochromate (32.3 g, 0.15 mol) and Celite (32.3 g) in CH2Cl2 (200 mL). The mixture was then diluted with CH2Cl2 and filtered through Celite. The filter cake was washed with CH2Cl2 (200 mL × 2). The extract was washed with water, dried over Na2SO4, and then concentrated. The residue was purified through a silica-gel column (eluent: 25:1 petroleum ether/ethyl acetate) to afford enal 11 as a colorless oil (13.3 g, 86%). 1H NMR (400 MHz, CDCl3) (δ, ppm): 9.79, 5.46 (m, 1H, Z–CHCH), 5.35 (m, 1H, Z–CHCH), 2.51 (m, 2H, CH2CHO), 2.39 (m, 2H, CH2CH2CHO), 2.06 (m, 2H, CHCH2–), 1.40–1.29 (m, 6H, –CH2–), 0.91 (t, 3H, J = 7.5 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 202.7, 132.1, 127.4, 44.2, 31.9, 29.6, 27.6, 23.0, 20.5, 14.5.
(Z)-1,1-Dibromoundeca-1,5-diene (12)
A solution of enal 11 (10.8 g, 80 mmol) in CH2Cl2 (240 mL) was cooled to 0 °C and triphenylphosphine (63 g, 240 mmol) was added to the reaction mixture. A solution of carbon tetrabromide (39.8 g, 120 mmol) in CH2Cl2 (120 mL) was added dropwise, then the reaction mixture was allowed to warm up slowly to room temperature, and stirred overnight. After removal of CH2Cl2 by evaporation, petroleum ether (200 mL × 2) was added to precipitate triphenylphosphine oxide. The mixture was filtered and the solvent was removed. The residue was purified through a silica gel column (eluent: petroleum ether) to afford diene 12 (21.6 g, 87%) as a colorless oil. 1H NMR (400 MHz, CDCl3) (δ, ppm): 6.44 (t, J = 6.83 Hz, 1H, CHCBr2), 5.49 (m, 1H, Z–CHCH), 5.37 (m, 1H, Z–CHCH), 2.23–2.19 (m, 4H, CH–CH2CH2–CH), 2.06 (m, 2H, CHCH2–), 1.43–1.32 (m, 6H, –CH2–), 0.92 (t, 3H, J = 7.5 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 138.6, 132.1, 127.9, 89.32, 33.5, 31.9, 29.9, 27.6, 25.9, 23.0, 14.5.
(Z)-Undec-5-en-1-yne (13)
A solution of diene 12 (15.5 g, 50 mmol) in dry THF (200 mL) was cooled to −70 °C under nitrogen. After a solution of n-BuLi (44 mL, 110 mmol, 2.5 M solution in hexane) was slowly added to the reaction mixture, the mixture was stirred at −70 °C for 2 h and then allowed to warm up to room temperature. The reaction mixture was quenched with a saturated solution of NH4Cl, and the organic phase was washed with brine, dried over Na2SO4, and then concentrated. The residue was purified through a silica gel column (eluent: petroleum ether) to afford enyne 13 as a colorless oil (6.83 g, 91%). 1H NMR (400 MHz, CDCl3) (δ, ppm): 5.53–5.42 (m, 2H, Z–CHCH), 2.34–2.25 (m, 4H, CH–CH2CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ), 2.08 (m, 2H, CHCH2–), 2.00 (s, 1H, CH), 1.41–1.30 (m, 6H, –CH2–), 0.93 (t, 3H, J = 7.5 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 132.1, 127.8, 84.6, 68.7, 31.9, 29.8, 27.7, 26.8, 23.0, 19.3, 14.5.
), 2.08 (m, 2H, CHCH2–), 2.00 (s, 1H, CH), 1.41–1.30 (m, 6H, –CH2–), 0.93 (t, 3H, J = 7.5 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 132.1, 127.8, 84.6, 68.7, 31.9, 29.8, 27.7, 26.8, 23.0, 19.3, 14.5.
(E)-5-Bromopent-4-en-1-ol (14)
A solution of enal 4a (16.3 g, 100 mmol) and sodium borohydride (2.27 g, 60 mmol) in methanol (500 mL) was stirred for 3 h at 0 °C. The reaction mixture was quenched with diluted HCl followed by evaporation of methanol. The reaction mixture was then extracted with EtOAc (100 mL × 2). The extract was washed with water and brine, dried over Na2SO4, and then concentrated. The residue was purified through a silica-gel column (eluent: 8:1 petroleum ether/ethyl acetate) to afford enol 14 as a colorless oil (14.2 g, 86%). 1H NMR (400 MHz, CDCl3) (δ, ppm): 9.76 (s, 1H, CHO), 6.15–6.12 (m, 2H, CHCH), 2.55–2.50 (m, 2H, CH2), 2.44 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3) (δ, ppm): 202.1, 131.5, 130.8, 42.9, 21.3.
(4E,10Z)-Hexadeca-4,10-dien-6-yn-1-ol (15)
A solution enol 14 (3.3 g, 20 mmol) and enyne 13 (3.0 g, 20 mmol) in dry diethylamine (40 mL) was degassed with a stream of nitrogen for 20 min. After subsequent addition of Pd(PPh3)4 (1155 mg, 1 mmol) and CuI (209 mg, 1.1 mmol), the reaction mixture was degassed for an additional 10 min. The reaction mixture was stirred overnight and filtered through Celite. The filter cake was washed with CH2Cl2 (50 mL × 2). After concentration of the combined organic phase, the residue was purified through a silica-gel column (eluent: 10:1 petroleum ether/ethyl acetate) to afford enynol 15 as a colorless oil (3.89 g, 83%). 1H NMR (400 MHz, CDCl3) (δ, ppm): 6.07 (td, 1H, J = 7.05 Hz, J = 16.0 Hz, CHCH–CH2), 5.51 (d, 1H, J = 16.0 Hz, CHCH), 5.48–5.40 (m, 2H, Z–CHCH), 3.66 (t, 2H, J = 6.4 Hz), 2.34–2.27 (m, 4H, CH–CH2CH2–C), 2.22–2.17 (m, 2H, CH2), 2.08–2.03 (m, 2H, CH2), 1.70–1.61 (m, 2H, CH2), 1.40–1.31 (m, 6H, –CH2–), 0.91 (t, 3H, J = 7.4 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 142.8, 131.9, 128.1, 110.9, 89.0, 79.5, 62.5, 32.1, 31.9, 29.8, 29.6, 27.7, 27.1, 23.0, 20.2, 14.5.
(4E,6Z,10Z)-Hexadeca-4,6,10-trien-1-ol (1a)
A mixture of Zn powder (13.8 g, 200 mmol) and 1,2-dibromoethane (1.88 g, 10 mmol) in ethanol (20 mL) was heated until a vigorous reaction occurred. The mixture was again treated with 1,2-dibromoethane (1.88 g, 10 mmol) and refluxed for 10 min. The mixture was then cooled to 50 °C followed by addition of a mixture of LiBr (4.34 g, 50 mmol) and CuBr (2.87 g, 20 mmol) in THF (10 mL) within 3 min. After addition of a solution of enynol 15 (1172 mg, 5 mmol) in THF (10 mL), the reaction mixture was refluxed for 4 h and then quenched with a saturated solution of NH4Cl. The organic phase was washed with brine, dried over Na2SO4, and concentrated. The residue was purified through a silica-gel column (eluent: 8:1 petroleum ether/ethyl acetate) to afford trienol 1a as a colorless oil (981 mg, 82%). 1H NMR (400 MHz, CDCl3) (δ, ppm): 6.30 (ddd, 1H, J = 15.0, 11.0, 1.0 Hz, CHCH), 5.95 (dd, 1H, J = 11.0, 10.7 Hz, CHCH), 5.62 (dt, 1H, J = 15.0, 7.5 Hz, CHCH), 5.26–5.43 (m, 3H, CHCH), 3.61 (t, 2H, J = 6.5 Hz, CH2OH), 2.19 (m, 4H, CH–CH2CH2–C), 2.14 (m, 2H, CH2), 2.05 (m, 2H, CH2), 1.66 (m, 2H, CH2), 1.18–1.40 (m, 6H, –CH2–), 0.90 (t, 3H, J = 7.3 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 134.5, 131.5, 129.9, 129.2, 128.9, 126.4, 62.8, 32.5, 31.8, 29.7, 29.5, 27.9, 27.5, 27.2, 23.0, 14.4.
(4E,6E,10Z)-Hexadeca-4,6,10-trien-1-ol (2a)
Lithium tetrahydroaluminate (950 mg, 25 mmol) was added in portions to an ice-cooled mixture of dry diglyme (12 mL) and THF (1.5 mL) under nitrogen atmosphere. When the vigorous foaming had subsided, the solution of enynol 15 (1172 mg, 5 mmol) in diglyme (3 mL) was added to the thick slurry. Following the initial foaming, the reaction mixture was heated at 117–121 °C for 24 h. The mixture was cooled to 0 °C and petroleum ether (100 mL) was added followed by a cautious and dropwise addition of water (2 mL). Then, 20% (w/w) NaOH solution (1.6 mL) and H2O (8 mL) were added to the reaction mixture. The mixture was stirred for 15 min to allow the precipitate to granulate, then the petroleum ether layer was decanted, and the solid residue was rinsed with petroleum ether (50 mL × 3). The combined organic phases were washed with water and brine, dried over Na2SO4, and then concentrated. The residue was purified through a silica-gel column (eluent: 8:1 petroleum ether/ethyl acetate) to afford trienol 2a as a colorless oil (875 mg, 74%). 1H NMR (400 MHz, CDCl3) (δ, ppm): 5.98–6.07 (m, 2H, E–CHCH), 5.53–5.63 (m, 2H, E–CHCH), 5.45–5.37 (m, 2H, Z–CHCH), 3.60 (t, 2H, J = 6.5 Hz, CH2OH), 2.21 (m, 4H, CH–CH2CH2–C), 2.15 (m, 2H, CH2), 2.04 (m, 2H, CH2), 1.66 (m, 2H, CH2), 1.18–1.40 (m, 6H, –CH2–), 0.90 (t, 3H, J = 7.3 Hz, –CH3); 13C NMR (100 MHz, CDCl3) (δ, ppm): 132.3, 131.7, 131.5, 130.2, 129.3, 126.7, 62.7, 32.8, 32.1, 31.6, 29.6, 29.4, 27.7, 27.3, 23.0, 14.3.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21362018 and 81460525), the Yunnan Applied Basic Research Project (No. 2014FB117), and the Science and Technology Foundation of Yunnan University (No. 2011YB12).Notes and references
- P. S. Beevor, A. Cork, D. R. Hall, B. F. Nesbitt, R. K. Day and J. D. Mumford, J. Chem. Ecol., 1986, 12, 1 CrossRef CAS PubMed.
- P. A. C. Ooi, L. G. Chan, K. K. Chong, T. C. Hai and M. J. Mamat, Management of the Cocoa Pod Borer, The Malaysian Plant Protection Society, Kuala Lumpur, 1987, p. 4 Search PubMed.
- Y. Yen, S. Lin and M. Suen, Synth. Commun., 1992, 22, 1567 CrossRef CAS.
- A. R. Pereira and J. A. Cabezas, J. Org. Chem., 2005, 70, 2594 CrossRef CAS PubMed.
- (a) Y. H. Tao, W. X. Cheng, Y. S. Zhang and K. Gu, Chem. Res. Chin. Univ., 2005, 26, 1072 CAS; (b) Z. Ma, X. Yang, Y. Zhang, X. Huang and Y. Tao, Synlett, 2012, 23, 581 CrossRef CAS; (c) Y. Tao, X. Yang, Y. Jin and Q. Wang, Synth. Commun., 2013, 43, 415 CrossRef CAS.
- Z. Wang, X. Lu, A. Lei and Z. Zhang, J. Org. Chem., 1998, 63, 3806 CrossRef CAS.
- Z. Zhang, X. Lu, Z. Xu, Q. Zhang and X. Han, Organometallics, 2001, 20, 3724 CrossRef CAS.
- (a) K. Kojima, M. Kimura, S. Ueda and Y. Tamaru, Tetrahedron, 2006, 62, 7512 CrossRef CAS; (b) X. Yang, Y. Zhang, Y. Yao and Y. Tao, Chin. J. Org. Chem., 2014, 34, 1458 CrossRef CAS.
- (a) N. A. Bergman and M. Jansson, J. Org. Chem., 1987, 52, 4449 CrossRef CAS; (b) V. Ragoussis, S. Perdikaris, A. Karamolegkos and K. Magkiosi, J. Agric. Food Chem., 2008, 56, 11929 CrossRef CAS PubMed.
- M. A. Leeuwenburgh, R. E. J. N. Litjens, J. D. C. Codee, H. S. Overkleeft, G. A. V. Marel and J. H. V. Boom, Org. Lett., 2000, 2, 1275 CrossRef CAS PubMed.
- K. M. Brummond, P. C. Sill and H. Chen, Org. Lett., 2004, 6, 149 CrossRef CAS PubMed.
- (a) W. Boland, N. Schroer and C. Sieler, Helv. Chim. Acta, 1987, 70, 1025 CrossRef CAS; (b) J. Mulzer and M. Berger, J. Org. Chem., 2004, 69, 891 CrossRef CAS PubMed.
- J. Grodner, Tetrahedron, 2009, 65, 1648 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |