
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of protein imprinted polymers via protein-catalyzed eATRP on 3D gold nanodendrites and their application in biosensors†
Yue Sun *,
Jiameng Zhang,
Juan Li,
Mengyuan Zhao and
Yutong Liu
*,
Jiameng Zhang,
Juan Li,
Mengyuan Zhao and
Yutong Liu
School of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian 116029, China. E-mail: yuesun@lnnu.edu.cn
First published on 30th May 2017
Abstract
Sensitive detection of metalloproteins is very essential in human pathologies. Hemoglobin (Hb) was represented as a metal-containing protein to prepare protein imprinted polymers (PIPs) and used for electrochemical biosensors. The PIPs were synthesized via electrochemically mediated atom transfer radical polymerization (eATRP) with protein as, both, catalyst and template on the surface of 3D Au nanodendrites. The nanodendrites modified with PIPs could determine Hb by differential pulse voltammetry (DPV) method. A linear response from 1.0 × 10−13 to 1.0 × 10−1 mg mL−1 with a detection limit of 3.2 × 10−14 mg mL−1 (S/N = 3) for Hb was achieved from the results of the experiments. Compared with other Hb sensors based on PIPs, the broader linear range and lower detection limit suggested the promising prospect of the biosensor. In a word, this study significantly reports the preparation of PIPs, catalysis of eATRP by proteins and the sensitive detection of metalloproteins.
1. Introduction
The metalloproteins, such as Fe-, Cu- and Ni-containing proteins, play an important role as cofactors in biological systems and are relevant in several pathophysiological processes in carcinogenesis or neurodegenerative diseases (such as Alzheimer's, Parkinson's and leukemia).1–3 Sensitive detection of metalloproteins is very essential in human pathologies. Various mass spectrometries and/or immunoassays are the common methods for detecting metalloproteins.4,5 However, mass spectrometry analyses are usually complicated and time-consuming and require expensive experimental equipment; at the same time, the antibody of the immunoassay is difficult to purify and not easy to maintain the activity.6 Therefore, it is necessary to explore a simple method for the detection of metalloproteins without an antibody.Protein imprinted polymers (PIPs) are synthetic antibody mimics capable of specific molecular recognition for the template protein. Compared with the biological antibodies, PIPs are more stable, easier to process and less expensive;7–10 thus they have versatile applications in biosensing,11 proteomics,12 enrichment of protein13 and so on. Most imprinted polymers are currently prepared using radical polymerization.14 Among these, “1iving”/controlled atom transfer radical polymerization (ATRP), which obtains narrow polydispersity index, architectures, functionalities and well-defined composition,15–18 can proceed in aqueous solution at ambient temperature, and thus is very suitable for imprinting proteins.19,20 Unfortunately, the catalysts of ATRP are usually low-valent transition metal complexes, which are poisonous, expensive, sensitive to air and often leave residue in the end polymers (hard to get rid of).21 As a result, several improved ATRPs have been developed to overcome these drawbacks.
Nowadays, the improved ATRP can be broadly divided into two types. One is changing the implementation of ATRP to reduce the amount of catalysts, such as reverse ATRP (RATRP), activators generated by electron transfer ATRP (AGET ATRP), and electrochemically mediated ATRP (eATRP). Among these, eATRP was one of the most powerful and versatile methods. As the low-valent catalysts of ATRP are generated through electrochemical reduction of high-valent transition metal, the catalyst concentration can be decreased to only several parts per million.22 The other type is replacing transition metal complexes with green and nontoxic materials. For example, Bruns et al. discovered that some metalloproteins, such as horseradish peroxidase (HRP), laccase and hemoglobin (Hb), are able to catalyze the ATRP of acrylate and acrylamide monomers using sodium ascorbate as reducing agents.23–25
Our group combined the advantages of the two types and developed the protein-catalyzed eATRP, in which polymerization was conducted by metals in proteins (Hb–Fe(III) to Hb–Fe(II)) after the electrochemical reduction.11 The protein-catalyzed eATRP needed neither transition metal complexes nor reducing agents, which eliminated all the negative effects of transition metal complexes and reducing agents on the polymers and showed broad prospect in functional biomaterial preparation particularly for PIPs. In spite of this, the reports about protein-catalyzed eATRP are very limited, and there were some problems when protein-catalyzed eATRP was used for PIPs. (1) The reduction of metal in protein was weak on the cathode of eATRP. The cathode was the core of the eATRP process, where the reduction place of high-valent metal was. The cathodes that had been reported include Pt, gold, iron, and nickel-chromium.26,27 These electrodes could act as a good electron carrier for reduction of high-valent transition metal complexes. Nevertheless, direct electron transfer of most redox proteins (including metalloproteins) on them was a great challenge due to the deeply buried redox-active center (such as the metal) inside the proteins.28 (2) The substrates reported for eATRP are usually planar; the imprinting receptors of PIPs on the plane surface are very limited, which hampers the further application of PIPs.
To solve those problems, in this study, Hb was used as both the protein catalyst and the template for PIPs because of its catalytic performance in ATRP and the important roles in clinical detection. The poly(toluidine blue) (PTB) modified glassy carbon electrode (GCE) coated with nano-Pt (GCE/PTB/nPt) was selected from different materials to be the cathode for eATRP. Gold nanodendrites (ND) were used as the substrate of PIPs to increase the number of imprinting receptors because they had hyperbranched three-dimensional (3D) architecture, substantially enlarging the available surface area of the electrode.29,30 The ND modified with PIPs could be used as the electrochemical biosensor to determine Hb by differential pulse voltammetry (DPV) method.
2. Materials and methods
2.1. Chemicals
Pt electrode (Φ = 2 mm), GCE (Φ = 3 mm) and Au electrode (Φ = 2 mm) were purchased from Chenhua Instruments Co. (Shanghai, China). Cu and Zn disk electrodes (Φ = 3 mm) were supplied from Gauss Union Instruments Co. (Wuhan, China). Graphite electrode (GE) was purchased from JiXing ShengAn Company (Beijing, China). Acrylamide (AM, functional monomer), N,N′-methylene bis-acrylamide (MBA, cross-linker), toluidine blue (TB), ammonium persulfate, sodium dodecylsulfate (SDS) and cupric sulfate (CuSO4·5H2O) were obtained from Kemiou Chemical Co. (Tianjin, China). Chloroauric acid (HAuCl4·4H2O) and chloroplatinic acid (HPtCl6·6H2O) were purchased from Beijing Chemical Plant (Beijing, China). Furthermore, 2-bromoisobutyryl bromide (BiBB, 98%) was from Alfa Aesar Chemical Co. and 4-hydroxythiophenol (HTP) was obtained from Acros Co. (Japan). Lysozyme (Lyz, MW 14.4 kDa), bovine serum albumin (BSA, MW 66 kDa), human serum albumin (HSA, MW 69 kDa) and Hb (MW 65 kDa) were supplied by Solarbio Inc. (Beijing, China). Bovine blood sample was obtained from Wafangdian farm in Dalian City, China. Phosphate buffered solution (PBS, pH 7.0) was prepared using 0.02 mol L−1 Na2HPO4 and 0.02 mol L−1 KH2PO4, and 0.02 mol L−1 acetate buffer solution (ABS, pH 5.0) was prepared using acetic acid and sodium acetate.All chemicals were of analytical reagent grade. All the water used in experiments was the hyperpure water (resistivity > 18 MΩ cm).
2.2. Apparatus
Cyclic voltammograms (CV), eATRP and DPV were carried out with a CHI660D electrochemical workstation (Chenhua, Shanghai, China).The electroreduction activities of different electrodes toward Hb were studied by CV. The available electrodes were used as the working electrode, and saturated calomel electrode (SCE) and platinum wire were used as the reference and the counter electrodes, respectively. CVs were recorded in the potential range between −0.8 and 0.2 V in PBS containing 10 mg mL−1 Hb or not. The scan rate was 50 mV s−1.
The CV characterization was carried out in a three-electrode system with Au or modified Au as the working electrode; SCE and platinum wires were used as the reference and the counter electrodes, respectively. CV characterization was performed in PBS containing 5 mmol L−1 Fe(CN)63−/4− and 0.1 mol L−1 KCl at a scan rate of 100 mV s−1. A sample interval of 0.001 V, quiet time of 2 s and a sensitivity of 1 × 10−4 A/V were set as the corresponding CV parameters.
The polymerization time was studied by CV, which was performed in the same condition with CV characterization.
The surface of the modified electrode was characterized by scanning electron microscopy (SEM) (SU8010, Hitachi, Japan) and energy-dispersive X-ray spectroscopy (EDX) (SU8010, Hitachi, Japan).
The surface chemical composition of the polymer prepared via eATRP was determined by X-ray photoelectron spectroscopy (XPS) using a Thermo ESCALAB 250Xi spectrometer with a monochromatic Al Kα radiation.
2.3. Preparation of GCE/PTB/nPt
GCE/PTB/nPt was prepared by a two-step process. First, the electropolymerization of TB on GCE (GCE/TB) was performed by CV. The clean GCE was used as the working electrode; SCE and platinum wires were used as the reference and counter electrodes, respectively. CVs were recorded in the potential range from −0.8 to 1.3 V for 50 scan segments at a scan rate of 50 mV s−1 in 10 mL ABS containing 5 × 10−5 mol L−1 TB. The other parameters were the same as those for CV characterization. Second, the nano-Pt was electrodeposited on GCE/TB by chronoamperometry at −0.7 V (vs. SCE) for 100 s in 0.01 mol L−1 HPtCl6·6H2O (in ABS) with a sample interval of 0.1 s and a sensitivity of 1 × 10−3 A/V.2.4. Preparation of 3D Au ND
The 3D Au NDs were fabricated by electrodeposition, which was performed by chronoamperometry at −0.9 V (vs. SCE) on the cleaned Au electrode in an aqueous electrolyte containing CuSO4·5H2O (0.02 mol L−1) and HAuCl4·4H2O (0.8 mol L−1). The electrodeposition process was maintained for 400 s at room temperature with a sample interval of 0.1 s and a sensitivity of 1 × 10−3 A/V. Then, the electrode modified with deposits was immersed in HNO3 solution (3 mol L−1) to remove the Cu layer. The Au electrode modified with 3D Au ND was obtained (Au/ND).2.5. Preparation of PIPs
The PIPs were fabricated and are schemed in Fig. 1. In order to find a suitable material for the electrochemical reduction of Hb, the cathode of eATRP and substrate of PIPs were achieved by two different electrodes. As shown in Fig. 1, 3D gold ND was first prepared on the clean Au electrode (Au/ND). Then, the thiol initiator was immobilized on the surface of Au/ND. The thiol initiator was prepared and used according to our previously published report.31 After the Au/ND, GCE/PTB/nPt, SCE and platinum wires were inserted into PBS containing Hb (2.0 mg mL−1), monomer (AM, 0.1 mol L−1) and cross-linker (MBA, 0.1 mol L−1); eATRP reaction could be carried out in PBS using chronoamperometry. For a typical eATRP, the GCE/PTB/nPt was used as working electrode (cathode), and SCE and platinum wires were used as the reference and counter electrodes, respectively. The Au/ND should be located as close as possible to the GCE/PTB/nPT. When a proper potential (Eapp) was applied to GCE/PTB/nPt, Hb–Fe(III) would be reduced to Hb–Fe(II) (catalyst for ATRP) and trigger the polymerization on the surface of the Au/ND.23 Except the Eapp, the other parameters included a sample interval of 0.1 s and a sensitivity of 1 × 10−4 A/V. | ||
| Fig. 1 Synthesis scheme of PIPs on 3D Au ND via eATRP with GCE/PTB/nPt as cathode. | ||
Finally, Hb was removed from the polymer by soaking it in a 10% (v/v) acetic acid solution containing 10% (w/v) SDS for 2 h. After being rinsed with PBS three times, the Au/ND modified with PIPs (Au/ND/PIPs) was prepared. The non-imprinted polymers (NIPs) were prepared on the surface of the Au/ND electrode (Au/ND/NIPs) by the traditional free radical polymerization method. In a typical procedure, ammonium persulfate (0.9 mol L−1) was used as initiator to polymerize AM (0.1 mol L−1) and MBA (0.1 mol L−1) at room temperature for 5 h.
2.6. Determination of Hb by DPV
The performance of the Au/ND/PIPs was studied by the DPV method. Prior to each test, the Au/ND/PIPs electrode was incubated in Hb solution for 5 min at room temperature and washed three times with PBS. The DPV was operated from −0.1 to 0.4 V in PBS containing 5 mmol L−1 Fe(CN)63−/4−, against the SCE reference electrode. An increment potential of 4 mV, amplitude of 50 mV, a pulse width of 0.2 s and a pulse period of 0.5 s were set as the corresponding DPV parameters.The signal response of Au/ND/PIPs toward the Hb was expressed as ΔI, where ΔI was the current reduction in response to the additional amount of the Hb. For example, when the Au/ND/PIPs electrode was prepared and incubated in the blank solution, its peak current of DPV was I0; after the electrode was incubated in Hb solution, its peak current of DPV was I1, ΔI = I − I1.
3. Results and discussion
3.1. Preparation of PIPs on 3D gold ND
The reduction of Fe(III) in Hb was the precondition and key factor to the successful implementation of eATRP. In order to select the proper cathode, the typical materials including noble metal (Pt and Au), base metal (Ni and Zn), nonmetal (graphite and GCE) and modified nonmetal (GCE/PTB and GCE/PTB/nPt) were used to study their electroreduction toward Hb by cyclic voltammetry (CV). As shown in Fig. 2, the CV of Pt (Fig. 2A) had no reduction peak in the Hb solution, indicating that Pt was not suitable for Hb catalyzed eATRP. The CV of Au (Fig. 2A) exhibited an irreversible reduction peak at approximately −0.409 V (Epc, vs. SCE) with the peak current of 1.808 μA. The peak position was consistent with the characteristics of electroactive center of heme Fe(III)/Fe(II) couples in the Hb molecules,32,33 suggesting the good electrochemical reduction of Au toward Hb. Moreover, neither Ni nor Zn, had a reduction peak in CV (Fig. 2B). The graphite electrode (GE) had no electrochemical reduction toward Hb, too, whereas GCE had a reduction peak at −0.406 V (Epc, vs. SCE) with the peak current of 1.819 μA (Fig. 2C). In order to further improve the electrochemical reduction activity, GCE was modified with PTB (GCE/PTB) or PTB coated with nano-Pt (GCE/PTB/nPt). The CV of GCE/PTB had no reduction peak (Fig. 2D). The CV of GCE/PTB/nPt (Fig. 2D) showed an irreversible reduction peak at −0.388 V (Epc, vs. SCE), which was a little more positive than that of Au and GCE. The peak current of GCE/PTB/nPt in Hb was 9.711 μA, which was much higher than that of Au and GCE. The more positive Epc and higher peak current indicated that the Fe(III) center of Hb was more easily reduced on GCE/PTB/nPt. The reason for this may be that nPt played the role of an efficient electron-conducting tunnel and had a high ratio of surface to volume. Consequently, Hb attached to the nPt surface had more spatial freedom in its orientation, which made it much easier for the electroactive center of Hb to unfold.34,35 From Fig. 2, we can observe that GCE/PTB/nPt had the best electroreduction activity toward Hb, and thus was used as the cathode for eATRP. | ||
| Fig. 2 The CV curves of typical noble metal (A), base metal (B), nonmetal (C) and modified nonmetal (D) materials in Hb solution (10 mg mL−1 Hb in PBS, pH 7.0) or blank (PBS, pH 7.0) between −0.8 and 0.2 V. The scan rate was 50 mV s−1. | ||
The surface morphology of the GCE/PTB/nPt was observed with scanning electron microscopy (SEM), and the results are shown in Fig. S1(A).† Spherical structure of nano-Pt with a diameter of ∼30 nm was visualized. Furthermore, energy-dispersive X-ray (EDX) spectroscopy results of GCE/PTB/nPt showed the significant presence of C and Pt, as presented in Fig. S1(B).† The good electroreduction of GCE/PTB/nPt may be due to the small size, high surface area and good biocompatibility of nPt.28,36
According to the literature,37 the applied potential (Eapp) of eATRP was determined by the equation Eapp = Epc − 120 mV; hence, a potential of −0.51 V was applied to conduct the polymerization. The polymerization time was examined by CV, which indicated the electron transfer ability of the polymer on the Au/ND electrode. Fig. S2(A)† shows the variation between anodic peak current of CV curves and the polymerization time. As can be seen, peak current decreased sharply with the time increasing from 0.17 to 3 h. When the polymerization time increased further, the peak current of the CV curves changed very slowly, and thus polymerization time was selected to be 3 h (as indicated by the arrow). The Hb concentration was selected by measuring the sensitivity (slope of calibration curve) of the Au/ND/PIPs electrode. As shown in Fig. S2(B),† increasing the concentration of Hb from 0.5 to 2.0 mg mL−1 improved the sensitivity of the Au/ND/PIPs electrode. However, too much of Hb (2.0 to 10.0 mg mL−1) caused the sensitivity to decrease. This may be because when there were too many proteins, they would possibly aggregate and result in nonideal imprinted cavities.38 From the results of Fig. S2(B),† Hb concentration for eATRP was selected to be 2.0 mg mL−1.
3.2. Characterization of PIPs
The modified electrodes were investigated by CV and are shown in Fig. 3. As can be seen, the bare Au electrode showed the characteristic quasi-reversible redox cycle. When the electrode was modified with gold ND (Au/ND), an evident increase in the anodic and cathodic peak current was obtained, showing that the ND had good electron transfer ability. After the polymer formed on the ND (Au/ND/polymer), a decrease in peak current was observed. This may be attributed to the polymer film, which acted as the inert electron and mass transfer blocking layer to hinder the diffusion of the electrochemical probe toward the electrode surface.39 After the Hb was removed from the polymer (Au/ND/PIPs), the current response of the CV increased, which suggested that the imprinted cavities were formed in the PIPs and left channels for the penetration of the probe to reach the electrode.40 | ||
| Fig. 3 The CV characterization of the stepwise modified electrodes (1, bare Au; 2, Au/ND; 3, Au/ND/polymer; 4, Au/ND/PIPs) in PBS (pH 7.0) containing 5 mmol L−1 Fe(CN)63−/4− and 0.1 mol L−1 KCl. The scan rate of CV was 100 mV s−1. | ||
XPS was used to investigate the chemical composition of Au/ND/polymer (Fig. 4). The figure shows the characteristic peaks of O 1s, N 1s, C 1s, Au 4f and Br 3d at 531.3, 399.6, 285.3, 84.2 and 70.1 eV, respectively.41,42 The active Br atoms were exposed at the surface, implying the successful implementation of ATRP.15,41,42 The inset of Fig. 4 showed the fine XPS of Fe 2p at 721.1 and 710.7 eV, corresponding to its spin doublet; because Fe was from Hb, the spectra of Fe thus indicated the existence of Hb in Au/ND/polymer.43
 | ||
| Fig. 4 The XPS spectra of Au/ND/polymer. | ||
The surface morphology of the modified electrodes was observed with SEM (Fig. 5). As can be seen, Au/ND (Fig. 5A), Au/ND/polymer (Fig. 5B) and Au/ND/PIPs (Fig. 5C) had similar dendritic structures at low magnification (top row in Fig. 5). When they were at higher magnification (bottom row in Fig. 5), the difference of surface morphology was clearly visualized. A long main trunk with short side branches appeared on the surface of Au/ND (Fig. 5D). The EDX spectroscopy confirmed that they were only made of Au (Fig. S3†). When NDs were modified with the polymer, the short side branches disappeared, and the “thick trunk” was observed (Fig. 5E); this could be ascribed to the polymer formed on the ND, which covered and connected the short side branches together. After the removal of protein, a porous topology was shown on the surface of the polymer (Fig. 5F), which may be attributed to the imprinted Hb structures present on the PIPs.44
 | ||
| Fig. 5 SEM images of Au/ND (A, D), Au/ND/polymer (B, E) and Au/ND/PIPs (C, F) at different magnification. | ||
From the results of CV, XPS and SEM data, polymer and PIPs were successfully synthesized on the ND via Hb catalyzed eATRP.
3.3. Responses of the Au/ND/PIPs toward Hb
The proposed Au/ND/PIPs were used as electrochemical biosensors and detected Hb by DPV measurement. Fig. 6A demonstrated the DPV curves of the Au/ND/PIPs after rebinding with a series of concentrations of Hb. With the concentration of Hb increasing, more imprinted cavities were rebound, and the peak currents reduced accordingly.45 The relation of current reduction (ΔI, signal response) with the Hb concentration logarithm is shown in Fig. 6B. ΔI increased linearly with increasing concentration of Hb from 1.0 × 10−13 to 1.0 × 10−1 mg mL−1. From the figure, the linear regression equation was obtained as ΔI (μA) = 0.280 log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C (mg mL−1) + 5.399 (R2 = 0.992). The detection limit (LOD) was 3.2 × 10−14 mg mL−1 Hb estimated at a signal-to-noise ratio of 3. In order to highlight the advantages of the biosensor, the analytical properties of the Au/ND/PIPs were compared with those of the similar sensors for Hb. From Table S1,† it could be seen that the prepared PIPs had lower detection limit and larger linear range.
C (mg mL−1) + 5.399 (R2 = 0.992). The detection limit (LOD) was 3.2 × 10−14 mg mL−1 Hb estimated at a signal-to-noise ratio of 3. In order to highlight the advantages of the biosensor, the analytical properties of the Au/ND/PIPs were compared with those of the similar sensors for Hb. From Table S1,† it could be seen that the prepared PIPs had lower detection limit and larger linear range.
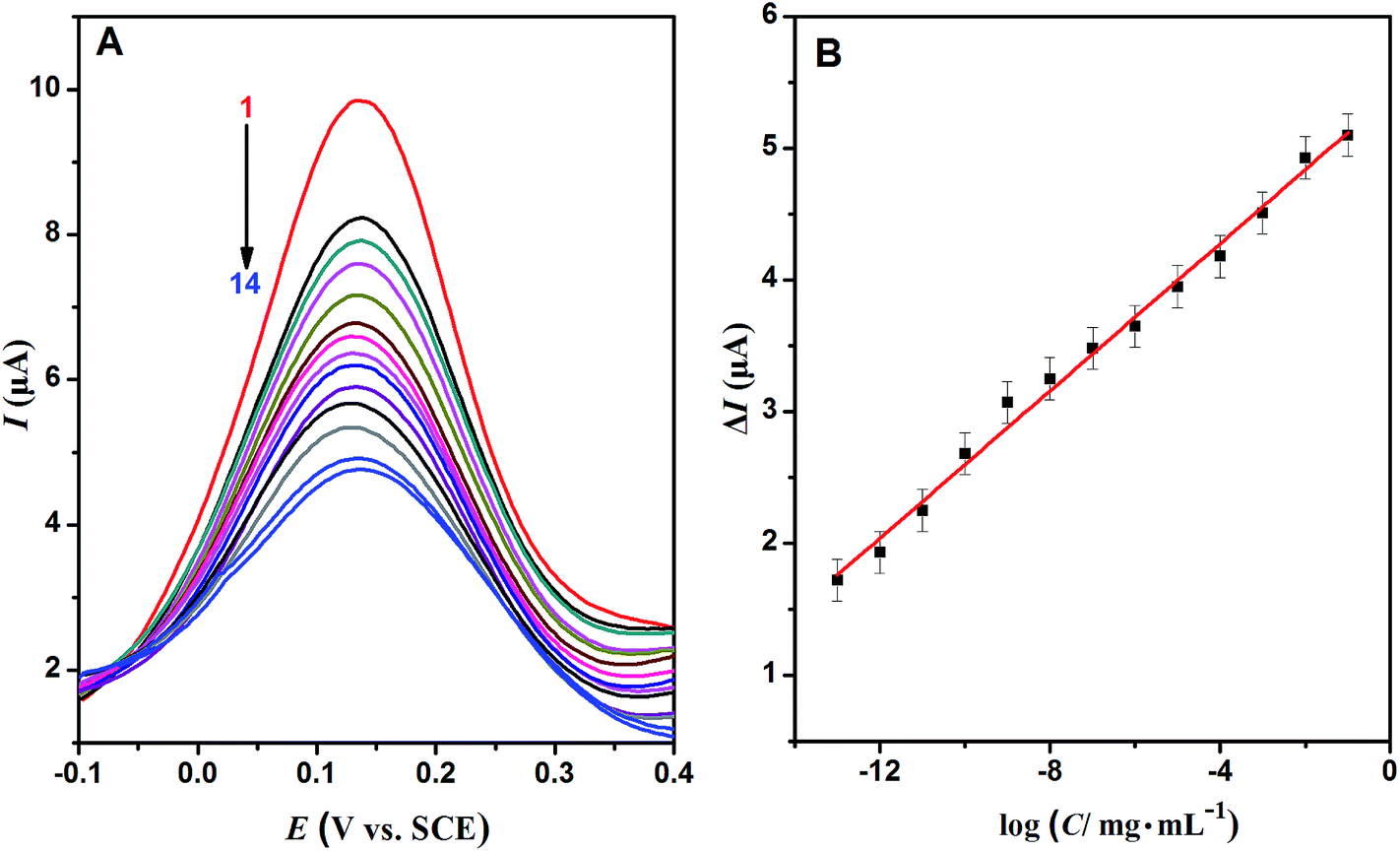 | ||
| Fig. 6 (A) The DPV curves of Au/ND/PIPs in PBS (pH 7.0) containing 5 mmol L−1 Fe(CN)63−/4− after rebinding with Hb (concentrations of Hb from curve 1 to curve 14 were 0, 10−13, 10−12, 10−11, 10−10, 10−9, 10−8, 10−7, 10−6, 10−5, 10−4, 10−3, 10−2, 10−1 mg mL−1). (B) The calibration plot of Au/ND/PIPs. | ||
3.4. Selectivity and application of the Au/ND/PIPs
The selectivity test of Au/ND/PIPs was carried out using Lyz, BSA and HSA as comparative proteins. The signal response of each protein on the Au/ND/PIPs and Au/ND/NIPs in the same concentration (10−1 mg mL−1) was determined by DPV method. As can be seen in Fig. 7, the ΔI value of the Au/ND/PIPs electrode toward Hb was 5.10 μA, which was 5.80, 9.27 and 22.17 times that toward Lyz, BSA and HSA, respectively. The results indicated good selectivity of Au/ND/PIPs for the determination of Hb. This could be interpreted by the specific integration of Hb into the imprinted cavities in the Au/ND/PIPs. The specific interaction may involve two roles, including multiple weak interactions (such as amino-containing, π–π bonds and van der Waals forces) within the imprinted cavities with the template proteins and the sterically complementary imprinted structure just fitting for the unique molecular structure of Hb.46 The two roles would have strong binding force for template proteins and steric effects hindering the adsorption of interfering protein. Thus, Hb could enter the imprinted cavities appropriately, which resulted in low diffusion of the probe and thus decreased the peak current of DPV, whereas those interfering proteins could not bind with the specific imprinted cavities. | ||
| Fig. 7 The selectivity of Au/ND/PIPs biosensor. | ||
The selectivity was also evaluated by the imprinting factor (k). The value of k was calculated using equation k = ΔI(PIPs)/ΔI(NIPs), where ΔI(NIPs) was the response of the Au/ND/NIPs for protein and ΔI(PIPs) was that of Au/ND/PIPs for protein.47 As shown in Fig. 7, the values of k were 1.96, 1.67, 1.77 and 11.09 for Lyz, BSA, HSA and Hb, respectively. The largest k value for Hb suggested that Au/ND/PIPs possessed a capacity toward Hb much higher than that of Au/ND/NIPs electrode. All the above results indicated good selectivity of Au/ND/PIPs toward the template protein.
The repeatability of the Au/ND/PIPs was examined by measuring Hb (10−1 mg mL−1) three times with the same electrode repeatedly. The results revealed that the electrode possessed a relative standard deviation (RSD) of 4.49%, suggesting that the Au/ND/PIPs had good repeatability. Three Au/ND/PIPs were prepared under the same conditions and used to detect 10−1 mg mL−1 Hb to determine the reproducibility. The RSD was 4.32%, implying good reproducibility of Au/ND/PIPs.
To evaluate the applicability of the Au/ND/PIPs, real sample analysis was carried out using standard addition method. The bovine blood sample was first treated according to a literature procedure and analyzed with a blood test meter.46 Then, the samples were diluted and assayed using the Au/ND/PIPs according to the proposed method. The results are listed in Table S2,† which showed that the Au/ND/PIPs sensor was capable of detecting Hb with a good recovery, indicating that the proposed Au/ND/PIPs sensor can be used for the analysis of real samples.
4. Conclusions
In this study, Hb imprinted polymers were successfully prepared via Hb catalyzed eATRP on the surface of 3D Au nanodendrites. The cathode of eATRP was prepared by deposition of nano-Pt on the GCE modified with PTB. The potential, time and Hb concentration for eATRP were determined to be −0.51 V (vs. SCE), 3 h and 2.0 mg mL−1, respectively. The PIPs prepared on nanodendrites could be used as a biosensor for detecting Hb. The resultant sensor exhibited a linear response from 1.0 × 10−13 to 1.0 × 10−1 mg mL−1 with a detection limit of 3.2 × 10−14 mg mL−1. This study significantly reports the preparation of PIPs, protein-catalyzed eATRP and the sensitive detection of metalloproteins.Acknowledgements
Authors gratefully acknowledge the financial support from Key Laboratory of Education Department of Liaoning Province (No. L201683656), the National Natural Science Foundation of China (No. 21304041) and the High Level Talent Innovation Support Project from Dalian (No. 2016RQ047).References
- N. Pourreza and H. Golmohammadi, RSC Adv., 2015, 5, 1712–1717 RSC.
- J. S. Becker, M. Zoriy, J. S. Becker, C. Pickhardt, E. Damoc, G. Juhacz, M. Palkovits and M. Przybylski, Anal. Chem., 2005, 77, 5851–5860 CrossRef CAS PubMed.
- J. S. Becker, S. Mounicou, M. V. Zoriy, J. S. Becker and R. Lobinski, Talanta, 2008, 76, 1183–1188 CrossRef CAS PubMed.
- J. S. Becker, M. Zoriy, M. Przybylski and J. S. Becker, J. Anal. At. Spectrom., 2007, 22, 63–68 RSC.
- A. S. Medel, M. M. Bayón, J. Bettmer, M. L. Sanchez and J. R. Encinar, TrAC, Trends Anal. Chem., 2012, 40, 52–63 CrossRef.
- Y. Wang, Q. Zhang, Y. Ren, L. Jing and T. Wei, Chem. Res. Chin. Univ., 2013, 30, 42–48 CrossRef.
- Z. Altintas, M. Gittens, A. Guerreiro, K. A. Thompson, J. Walker, S. Piletsky and I. E. Tothill, Anal. Chem., 2015, 87, 6801–6807 CrossRef CAS PubMed.
- L. P. Gomez, A. Spangenberg, X. A. Ton, Y. Fuchs, F. Bokeloh, J. P. Malval, B. Tse Sum Bui, D. Thuau, C. Ayela, K. Haupt and O. Soppera, Adv. Mater., 2016, 28, 5931–5937 CrossRef CAS PubMed.
- S. Nestora, F. Merlier, S. Beyazit, E. Prost, L. Duma, B. Baril, A. Greaves, K. Haupt and B. T. S. Bui, Angew. Chem., Int. Ed., 2016, 55, 6252–6256 CrossRef CAS PubMed.
- J. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Angew. Chem., Int. Ed., 2003, 115, 5494–5496 CrossRef.
- Y. Sun, H. Du, Y. Lan, W. Wang, Y. Liang, C. Feng and M. Yang, Biosens. Bioelectron., 2016, 77, 894–900 CrossRef CAS PubMed.
- X. Li, J. Zhou, L. Tian, W. Li, Z. Ali, N. Ali, B. Zhang, H. Zhang and Q. Zhang, Sens. Actuators, B, 2016, 225, 436–445 CrossRef CAS.
- J. Cao, X. Zhang, X. He, L. Chen and Y. Zhang, Chem.–Asian J., 2014, 9, 526–533 CrossRef CAS PubMed.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- T. Du, B. Li, X. Wang, B. Yu, X. Pei, W. T. Huck and F. Zhou, Angew. Chem., Int. Ed., 2016, 55, 4260–4264 CrossRef CAS PubMed.
- Y. Tokura, Y. Jiang, A. Welle, M. H. Stenzel, K. M. Krzemien, J. Michaelis, R. Berger, C. B. Kowollik, Y. Wu and T. Weil, Angew. Chem., Int. Ed., 2016, 55, 5692–5697 CrossRef CAS PubMed.
- J. Yan, B. Li, B. Yu, W. T. Huck, W. Liu and F. Zhou, Angew. Chem., Int. Ed., 2013, 52, 9125–9129 CrossRef CAS PubMed.
- M. Ding, X. Jiang, J. Peng, L. Zhang, Z. Cheng and X. Zhu, Green Chem., 2015, 17, 271–278 RSC.
- Q. Q. Gai, F. Qu, Z. J. Liu, R. J. Dai and Y. K. Zhang, J. Chromatogr. A, 2010, 1217, 5035–5042 CrossRef CAS PubMed.
- H. J. Wang, W. H. Zhou, X. F. Yin, Z. X. Zhuang, H. H. Yang and X. R. Wang, J. Am. Chem. Soc., 2006, 128, 15954–15955 CrossRef CAS PubMed.
- D. J. Li, Y. J. Fu and M. H. Qin, Adv. Mater. Res., 2013, 734, 2108–2112 CrossRef.
- A. J. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- T. B. Silva, M. Spulber, M. K. Kocik, F. Seidi, H. Charan, M. Rother, S. J. Sigg, K. Renggli, G. Kali and N. Bruns, Biomacromolecules, 2013, 14, 2703–2712 CrossRef CAS PubMed.
- C. Fodor, B. Gajewska, O. R. Graham, E. A. Apebende, J. Pollard and N. Bruns, Polym. Chem., 2016, 7, 6617–6625 RSC.
- S. J. Sigg, F. Seidi, K. Renggli, T. B. Silva, G. Kali and N. Bruns, Macromol. Rapid Commun., 2011, 32, 1710–1715 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2016, 7, 5357–5365 RSC.
- M. Fantin, F. Lorandi, A. A. Isse and A. Gennaro, Macromol. Rapid Commun., 2016, 37, 1318–1322 CrossRef CAS PubMed.
- C. Guo, F. Hu, C. M. Li and P. K. Shen, Biosens. Bioelectron., 2008, 24, 819–824 CrossRef CAS PubMed.
- A. K. Das, J. Samdani, H. Y. Kim and J. H. Lee, Electrochim. Acta, 2015, 158, 129–137 CrossRef CAS.
- F. Li, D. Pan, M. Lin, H. Han, X. Hu and Q. Kang, Electrochim. Acta, 2015, 176, 548–554 CrossRef CAS.
- Y. Sun, H. Du, Y. Deng, Y. Lan and C. Feng, J. Solid State Electrochem., 2015, 20, 105–113 CrossRef.
- S. Chen, R. Yuan, Y. Chai, L. Zhang, N. Wang and X. Li, Biosens. Bioelectron., 2007, 22, 1268–1274 CrossRef CAS PubMed.
- Y. Xu, J. Liang, C. Hu, F. Wang, S. Hu and Z. He, J. Biol. Inorg Chem., 2007, 12, 421–427 CrossRef CAS PubMed.
- W. Sun, D. Wang, G. Li, Z. Zhai, R. Zhao and K. Jiao, Electrochim. Acta, 2008, 53, 8217–8221 CrossRef CAS.
- H. Gu, A. Yu and H. Chen, J. Electroanal. Chem., 2001, 516, 119–126 CrossRef CAS.
- S. George and H. K. Lee, J. Phys. Chem. B, 2009, 113, 15445–15454 CrossRef CAS PubMed.
- P. Chmielarz, S. Park, A. Simakova and K. Matyjaszewski, Polymer, 2015, 60, 302–307 CrossRef CAS.
- A. Fatoni, A. Numnuam, P. Kanatharana, W. Limbut and P. Thavarungkul, Analyst, 2014, 139, 6160–6167 RSC.
- Y. Liu, J. Liu, H. Tang, J. Liu, B. Xu, F. Yu and Y. Li, Sens. Actuators, B, 2015, 206, 647–652 CrossRef.
- X. Tan, Q. Hu, J. Wu, X. Li, P. Li, H. Yu, X. Li and F. Lei, Sens. Actuators, B, 2015, 220, 216–221 CrossRef CAS.
- K. Pan, R. Ren, Y. Dan and B. Cao, J. Appl. Polym. Sci., 2011, 122, 2047–2053 CrossRef CAS.
- J. H. Seo, R. Yang, J. Z. Brzezinski, B. Walker, G. C. Bazan and T. Q. Nguyen, Adv. Mater., 2009, 21, 1006–1011 CrossRef CAS.
- Y. Wu, W. Xu, L. Bai, Y. Yuan, H. Yi, Y. Chai and R. Yuan, Biosens. Bioelectron., 2013, 50, 50–56 CrossRef CAS PubMed.
- A. Tiwari, S. R. Deshpande, H. Kobayashi and A. P. Turner, Biosens. Bioelectron., 2012, 35, 224–229 CrossRef CAS PubMed.
- S. Wu, W. Tan and H. Xu, Analyst, 2010, 135, 2523–2527 RSC.
- J. Luo, S. Jiang and X. Liu, Sens. Actuators, B, 2014, 203, 782–789 CrossRef CAS.
- X. Kan, Z. Xing, A. Zhu, Z. Zhao, G. Xu, C. Li and H. Zhou, Sens. Actuators, B, 2012, 168, 395–401 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03772d |
| This journal is © The Royal Society of Chemistry 2017 |