DOI:
10.1039/C7RA03735J
(Paper)
RSC Adv., 2017,
7, 28802-28818
Investigation on a smart nanocarrier with a mesoporous magnetic core and thermo-responsive shell for co-delivery of doxorubicin and curcumin: a new approach towards combination therapy of cancer
Received
31st March 2017
, Accepted 20th May 2017
First published on 31st May 2017
Abstract
In this work, we report on the synthesis and characterization of a novel and smart nanocarrier with a mesoporous magnetic core and thermo-responsive shell for co-delivery of hydrophilic doxorubicin (Dox) and hydrophobic curcumin (Cur) as a combinational therapy for cancer treatment. The P(NIPAM-MAm) coated mesoporous Fe3O4 (MIO-P(NIPAM-MAm)) nanocomposite was prepared by in situ cross linked polymerization of NIPAM and MAm on the surface of pre-synthesized mesoporous Fe3O4 nanoparticles (MIO NPs) in the presence of an oxidizer and cross linker. MIO NPs were synthesised by co-precipitation method using CTAB as the sacrificial soft template. Different characterization techniques have been used to study the physicochemical properties of MIO NPs and the MIO-P(NIPAM-MAm) nanocomposite. Particle sizes of the MIO-P(NIPAM-MAm) nanocomposite estimated by TEM were found to be in between 200–500 nm. VSM results show MIO and MIO-P(NIPAM-MAm) nanocomposites to be superparamagnetic in nature. MIO-P(NIPAM-MAm) nanocomposites exhibited a lower critical solution temperature (LCST) of 41 °C, which is suitable for controlled drug delivery applications unlike pure PNIPAM based nanocarriers. The encapsulation efficiency of Dox and Cur were found to be 96% and 90% respectively. Temperature dependent release studies from MIO-P(NIPAM-MAm)-Cur-Dox indicated a slower release of drugs (both Dox and Cur) below LCST and a sustained release above LCST. Different mathematical models (such as zero order, first order, Higuchi and Korsmeyer–Peppas) were used to fit the experimental release profiles of both drugs. MTT assays on normal and HeLa cells demonstrated the non-toxic nature of the MIO-P(NIPAM-MAm) nanocomposite. The co-loaded MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite exhibited higher in vitro anti-cancer activity compared to free Dox, free Cur, and a free Dox + free Cur mixture. Such a co-loaded smart delivery system could have potential for controlled and targeted drug delivery in cancer diagnosis.
1. Introduction
Nanocarrier based drug delivery systems are playing an important role in cancer treatment.1,2 They offer several advantages over the administration of free drug molecules, such as low clearance (renal and liver clearance), long blood circulation time, low toxicity and enhanced tumour cell death.1 The last decade has witnessed tremendous advances in the design and development of various kinds of organic and inorganic nanocarriers including polymeric nanoparticles, liposomes, nanogels, nanocapsules, and mesoporous nanoparticles.3–7 Delivering anticancer drugs at a targeted site in a controlled manner, while retaining their activity is the primary aim of all nanocarriers. Liposomes have been applied as drug carriers due to their abilities such as loading both hydrophilic and hydrophobic drugs, preventing degradation of drugs and improving internalization. But, the liposome poses several limitations including non-targeting ability, low encapsulation efficiency, high production cost, rapid leakage of water soluble drug in the presence of blood components and poor storage stability.8 Polymer based nanocarriers, therefore, are getting increasing attention because of their excellent biocompatibility, biodegradability, low cost, sustained release behaviour and good storage stability. Non-targeting ability, low encapsulation capacity and leaking of encapsulated drugs are drawbacks of pure polymer based nanocarriers. Targeting ability in these nanocarriers can be introduced by incorporating magnetic nanoparticles (MNPs) within nanocarriers, which is simpler than ligand based targeting. Recently, organic–inorganic based hybrid nanocarriers have been adopted as novel biomaterials in a broad range of biomedical applications, due to their biocompatibility, water dispersibility, controllable size/shape, large loading capacity and combinational drug delivery ability.9,10 Fe3O4 NPs have attracted increasing attention for biomedical applications due to their inherent biocompatibility and superparamagnetism. For instance, they are being used in controlled-targeted drug delivery, hyperthermia, MRI, and imaging.11,12 Fe3O4 NPs are generally used as one component (such as core in mesoporous silica or polymeric NPs) in nanocomposite nanocarriers to provide magnetic controllability but this requires complex synthesis procedure.13 Till date most of the investigations in this field were concentrated on the use of solid nonporous Fe3O4 NPs, which offer limited surface area and provide no porosity for drug loading and molecular-targeting conjugation. This will ultimately results in lowering of targeting and therapeutic efficiency of Fe3O4 NPs. To overcome this limitation, a new form of magnetic nanoparticles such as, mesoporous magnetic nanoparticle with high magnetic moment, high surface area, high drug loading and adequate biocompatibility have been developed.13–15 Creating porosity in magnetic nanoparticles not only simplifies the synthesis process of mesoporous magnetic nanocarrier but also provide magnetic property and loading ability. The magnetic property of mesoporous magnetic nanocarrier could facilitate magnetic targeting ability as well as hyperthermia based heating property. But, the main disadvantages of these mesoporous magnetic NPs (if used in its pure form without any modification) for drug delivery applications are their rapid clearance by the reticulo-endothelial system (RES) and fast/uncontrolled drug release.16 Core–shell type nanocomposite of mesoporous magnetic NPs and stimuli responsive polymer can avoid fast/uncontrolled drug release and provide targeting ability.17 Among various stimuli (such as, light, temperature, pH, magnetic and electrical field) responsive polymers, temperature responsive polymers are very popular because for them slight rise in temperature of the body act as triggering stimuli for drug release.18 Poly(N-isopropylacrylamide) (PNIPAM) is the well-known thermo-responsive polymer having lower critical solution temperature (LCST) ∼32 °C. Above LCST they undergo reversible volume shrinkage due to expulsion of hydrophobically bounded water molecules from the polymer chains and below LCST the polymer absorb water molecules and re-swells.19 Due to its LCST lower than human body temperature, PNIPAM cannot be suitable for controlled drug delivery applications in its intrinsic form. To make it suitable candidate for drug delivery application LCST of PNIPAM can be raised above the normal body temperature by incorporating suitable co-monomer units (hydrophilic moieties). Till date, several successful researches have been performed in order to increase the LCST of PNIPAM by incorporating hydroxyethyl methacrylate (HEMA),20 acrylic acid,21 2-carboxy isopropylacrylamide,22 styrene,23 Maleic acid,24 acrylamide25 and N,N-dimethylacrylamide26 as comonomer for controlled drug release in the body. As of now, most of the reported thermoresponsive polymer based nanocarriers can only load and deliver single type of drug at a time (Dox, sorafenib, 5-fluorouracil, curcumin and cisplatin) i.e. either hydrophilic or hydrophobic drug.25,27,28 Combinational therapy (i.e. giving dose of more than one type of drug at the same time) is getting increasing attention in cancer therapy because of the synergistic effect of enhanced tumour cell death and low side effect. It would be appropriate, therefore, to develop a nanocarrier, which can load and deliver multiple types of drugs (both hydrophobic and hydrophilic) simultaneously. Very limited such type of nanocarriers have recently been reported.29,30
Increased therapeutic efficacy and reduced side effects of Dox have been reported using nanocarriers based combinational therapy of Dox and Curcumin.31–33 For example, co-delivery of Dox and Cur using polymeric NPs and liposome have shown enhanced anticancer activity in chronic myelogenous leukaemia, lung, breast and liver cancers.31,32,34 B. L. Wang et al. reported Cur-Dox/MPEG-PCL micelles for co-delivery of Cur and Dox. They have found better inhibition of LL/2 lung cancer growth both in vivo and in vitro.35 Recently, X Zhao et al. has successfully prepared lipid nanoparticles using a high-pressure microfluidics technique to deliver Dox and Cur simultaneously. Lipid nanoparticles were found to be non-toxic and sustained released pattern of Dox has been reported.36 The preparations of these micelles and lipid based nano-formulations are very complex and highly expensive, which limit their large scale applicability. It was hypothesized that an ideal nanocarrier can be developed by combining hydrophilic and hydrophobic compartments in a single nano system and such nanocarrier would have the capability to load hydrophilic and hydrophobic drugs simultaneously. Thus, the objective of this research was, to design and develop a novel, biocompatible, inexpensive and magnetically guidable thermoresponsive nanocarrier (thermoresponsive polymer coated mesoporous magnetic nanocarrier having LCST greater than body temperature), by a simple method, which can load and deliver hydrophilic (Dox) and hydrophobic (Cur) drugs simultaneously. In this work thermoresponsive polymer-P(NIPAM-MAm) coated mesoporous Fe3O4 nanoparticles-MIO-P(NIPAM-MAm) nanocomposites have successfully been prepared and characterized using various techniques. Further, MIO-P(NIPAM-MAm) nanocomposites were loaded with Cur and Dox. The drugs released behaviour and anticancer activity of MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite were studied.
2. Materials and methods
2.1 Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O, >98%), iron(II) chloride tetrahydrate (FeCl2·4H2O, >98%), N-isopropyl acrylamide (NIPAM, >99%), methacrylamide (MAm, 98%), N,N′-methylenebisacrylamide (MBA, 99%), methylthiazolyldiphenyl-tetrazolium bromide (MTT, >99.9%) and doxorubicin (DOX, >98%) purchased from Sigma Aldrich were used as reagents. Curcumin was purchased from Molychem, India. Potassium persulphate (KPS), cetyltrimethyl ammonium bromide (CTAB) and sodium lauryl sulphate (SDS) were purchased from SRL, India. NaOH was purchased from SDFCL, India. All reagents were of analytical grade and were used directly without further purification. Milli-Q water was used throughout the experiment. RAW 264.7 and HeLa cells were obtained from National Center for Cell Science (NCCS) Pune, India. Dulbecco's modified Eagle medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Life technologies).
2.2 Preparation of mesoporous Fe3O4 nanoparticles (MIO NPs)
MIO NPs were synthesized by co-precipitation method using CTAB as soft template. In typical synthesis procedure, first 0.5 gm CTAB and 4 gm NaOH were added to 230 ml of water. Resulting solution was subjected to magnetic stirring and stirred for 30 min at 80 °C. Solution of FeCl3 and FeCl2 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio) in 20 ml of water was prepared separately. Iron chloride solution was then drop-wise added to the above alkaline miceller solution while stirring at 80 °C. The reaction temperature was maintained at 80 °C for 6 hours with a continuous stirring at 500 rpm. Resulting black precipitate was then isolated by magnetic separation and washed twice with ethanol and water. There after synthesized sample was dried overnight at 100 °C and termed as prepared MIO NPs. Finally, the product was calcined at 400 °C for 2 hours for the decomposition or removal of remaining CTAB and complete crystallization of Fe3O4. Finally obtained black coloured MIO NPs was ground in a mortar and pestle and stored in closed glass vial at room temperature for subsequent use.
:1 mole ratio) in 20 ml of water was prepared separately. Iron chloride solution was then drop-wise added to the above alkaline miceller solution while stirring at 80 °C. The reaction temperature was maintained at 80 °C for 6 hours with a continuous stirring at 500 rpm. Resulting black precipitate was then isolated by magnetic separation and washed twice with ethanol and water. There after synthesized sample was dried overnight at 100 °C and termed as prepared MIO NPs. Finally, the product was calcined at 400 °C for 2 hours for the decomposition or removal of remaining CTAB and complete crystallization of Fe3O4. Finally obtained black coloured MIO NPs was ground in a mortar and pestle and stored in closed glass vial at room temperature for subsequent use.
2.3 Preparation of MIO-P(NIPAM-MAm) nanocomposite
Core shell type nanocomposite of MIO and P(NIPAM-MAm) was synthesised by in situ precipitation polymerization of NIPAM and MAm on MIO NPs in presence of oxidizer and cross linker. In the typical procedure, a solution containing 441.0 mg of NIPAM, 58.3 mg of MAm, 30.8 mg of MBA and 25 mg of SDS was prepared in 200 ml of water and kept for stirring at temperature of 70 °C. 250 mg of MIO NPs suspended in 30 ml of water was then added to the above monomer mixture. The mixture was then stirred at 70 °C for another 30 minutes for proper mixing. 75 mg of potassium persulphate (KPS) in 10 ml of water was then added and the reaction was allowed to proceed for 7 hours at temperature of 70 °C. Obtained nanocomposite was collected by centrifugation and washed with water and then dialyzed for 48 hours using a dialysis bag of 14000 kDa. Finally obtained magnetic-polymeric nanocarrier was named as MIO-P(NIPAM-MAm).
2.4 Preparation of MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite
As prepared MIO-P(NIPAM-MAm) was loaded with two kinds of anticancer drug, Dox and Cur by the following method. First, Cur (5 mg) and MIO-P(NIPAM-MAm) were dispersed in 5 ml solvent (mixture of 2.5 ml ethanol & 2.5 ml water). The mixture containing nanocomposites and Cur was stirred at room temperature for 1 hour for uniform mixing. After that, the medium temperature was increased above the LCST of polymer (to incorporate Cur into MIO NPs) and the mixture was stirred for 6 hours at the same temperature. Thereafter mixture was cooled down to room temperature to obtain Cur loaded MIO-P(NIPAM-MAm), which was termed as MIO-P(NIPAM-MAm)-Cur. Curcumin loaded particles were centrifuged and washed repeatedly to remove physically adsorbed Cur from the surface of the particles. Supernatant having unloaded free Cur was collected and stored in dark. To load Dox in MIO-P(NIPAM-MAm)-Cur, 1 mg of Dox in 5 ml of water was added into MIO-P(NIPAM-MAm)-Cur. The mixture was stirred at ∼4 °C for 24 hours to incorporate Dox within the polymeric matrix. The mixture was centrifuged at 6000 rpm for 10 min and obtained Dox loaded MIO-P(NIPAM-co-MAm)-Cur-Dox was washed thrice with water to remove any loosely adsorbed Dox molecules. Finally obtained MIO-P(NIPAM-co-MAm)-Cur-Dox and the supernatant having free Dox were stored in dark for subsequent use.
2.5 Encapsulation efficiency
The amount of drug loaded in the nanocomposites has been quantified using UV-Vis spectrophotometer. To estimate the amount of loaded drug in the nanocomposites amount of free Dox and Cur present in the corresponding supernatants was determined. The absorbances of the supernatants were measured at wavelength ∼480 nm and 420 nm for Dox and Cur, respectively. Standard calibration curves of concentration vs. absorbance of both the drugs, Dox and Cur were plotted. Drug loading efficiency and loading capacity of MIO-P(NIPAM-MAm) nanocomposites were estimated from the following equations,| | |
Encapsulation efficiency = total amount of loaded drug × 100/total amount of drug taken in the loading experiment
| (1) |
| | |
Encapsulation capacity = total amount of loaded drug × 100/amount of nanoparticles
| (2) |
where, | | |
total amount of loaded drug = total amount of drug taken in the loading experiment − amount of free drug obtained in the supernatant
| (3) |
2.6 In vitro release study
In vitro release study of drugs (Dox and Cur) from MIO-P(NIPAM-MAm)-Cur-Dox was carried out at two different temperatures, 37 °C and 42 °C in phosphate buffer saline (PBS) of pH 7.4 by using dialysis bag diffusion method. In typical procedure, 5 mg of MIO-P(NIPAM-MAm)-Cur-DOX nanoparticle in 0.5 ml of buffer was placed in dialysis bag (molecular weight cut off of 14 kDa.) sealed from one end. Other end of the dialysis bag was also sealed. Dialysis bag with MIO-P(NIPAM-MAm)-Cur-Dox was then soaked in 40 ml of buffer solution (0.1 M) containing 0.2 wt% SDS and continuously stirred at 150 rpm. 3 ml of aliquots were withdrawn from release medium after fixed regular time intervals and equal amount of fresh buffers were added to maintain the sink condition throughout the experiment. The concentrations of the samples were determined by measuring the absorbance at the λmax of Dox (480 nm) and Cur (420 nm).
2.7 MTT assay
To determine cell biocompatibility of the MIO-P(NIPAM-MAm) nanoparticles MTT metabolic activity assay against RAW 264.7 cells was used. Cells were cultured in DMEM media supplemented with 10% FBS, 1% L-glutamine, and 100 U ml−1 penicillin. In typical procedure, 100 μl media with RAW 264.7 cells was seeded in 96 well plates at a density of ∼5 × 103 cells per wall and incubated for 24 hours at 37 °C in humidified air containing 5% CO2. To determine the cytotoxicity effect of the nanoparticles on cells, they were treated with MIO-P(NIPAM-MAm) nanocomposite for 24 h. For this, different concentrations of MIO-P(NIPAM-MAm) nanoparticles suspension in media (6.25, 12.5, 25, 50, 100, 200, 500, and 1000 μg ml−1) were added with cells and incubated for 24 hours at 37 °C and 5% CO2. 20 μl of MTT dye (stock 5 mg ml−1 in 1X PBS) was added to each well and incubated for another 4 hours at 37 °C. After that the media were discarded and 100 μl of DMSO was added to each well of plates. The plates were then slowly shaken to dissolve the formazan crystals formed inside and absorbance was recorded with a microplate absorbance reader at a wavelength of 570 nm. Each experiment was performed in triplet in order to minimize the experimental errors. Untreated cells in the medium was considered as control. Cell viability (%) was calculated using the following equation,| | |
Cell viability (%) = absorbance of treated culture × 100/absorbance of control culture
| (4) |
In vitro cell cytotoxicity of the blank MIO-P(NIPAM-MAm), free Dox, free Cur, free Dox + free Cur mixture and MIO-P(NIPAM-MAm)-Cur-DOX nanocomposite against HeLa cells was also carried out by MTT assay in similar way. Dose concentrations of free and loaded Dox were 0.1, 1, 5, 10, 20 and 40 μg ml−1. Dose concentrations of free and loaded Cur were 0.5, 5, 25, 50, 100, and 200 μg ml−1. Since the curcumin has a very low aqueous solubility at room temperature. Thus, aqueous dispersion of curcumin (>100 μg ml−1) was prepared by ultrasonication of curcumin in heated aqueous solvent (at ∼70–80 °C). Dose concentrations of blank MIO-P(NIPAM-MAm) nanocomposite were 2.5, 25, 125, 250, 500 and 1000 μg ml−1. Same dose as of MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite has been tested to ensure contribution (towards cell inhibition) from MIO-P(NIPAM-MAm) nanocomposite.
2.8 Characterizations
The crystal structures of the as prepared MIO NPs, MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were investigated by powder XRD (Bruker D8 Advance X-ray Diffractometer) using Cu Kα radiation in the 2θ range 20 to 80°. To obtain the surface area and pore size of as-prepared MIO NPs and MIO NPs, samples were characterized for Brunauer–Emmett–Teller (BET) surface area (Micromeritics 2030). Transmission electron microscopy (FEI Tecnai T20G2 S TWIN TEM) and scanning electron microscopy (Hitachi S-3400N) were used to determine the particle sizes and morphology of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite. Phase composition of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite was studied by selected area electron diffraction (SAED) pattern. Hydrodynamic particle size distribution and Zeta potential measurement were carried out using Malvern Zetasizer. The chemical structure and major functional groups of the MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were studied using Fourier transformed infrared (Perkin-Elmer 2000 FT-IR spectrometer) spectroscopy in the wavelength range of 400 to 4000 cm−1 by KBr pellet method. Phase transition phenomena (LCST measurement) of linear PNIPAM and MIO-P(NIPAM-MAm) was studied by Differential scanning calorimeter (Mettler Toledo DSC 822e module) under N2 atmosphere from 25 to 60 °C at a heating rate of 10 °C min−1. Thermo gravimetric analysis (TGA) was performed by Mettler Toledo TGA/SDTA 851e module under N2 atmosphere from 50 to 1000 °C at a heating rate of 10 °C min−1 to investigate the thermal stability of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite. Magnetic properties of MIO NPs and MIO-P(NIPAM-MAm) samples were analysed using a Lakeshore (Model 7407) Vibrating Sample Magnetometer (VSM) in magnetic fields up to 1.5 T at ambient temperature (298 K). Drug loading and release studies of MIO-P(NIPAM-MAm) were performed by UV-Vis-NIR spectrometer (Jasco V-670).
3. Results and discussion
3.1 Preparation of MIO NPs, MIO-P(NIPAM-MAm) and MIO-P(NIPAM-MAm)-Cur-Dox nanocomposites
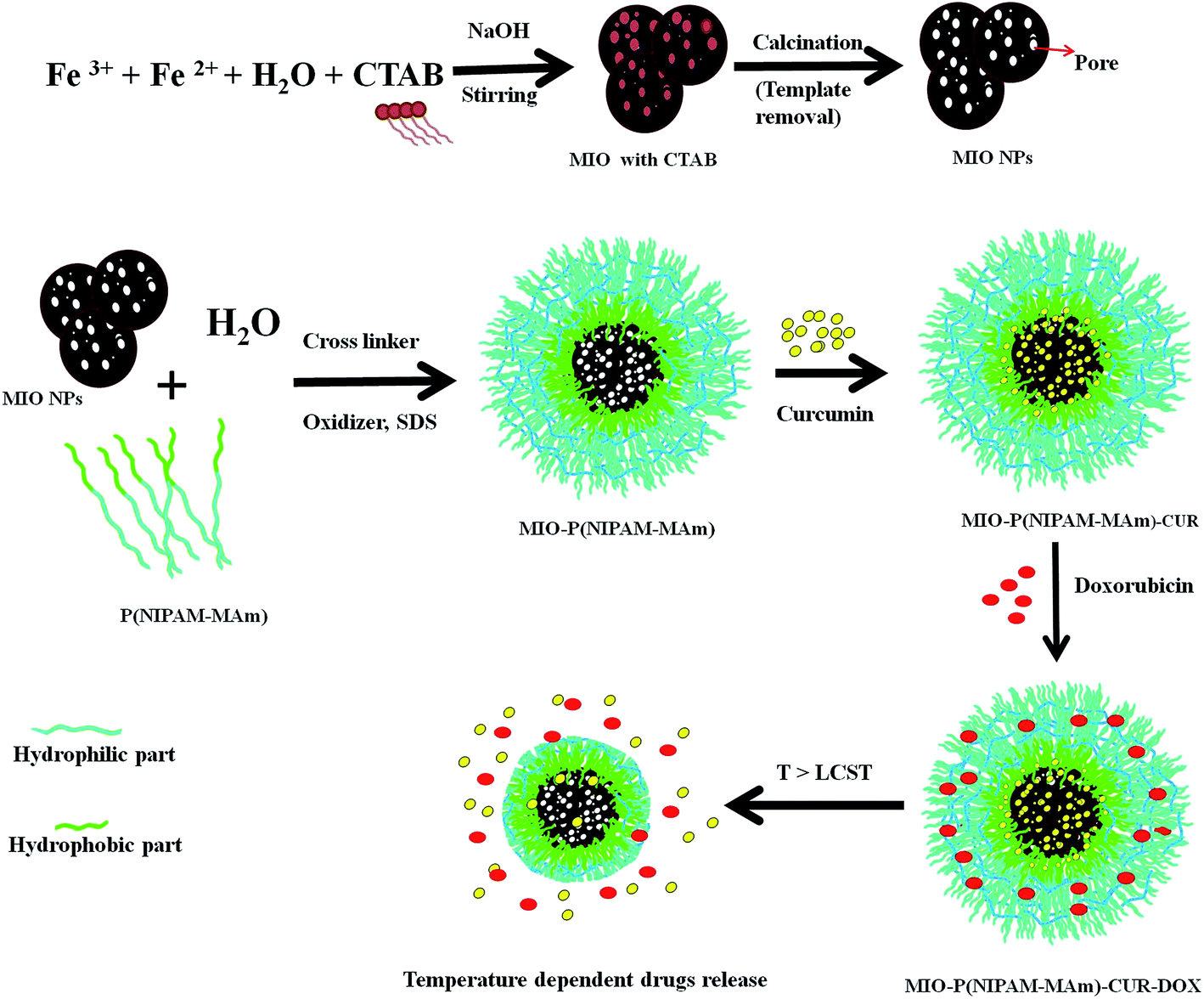
MIO-P(NIPAM-MAm) nanocomposite was synthesized by two step process as shown in Scheme 1. First MIO NPs was synthesized by co-precipitation method using CTAB as the sacrificial template. Co-precipitation of Fe(II) and Fe(III) on CTAB template and further removal of CTAB have resulted in the formation of MIO NPs. Dispersion of CTAB in water (above its critical miceller concentration) resulted in the formation of miceller rod due to the hydrophobic interaction among the non-polar part of the CTAB molecule, which acted as soft template. Generally, removal of the template is carried out either by washing with suitable solvent or by burning/calcination of as prepared sample. Here, removal of CTAB was carried out by calcination process as it provides complete removal of CTAB and good crystallization of Fe3O4. MIO-P(NIPAM-MAm) nanocomposites were synthesized by in situ coating/deposition of polymer on MIO NPs in presence of oxidizer and cross linker. Hydrophobic interaction between the polymer and MIO NPs has facilitated the deposition of polymer onto MIO NPs. MIO NPs may have played the role of heterogeneous nucleation sites for the deposition and precipitation of polymer.37 Prior to the loading studies, prepared MIO NPs and MIO-P(NIPAM-MAm) nanocomposites were systematically characterized for different properties for biomedical application. Cur was loaded in the nanocomposites above the LCST temperature of MIO-P(NIPAM-MAm) nanocomposites. Above LCST temperature was chosen to load Cur in nanocomposite with anticipation that it would facilitate the incorporation/entrapment of hydrophobic Cur to the interior of MIO NPs through its available open pores. In addition, above LCST temperature of MIO-P(NIPAM-MAm) nanocomposites increased adsorption rate of Cur on particles is expected through hydrophobic interaction between Cur and polymer (PNIPAM-MAm was hydrophobic in nature above the LCST). Also, reduced diffusion length of Cur through polymer would facilitate loading of Cur in the interior of nanocomposite, i.e. in the MIO NPs and in the hydrophobic region of polymer. Further, cooling of medium below LCST temperature will leave cur trapped inside the MIO and/hydrophobic region of polymer due to the formation of a hydrophilic swollen shield of polymer as barrier at outer side of the nanoparticle. Thus, this whole phenomenon will result in the complete incorporation of Cur toward core region of nanocomposite loading of hydrophilic Dox in the hydrophilic polymeric matrix could have taken place through electrostatic and or hydrogen bonding between polymer and Dox molecules.38 Positively charged amine groups of Dox (pKa 8.2) would have electrostatically bound with the negatively charged polymer matrix. Since there are different types of functional groups available both in polymer (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –N–H, etc) and Dox molecules (–OH, –CO, –C–OH, and –NH2), hydrogen bonding between partially negative charged oxygen and partially positive charged hydrogen of these groups also likely to contribute toward loading of Dox in hydrophilic polymer. Loading of Dox in NIPAM through hydrogen bonding between Dox and amide group of PNIPAM has also been reported earlier.39
O, –N–H, etc) and Dox molecules (–OH, –CO, –C–OH, and –NH2), hydrogen bonding between partially negative charged oxygen and partially positive charged hydrogen of these groups also likely to contribute toward loading of Dox in hydrophilic polymer. Loading of Dox in NIPAM through hydrogen bonding between Dox and amide group of PNIPAM has also been reported earlier.39
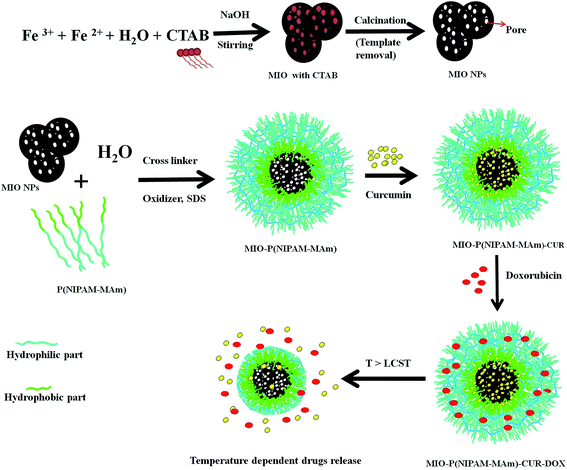 |
| | Scheme 1 Schematic illustration of the formation process of MIO-P(NIPAM-MAm) nanocomposite and its stimuli responsive behaviour. | |
3.2 Structural, thermal and magnetic characterizations of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite
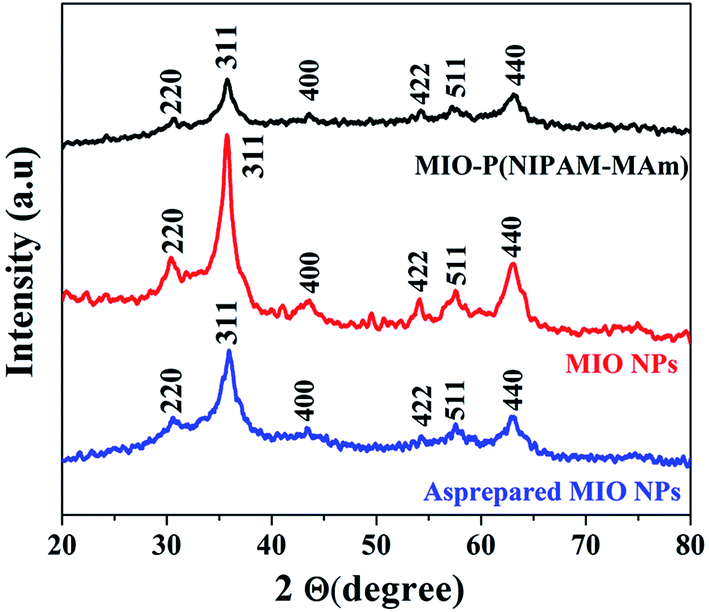
3.2.1. X-ray diffraction analysis. The phase and crystalline structure of as prepared MIO NPs, MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were studied by powder XRD technique. XRD patterns of as prepared MIO NPs, MIO NPs and MIO-P(NIPAM-MAm) nanocomposite recorded in the 2θ range 20 to 80° are shown in Fig. 1. All the samples have shown sharp diffraction peaks at 2θ, 30.29°, 35.7°, 43.36°, 53°, 57.2° and 62.7° corresponding to reflections obtained from (220), (311), (400), (422), (511) and (440) crystal planes of MIO NPs.40,41 These observed diffraction peaks of all the three samples agreed well with the standard Fe3O4 powder diffraction data (JCPDS cards no. 88-315) having spinal cubic structure. Thus, XRD pattern confirmed successful formation of spinal cubic MIO NPs. In addition, no additional diffraction peak of impurities was observed confirming the purity of the spinal MIO NPs. It was observed that in comparison to as prepared MIO NPs the peaks intensities of the MIO NPs were increased which indicated that calcination of sample has improved the crystallinity of MIO NPs due to increase in the crystalline volume and decreased amorphous phases in the sample.42,43 In other word, at calcination temperature more and more atoms arranged or ordered themselves in the lattice of spinel cubic MIO and thus results more periodicity or crystallinity.44 In case of MIO-P(NIPAM-MAm) nanocomposite, all the diffraction peaks of MIO were present in the XRD spectra. This indicated that the crystal structure of MIO NPs was not destroyed by the polymer synthesis procedure and coating of polymers. However, the intensities of the peaks for MIO-P(NIPAM-MAm) nanocomposites were reduced, which can be attributed to the coating of amorphous polymer matrix on the surface of MIO NPs.45 In other word presence of relatively less amount of MIO NPs in MIO-P(NIPAM-MAm) nanocomposites (than pure MIO NPs) could have resulted in reduced XRD peak intensity.40 The crystallite sizes of as prepared MIO and MIO NPs were estimated from the most intense peak (311) by using the Debye–Scherer formula. The crystallite size of as prepared MIO and MIO NPs were found to be ∼6.2 and 10.2 nm, respectively. The lattice constant of the MIO NPs, estimated from the PowderX software, was found to be 8.32 Å, which also confirmed successful formation of spinal cubic MIO NPs. Appearance of characteristic peaks of MIO in XRD pattern of nanocomposite confirmed successful formation of MIO-P(NIPAM-MAm) nanocomposite. No sharp diffraction peak of polymer has been observed in the XRD pattern of MIO-P(NIPAM-MAm) nanocomposite. Similar XRD patterns for Fe3O4-polymer systems have also been reported earlier.40,46,47
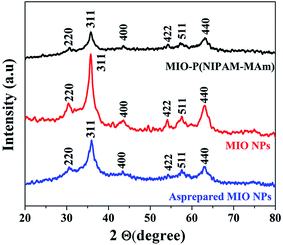 |
| | Fig. 1 XRD patterns of as prepared MIO NPs, MIO NPs and MIO-P(NIPAM-MAm) nanocomposite. | |
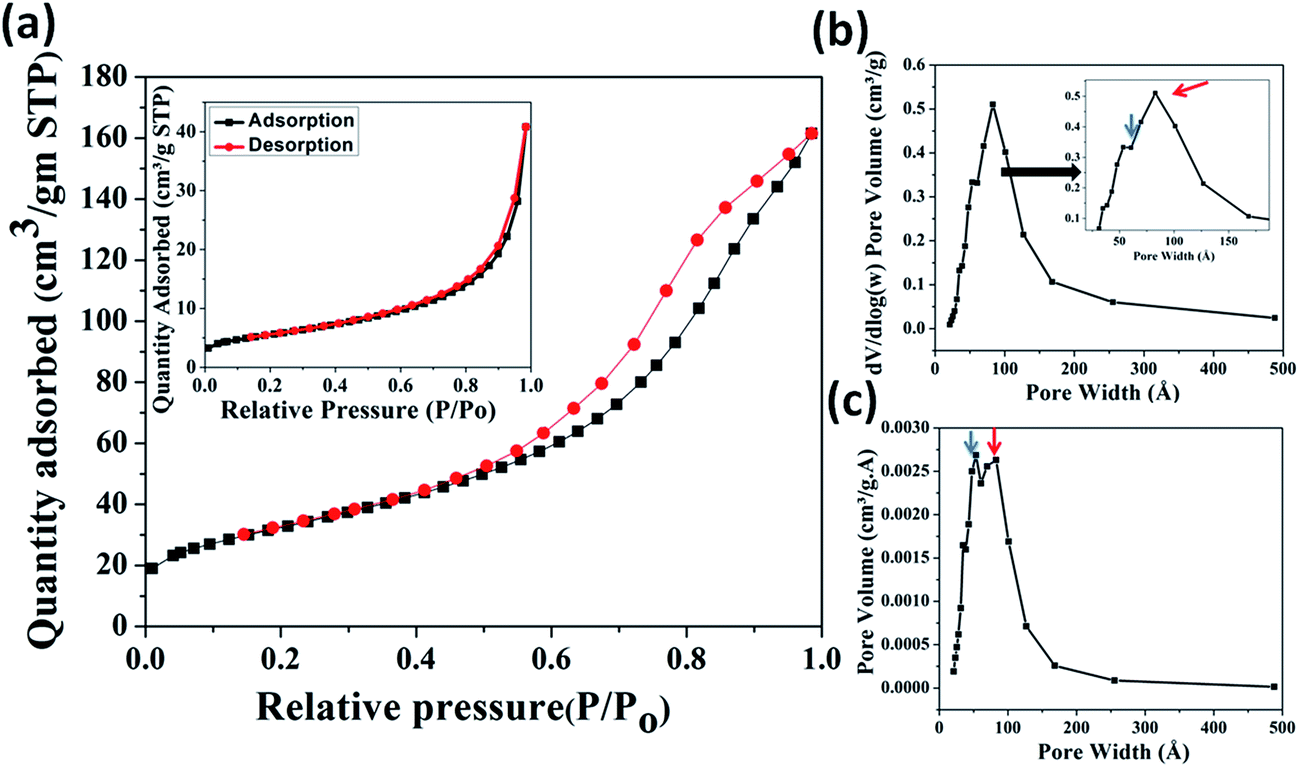
3.2.2. BET analysis. The surface area and porosity of MIO NPs were characterized using N2 adsorption/desorption isotherm measurement. The obtained N2 isotherm of MIO NPs is shown in Fig. 2(a). Presence of hysteresis in the recorded isotherm indicated mesoporous nature of MIO NPs. The removal of soft template of CTAB on calcinations at 400 °C have resulted porosity in MIO NPs. The isotherm displayed the characteristic of a type IV isotherm with H1 hysteresis loop in accordance with the IUPAC classification,48 suggesting presence of mesopores in MIO NPs. The surface area was estimated using the linear region of Brunauer–Emmett–Teller (BET) data at lower relative pressure (P/Po) range of 0.05 to 0.3. The BET surface area was found to be 117.4 m2 g−1. Single point surface area (at P/Po = 0.27) and Langmuir surface area were calculated to be 114.1 and 176.6 m2 g−1, respectively. The obtained BET surface area value is relatively larger or comparable with other reported values for mesoporous Fe3O4 system.49,50 Simplicity of the present synthesis procedure with high surface area and porosity of prepared MIO NPs make this present system superior to others.
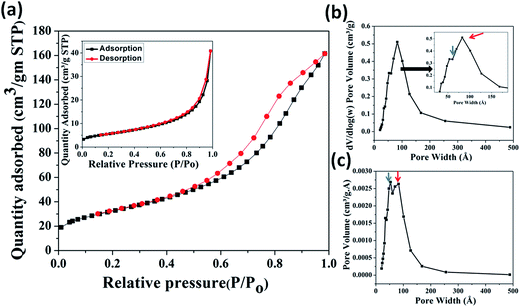 |
| | Fig. 2 BET N2 adsorption (black) and desorption (red) isotherms (a), differential logarithmic mesopore size distribution (b) and differential mesopore size distribution (c) of MIO NPs. N2 adsorption/desorption isotherm measurement of as-prepared MIO (MIO before CTAB removal) is shown in inset of (a). Inset of (b) shows enlarge portion of indicated region. | |
The pore size and distribution were calculated from the desorption branch of the isotherm using the Barrett–Joyner–Halenda (BJH) method. Fig. 2(b) and (c) show the differential logarithmic mesopore size distribution and differential mesopore size distribution of MIO NPs. Both the distribution plots have showed presence of two coupled peaks which are marked with blue (centred at ∼4 nm) and red arrows (centred at ∼8 nm). These two peaks are more clearly visible in figure (c). Thus the pore size distribution (Fig. 2(b) and (c)) data reveals that two types of pores, one of size ∼4 nm and second of size ∼8 nm were present in the sample. It is believed that the pores of diameter ∼4 nm could have generated by removal of CTAB and the bigger pores of diameter ∼8 nm can be related with the interparticle pores formed during aggregation of smaller MIO NPs. The pore size distribution data also indicated the presence of nearly uniform size pores. Single point adsorption total pore volume was estimated to be 0.52 cm3 g−1 at P/Po = 0.98. Slow and linear increase in the adsorption isotherm, in the lower pressure range (till P/Po 0.3) can be attributed to the monolayer formation by N2 gas molecules. The rapid increase in the adsorbed quantity, in the pressure range 0.45 < P/Po < 1, is probably due to the capillary condensation of N2 gas in the mesopores.48 To confirm, the generation of mesopores on the surface of MIO NPs via the removal of CTAB, N2 adsorption/desorption isotherm measurement of as-prepared MIO (MIO before CTAB removal) was also carried out and is shown in inset of Fig. 2(a). The isotherm displayed very less area of hysteresis loop than calcined MIO NPs which could be attributed to less porosity due to non-removal of CTAB template. The BET surface area was found to be 19.8 m2 g−1. Single point surface area (at P/Po = 0.27) and Langmuir surface area were calculated to be 19.4 and 29.9 m2 g−1, respectively. Single point adsorption total pore volume was estimated to be 0.06 cm3 g−1 at P/Po = 0.98. Lower surface area and pore volume of as-prepared MIO NPs (before CTAB removal) than the calcined MIO NPs (after CTAB removal) suggested that mesopores (∼4 nm) are generated due to the removal of CTAB.
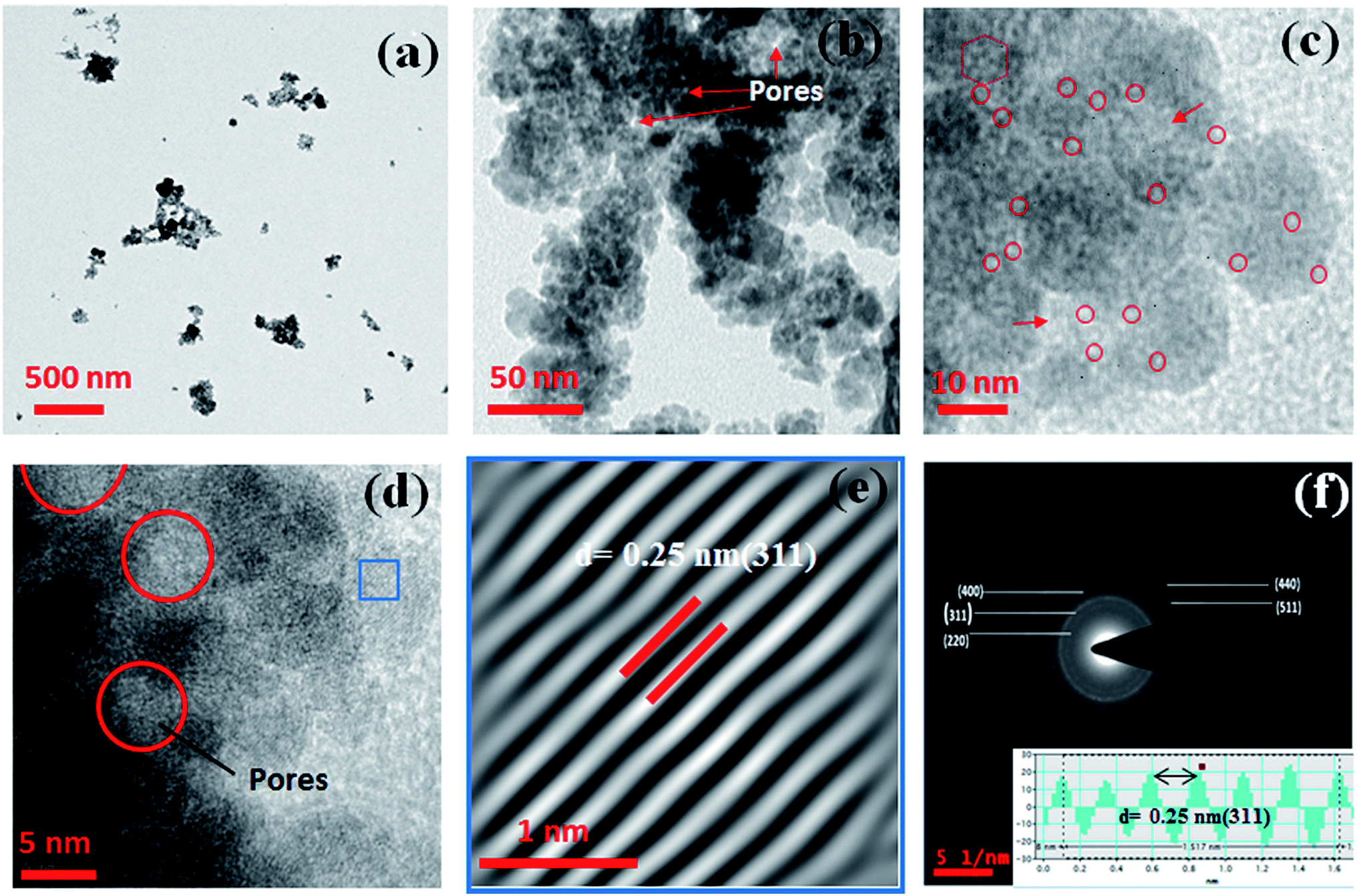
3.2.3. TEM, FESEM and DLS analysis. The size and morphology of the synthesized MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were studied using TEM and FESEM. Fig. 3(a–c) shows the TEM micrographs of MIO NPs at different magnifications viz., (a) scale bar at 500 nm (b) scale bar at 50 nm and (c) scale bar at 10 nm. From Fig. 3(a) and (b), it was observed that particles of size 15–30 nm aggregated to form a bigger mesoporous cluster of size 50–200 nm in diameter. The average particle size of single MIO NPs was found to be ∼23 nm. It is observed that these single MIO NPs itself have porosity on its surface with the average pore diameter of ∼3 nm (marked as red circle), as shown in figure (c). The marked red hexagon region in figure (c) clearly shows presence of six pores together. These observations indicated that CTAB template had been removed on calcination leaving pores on the MIO NPs. Presence of interparticle pores (indicated by red arrow in figure (b) and (c)) of diameter ∼5–8 nm were also observed in TEM micrograph. These interparticle pores could have formed during the aggregation of smaller MIO NP to form bigger mesoporous cluster. Further, confirmation of presence of pores was done by HRTEM analysis and HRTEM micrograph of MIO NPs is shown in Fig. 3(d). Absence of atomic lattice planes in the pores locations (red circle in figure (d)) on MIO NPs in HRTEM image confirmed the presence of inter-particle pores of 5–8 nm in diameter on MIO nanoclusters. Thus, it was observed from TEM analysis that there are two types of pores present in the MIO NPs, one of diameter ∼3 nm and another of ∼5–8 nm. Similar observations were made by BET analysis and pore size distributions have shown presence of two peaks. Fig. 3(e) shows the inverse fast Fourier transform (IFFT) image of a selected region (blue frame) of HRTEM image (Fig. 3(d)). IFFT image clearly shows the presence of array of ordered planes with d spacing of 0.25 nm, which corresponds to (311) plane of cubic Fe3O4. The SAED pattern shown in Fig. 3(f) reveals the polycrystalline nature of MIO NPs. Obtained SAED diffraction rings of MIO NPs match well with the crystal planes of cubic Fe3O4 and have been assigned to (220), (311), (400), (511), and (440) planes, respectively, from inner to outer side of the diffraction pattern (JCPDS cards no. 88-315). The inter planer spacing measured from the line profile (inset of (f)) was found to be ∼0.25 nm, which corresponds to (311) lattice plane of Fe3O4. Thus, HRTEM and SAED results confirmed that the prepared MIO NPs belong to cubic spinal Fe3O4.
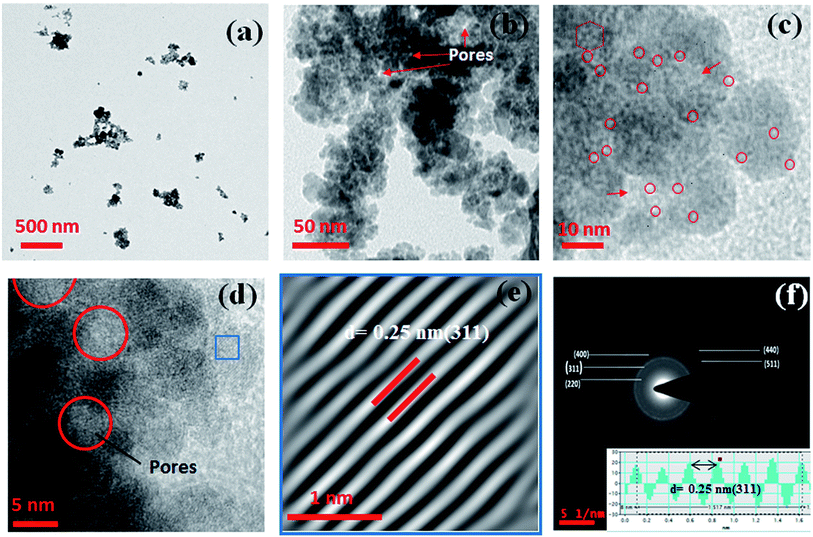 |
| | Fig. 3 TEM micrographs at different magnifications viz. (a) scale bar at 500 nm (b) scale bar at 50 nm and (c) scale bar at 10 nm. (d) HRTEM micrograph (e) IFFT image and (f) SAED pattern of MIO NPs. Inset of figure (f) shows line profile through figure (e). | |
Fig. 4(a–c) shows the TEM micrographs of MIO-P(NIPAM-MAm) nanocomposite at different magnifications viz., (a) scale bar at 500 nm (b) scale bar at 100 nm and (c) scale bar at 50 nm. TEM micrographs show formation of spherical shape core shell particles. It can be observed from TEM images 4(a and b) that MIO nanoclusters (black colour) are well coated with polymer (light grey). The size of these spherical shape core shell type of particles was found to be 200–500 nm (inset of Fig. 4(a) and (b)). HRTEM analysis also confirmed the formation of core shell structure, with core of crystalline MIO nanoclusters in the centre of the amorphous polymeric shell, as shown in Fig. 4(c and d). Appearance of very clear ordered lattice planes of cubic Fe3O4 in the HRTEM micrograph of the core region of MIO-P(NIPAM-MAm) confirms presence of crystalline MIO core in the nanocomposites. For better understanding and clear visualization, IFFT of selected area (blue frame of (d)) has been carried out and is shown in Fig. 4(e). The d spacing of the observed planes is found to be 0.3 nm, which corresponds to the (220) plane of MIO NPs. Further confirmation of presence of crystalline MIO in the nanocomposites was made by SAED pattern of the nanocomposites. Indexed SAED pattern of MIO-P(NIPAM-MAm) nanocomposite is shown in Fig. 4(f) and obtained ring pattern suggests the presence of polycrystalline MIO in the nanocomposite. Thus, HRTEM image, IFFT image and SAED patterns confirm the presence of crystalline core in the prepared core shell nanocomposites. The presence of crystalline MIO core also indicates that the coating of polymer or coating procedure of polymer on the surface of MIO does not alter the crystal structure of the magnetic component in the core–shell MIO-P(NIPAM-MAm) nanocomposite.
 |
| | Fig. 4 TEM micrographs at different magnifications viz. (a) scale bar at 500 nm (b) scale bar at 100 nm and (c) scale bar at 50 nm. Inset of (a) shows enlarge single MIO-P(NIPAM-MAm) nanocarrier. (d) HRTEM micrograph (e) IFFT image and (f) SAED pattern of MIO-P(NIPAM-MAm). | |
Fig. 5(a) and (b) shows the FESEM images of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite, respectively. It can be observed that nearly spherical cluster sizes of 50–200 nm were formed due to the aggregation of many small size particles of MIO NPs, as shown in Fig. 5(a). From the FESEM image of MIO-P(NIPAM-MAm) nanocomposite it was observed that particles had wide size distribution and spherical shape (inset of Fig. 5(b)). The particle size of MIO-P(NIPAM-MAm) nanocomposite was found to 200–500 nm. The increased particle size of the nanocomposites confirmed coating of polymer on the surface of MIO nanoparticles. Thus, FESEM analysis also confirmed successful formation of MIO-P(NIPAM-MAm) nanocomposite.
 |
| | Fig. 5 FESEM micrographs of (a) MIO NPs and (b) MIO-P(NIPAM-MAm) nanocomposite. Inset of figure (b) shows enlarge image of single MIO-P(NIPAM-MAm) nanocomposite particle. | |
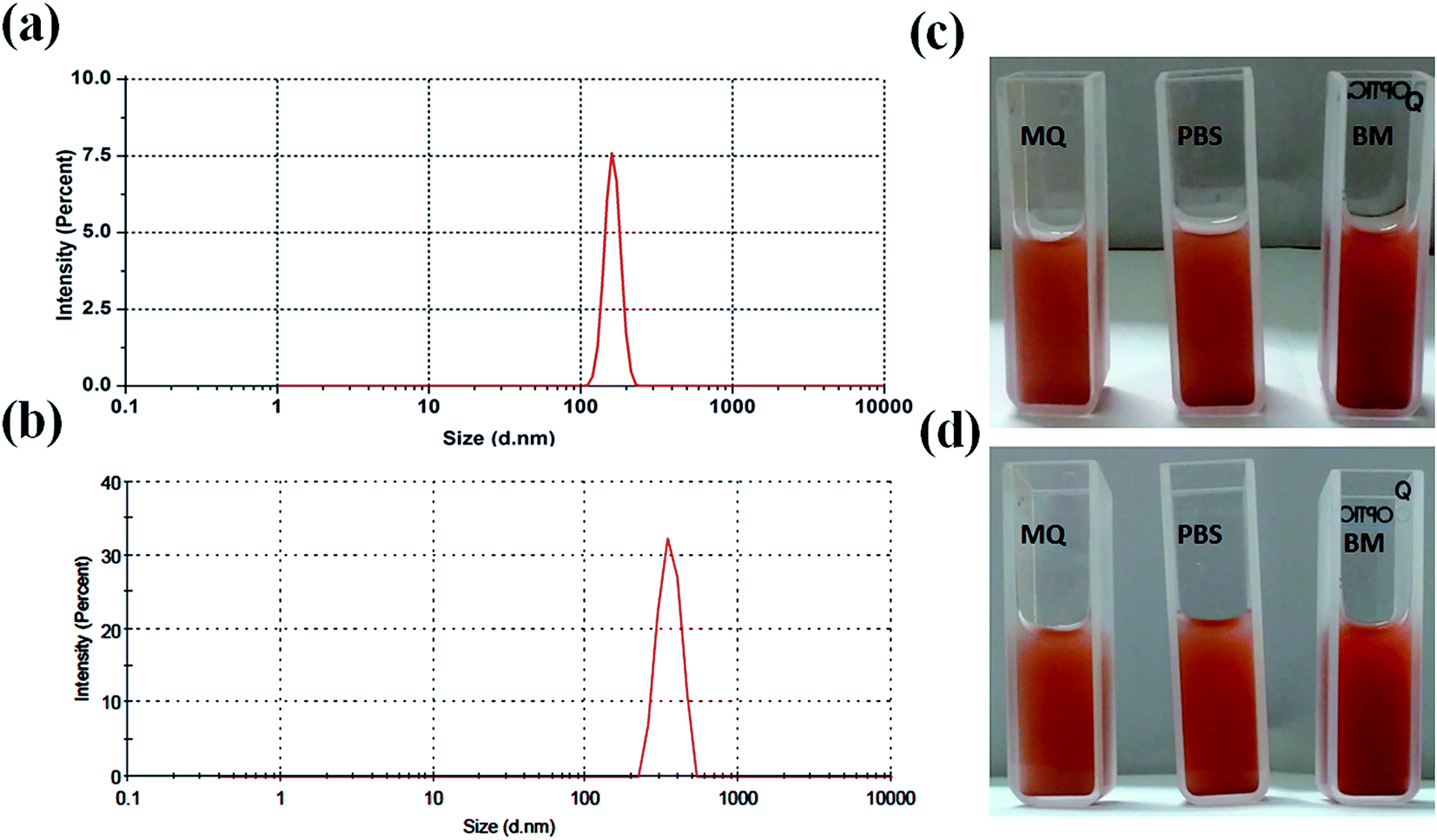
For biological applications, it is important to study the hydrodynamic particles size and particles size distribution of nanoparticle in liquid medium. Thus, particle size distribution of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were estimated in liquid suspension (in PBS), by the dynamic light scattering (DLS) technique and shown in Fig. 6(a) and (b), respectively. Both the samples have shown single distribution peak with narrow particles size distribution. Average hydrodynamic particles size of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were found to be ∼163 and ∼548 nm. For biological application nanocarrier should have good aqueous dispersibility and stability. Dispersibility or stability of nanocarrier in different solvent such as water, PBS and biological media (BM) have been studied. Fig. 6(c) and (d) are the digital images of the MIO-P(NIPAM-MAm) nanocomposite dispersioned in MQ water, PBS, and biological media taken at t = 0 and 48 hours. These images suggested fairly stable nature of the MIO-P(NIPAM-MAm) nanocomposite in all the tested solvent even after 48 hours. Colloidal stability and surface charge of MIO-P(NIPAM-MAm) nanocomposite in PBS have also been studied through Zeta potential measurement. The zeta potential value of MIO-P(NIPAM-MAm) nanocomposite is found to be −13 mV.
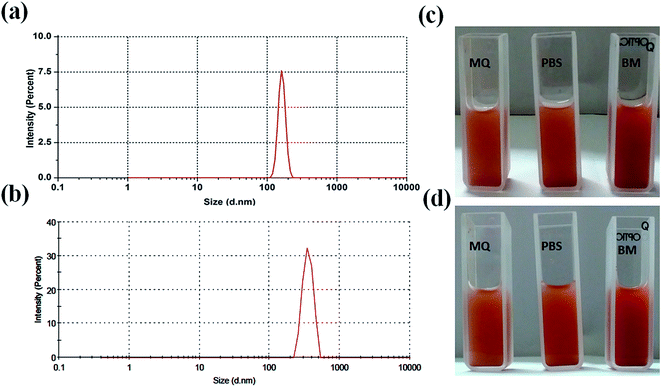 |
| | Fig. 6 Particle size distribution of MIO NPs (a) and MIO-P(NIPAM-MAm) nanocomposite (b) in PBS, measured by the DLS technique. Digital images of MIO-P(NIPAM-MAm) nanocomposite dispersioned in MQ water, PBS, and biological media taken at t = 0 (c) and 48 hours (d). | |
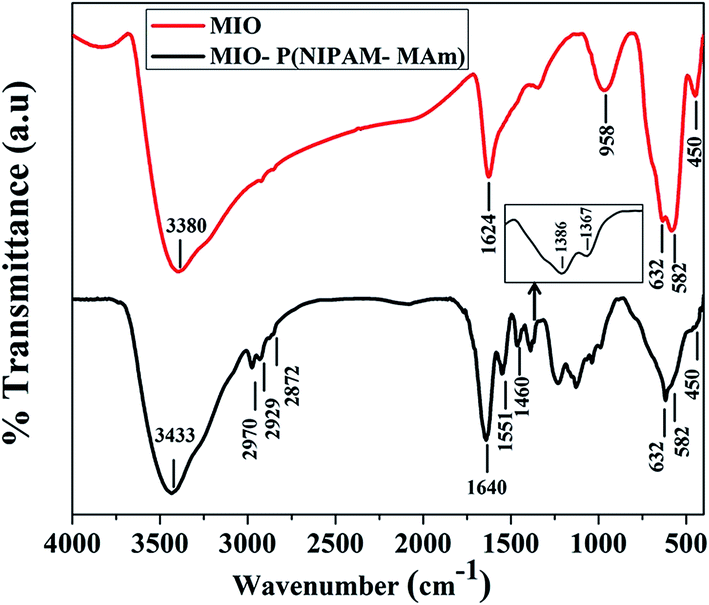
3.2.4. FTIR analysis. FTIR is a reliable method to characterize the functional groups and types of chemical bonds present in a chemical compound. Fig. 7 shows the FTIR spectra of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite. The IR spectra of MIO NPs show all characteristic absorption bands of Fe3O4 at 450 cm−1, 582 cm−1 and 632 cm−1.51 The bands at ∼450 and 582 to 632 cm−1 arise due to the stretching vibration of octahedral and tetrahedral coordination complexes, respectively, in the crystalline lattice of Fe3O4. Presences of these characteristic bands of Fe3O4 are indicative of successful formation of MIO NPs. The additional absorption bands at 3380 and 1624 cm−1 could be assigned to the absorbed moisture on the surface of sample. The FTIR spectra of MIO-P(NIPAM-MAm) nanocomposite exhibited characteristic absorption peaks of both MIO NPs and P(NIPAM-MAm), which is indicative of successful formation of the nanocomposite. The characteristic absorption peaks of P(NIPAM-MAm) were observed at 1640, 1551 and 1460 cm−1. The IR spectra at 1640 cm−1 is due to the stretching (amide I) vibration of CO.52 Absorption at 1551 and 1460 cm−1 were attributed to –NH (present in NIPAM) and –NH2 (present in MAm) plane bending (amide II) modes.53 The peaks at 2970, 2929 and 2872 cm−1 are due to symmetric and asymmetric stretching of CH, CH2 and CH3, respectively. In addition, doublet absorption bands observed at 1386 and 1367 cm−1 are due to the –CH vibration modes of isopropyl group of NIPAM.52 A broad absorption peak at 3433 cm−1 was assigned to stretching vibration of –NH in NIPAM and MAm.53 Thus, appearance of bands both from NIPAM and MAm in the FTIR spectra of MIO-P(NIPAM-MAm) suggested copolymerization of NIPAM and MAm monomer.53 Presence of absorption bands at 450 cm−1, 582 cm−1 and 632 cm−1 in the FTIR spectra of MIO-P(NIPAM-MAm) nanocomposite clearly indicated the presence of MIO NPs in the MIO-P(NIPAM-MAm) nanocomposite. However, the peak intensities of Fe–O stretching bonds were relatively weak, which could be attributed to complete encapsulation of P(NIPAM-MAm) polymer around the MIO NPs and also due to lesser concentration of MIO NPs, than in pure MIO NPs, in the nanocomposite sample as per Lambert Beer's law. According to Lambert Beer's law, absorbance in IR spectroscopy is directly proportional to the concentration and thickness of the sample. The obtained FTIR data confirmed the successful coating of P(NIPAM-MAm) polymers on to the MIO NPs core.
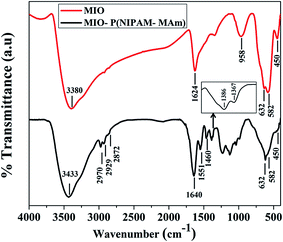 |
| | Fig. 7 FTIR spectra of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite. | |
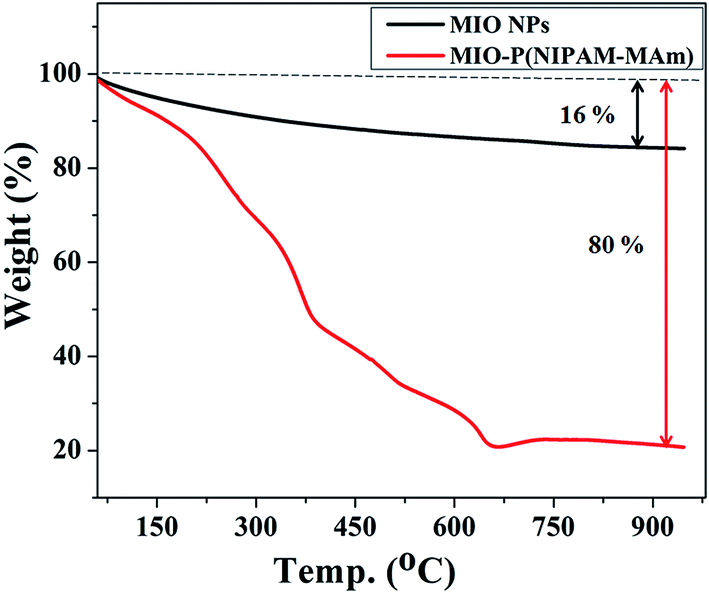
3.2.5. Thermo gravimetric (TGA) analysis. TGA was carried out to determine the relative composition of the MIO NPs and P(NIPAM-MAm) in the MIO-P(NIPAM-MAm) nanocomposite. Fig. 8 shows the TGA curves of MIO NPs and MIO-P(NIPAM-MAm) nanocomposites. The TGA curve of MIO NPs shows total weight loss of about 16%, which can be attributed to the removal of adsorbed moisture on the sample. The TGA curve of MIO-P(NIPAM-MAm) nanocomposite exhibited a multi stage degradation behaviour. The initial weight loss of ∼10% upto 200 °C was due to the evaporation of water molecules from the sample. It is observed that degradation of polymers began at 200 °C and completed at 650 °C with an approximate weight loss of about 70%. Thus, MIO-P(NIPAM-MAm) nanocomposite exhibited the total weight loss of ∼80%. It is observed that there was no significant weight loss after 650 °C, which confirmed that the nanocomposite consists of ∼20% of MIO NPs. Thus, TGA analysis also confirmed formation of MIO-P(NIPAM-MAm) nanocomposite.
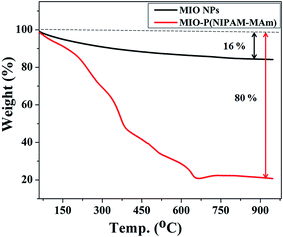 |
| | Fig. 8 TGA curves of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite. | |
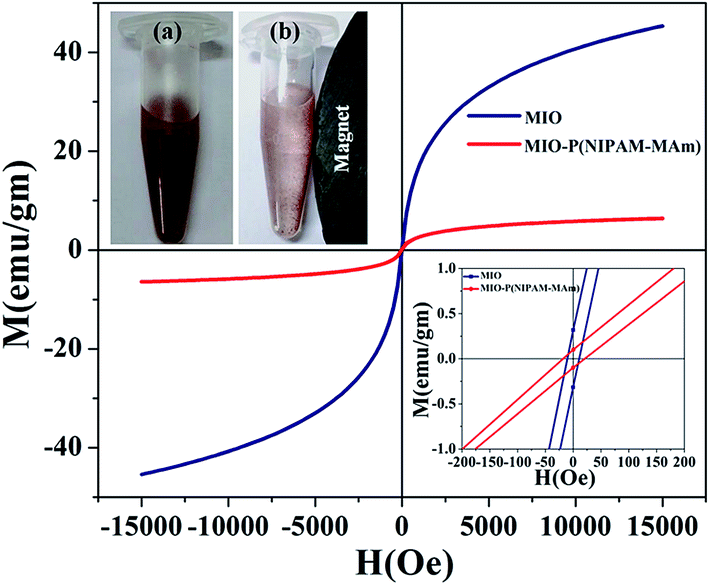
3.2.6. VSM analysis. The magnetic properties of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were examined by vibrating sample magnetometer. The magnetization (M) versus applied magnetic field (H) plots of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite measured at an applied field between −1.5 and 1.5 T at 300 K are shown in Fig. 9. The absence of hysteresis loop demonstrated the superparamagnetic nature of both the samples with negligible coercivity and remnant magnetization.54 The saturation magnetization (Ms) values for MIO NPs and MIO-P(NIPAM-MAm) nanocomposite were found to be 45 emu g−1 and 6.5 emu g−1, respectively. Ms value of the sample represents its magnetic controllability by an external applied magnetic field. Obtained Ms value of MIO-P(NIPAM-MAm) nanocomposites is sufficient for magnetically guided drug delivery applications. The reduced saturation magnetization of the MIO-P(NIPAM-MAm) nanocomposite is due to the introduction of non-magnetic polymer in the sample.25 The obtained Ms value of the MIO-P(NIPAM-MAm) nanocomposite indicates that it is made up of ∼14.5% magnetic component and ∼85.5% nonmagnetic component. These values are nearly similar with the results obtained by TGA data. Remnant magnetization values were found to be 0.3 and 0.9 emu per gram for MIO NPs and MIO-P(NIPAM-MAm) nanocomposite, respectively (inset of Fig. 9). Coercivities were found to be ∼10 and 18 Oe for MIO NPs and MIO-P(NIPAM-MAm) nanocomposite, respectively. Magnetic controllability of aqueous MIO-P(NIPAM-MAm) nanocomposite under external magnetic field has also been demonstrated. Digital photographs of aqueous MIO-P(NIPAM-MAm) nanocomposite without (a) and with permanent magnet (b) are shown in the inset of Fig. 9 (top). Image (a) shows aqueous dispersion of MIO-P(NIPAM-MAm) nanocomposite after storage of ∼1 weak. It is observed from image (a) that MIO-P(NIPAM-MAm) nanocomposite has good dispersibility and high stability in water due to its hydrophilic nature. On application of permanent magnet near to above dispersion, MIO-P(NIPAM-MAm) nanocomposite in water rapidly attracted toward the magnet, indicating its magnetic guidability (b).
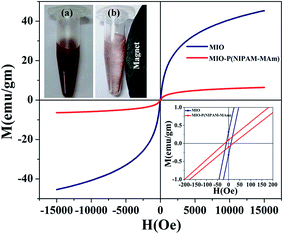 |
| | Fig. 9 Room temperature magnetization behaviour (M–H) of MIO NPs and MIO-P(NIPAM-MAm) nanocomposite in an applied field upto15000 Oe. The inset at the bottom corresponds to a zoom of M–H behaviour to help identifying the remnant magnetization and coercivity fields. The inset at the top shows digital photographs of aqueous MIO-P(NIPAM-MAm) nanocomposite (10 mg ml−1), (a) without magnet and (b) near the magnet. | |
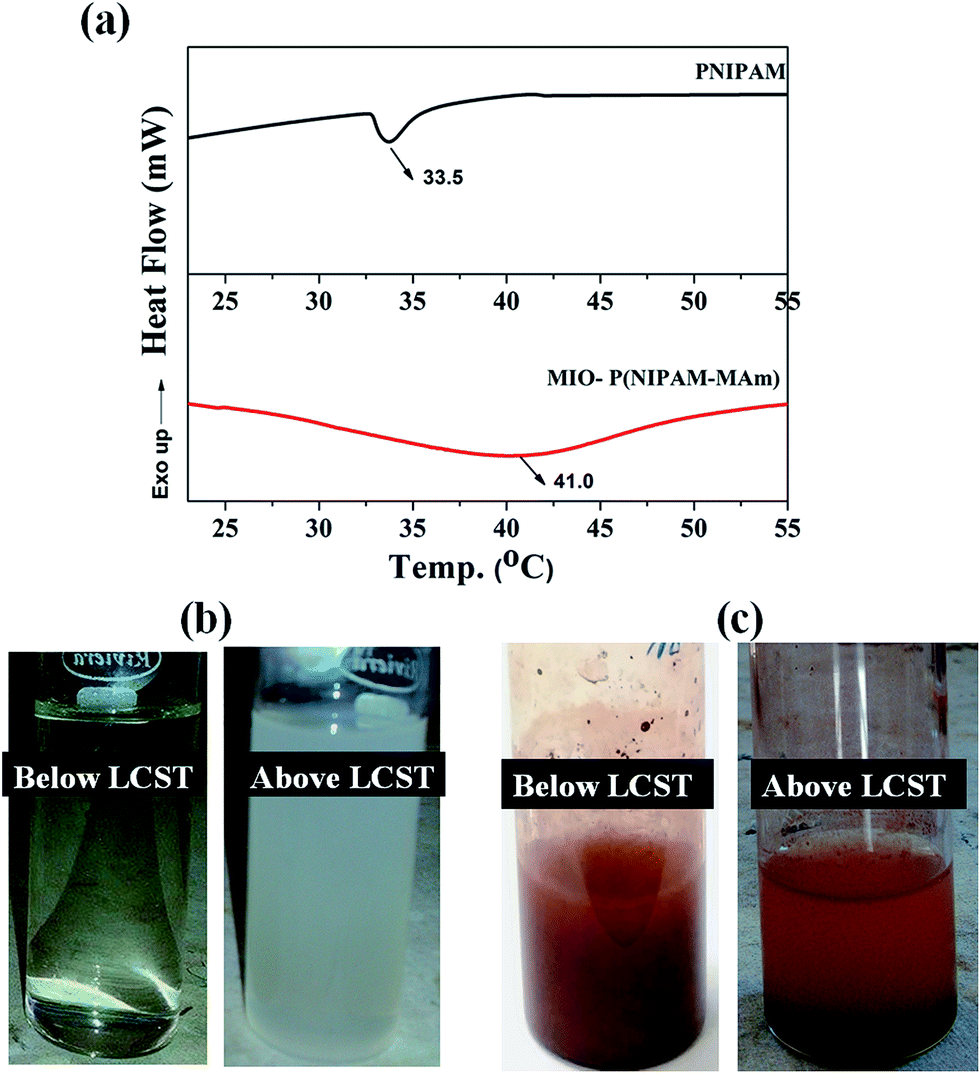
3.2.7. Determination of LCST of MIO-P(NIPAM-MAm) nanocomposite. LCST of PNIPAM is below human body temperature (∼32–33 °C), thus in its intrinsic form it is not suitable to be used as thermo sensitive drug delivery carrier. It is well known that LCST of PNIPAM can be tailored by incorporation of suitable monomer/co-monomers. It has been observed that LCST of PNIPAM increases by introducing hydrophilic co-monomers and decreases by introducing hydrophobic co-monomers.55,56 DSC is a well-established method to investigate the phase transition of PNIPAM and PNIPAM copolymers in solution. LCST of pure PNIPAM and MIO-P(NIPAM-MAm) nanocomposite were estimated by DSC measurement at a heating rate of 10 °C min−1 and is shown in Fig. 10(a). In the DSC curve of pure PNIPAM a sharp endothermic peak, characteristic of PNIPAM, was observed at ∼33 °C. In case of MIO-P(NIPAM-MAm) nanocomposite, a broad endothermic peak centred at ∼41 °C was observed. These endothermic peaks at the transition temperatures can be attributed to the heat absorption associated with the breaking of hydrogen bonds between polymer and water molecules.57 The increased LCST of nanocomposite can be explained in terms of thermodynamics. The phase transition behaviour of PNIPAM around LCST is an entropy controlled process with ΔH > 0 and ΔS > 0, resulting an endothermic peak around 33 °C in the DSC curve.58 For MIO-P(NIPAM-MAm) hydrogels, due to incorporation of hydrophilic monomer (MAm) the changes in ΔH and ΔS around LCST is decreased, which can be attributed to the formation of hydrogen bonds in between the polymers or polymer and water. This caused the shifting in hydrogel LCST to higher temperature. As shown in Fig. 10, LCST of P(NIPAM-MAm) is shifted to 41 °C and the endothermic peak covered wider temperature range. It was observed that the endothermic peak for PNIPAM is much sharper than the peak for MIO-P(NIPAM-MAm) nanocomposite. The presence of cross linker in the polymer and its gradual dehydration with increasing temperature resulted in broad endothermic peak for MIO-P(NIPAM-MAm) nanocomposite, whereas for PNIPAM due to absence of cross linker exhibited sharp dehydration at its LCST resulting in narrow endothermic peak.59 Figure (b) shows the digital images of aqueous dispersion of PNIPAM below and above LCST. It can be observed that aqueous dispersion of PNIPAM, which was transparent below LCST turned to turbid milky above LCST. This can be attributed to the transition of PNIPAM from hydrophilic (below LCST) to hydrophobic above LCST. Figure (c) shows the digital images of aqueous dispersion of MIO-P(NIPAM-MAm) nanocomposite below and above LCST. It was observed that MIO-P(NIPAM-MAm) nanocomposite mostly settled down in medium above LCST (41 °C) within minute of its dispersion. This is due to the weakened hydrophilicity of MIO-P(NIPAM-MAm) nanocomposite at 41 °C. Thus, observed result suggests LCST of MIO-P(NIPAM-MAm) is ∼41 °C.
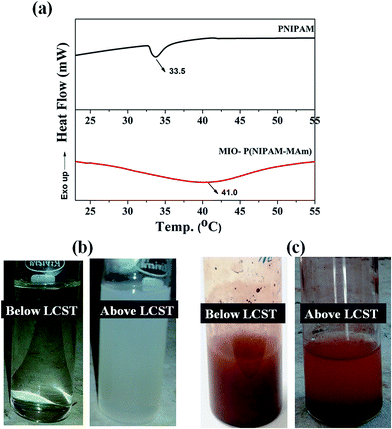 |
| | Fig. 10 DSC thermograms of aqueous solutions of (a) PNIPAM and MIO-P(NIPAM-MAm) nanocomposite. Digital images of aqueous dispersion of (b) PNIPAM and (c) MIO-P(NIPAM-MAm) nanocomposite below and above LCST respectively. | |
3.3 Drug loading studies
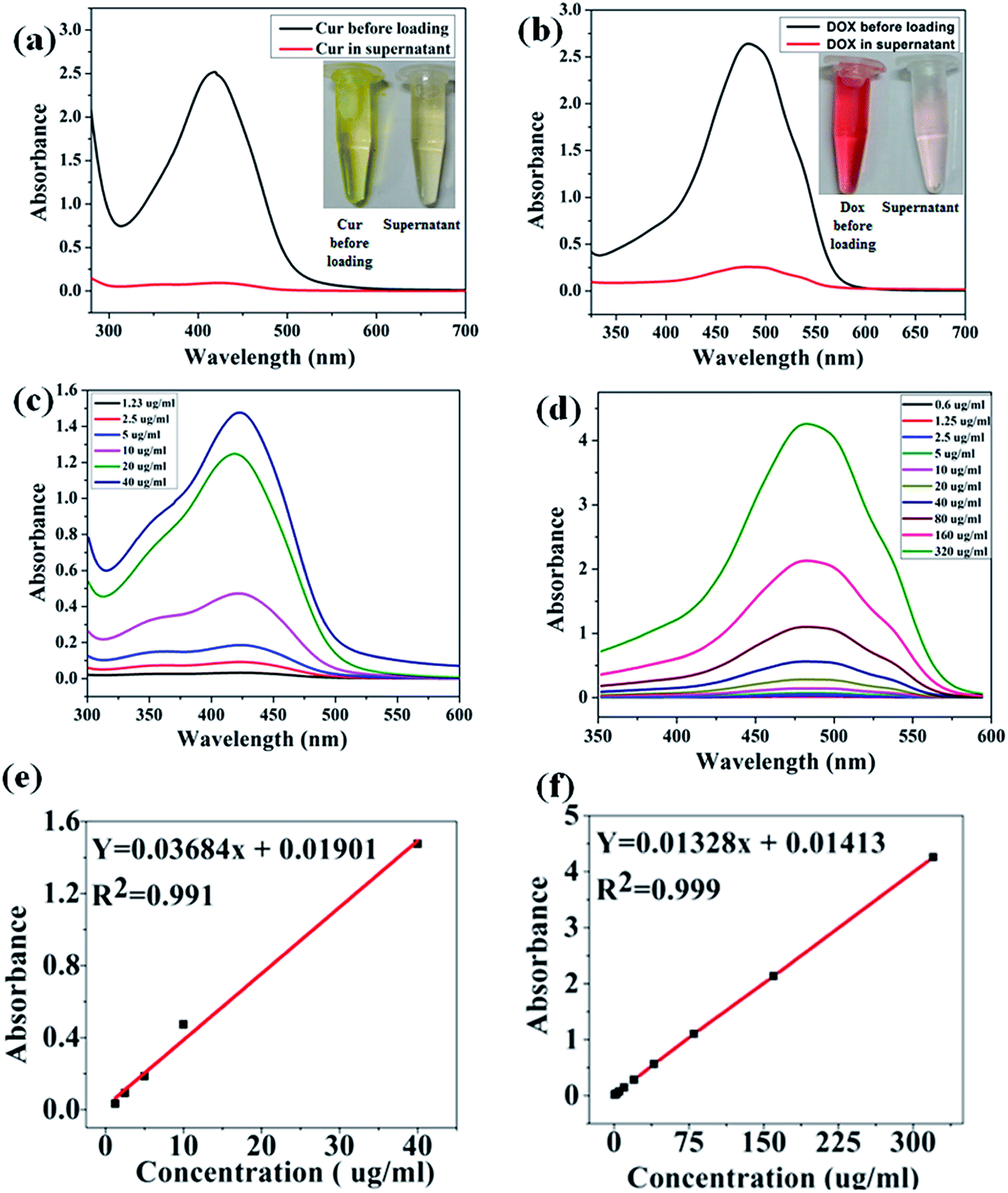
Owing to the hydrophilic nature of the polymer coating and hydrophobic nature of the mesoporous ferrite core, prepared MIO-P(NIPAM-MAm) core–shell nanocomposite could be used to carry both hydrophilic and hydrophobic drug. In this study, hydrophilic and hydrophobic anticancer drugs such as, Dox and Cur, respectively, were chosen as the model drugs and their loading and release behaviour were studied using UV-Vis spectrophotometer. The amount of drug loaded was quantified by subtracting the amount of free unloaded drug from the total drug taken in the experiment. The comparative absorption spectra with the corresponding digital images of supernatant and total drugs taken for both Cur and Dox are shown in Fig. 11(a) and (b) respectively. Presence of very small intense absorption peaks of both free Cur and Dox in supernatant suggested their high loading. Appearances of very light yellow and red colours of unloaded Cur and Dox, relatively, in the supernatant also suggested their high loading in the nanocomposite. Standard calibration curves of both Cur (Fig. 11(c)) and Dox (Fig. 11(d)) were plotted and used for quantification of drugs loading. A linear fitting of the standard curves both for Cur (figure e) and Dox (figure f) were obtained. The drug encapsulation capacity and encapsulation efficiency were calculated by using eqn (1) and (2). The Drug loading capacity and encapsulation efficiency of Cur were found to be ∼25% (250 ng μg−1 particle) and ∼96.8%, respectively. The drug encapsulation capacity and encapsulation efficiency of Dox were found to be ∼5% (50 ng μg−1 particle) and ∼90%, respectively. Co-loading of Cur and Dox in nanocomposite was confirmed by obtaining absorption spectra of drug loaded (MIO-P(NIPAM-MAm)-Cur-Dox) sample.
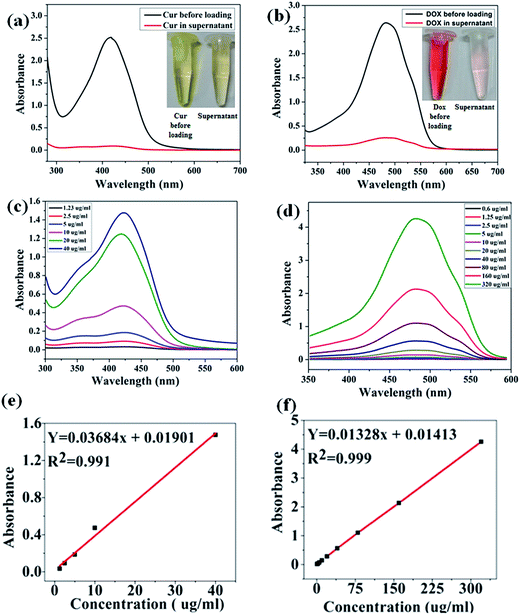 |
| | Fig. 11 UV-Vis absorption spectra and digital photographs of (a) Cur before loading and free Cur in supernatant (b) Dox before loading and free Dox in supernatant. UV-Vis absorption spectra of different known concentration of (c) Cur and (d) Dox with corresponding standard calibration ((e) and (f)). | |
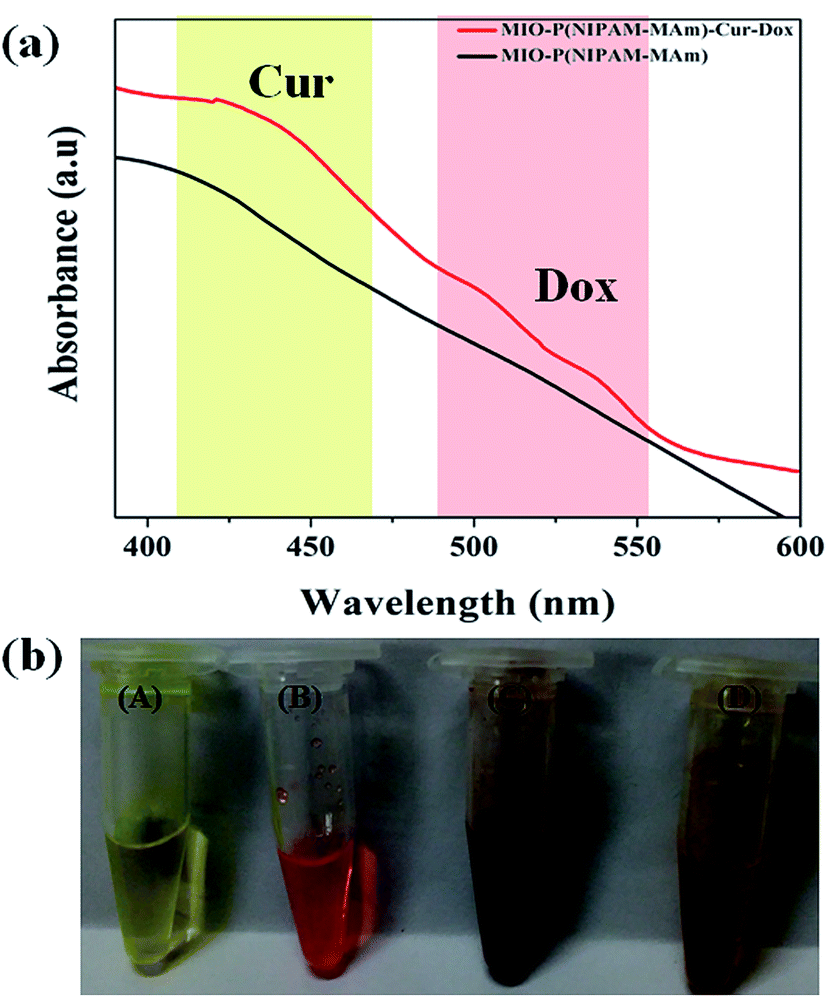
Fig. 12(a) shows the absorption spectra of MIO-P(NIPAM-MAm) and MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite. Simultaneous appearances of characteristic absorption peaks of both Cur (at ∼420 nm) and Dox (at ∼500–536 nm) in MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite strongly confirmed loading of Cur and Dox in the nanocomposite. Digital images in Fig. 12(b) show the physical appearances of the free Cur, free Dox, MIO-P(NIPAM-MAm) and MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite dispersion. It can be observed that blackish brown MIO-P(NIPAM-MAm) nanocomposite turned into reddish brown after loading with red coloured Dox and yellow coloured Cur, which also suggests formation of MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite.
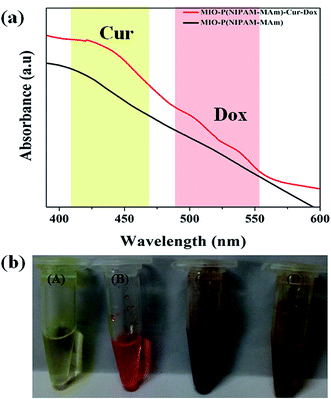 |
| | Fig. 12 (a) UV-Vis absorption spectra of MIO-P(NIPAM-MAm) and MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite (b) digital images of (A) free Cur, (B) free Dox, (C) MIO-P(NIPAM-MAm) nanocomposite and (D) MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite dispersion. | |
3.4 Drug release studies
In vitro release study of Dox and Cur was carried out at two different temperatures (above and below LCST) to evaluate the thermo responsive nature of the MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite in PBS solution (pH 7.4) for 68 hours. Fig. 13(a) shows the release profile of Cur from the MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite at 37 °C (below LCST) and 42 °C (above LCST) as a function of time. It can be observed that the release profile at two different temperatures was almost same, but above LCST release rate of cur was faster than that at 37 °C. Upto 24 hours 14% Cur has been released at temperature above LCST of the system, while at temperature below LCST, only 6% drug release was observed, which enhances to 27% (337.5 μg) and 8% (100 μg) at 42 °C and 37 °C, respectively, in 68 h. The observed release behaviour suggests the drug release mechanism to be associated with the LCST of the polymeric carrier system. Fig. 13(b) shows the release profile of the Dox from the MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite at 37 °C (below LCST) and 42 °C (above LCST) as a function of time. Upto 24 hours 80% of loaded Dox has been released at temperature above LCST of the system, while at temperature below LCST, only 45% drug release is observed. The drug release significantly increases to 97% (242.5 μg) and 58% (145 μg) at 42 °C and 37 °C, respectively, in 68 hours. Because of stable nature of drug carrier, only a small amount of Dox is released at 37 °C from the MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite and this slow release below the LCST could most likely be attributed to the concentration difference of the drug inside and outside the nanocomposite. However, when the temperature is above the LCST, a rapid release of Dox is observed, which may be due to the deswelling of the thermoresponsive P(NIPAM-MAm) polymer i.e. the volume of the MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite was contracted resulting in rapid release of Dox in first 24 hour. After 24 hours, a slow and sustained release of Dox was observed. This slow release behaviour is attributed jointly to thermal response of P(NIPAM-MAm) and concentration gradient based diffusion of Dox in the medium. Decrease in the concentration gradient of Dox with time could be the reason for slow release of Dox. Compared to Dox relatively less release of Cur may be related to poor solubility of Cur in water. Increased diffusion path length, (since it was loaded in the interior hydrophobic core of the nanocomposite) could also be a reason for less release of Cur from MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite.
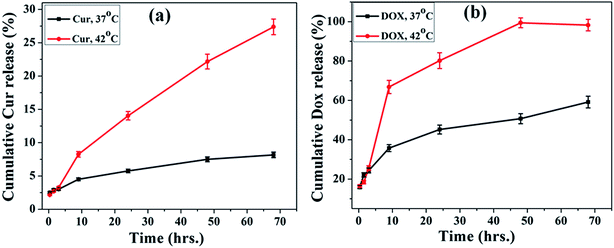 |
| | Fig. 13 The cumulative release of (a) Cur and (b) Dox from the MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite at temperature 37 °C and 42 °C. | |
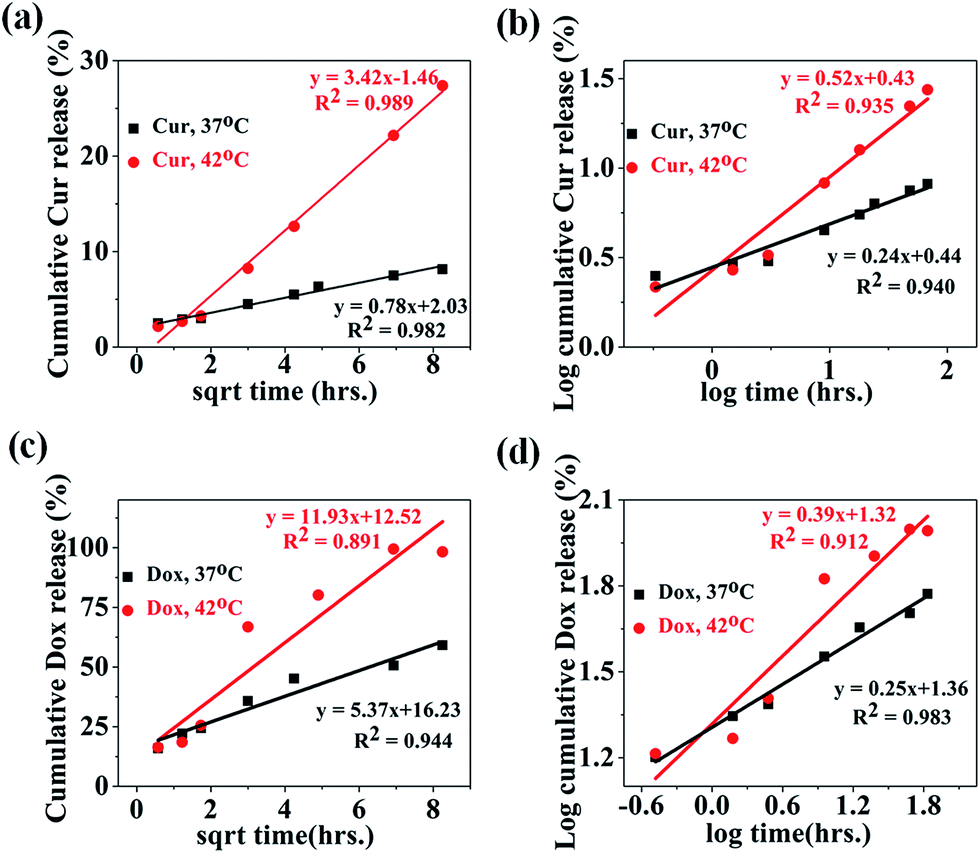
In the literature, a number of mathematical models have been used to interpret the drug release data and associated release mechanism. Thus, different mathematical models such as zero order, first order, Higuchi and Korsmeyer–Peppas were used to fit the experimental released profile of both drugs (Cur and Dox) obtained at 37 °C & 42 °C. The release kinetics and mechanism of Dox and Cur releases from the MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite were thus determined by fitting the release data with these models. Obtained fitting parameters (such as K0, K1, KH, n and R2) for all the models are mentioned in Table 1. The model which fitted best with the release profile data was identified by high correlation coefficient (R2). The release profiles of Cur from MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite have shown best fit with Higuchi's equation, with the R2 values of 0.982 and 0.989 for temperatures 37 °C and 42 °C, respectively (Fig. 14(a)). This suggests that Cur release was most probably diffusion controlled. The zero and first order kinetic models does not fit well with all the experimental data except for Cur release data at 42 °C, where both (zero and first order kinetic) have shown relatively better fitting but certainly not better than Huguchi model. Further confirmation of the diffusion controlled released mechanism of Cur was made by fitting the experimental release data with Korsmeyer–Peppas model with fitting coefficient, R2 = 0.940 and 0.935 at temperatures of 37 °C and 42 °C, respectively (Fig. 14(b)). The n values were found to be 0.24 and 0.52 for Cur released profile at 37 °C and 42 °C respectively. It is known that n < 0.5 indicates Fickian diffusion, whereas n > 0.5 indicates anomalous diffusion. Thus, at 37 °C, Cur release mechanism followed Fickian diffusion but at 42 °C, both diffusion and swelling-controlled release were observed.60,61 Release profiles of Dox from MIO-P(NIPAM-MAm) nanocomposite have shown poor fitting with Higuchi model with lower correlation coefficient, R2 = 0.944 and 0.891 at 37 °C and 42 °C, respectively (Fig. 14(c)). The Dox release data at temperatures 37° C and 42 °C gave better fitting with Korsmeyer–Peppas model (Fig. 14(d)). The Korsmeyer–Peppas fittings were better than Huguchi model fittings for both release data at 37 °C (R2 = 0.983) and 42 °C (R2 = 0.912). The n values, 0.25 and 0.39 at temperatures 37 °C and 42 °C, respectively, indicated a diffusion control mechanism for Dox release from MIO-P(NIPAM-MAm) nanocomposite.
Table 1 Release kinetic parameters of Dox and Cur released from MIO-P(NIPAM-MAm)-Cur-Dox at 37 and 42 °C
| Drug, temp. |
Zero-order |
First-order |
Higuchi |
Korsemeyer–Peppas |
| K0 |
R2 |
K1 |
R2 |
KH |
R2 |
n |
R2 |
| Cur, 37 °C |
0.0833 |
0.87 |
0.0008 |
0.88 |
0.78 |
0.98 |
0.24 |
0.94 |
| Cur, 42 °C |
0.3749 |
0.97 |
0.0044 |
0.98 |
3.42 |
0.99 |
0.52 |
0.93 |
| Dox, 37 °C |
0.557 |
0.80 |
0.0095 |
0.87 |
5.37 |
0.94 |
0.25 |
0.98 |
| Dox, 42 °C |
1.2303 |
0.73 |
0.0700 |
0.82 |
11.93 |
0.89 |
0.39 |
0.91 |
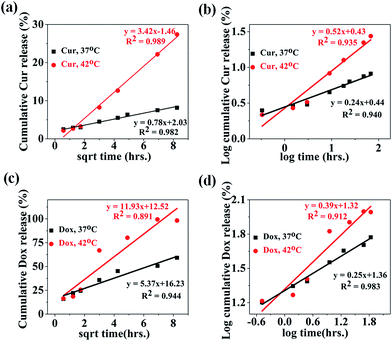 |
| | Fig. 14 Cur release data fitted to (a) Higuchi and (b) Korsmeyer–Peppas kinetics model at the temperature 37 °C and 42 °C. Dox release data fitted to (c) Higuchi and (d) Korsmeyer–Peppas kinetics model at the temperature 37 °C and 42 °C. | |
3.5 In vitro cytotoxicity studies
To evaluate the in vitro biocompatibility and non-toxicity of blank MIO-P(NIPAM-MAm) nanocomposite, MTT assay was performed against non-cancerous normal cells (RAW 264.7).62,63 Fig. 15(a) demonstrates the dose dependent cell viability of RAW 264.7 cells treated with MIO-P(NIPAM-MAm) nanocomposite in concentration of 6.25 to 1000 μg ml−1. It was observed that MIO-P(NIPAM-MAm) nanocomposite has no cytotoxicity on the RAW 264.7 cells even at high dose of 100 μg ml−1 after incubation for 24 h. A high cell viability (∼90%) was observed at 100 μg ml−1. However, when the dose of MIO-P(NIPAM-MAm) nanocomposite increased to 200 μg ml−1, slightly reduced cell viability (∼70%) was observed. When the dose of nanoparticles increased to 1000 μg ml−1 the cell viability was slightly reduced to ∼65%. These results have shown that the synthesized MIO-P(NIPAM-MAm) nanocomposite has less toxicity to RAW 264.7 cells even at high dose of 1000 μg ml−1. Thus, MTT assay results confirmed that the synthesized MIO-P(NIPAM-MAm) nanocomposite was biocompatible to tested normal cells and could potentially be applied for drug delivery application.
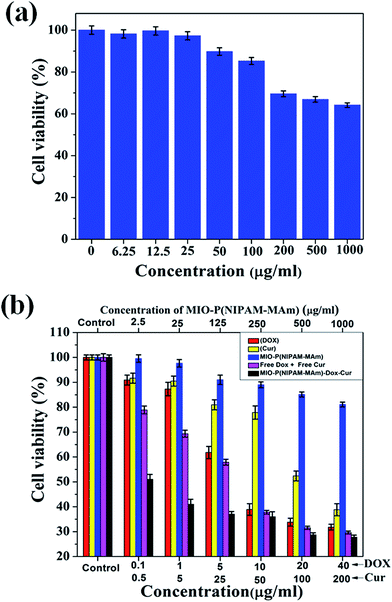 |
| | Fig. 15 Cell viabilities measured by MTT assay of (a) RAW 264.7 cells incubated with different concentration of the MIO-P(NIPAM-MAm) nanocomposite and (b) HeLa cell incubated with free Dox, free Cur, blank MIO-P(NIPAM-MAm) nanocomposite and MIO-P(NIPAM-MAm)-Cur-Dox at different concentrations for 24 h. | |
In order to determine the enhanced anticancer activities of loaded Cur and Dox, MTT assay of MIO-P(NIPAM-MAm) nanocomposite, free Dox, free Cur, free Dox + free Cur mixture, and MIO-P(NIPAM-MAm)-Cur-Dox against cancerous HeLa cells was performed. Fig. 15(b) demonstrates the cytotoxicity profile of blank MIO-P(NIPAM-MAM) nanocomposite, free DOX, free Cur, free Dox + free Cur mixture, and MIO-P(NIPAM-MAm)-Cur-Dox against HeLa cells after 24 hours of incubation. All the materials exhibited a dose-dependent cytotoxic effect on HeLa cells. It can be observed that the blank MIO-P(NIPAM-MAm) nanocomposite has less toxic effect on the viability of HeLa cell. The cell viability of HeLa cells treated with blank MIO-P(NIPAM-MAm) nanocomposite remained more than 80% even at maximum concentration of 1000 μg ml−1. The excellent biocompatible nature of MIO-P(NIPAM-MAm) nanocomposite is ideal for cancer treatments. Free Dox shows slow cell inhibition rate upto 1 μg ml−1 (12.5%) and then exhibited rapid killing rate till 10 μg ml−1 resulting in ∼60% inhibition followed by rapid decrease in inhibition rate. In case of free Dox maximum cell killing was found to be 67.8% at highest dose of 40 μg ml−1. IC50 for free Dox was found to be 10 μg ml−1. Free Cur shows slow cell inhibition rate upto 50 μg ml−1 (22%) followed by fast killing rate till the highest dose of 200 μg ml−1 resulting in ∼62% inhibition. IC50 for free Cur was found to be 100 μg ml−1. The free Cur has shown lesser cytotoxicity than free Dox. However, Cur has been demonstrated as an active agent that can induce synergetic effect with other chemotherapeutic drug (such as Dox) against different cancer cell lines.35 Free Dox + free Cur mixture shows ∼30% cell inhibition upto 1 μg ml−1 dose in term of Dox concentration. The IC50 value of free Dox + free Cur mixture was similar to the free Dox.32 In case of free Dox + free Cur mixture, the maximum cell killing was found to be ∼29% at highest dose. MIO-P(NIPAM-MAm)-Cur-Dox shows rapid decrease in cell viability (upto 52%) for very low dose (of co-loaded drugs), i.e. 0.1 μg ml−1 of loaded Dox and 0.5 μg ml−1 of loaded Cur. There after inhibition rate becomes slightly slow. MIO-P(NIPAM-MAm)-Cur-Dox shows maximum killing of 73% at highest dose of 40 μg ml−1 of loaded Dox and 200 μg ml−1 of loaded Cur. The cell inhibition of MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite was 48% (at dose of 0.1 μg ml−1 of loaded Dox and 0.5 μg ml−1 of loaded Cur), which was significantly higher than those obtained for free Dox (9%),free Cur (8%) and free Dox + free Cur mixture (21%) for the same dose. IC50 of MIO-P(NIPAM-MAm)-Cur-Dox was found to be 1 μg ml−1 in terms of loaded Dox. Observed IC50 value is better than previously reported value for similar system.32 The higher inhibition rate of MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite indicates that co-loading of Dox and Cur enhances the in vitro efficacy of the nanocomposite. Recently, Yumin Zhang et al. have also observed enhanced cell inhibition of HeLa cells for PEG-DOX-CUR NPs than free Dox, free Cur and free Dox-Cur mixture. They have explained this phenomenon as follow, first PEG-DOX-CUR NPs were internalized within cells, then Dox and Cur release by NPs were accumulated within cells and played a key role in killing cancerous cells.32 Similarly, in the present study, the superior cytotoxicity of MIO-P(NIPAM-MAm)-Cur-Dox may be due to the increased internalization as well as accumulation of Dox and Cur in HeLa cell.32,34 Thus, prepared MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite with enhanced anticancer activity, magnetic guidability, and dual drug loading ability could be an ideal nano-carrier for cancer therapy.
4. Conclusion
In this work, P(NIPAM-MAm) coated mesoporous Fe3O4 (MIO-P(NIPAM-MAm)) nanocomposite, a novel and smart nanocarrier was successfully been prepared for co-delivery of Dox and Cur in combinational therapy for cancer treatment. Prepared MIO NPs and MIO-P(NIPAM-MAm) nanocomposites were characterized for different physiochemical properties using XRD, SEM, TEM, TGA, DSC, VSM, and BET. Prepared MIO-P(NIPAM-MAm) nanocomposite is biocompatible, magnetically guidable and well dispersible in water due to its hydrophilic nature. VSM results showed prepared MIO and MIO-P(NIPAM-MAm) nanocomposite were superparamagnetic in nature. MIO-P(NIPAM-MAm) nanocomposite exhibited a LCST of 41 °C, which is suitable for use as thermo-responsive drug delivery nanocarrier, unlike pure PNIPAM based nanocarriers. To investigate the dual drug loading ability of MIO-P(NIPAM-MAm) nanocomposite, Dox and Cur were used as hydrophilic and hydrophobic model drugs, respectively. Loading efficiency of Dox and Cur were found to be 96% and 90% respectively. Temperature dependent drug release behaviour was observed for both the drugs. The release rates of Dox and Cur from MIO-P(NIPAM-MAm) nanocomposite were relatively faster at temperature above LCST (41 °C) than below LCST (body temperature-37 °C) in PBS 7.4. MTT assays on normal and HeLa cells demonstrated the non-toxic nature of the MIO-P(NIPAM-MAm) nanocomposite. MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite exhibited higher anti-cancer activity than free Dox, free Cur and free Dox-free Cur mixture. Obtained results suggest that prepared novel, smart, and biocompatible MIO-P(NIPAM-MAm)-Cur-Dox nanocomposite is ideal nano-formulation for simultaneous delivery of hydrophobic and hydrophilic anticancer drugs for combinational therapy of cancer.
Acknowledgements
Khushnuma Asghar greatly acknowledges the financial support obtained from University Grants Commission (UGC) in the form of MANF fellowship (201415-MANF-2014-15-MUS-BIH-42040) in carrying out this research work. The technical support received from the School of Engineering Sciences & Technology (SEST), Central Instruments Laboratory (CIL) and Centre for Nanotechnology at the University of Hyderabad is greatly appreciated.
References
- L. Kang, Z. Gao, W. Huang, M. Jin and Q. Wang, Acta Pharm. Sin. B, 2015, 5, 169–175 CrossRef PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- J. k. oh, D. I. Lee and J. M. Park, Prog. Polym. Sci., 2009, 34, 1261–1282 CrossRef CAS.
- A. R. O. Rodrigues, I. T. Gomes, B. G. Almeida, J. P. Araujo, E. M. S. Castanheira and P. J. G. Coutinho, Phys. Chem. Chem. Phys., 2015, 17, 18011–18021 RSC.
- D. Calle, V. Negri, P. Ballesteros and S. Cerdan, Theranostics, 2015, 5, 489–503 CrossRef CAS PubMed.
- H. Dai, Acc. Chem. Res., 2002, 35, 1035–1044 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- J. L. Vivero-Escoto and Y.-T. Huang, Int. J. Mol. Sci., 2011, 12, 3888–3927 CrossRef CAS PubMed.
- J. Liu, C. Detrembleur, S. Mornet, C. Jerome and E. Duguet, J. Mater. Chem. B, 2015, 3, 6117–6147 RSC.
- D. Shi, H. Sung Cho, Y. Chen, H. Xu, H. Gu, J. Lian, W. Wang, G. Liu, C. Huth, L. Wang, R. C. Ewing, S. Budko, G. M. Pauletti and Z. Dong, Adv. Mater., 2009, 2, 2170–2173 CrossRef.
- M. Rahimi, A. Wadajkar, K. Subramanian, M. Yousef, W. Cui, J. Hsieh and K. Nguyen, Nanomedicine, 2010, 6, 672–680 CrossRef CAS PubMed.
- S. Xuan, F. Wang, J. M. Y. Lai, K. W. Y. Sham, Y.-X. J. Wang, S.-F. Lee, J. C. Yu, C. H. K. Cheng and K. C.-F. Leung, ACS Appl. Mater. Interfaces, 2011, 3, 237–244 CAS.
- X. Liu, Q. Hu, Z. Fang, Q. Wu and Q. Xie, Langmuir, 2009, 25, 7244–7248 CrossRef CAS PubMed.
- S. Zhang, W. Wu, X. Xiao, J. Zhou, F. Ren and C. Jiang, Nanoscale Res. Lett., 2011, 6(89), 1–9 Search PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- J. Zhu, Y. Niu, Y. Li, Y. Gong, H. Shi, Q. Huo, Y. Liu and Q. Xu, J. Mater. Chem. B, 2017, 5, 1339–1352 RSC.
- S. Patra, E. Roy, P. Karfa, S. Kumar, R. Madhuri and P. K. Sharma, ACS Appl. Mater. Interfaces, 2015, 7, 9235–9246 CAS.
- G. Fundueanu, M. Constantin, I. Oanea, V. Harabagiu, P. Ascenzi and B. C. Simionescu, Acta Biomater., 2012, 8, 1281–1289 CrossRef CAS PubMed.
- T. M. Quynh, M. Yoneyamab, Y. Maki and T. Dobashi, J. Appl.
Polym. Sci., 2012, 123, 2368–2376 CrossRef CAS.
- B. Chang, X. Sha, J. Guo, Y. Jiao, C. Wang and W. Yang, J. Mater. Chem., 2011, 21, 9239–9247 RSC.
- H. Wakamatsu, K. Yamamoto, A. Nakao and T. Aoyagi, J. Magn. Magn. Mater., 2006, 302, 327–333 CrossRef CAS.
- A. Papagiannopoulos, J. Zhao, G. Zhang, S. Pispas and A. Radulescu, Eur. Polym. J., 2014, 56, 59–68 CrossRef CAS.
- S. Dhanya, D. Bahadur, G. C. Kundu and R. Srivastava, Eur. Polym. J., 2013, 49, 22–32 CrossRef CAS.
- L. Aihua, M. Hongjing, F. Shengyu and L. Jingquan, RSC Adv., 2016, 6, 33138–33147 RSC.
- K. Bauri, S. G. Roy, S. Arora, R. K. Dey, A. Goswami, G. Madras and P. De, J. Therm. Anal. Calorim., 2013, 111, 753–761 CrossRef CAS.
- B. Seonmi, K. S. Rajendra, H. K. Tae, W. S. Jae, S. S. Ueon, C. Wojciech and W. K. Hae, ACS Appl. Mater. Interfaces, 2016, 8, 8967–8979 Search PubMed.
- M. J. Moura, M. H. Gil and M. M. Figueiredo, Eur. Polym. J., 2013, 49, 2504–2510 CrossRef CAS.
- H. Ma, C. He, Y. Cheng, Z. Yang, J. Zang, J. Liu and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 27040–27048 CAS.
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044–1052 CrossRef CAS PubMed.
- R. Misra and S. K. Sahoo, Mol. Pharmaceutics, 2011, 8, 852–866 CrossRef CAS PubMed.
- Y. Zhang, C. Yang, W. Wang, J. Liu, Q. Liu, F. Huang, L. Chu, H. Gao, C. Li, D. Kong, Q. Liu and J. Liu, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed , 21225.
- J. Duan, H. M. Mansour, Y. Zhang, X. Deng, Y. Chen, J. Wang, Y. Pan and J. Zhao, Int. J. Pharm., 2012, 426, 193–201 CrossRef CAS PubMed.
- L. Lv, K. Qiu, X. Yu, C. Chen, F. Qin, Y. Shi, J. Ou, T. Zhang, H. Zhu, J. Wu, C. Liu and G. Li, J. Biomed. Nanotechnol., 2016, 12, 973–985 CrossRef CAS PubMed.
- B.-L. Wang, Y.-M. Shen, Q.-W. Zhang, Y.-L. Li, M. Luo, Z. Liu, Y. Li, Z.-y. Qian, X. Gao and H.-S. Shi, Int. J. Nanomed., 2013, 8, 3521–3531 Search PubMed.
- X. Zhao, Q. Chen, W. Liu, Y. Li, H. Tang, X. Liu and X. Yang, Int. J. Nanomed., 2015, 10, 257–270 Search PubMed.
- H. Mackova, D. Kralova and D. Horak, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5884–5898 CrossRef CAS.
- J. Zeng, P. Du, L. Liu, J. Li, K. Tian, X. Jia, X. Zhao and P. Liu, Mol. Pharmaceutics, 2015, 12, 4188–4199 CrossRef CAS PubMed.
- S. Purushotham and R. V. Ramanujan, Acta Biomater., 2010, 6, 502–510 CrossRef CAS PubMed.
- M. Ghorbani, H. Hamishehkar, N. Arsalani and A. A. Entezami, Int. J. Polym. Mater. Polym. Biomater., 2016, 65, 683–694 CrossRef CAS.
- M. Qasim, K. Asghar, B. R. Singh, S. Prathapani, W. Khan, A. H. Naqvi and D. Das, Spectrochim. Acta, Part A, 2015, 137, 1348–1356 CrossRef CAS PubMed.
- S. Liu, K. Yao, L.-H. Fu and M.-G. Ma, RSC Adv., 2016, 6, 2135–2140 RSC.
- M. Gharagozlou, Chem. Cent. J., 2011, 5(19), 1–7 Search PubMed.
- J. Singh, H. Singh and U. Batra, in Magnesium Doped Hydroxyapatite: Synthesis, Characterization and Bioactivity Evaluation, ed. R. Narayan, S. Bose and A. Bandyopadhyay, John Willy & Sons, Pennsylvania, 2015, vol 254, pp. 161–174 Search PubMed.
- J. A. Khan, M. Qasim, B. R. Singh, S. Singh, M. Shoeb, W. Khan, D. Das and A. H. Naqvi, Spectrochim. Acta, Part A, 2013, 109, 313–321 CrossRef CAS PubMed.
- M. Sun, A. Zhu, Q. Zhang, M. Ye and Q. Liu, Eur. Polym. J., 2015, 66, 569–576 CrossRef CAS.
- J. Wang, X. He, L. Chen and Y. Zhang, RSC Adv., 2016, 6, 47055–47061 RSC.
- M. Qasim, B. R. Singh, A. H. Naqvi, P. Paik and D. Das, Nanotechnology, 2015, 26, 285102 CrossRef CAS PubMed.
- X. Liu, Q. Hu, Z. Fang, Q. Wu and Q. Xie, Langmuir, 2009, 25, 7244–7248 CrossRef CAS PubMed.
- Z. Huang and F. Tang, J. Colloid Interface Sci., 2005, 281, 432–436 CrossRef CAS PubMed.
- F. Chen, Q. Chen, S. Fang, Y. Sun, Z. Chen, G. Xie and Y. Du, Dalton Trans., 2011, 40, 10857–10864 RSC.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. C. Wang, G. Zou and X.-J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- A. Ramazani, N. Arsalani, H. Khaksar Khiabani and A. M. Goganian, J. Biomimetics, Biomater., Tissue Eng., 2013, 18, 1000111 Search PubMed.
- R. Gui and H. Jin, RSC Adv., 2014, 4, 2797–2806 RSC.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–2124 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- M. Scholnhoff, A. Larsson, P. B. Welzel and D. Kuckling, J. Phys. Chem. B, 2002, 106, 7800–7808 CrossRef.
- J. Cheng, G. Shan and P. Pan, RSC Adv., 2015, 5, 62091–62100 RSC.
- Md. Ashraful Alam, M. Takafuji and H. Ihara, J. Colloid Interface Sci., 2013, 405, 109–117 CrossRef CAS PubMed.
- R. Das, D. Das, P. Ghosh, S. Dhara, A. B. Panda and S. Pal, RSC Adv., 2015, 5, 27481–27490 RSC.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS PubMed.
- K. Kumar, S. Kumar Doddi, M. Kalle Arunasree and P. Paik, Dalton Trans., 2015, 44, 4308–4317 RSC.
- M. I. Majeed, Q. Lu, W. Yan, Z. Li, I. Hussain, M. N. Tahir, W. Tremel and B. Tan, J. Mater. Chem. B, 2013, 1, 2874–2884 RSC.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Mohd Qasim
a,
Mohd Qasim