
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew triterpene saponins from the aerial parts of Androsace umbellata†
Zhi-Qi Yin *a,
Lei Wangab,
Cheng-Hua Lia,
Dong-Mei Zhangb,
Wei Zhangab,
Ying Wangb,
Ming Zhaoc,
Chun-Tao Chec and
Wen-Cai Yeb
*a,
Lei Wangab,
Cheng-Hua Lia,
Dong-Mei Zhangb,
Wei Zhangab,
Ying Wangb,
Ming Zhaoc,
Chun-Tao Chec and
Wen-Cai Yeb
aDepartment of Natural Medicinal Chemistry & State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, P.R. China. E-mail: cpu-yzq@cpu.edu.cn; Tel: +86-025-86185371
bInstitute of Traditional Chinese Medicine and Natural Products, College of Pharmacy, Jinan University, Guangzhou 510632, P.R. China
cDepartment of Medicinal Chemistry and Pharmacognosy, WHO Collaborating Center for Tradition Medicine, College of Pharmacy, University of Illinois at Chicago, Chicago, IL 60612, USA
First published on 15th May 2017
Abstract
Six new oleanane-type triterpene saponins, androsides A–F (1–6), along with two known compounds (7–8), were isolated from the aerial parts of Androsace umbellata. The structures were determined on the basis of spectroscopic data and acid hydrolysis. The aglycones in 1–5 are novel structures. Cytotoxicity assays using HepG2, HepG2/ADM, MCF-7, MCF-7/ADR and MDA-MB-231 cell lines indicated that 7 and 8, both bearing a 13β,28-epoxy group, were active. Compound 8 was shown to induce apoptosis in the HepG2/ADM cells.
Introduction
Plants of the genus Androsace (Primulaceae) include 120 species, of which about 73 species are distributed mainly in China.1 Pharmacological studies have demonstrated that Androsace plants possess anti-tumor, anti-inflammatory and anti-virus properties,2 and phytochemical investigations revealed triterpene saponins and flavonoids as major ingredients.3 Androsace umbellata is distributed in Asia, especially China, Japan and India. The whole plant of A. umbellata has been used in Chinese folk medicine for the treatment of laryngopharyngitis, tonsillitis and hydropsia.4 Recently, triterpene saponins isolated from A. umbellata were found to show cytotoxic activity against human tumor cell lines.5 Saxifragifolin D could inhibit breast cancer cell growth and induce interplay between apoptosis and autophagy.6In the search for bioactive constituents from A. umbellata, we have reported several triterpene and phenolic glycosides from this plant.7,8 Continuing phytochemical studies now led to the isolation of six new triterpenesaponins, androsides A–F (1–6) (Fig. 1), along with two known saponins (7–8).
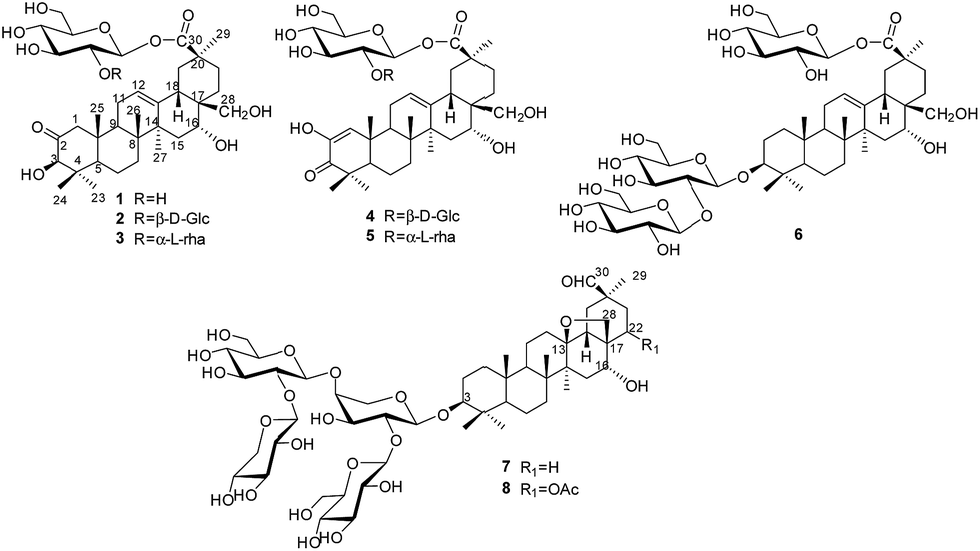 | ||
| Fig. 1 Chemical structures of 1–8. | ||
Results and discussion
Structural elucidation
An ethanol extract of the aerial parts of A. umbellata was subjected to repeated column chromatography over silica gel, ODS silica gel and Sephadex LH-20 to afford six new triterpene saponins, androsides A–F (1–6), along with saxifragifolin B (7) and saxifragifolin A (8).9Androside A (1) was obtained as an amorphous powder. Positive results from both Liebermann–Burchard and Molisch reactions indicated a triterpene glycoside structure. The molecular formula of 1 was determined to be C36H56O11 by the quasi-molecular ion [M + Na]+at m/z 687.3709 (calcd for C36H56O11Na: 687.3714) in HR-ESI-MS. The IR spectrum showed characteristic absorptions for hydroxyl (3442 cm−1), carboxyl (1726 cm−1) and olefinic bonds (1646 cm−1). Acid hydrolysis of 1 afforded D-glucose, which were detected by derivatization and HPLC analysis.
The 1H NMR spectrum of 1 (Table 1) displayed six tertiary methyl signals at δH 0.85, 0.85, 0.86, 1.29, 1.38, and 1.80 (each 3H, s), an olefinic proton at δH 5.62 (1H, br s), as well as an anomeric protons at δH 6.46 (1H, d, J = 8.0 Hz). The 13C NMR and DEPT spectra (Table 1) exhibited 36 carbon signals, 30 of which could be attributed to the aglycone. The 13C NMR resonances at δC 122.5 and 144.7 further suggested the presence of a Δ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12,13 double bond on an oleanane skeleton.10 When the 13C NMR spectrum of 1 was compared with that of ardisicrenoside C,11 signals for carbons of rings B–E were almost identical (Table 1). On the other hand, a ketone carbon signal resonating at δC 211.1 was observed, which was assigned to C-2 by the HMBC correlations between δC 211.1 and δH 2.23 and 2.48 (H-1), as well as between δC 211.1 and δH 4.19 (H-3) (Fig. 2). Thus, the aglycone of 1 was determined to be 2-oxo-3β,16α, 28-trihydroxy-olean-12-en-30-oic acid, which was further confirmed by comparison of the NMR data for ring A of 1 with those of dillenic acid B.10
12,13 double bond on an oleanane skeleton.10 When the 13C NMR spectrum of 1 was compared with that of ardisicrenoside C,11 signals for carbons of rings B–E were almost identical (Table 1). On the other hand, a ketone carbon signal resonating at δC 211.1 was observed, which was assigned to C-2 by the HMBC correlations between δC 211.1 and δH 2.23 and 2.48 (H-1), as well as between δC 211.1 and δH 4.19 (H-3) (Fig. 2). Thus, the aglycone of 1 was determined to be 2-oxo-3β,16α, 28-trihydroxy-olean-12-en-30-oic acid, which was further confirmed by comparison of the NMR data for ring A of 1 with those of dillenic acid B.10
| Position | 1 | 2 | 4 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a 1H NMR Spectrum was measured at 500 MHz; 13C NMR Spectrum was measured at 125 MHz.b Overlapping signals were reported without designating multiplicity. | ||||||
| 1 | 2.23, br d (12.0) 2.48, m | 53.8, CH2 | 2.21, d (12.5) 2.47, m | 53.8, CH2 | 6.52, s | 129.5, CH |
| 2 | — | 211.1, C | — | 211.1, C | — | 146.3, C |
| 3 | 4.19, s | 83.3, CH | 4.17, s | 83.3, CH | — | 201.1, C |
| 4 | — | 45.7, C | — | 45.7, C | — | 44.6, C |
| 5 | 1.55, br d (10.8) | 54.8, CH | 1.56, br d (11.0) | 54.8, CH | 1.65, br d (9.2) | 54.1, CH |
| 6 | 1.36.1.54, m | 18.9, CH2 | 1.36, 1.56, m | 18.9, CH2 | — | 19.1, CH2 |
| 7 | 1.35, 1.68, m | 32.8, CH2 | 1.34, 1.69, m | 32.8, CH2 | 1.34, 1.68, m | 33.0, CH2 |
| 8 | — | 40.3, C | — | 40.3, C | — | 40.6, C |
| 9 | 2.02, m | 46.7, CH | 2.02, m | 46.7, CH | 2.04, m | 42.8, CH |
| 10 | — | 43.2, C | — | 43.6, C | — | 38.4, C |
| 11 | 1.70, 2.01, m | 23.6, CH2 | 1.76, m | 23.7, CH2 | 1.90, 2.05, m | 23.8, CH2 |
| 12 | 5.62, s | 122.5, CH | 5.79, br s | 122.4, CH | 5.74, br s | 122.6, CH |
| 13 | — | 144.7, C | — | 144.8, C | — | 145.0, C |
| 14 | — | 41.9, C | — | 42.0, C | — | 42.2, C |
| 15 | 1.63, m 2.20, br d (13.4) | 34.9, CH2 | 1.63, m 2.19, dd (6.8.10.8) | 34.8, CH2 | 1.60, m 2.21, br d (11.1) | 34.8, CH2 |
| 16 | 4.66, s | 74.0, CH | 4.72, br s | 74.0, CH | 4.72, br s | 74.1, CH |
| 17 | — | 40.4, C | — | 40.4, C | — | 40.6, C |
| 18 | 2.78, m | 43.4, CH | 2.65, m | 43.4, CH | 2.86, dd (10.8, 11.6) | 43.8, CH |
| 19 | 2.35, br d (12.7) 2.85, d (12.2) | 44.7, CH2 | 2.41, m 2.88, t (13.0) | 44.2, CH2 | 2.42, 2.88, m | 44.1, CH2 |
| 20 | — | 44.6, C | — | 44.6, C | — | 44.6, C |
| 21 | 2.48, m | 33.6, CH2 | 2.53, m | 33.4, CH2 | 2.50, m | 33.9, CH2 |
| 22 | 2.35, br d (12.7) 2.57, m | 31.8, CH2 | 2.46, m | 31.8, CH2 | 2.43, m | 31.8, CH2 |
| 23 | 1.29, s | 29.4, CH3 | 1.28, s | 29.4, CH3 | 1.19, s | 27.8, CH3 |
| 24 | 0.85, s | 17.3, CH3 | 0.84, s | 17.2, CH3 | 1.11, s | 20.2, CH3 |
| 25 | 0.85, s | 16.6, CH3 | 0.85, s | 16.6, CH3 | 1.10, s | 22.1, CH3 |
| 26 | 0.86, s | 16.5, CH3 | 0.85, s | 16.5, CH3 | 0.92, s | 17.4, CH3 |
| 27 | 1.80, s | 27.3, CH3 | 1.80, s | 27.2, CH3 | 1.76, s | 27.1, CH3 |
| 28 | 3.65, dd (8.2, 2.0) | 70.0, CH2 | 3.52, 3.68, dd (10.8) | 70.1, CH2 | 3.53, 3.70, dd (9.5) | 70.1, CH2 |
| 29 | 1.38, s | 28.6, CH3 | 1.48 s | 28.5, CH3 | 1.49, s | 28.4, CH3 |
| 30 | — | 177.1, C | — | 177.1, C | — | 177.2, C |
| Glc1′ | 6.46, d (8.0) | 95.9, CH | 6.38, d (8.0) | 93.7, CH | 6.39, d (8.0) | 93.7, CH |
| 2′ | 4.21, t (8.1) | 74.5, CH | 4.46, d (8.7) | 81.3, CH | 4.46, t (8.5) | 81.3, CH |
| 3′ | 4.28, t (8.1) | 78.6, CH | 4.20, t (8.9) | 78.5, CH | 4.20, t (8.9) | 79.7, CH |
| 4′ | 4.30, br d (9.1) | 71.3, CH | 4.29, m | 71.0, CH | 4.29, t (9.0) | 71.0, CH |
| 5′ | 4.01, m | 79.3, CH | 3.94, m | 79.0, CH | 3.94, m | 79.0, CH |
| 6′ | 4.34, dd (4.5, 11.6) 4.42, d (9.8) | 62.4, CH2 | 4.30, dd (5.3, 9.5) 4.41, dd (4.5.11.6) | 62.3, CH2 | 4.30, br d (9.0) 4.41, br d (10.8) | 62.3, CH2 |
| Glc1′′ | 5.53 d (8.0) | 105.6, CH | 5.55 d (8.0) | 105.6, CH | ||
| 2′′ | 4.73, br s | 76.4, CH | 4.72, br s | 76.4, CH | ||
| 3′′ | 4.33, t (8.7) | 78.2, CH | 4.33, t (9.0) | 78.2, CH | ||
| 4′′ | 4.32, t (8.7) | 71.4, CH | 4.32, t (9.0) | 71.4, CH | ||
| 5′′ | 3.89, m | 78.4, CH | 3.89, m | 78.4, CH | ||
| 6′′ | 4.30, 4.41, m | 62.6, CH2 | 4.30, 4.41, m | 62.6, CH2 | ||
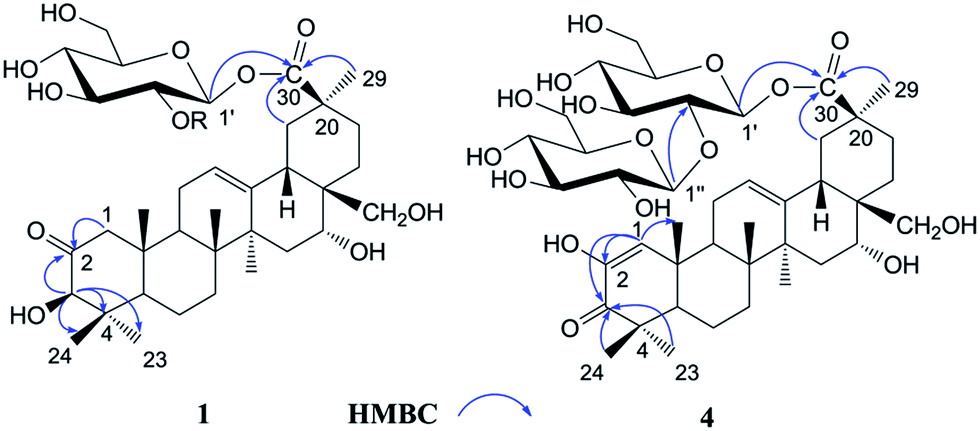 | ||
| Fig. 2 Selected key HMBC correlations of 1 and 4. | ||
For the glycone part, the anomeric carbon signal at δC 95.9 showed correlation with proton at δH 6.46 (d, J = 8.0 Hz) in the HSQC spectrum. Detailed analysis of the HSQC, HMBC and ROESY data of 1 led to the assignment of a glucopyranosyl unit (Table 1). The β-configuration of the anomeric proton was confirmed by the large 3JH1-H2 coupling constant. The linkage position of the sugar unit was determined with the aid of HMBC data (Fig. 2), in which correlation between H-1 of glucose (δH 6.46) and C-30 of the aglycone (δC 177.1) was clearly observed. In conclusion, all available evidence suggested the structure of 2-oxo-3β, 16α, 28-trihydroxy-olean-12-en-30-oic acid 30-O-β-D-glucopyranosyl ester for 1. It is noteworthy that the aglycone of 1 is reported for the first time.
Androside B (2) was isolated as an amorphous powder. The high resolution ESI-MS spectrum showed a quasi-molecular ion at m/z 849.4259 [M + H]+ (calcd for C42H67O16: 849.4243), consistent with the molecular formula C42H66O16. The 1H NMR spectrum of 2 (Table 1) displayed six tertiary methyl signals at δH 0.84, 0.85, 0.85, 1.28, 1.48, and 1.80 (each s), an olefinic proton at δH 5.79 (br s), as well as two anomeric protons at 6.38 (d, J = 8.0 Hz) and 5.53 (d, J = 8.0 Hz). The 13C NMR data (Table 1) were similar to those of 1 except for the appearance of an additional glucopyranosyl unit (δC 105.6, 76.4, 78.2, 71.4, 78.4, 62.6). Acid hydrolysis of 2 afforded D-glucose only, which was identified by derivatization and HPLC analysis. Comparison of the NMR data of 2 with those of 1 revealed the downfield shift (+6.8 ppm) for C-2′ of glucose, suggesting an additional glucopyranosyl residue at C-2′. This conclusion was supported by the HMBC correlation between H-1′′ (δH 5.53) of the terminal glucose and C-2′ (δC 81.3) of the inner glucose. Based on the above evidence, 2 was determined to be 2-oxo-3β,16α,28-thihydroxy-olean-12-en-30-oic acid 30-O-β-D-glucopyranosyl(1→2)-glucopyranosyl ester.
The molecular formula of androside C (3) was determined to be C42H66O15 by HR-ESI-MS (833.4288 [M + Na]+; calcd for C42H66O15Na: 833.4293). The 1H and 13C NMR spectral features of the aglycone (Table 2) were similar to those of 2, suggesting the same aglycone structure. For the glycone part of 3, the presence of two sugar units was implied by the observation of two anomeric proton signals at δH 6.32 (d, J = 8.0 Hz) and 6.56 (br s), as well as two anomeric carbon signals at δC 101.5 and 94.5. Indeed, acid hydrolysis of 3 yielded L-rhamnose and D-glucose. The anomeric configurations of D-glucose and L-rhamnose were determined to be β and α, respectively, based on the coupling constants of the anomeric protons and 13C NMR data.12
| Position | 3 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a 1H NMR Spectrum was measured at 500 MHz; 13C NMR Spectrum was measured at 125 MHz.b Overlapping signals were reported without designating multiplicity. | ||||||
| 1 | 2.17, 2.46, m | 53.8, CH2 | 6.54, s | 129.5 d | 0.88, 1.41, m | 38.9, CH2 |
| 2 | — | 211.1, C | — | 146.3, C | 2.18, 1.78, m | 26.6, CH2 |
| 3 | 4.18, s | 83.3, CH | — | 201.1, C | 3.27, dd (8.0, 2.0) | 88.9, CH |
| 4 | — | 45.7, C | — | 44.6, C | — | 39.5, C |
| 5 | 1.56, m | 54.8, CH | 1.65, m | 54.1, CH | 0.71, d (11.7) | 55.8, CH |
| 6 | — | 18.9, CH2 | — | 19.1, CH2 | 1.42, 1.29, m | 18.4, CH2 |
| 7 | 1.34, 1.68, m | 32.8, CH2 | 1.34, 1.68, m | 33.0, CH2 | 1.56, 1.27, m | 33.2, CH2 |
| 8 | — | 40.3, C | — | 40.6, C | — | 40.0, C |
| 9 | 2.02, t (9.8) | 46.8, CH | 2.04, m | 42.8, CH | 1.66, t (8.9) | 47.0, CH |
| 10 | — | 43.4, C | — | 38.4, C | — | 36.8, C |
| 11 | 1.76, m | 23.7, CH2 | 1.9, 2.05, m | 23.8, CH2 | 1.78, m | 23.7, CH2 |
| 12 | 5.79, br s | 122.4, CH | 5.84, br s | 122.6, CH | 5.64, br s | 123.1, CH |
| 13 | — | 144.8, C | — | 145.0, C | — | 144.7, C |
| 14 | — | 41.9, C | — | 42.2, C | — | 41.7, C |
| 15 | 1.63, br d (14.2) 2.18, br d (12.2) | 34.9, CH2 | 1.60, 2.18, m | 34.8, CH2 | 1.61, 2.19, | 34.9, CH2 |
| 16 | 4.62, br s | 74.0, CH | 4.62, br s | 74.1, CH | 4.66, br s | 74.2, CH |
| 17 | 40.5, C | — | 40.6, C | — | 40.4, C | |
| 18 | 2.88, m | 42.8, CH | 2.88, m | 43.0, CH | 2.74, m | 43.2, CH |
| 19 | 2.49, 2.91, m | 44.1, CH2 | 2.45, 2.91, m | 44.0 t | 2.34, 2.78, m | 44.7, CH2 |
| 20 | — | 44.7, C | — | 44.6, C | — | 44.7, C |
| 21 | 2.50, m | 33.9, CH2 | 2.50, m | 33.9, CH2 | 2.49, 1.58, m | 33.6, CH2 |
| 22 | 2.35, 2.43, m | 31.8, CH2 | 2.35, 2.43, m | 31.8, CH2 | 2.58, 2.34, m | 31.8, CH2 |
| 23 | 1.29, s | 29.4, CH3 | 1.20, s | 27.8, CH3 | 1.25, s | 28.1, CH3 |
| 24 | 0.84, s | 17.3, CH3 | 1.10, s | 20.2, CH3 | 1.09, s | 16.9, CH3 |
| 25 | 0.85, s | 16.6, CH3 | 1.12, s | 22.1, CH3 | 0.82, s | 15.7, CH3 |
| 26 | 0.86, s | 16.5, CH3 | 0.93, s | 17.4, CH3 | 0.87, s | 16.8, CH3 |
| 27 | 1.81, s | 27.3, CH3 | 1.77, s | 27.1, CH3 | 1.81, s | 27.4, CH3 |
| 28 | 3.62, dd (8.2, 2.0) | 69.8, CH2 | 3.62, dd (8.2, 2.0) | 70.1, CH2 | 3.65, dd (8.2, 2.0) | 70.0, CH2 |
| 29 | 1.54, s | 28.4, CH3 | 1.54, s | 28.4, CH3 | 1.37, s | 28.6, CH3 |
| 30 | — | 177.3, C | — | 177.2, C | — | 177.1, C |
| Glc1′ | 6.32, d (8.0) | 94.5, CH | 6.34, d (8.0) | 94.5, CH | 4.88, d (7.5) | 105.0, CH |
| 2′ | 4.51, t (8.8) | 76.1, CH | 4.53, m | 76.2, CH | 4.21, dd (2.8.8.8) | 83.4, CH |
| 3′ | 4.32, t (9.0) | 78.6, CH | 4.33, m | 79.7, CH | 4.25, dd (4.0.9.0) | 77.9, CH |
| 4′ | 4.23, m | 71.7, CH | 4.24, m | 71.8, CH | 4.11, t (9.3) | 71.7, CH |
| 5′ | 3.96, m | 79.7, CH | 3.97, m | 78.6, CH | 3.88, m | 78.3, CH |
| 6′ | 4.26, m 4.36, dd (2.5.11.8) | 62.4, CH2 | 4.26, br d (9.4) 4.36, br d (11.4) | 62.6, CH2 | 4.42, br d (10.0) 4.51, dd (2.3.11.7) | 62.7, CH2 |
| Glc1′′ | 5.34, d (7.5) | 106.0, CH | ||||
| 2′′ | 4.09, t (9.0) | 77.1, CH | ||||
| 3′′ | 4.21, dd (2.8.8.8) | 78.2, CH | ||||
| 4′′ | 4.11, t (9.3) | 71.6, CH | ||||
| 5′′ | 3.88, m | 77.9, CH | ||||
| 6′′ | 4.42, br d (10.0) 4.45, dd (3.2.11.5) | 62.8, CH2 | ||||
| Rha 1′′ | 6.56, s | 101.5, CH | 6.56, br s | 101.5, CH | ||
| 2′′ | 4.73, m | 72.5, CH | 4.74, m | 72.5, CH | ||
| 3′′ | 4.52, t (8.8) | 72.5, CH | 4.53, m | 72.5, CH | ||
| 4′′ | 4.28, m | 74.1, CH | 4.28, br d (7.9) | 73.9, CH | ||
| 5′′ | 4.66, dd (6.2, 9.4) | 70.1, CH | 4.66, dd (6.2, 9.9) | 69.8, CH | ||
| 6′′ | 1.73, d (6.1) | 18.8, CH3 | 1.73, d (6.1) | 18.8, CH3 | ||
| Glc 1′′′ | 6.45, d (8.0) | 95.9, CH | ||||
| 2′′′ | 4.21, dd (2.8.8.8) | 74.4, CH | ||||
| 3′′′ | 4.29, t (7.5) | 78.6, CH | ||||
| 4′′′ | 4.30, t (6.9) | 71.2, CH | ||||
| 5′′′ | 3.99, m | 79.3, CH | ||||
| 6′′′ | 4.33, dd (4.8.11.8) 4.42, br d (10.0) | 62.4, CH2 | ||||
The sequence and linkage of the sugars were then determined by 2D NMR analysis. Thus, HMBC cross peaks between H-1′′ (δH 6.56) of rhamnose and C-2′ (δC 76.1) of glucose, and between H-1′ of glucose (δH 6.32) and C-30 of the aglycone (δC 177.3), were clearly observed. The above findings led to the assignment of 3 as 2-oxo-3β, 16α,28-thihydroxy-olean-12-en-30-oic acid 30-O-α-L-rhamnopyrano syl(1→2)-β-D-glucopyranosyl ester.
Androside D (4) was shown to possess a molecular formula C42H64O16 from its HR-ESI-MS data, m/z 847.4088 [M + Na]+ (calcd for C42H64O16Na: 847.4092). The IR spectrum displayed characteristic absorptions of hydroxyl (3424 cm−1), carboxyl (1728 cm−1) and olefinic bonds (1642 cm−1). In the UV spectrum, an absorption maximum at 270 nm indicated the presence of an α, β-unsaturated ketone. The 1H NMR spectrum of 4 (Table 1) displayed signals for six tertiary methyl groups [δH 0.92, 1.10, 1.11, 1.19, 1.49 and 1.76 (each s)], two olefinic protons [δH 6.52 (s) and 5.74 (br s)], and two anomeric protons [δH 6.39 (d, J = 8.0 Hz) and 5.55 (d, J = 8.0 Hz)]. In the 13C NMR and DEPT spectra (Table 1), 42 carbon signals (Table 1) were observed, of which 30 signals could be assigned to the aglycone. Comparison of the NMR data of 4 with those of 2 revealed the signals were almost identical, except for differences in ring A. The HMBC correlations between C-3 (δC 201.1) and H-1 (δH 6.52)/H-23 (δH 1.19)/H-24 (δH 1.11) led to the assignment of a carbonyl at the C-3 position (Fig. 2). Furthermore, HMBC cross peaks between H-1 (δH 6.52) and C-2 (δC 146.3)/C-5 (δC 54.1)/C-9 (δC 42.8)/C-25 (δC 22.1) revealed a double bond between C-1 and C-2, as well as a hydroxyl group at C-2 (Fig. 2). These conclusions could be further confirmed by the ROESY correlations between H-1(δH 6.52) and H-9 (δH 2.04), and between H-1 and H-25 (δH 1.10). Therefore, the aglycone of 4 was identified to be 3-oxo-2, 16α, 28-trihydroxy-olean-1, 12-dien-30-oic acid.
For the glycone part, D-glucose was identified by acid hydrolysis and HPLC analysis. The NMR data for the sugar chain of 4 were identical to those of 2, indicating the presence of 30-O-β-D-glucopyranosyl(1→2)-glucopyranosyl ester, which could be further verified by an HMBC experiment (Fig. 2). Thus, HMBC correlations between H-1 (δH 6.52) of the outer glucose and C-2 (δC 81.3) of the inner glucose, as well as between H-1 (δH 6.39) of the inner glucose and C-30 (δC 177.2) of aglycone, were observed. Based on the above evidence, the structure of 4 was established to be 3-oxo-2,16α,28-trihydroxy-olean-1,12-dien-30-oic acid 30-O-β-D-glucopyranosyl(1→2)-glucopyranosyl ester.
The molecular formula of androside E (5) was assigned as C42H64O15 on the basis of its HR-ESI-MS data (m/z 831.4143 [M + Na]+; calcd for C42H64O15Na: 831.4137). The 1H and 13C NMR spectra (Table 2) indicated that 5 was also a triterpene saponin bearing two sugar units. The NMR data of 5 were similar to those of 4, except the signals of glucopyranosyl in 4 (δC 105.6, 76.4, 78.2, 71.4, 78.4 and 62.5) were now replaced by those of a rhamnopyranosyl unit (101.5, 72.5, 72.5, 73.9, 69.8 and 18.8). Acid hydrolysis of 5 yielded D-glucose and L-rhamnose. The chemical shifts and J values of the anomeric protons indicated the β-configuration of glucopyranosyl and α-configuration of rhamnopyranosyl units. In the HMBC spectrum, cross-peaks were observed between H-1 (δH 6.58) of rhamnose and C-2 (δC 76.2) of glucose, as well as between H-1 (δH 6.34) of glucose and C-30 (δC 177.2) of aglycone. Thus, the structure of 5 was determined to be 3-oxo-2, 16α,28-trihydroxy-olean-1,12-dien-30-oic acid 30-O-α-L-rhamnopy ranosyl(1→2)-β-D-glucopyranosyl ester. It is noteworthy that the new aglycone structure in 4 and 5 is reported for the first time.
The molecular formula of androside F (6) was determined to be C48H78O20 by its HR-ESI-MS spectrum (m/z 1009.4775 [M + Cl]−; calcd for C48H78O20Cl, 1009.4775). The 1H and 13C NMR spectroscopic data of the aglycone (Table 2) were in good agreement with those of ardisicrenoside C11 The β-configuration of 3-OH and α-configuration of 16-OH were deduced by the coupling constants of H-3 (dd, J = 8.0, 2.0 Hz) and H-16 (br s), respectively. Thus, the aglycone of 6 was determined to be jacquinic acid (3β, 16α, 28-trihydroxy-olean-12-en-30-oic acid). The NMR data for the sugar moiety and GC analysis of the derivatives of its acid hydrolysateled to the assignment of three β-D-glucopyranose units. Connectivity of the sugars was determined on the basis of HMBC data. Thus, correlations between H-1 of the inner glucose [δH 4.88 (1H, d, J = 7.5 Hz)] and C-3 of the aglycone (δC 88.9), between H-1 of the outer glucose [δH 5.34 (1H, d, J = 7.5 Hz)] and C-2 of the inner glucose (δC 83.4), as well as between H-1 of the third glucose [δH 6.45 (1H, d, J = 8.0 Hz)] and C-30 (δC 177.1) of the aglycone, were observed. These data revealed the structure of sugar chain as shown in Fig. 1, which was further confirmed by comparison of the NMR data with those of asteryunnanoside I.13 Consequently, the structure of 6 was elucidated to be 3-O-β-D-glucopyranosyl(1→2)-β-D-glucopyranosyl 3β,16α,28-trihydroxy-olean-12-en-30-oic acid 30-O-β-D-glucopyranosyl ester.
Cytotoxicity
The cytotoxicity of 1–8 was tested in human hepatoma carcinoma cell lines (HepG2 and HepG2/ADM) and human breast cancer cell lines (MCF-7, MCF-7/ADR and MDA-MB-231) by MTT assay. As shown in Table 3, among the tested compounds, only 7 and 8 bearing a 13β,28-epoxy group exhibited inhibitory activity in these cancer cell lines, with IC50 values in the range of 36.19–51.86 μM and 9.29–17.71 μM, respectively. It was observed that the drug resistant cancer cell line HepG2/ADM was more sensitive to compound 8 than its parental cell line HepG2, while the sensitivities of MCF-7, MCF-7/ADR and MDA-MB-231 towards compound 8 were almost equivalent. Compound 7 exhibited similar cytotoxic activities in all tested cell lines.| Compounds | IC50 (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ± SD) μMb ± SD) μMb |
||||
|---|---|---|---|---|---|
| HepG2 | HepG2/ADM | MCF-7 | MCF-7/ADR | MDA-MB-231 | |
| a Cytotoxic activities of compounds 1–8 were tested by using MTT assay. All data are presented as means ± standard deviation of at least three independent experiments.b IC50: concentration of the tested compound inhibiting 50% cell growth.c DOX: doxorubicin was used as positive control. | |||||
| DOXc | 0.18 ± 0.03 | 143.62 ± 5.12 | 0.85 ± 0.24 | 37.86 ± 5.56 | 21.13 ± 0.15 |
| 1 | >200 | >200 | >200 | >200 | >200 |
| 2 | >200 | >200 | >200 | >200 | >200 |
| 3 | >200 | >200 | >200 | >200 | >200 |
| 4 | >200 | >200 | >200 | >200 | >200 |
| 5 | >200 | >200 | >200 | >200 | >200 |
| 6 | >200 | >200 | >200 | >200 | >200 |
| 7 | 40.34 ± 2.51 | 36.19 ± 2.57 | 42.57 ± 3.75 | 43.86 ± 2.50 | 51.86 ± 5.86 |
| 8 | 17.71 ± 0.62 | 9.97 ± 0.46 | 10.32 ± 0.13 | 10.52 ± 1.13 | 9.29 ± 0.32 |
In the presence of compound 8, an increase in sub-G1 DNA content was observed in HepG2/ADM cells after 72 h treatment in a dose-dependent manner (Fig. 3A). Apoptosis was analyzed by Annexin V-FITC/PI double staining assay and the population of apoptotic cells was quantified. As shown in Fig. 3B, the population of early and late apoptotic cells was increased from 19.1% to 44.5% in a dose-dependent manner. Compound 8 also induced PARP cleavage (Fig. 3C), a marker of apoptosis.14 Taken together, the findings suggested that compound 8 induced apoptosis in HepG2/ADM cells.
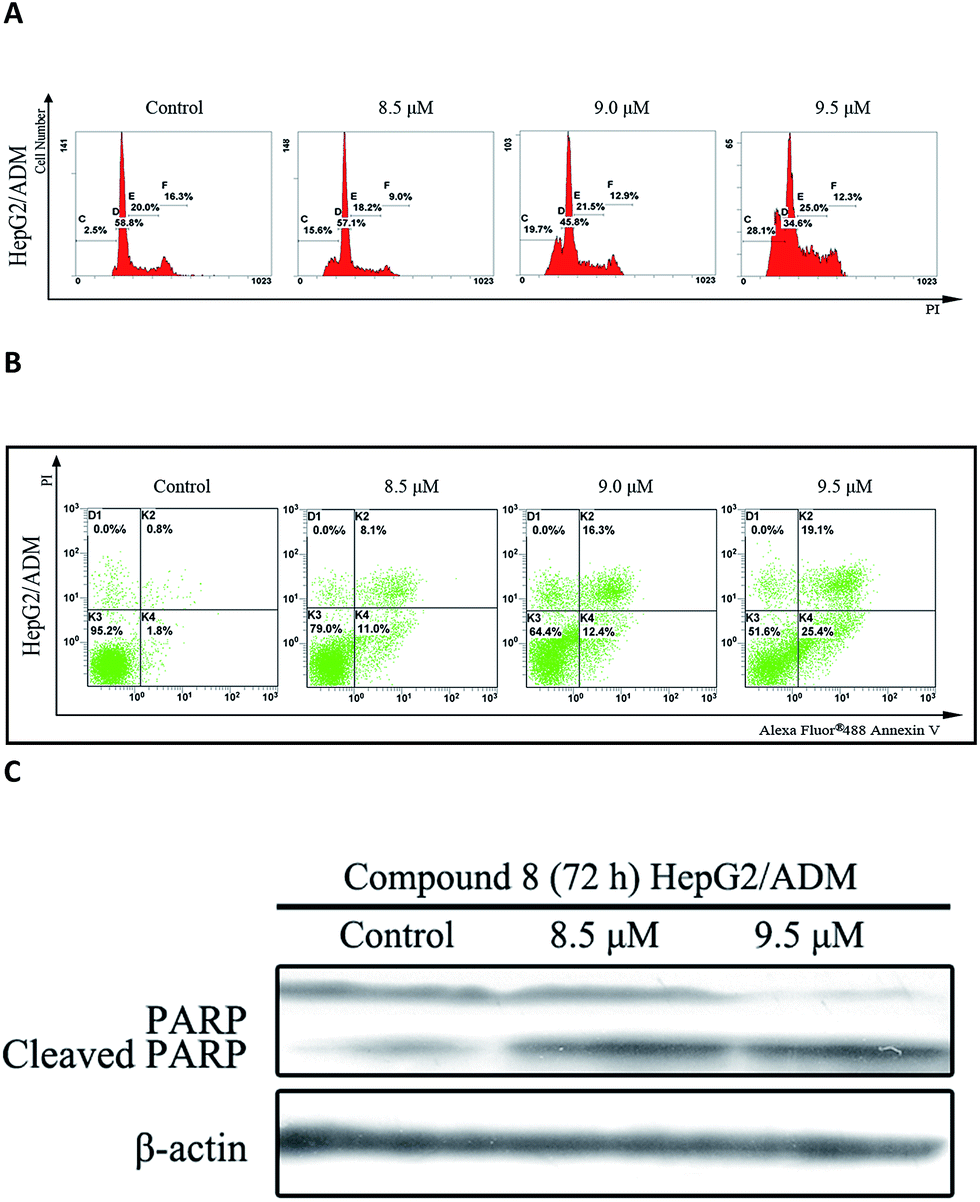 | ||
| Fig. 3 Apoptosis induced by 8 in HepG2/ADM cells. (A) Flow cytometric analysis of DNA content of HepG2/ADM cells treated with 8 (8.5 μM, 9.0 μM and 9.5 μM) for 72 h. (B) Flow cytometry analysis of apoptotic rate of HepG2/ADM cells induced by 8 (8.5 μM, 9.0 μM and 9.5 μM) for 72 h. (C) Western blot analysis of cleaved PARP in HepG2/ADM cells after treatment with 8 (8.5 μM and 9.5 μM) for 72 h. β-actin served as a loading control. | ||
Experimental section
General experimental procedures
Melting points were measured on an X-4 micro melting point apparatus (without correction). Optical rotations were obtained by a Jasco P-1020 digital polarimeter in a 0.1 dm length cell. IR spectra were determined on a Nicolet Impact 410 plus infrared spectrometer with KBr disc. 1D and 2D NMR experiments were performed on a Bruker AV-500 spectrometer using pyridine-d5 as solvent with tetramethylsilane (TMS) as internal reference. ESI-MS data were carried out on a HP-1100 LC/EST mass spectrometer. HR-ESI-MS data were measured on an Agilent 6210 ESI/TOF mass spectrometer. TLC was performed on precoated silica gel GF254 (Yantai Chemical Industry Research Institute, P. R. China) and precoated RP-18 F254 S plates (Merck). Silica gel (200–300 mesh; Qingdao Marine Chemical Factory, P. R. China), octadecylsilanized silica gel (ODS, YMC Co. Ltd.) and Sephadex LH-20 (Pharmacia Biotec AB) were used for column chromatographies. D-Glucose, L-glucose, D-rhamnose, and L-rhamnose were obtained from Sigma-Aldrich (USA).Plant material
The fresh aerial parts of Androsace umbellata (Lour.) Merr. were collected in Nanjing city, Jiangsu Province of P. R. China, in March of 2004, and were authenticated by Prof. Min-Jian Qin (China Pharmaceutical University). A voucher specimen (no. 20040316) was deposited in the herbarium of China Pharmaceutical University, Nanjing, P. R. China.Extraction and isolation
The fresh aerial parts of A. umbellata (2.5 kg) were pulverized and extracted with 70% EtOH three times (each 2 h) under reflux. The extract was concentrated under vacuum and suspended in H2O, then successively extracted with petroleum ether, EtOAc and n-BuOH. The n-BuOH extract (100 g) was separated by silica gel column chromatography eluted with gradient CHCl3–CH3OH (85:15 → 1:1, v/v) to give six fractions (A–E). Fraction A (10 g) was chromatographied on silica gel eluting with CHCl3–CH3OH (95:5 → 85:15, v/v) and purified by ODS column chromatography [CH3OH–H2O (40:60 → 75:25, v/v)] to afford 1 (22 mg), 2 (12 mg) and 4 (13 mg). Fraction B (5 g) was subjected to silica gel eluting with CHCl3–CH3OH (95:5 → 80:20, v/v), then separated by ODS column chromatography [CH3OH–H2O (45:55 → 80:20, v/v)] to obtain 3 (16 mg) and 5 (10 mg). Fraction C (3.5 g) was subjected to ODS column chromatography [CH3OH–H2O (45:55 → 80:20, v/v)], and purified by Sephadex LH-20 (CH3OH) to yield 6 (21 mg). Fraction D (15 g) was separated by silica gel and eluted with CHCl3–CH3OH (90:10 → 1:1, v/v) to give five subfractions (D1–D5). Compounds 7 (23 mg) and 8 (48 mg) were obtained from subfraction D3 by ODS column chromatography [CH3OH–H2O (35:65 → 77:25, v/v)].
Characterization of new compounds
HPLC analysis for sugar residues
Each compound (2 mg) was dissolved in 4 mol L−1 HCl (10 mL) and heated at 90 °C in water bath for 6 h. After reaction product was dissolved in H2O. The mixture was extracted with EtOAc for three times. The aqueous layer containing sugars was concentrated to dryness, mixed with L-cysteine methyl ester hydrochloride, and heated at 60 °C in an oven in the presence of anhydrous pyridine (1 mL) for 1 h. Isothiocyanate (2 mg) was then added to the mixture and heated at 60 °C for another hour. Each reaction mixture was analyzed by HPLC under the following conditions: an Agilent 1200 chromatograph equipped with a Cosmosil 5C18-MS-II column (4.6 × 250 mm i.d., Nacalai Tesque Inc.); mobile phase: isocratic elution of 25% CH3CN–H2O in 50 mmol L−1 HCl; flow rate: 0.8 mL min−1; injection volume: 10 μL; column temperature: 35 °C; UV detection wavelength: 250 nm. The standard L-rhamnose, D-rhamnose, D-glucose, L-glucose, D-xylose, L-xylose, L-arabinose and D-arabinose were run under the same conditions. Comparison of the retention time of the monosaccharide derivatives led to the determination of D-glucose (19.2 min) and L-rhamnose (32.6 min).Cell cytotoxicity assay
:1) and tested by Epics XL flow cytometry (Beckman Coulter, Brea, CA, USA). Data were analyzed quantitatively with an EXPO32 ADC software (Beckman Coulter, Brea, CA, USA).200 × g at 4 °C for 15 min, the supernatant was gathered as total protein lysates and stored at −80 °C until use. Protein concentration was tested by the BCA protein assay kit. Electrophoresis and immunoblotting analysis was carried out as previously described.6Conclusions
In this work, six new oleanane-type triterpene saponins and two known compounds were isolated from the aerial parts of Androsace umbellata. The structures were determined on the basis of spectroscopic data and acid hydrolysis. All new compounds showed no obvious cytotoxicity against the test cells. However, cytotoxicity assays indicated that 7 and 8, both bearing a 13β,28-epoxy group, were active. Compound 8 was shown to induce apoptosis in the HepG2/ADM cells. These results provide a basis for evaluating the structure–activity relationships of other oleanane-type triterpene saponins, as well as for developing the compound 8 as potential anti-hepatoma drug.Acknowledgements
The authors thank Prof. Min-Jian Qin (China Pharmaceutical University) for authenticating the plant material and Prof. Wen-Bin Shen (China Pharmaceutical University) for NMR measurements. This work was partially supported by the National Natural Science Foundation of China (No. 81001379), and sponsored by the Ninth Batch of “Six Talent Peaks” Project of Jiangsu Province (No. 2012-YY-008), and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- J. Y. Wang, X. J. Li, G. Hao and J. Q. Liu, Acta Phytotaxon. Sin., 2014, 42, 481–499 Search PubMed.
- R. X. He, G. F. Wei, H. Yao, Y. Chang, L. Xu and C. L. Zhang, Chin. J. Exp. Tradit. Med. Formulae, 2012, 18, 296–299 Search PubMed.
- J. Lei, Y. C. Xiao, J. Huang, M. Tang and P. C. Deng, Helv. Chim. Acta, 2009, 92, 1439–1444 CrossRef CAS.
- L. R. Song, Chinese Bencao, Shaihai Science and Technology Press, Shanghai, 1998, vol. 6, pp. 87–89 Search PubMed.
- J. H. Park, J. H. Kwak, J. H. Khoo, S. H. Park, D. U. Kim, D. M. Ha, S. U. Choi, S. C. Kang and O. P. Zee, Arch. Pharmacal Res., 2010, 33, 1175–1180 CrossRef CAS PubMed.
- J. M. Shi, L. L. Bai, D. M. Zhang, A. Yiu, Z. Q. Yin, W. L. Han, J. S. Liu, Y. Li, D. Y. Fu and W. C. Ye, Biochem. Pharmacol., 2013, 85, 913–926 CrossRef CAS PubMed.
- Z. Q. Yin, C. H. Li, Y. Wang, W. C. Ye and J. Zhang, Chin. Chem. Lett., 2009, 20, 836–838 CrossRef CAS.
- Y. Wang, D. M. Zhang, W. C. Ye, Z. Q. Yin, K. P. Fung, S. X. Zhao and X. S. Yao, Planta Med., 2008, 74, 1280–1284 CrossRef CAS PubMed.
- J. P. Waltho, D. H. Williams and S. B. Mahato, J. Chem. Soc., Perkin Trans. 1, 1986, 1527–1531 RSC.
- N. Andre, D. W. Anthony, S. Otto and R. Topul, J. Nat. Prod., 1994, 57, 1245–1250 CrossRef.
- K. Koike, Z. H. Jia, S. Ohura and S. Mochida, Chem. Pharm. Bull., 1999, 47, 434–435 CrossRef CAS PubMed.
- A. Yokosuka, T. Sano, K. Hashimoto, H. Sakagami and Y. Mimaki, Chem. Pharm. Bull., 2009, 57, 1425–1430 CrossRef CAS PubMed.
- Y. Shao, B. N. Zhou and J. H. Gao, Phytochemistry, 1995, 38, 675–680 CrossRef CAS PubMed.
- D. M. Zhang, J. S. Liu, M. K. Tang, A. Yiu, H. H. Cao, L. Jiang, J. Y. Chan, H. Y. Tian, K. P. Fung and W. C. Ye, Eur. J. Pharmacol., 2012, 692, 19–28 CrossRef CAS PubMed.
- J. S. Liu, D. M. Zhang, Y. Li, W. M Chen, Z. X. Ruan, L. J. Deng, L. W. Wang, H. Y. Tian, A. Yiu, C. L. Fan, H. Luo, S. W. Liu, Y. Wang, G. K. Xiao, L. X. Chen and W. C. Ye, J. Med. Chem., 2013, 56, 5734–5743 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and other relevant information for compounds 1–8. See DOI: 10.1039/c7ra03948d |
| This journal is © The Royal Society of Chemistry 2017 |