
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Factors affecting formation of deethyl and deisopropyl products from atrazine degradation in UV/H2O2 and UV/PDS
Congwei Luoa,
Jin Jiang *a,
Chaoting Guana,
Jun Ma*a,
Suyan Pangb,
Yang Songa,
Yi Yanga,
Jianqiao Zhangc,
Daoji Wud and
Yinghong Guane
*a,
Chaoting Guana,
Jun Ma*a,
Suyan Pangb,
Yang Songa,
Yi Yanga,
Jianqiao Zhangc,
Daoji Wud and
Yinghong Guane
aState Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin, 150090, China. E-mail: jiangjinhit@126.com; majun@hit.edu.cn; Fax: +86-451-86283010; Tel: +86-451-86283010
bKey Laboratory of Green Chemical Engineering and Technology of College of Heilongjiang Province, College of Chemical and Environmental Engineering, Harbin University of Science and Technology, Harbin 150040, China
cLuohu District Environment Protection and Water Affairs Bureau, China
dSchool of Municipal and Environmental Engineering, Shandong Jianzhu University, Jinan, 250010, China
eSchool of Water Conservancy and Civil Engineering, Northeast Agricultural University, Harbin 150040, China
First published on 5th June 2017
Abstract
In this study, the formation of deethyl products (DEPs) (i.e., atrazine amide (Atra-imine) and deethylatrazine (DEA)) and deisopropyl product (i.e., deisopropylatrazine (DIA)) from parent atrazine (ATZ) degraded in UV/H2O2 and UV/PDS processes under various conditions was monitored. It was found that SO4˙− displayed a more distinctive preference to the ethyl function group of ATZ than HO˙, leading to the higher ratio of DEPs/DIA in UV/PDS system than that in UV/H2O2 system in pure water. The effects of water matrices (i.e., natural organic matter (NOM), carbonate/bicarbonate (HCO3−/CO32−), and chloride ions (Cl−)) on ATZ degradation as well as formation of DEPs and DIA were evaluated in detail. The degradation of ATZ by UV/PDS was significantly inhibited in the presence of NOM, HCO3−/CO32− or Cl−, because these components could competitively react with SO4˙− and/or HO˙ to generate lower reactive secondary radicals (i.e., organic radicals, carbonate radicals (CO3˙−) or reactive chlorine radicals (RCs)). The yields of these DEPs and DIA products from ATZ degradation were not impacted by NOM or HCO3−/CO32−, possibly due to the low reactivity of organic radicals and CO3˙− toward the side groups of ATZ. Howbeit, the increase of DIA yield companied with the decrease of DEPs yield was interestingly observed in the presence of Cl−, which was attributed to the promotion of Cl− at moderate concentration (mM range) for the conversion of SO4˙− into HO˙. Comparatively, in the UV/H2O2 process, NOM and HCO3−/CO32− exhibited a similar inhibitory effect on ATZ degradation, while the influence of Cl− was negligible. Differing from UV/PDS system, all these factors did not change DEPs and DIA yields in UV/H2O2 process. Moreover, it was confirmed that RCs had a greater selectivity but a lower reactivity on attacking the ethyl function group than that of SO4˙−. These findings were also confirmed by monitoring the degradation of ATZ as well as the formation of DEPs and DIA in three natural waters.
Introduction
Atrazine (6-chloro-N-ethyl-N′-(1-methylethyl)-1,3,5-triazine-2,4-diamine; ATZ), which has been most widely used as a synthetic herbicide to kill broadleaf weeds over the last half century,1,2 has been frequently detected in ground and surface water.2–4 Since many studies reported the high toxicity and endocrine disruption potentials of ATZ even at trace levels, the effective removal of ATZ in drinking water treatment has attracted great concerns in the protection of aquatic ecosystems.5 Numerous technologies have been developed to remove ATZ, including biological treatment, physical absorption, and chemical oxidation.6–10 Among these, chemical degradation has been generally recognized as a promising and decisive step.Recently, advanced oxidation processes (AOPs) based on hydroxyl radicals (HO˙, E0 = 1.89–2.72 V vs. NHE) and sulfate radicals (SO4˙−, E0 = 2.5–3.1 V vs. NHE) have been of increasing interest as an option of ATZ degradation.1,11,12 The reaction rates of these two radicals with ATZ have been reported to be similar and very fast (i.e., k = 3.0 × 109 M−1 s−1).13,14 The effects of water matrices, including carbonate/bicarbonate (CO32−/HCO3−), chloride (Cl−), and natural organic matters (NOM) on the degradation kinetics of ATZ in these AOPs have been evaluated as well. It has been suggested that these ubiquitous species can react with HO˙ and/or SO4˙− to different degrees, thus influencing the degradation rate of ATZ. For instance, it was reported that the presence of CO32−/HCO3− (>4 mM) inhibited ATZ oxidation by HO˙ and SO4˙− generated from magnetic porous copper ferrite catalyzed peroxymonosulfate.15 In our previous study, it was found that Cl− over the concentration range of 0.5 to 10 mM had a slight scavenging effect on ATZ removal in UV/hydrogen peroxide (UV/H2O2) process but significantly suppressed the degradation of ATZ in UV/persulfate (UV/PDS) process.16 Additionally, ATZ was degraded more efficiently by SO4˙− than by HO˙ in the presence of natural organic matter (NOM), which was attributed to the lower SO4˙− reaction rates with NOM than HO˙ (kSO4−˙,ATZ = 6.8 × 103 mg C−1 s−1 vs. kHO˙,ATZ = 1.4 × 104 mg C−1 s−1).12
Moreover, the mechanisms of ATZ transformation in HO˙ and/or SO4˙− based oxidation processes have been extensively investigated,6,17–19 and dealkylation is suggested as a preferred reaction pathway.20–23 HO˙ and/or SO4˙− abstract H-atom from the carbon adjacent to nitrogen to produce a carbon-centered radical intermediate, and the addition of O2 to this intermediate results in generation of a peroxide radical. Then, followed by the loss of perhydroxyl radical, atrazine amide (Atra-imine) can be produced, and its subsequent hydrolysis leads to the formation of deethylatrazine (DEA).1 A similar mechanism is expected for the formation of simazine amide and its hydrolysis product deisopropylatrazine (DIA). The hydrolysis of simazine amide is much faster than Atra-imine.24 In addition, another two pathways including alkyl chain oxidation and dechlorination-hydroxylation can also occur during ATZ oxidation.25
Further, the percentages of deethyl products (DEPs) and desisopropyl product (i.e., DIA) were calculated, and their total yields accounted for about 70% degradation of ATZ.1,26 The total yield of DEPs was determined to be higher than the yield of DIA. For instance, the molar ratio of DEPs to DIA was calculated to be 3 in the case of HO˙, while for SO4˙− it was 10.12,27 The higher yield of DEPs than DIA is generally explained by the facts that: (i) the N–H bond at the N-ethyl group is more acidic than that at the N-isopropyl group, and the deprotonation would occur more preferably at the H–N-ethyl site,12 (ii) both DEPs and DIA are unstable toward the further attack of reactive radicals, and the rate constant for the reaction of SO4˙− and/or HO˙ with DIA was slightly higher than that with DEPs (i.e., a slightly faster consumption of DIA),12,27 and (iii) steric hindrance effect of isopropyl group is much more significant than that of ethyl group, thus limiting the formation of DIA.28
Generally, secondary reactive organic or inorganic radicals (e.g., carbonate radical CO3˙−), reactive chlorine radicals (RCs) can be generated by HO˙ and/or SO4˙− reacting with these water matrices.29–31 CO3˙− as a highly selective one-electron oxidant (E0 = 1.78 V vs. NHE) can oxidize some electron-rich compounds (e.g., S-containing organics, N-containing organics, and phenols) through electron transfer or hydrogen abstraction.32–34 Chlorine atoms (Cl˙, E0 = 2.4 V vs. NHE) and dichloride radical (Cl2˙−, E0 = 2.0 V vs. NHE) can also react with various organic compounds, and the pathways mainly involve one-electron oxidation, H-abstraction, and addition to unsaturated C–C bond.35,36 The steady-state concentrations of CO3˙− and RCs have been estimated to be higher than those of HO˙ and SO4˙− for both UV/H2O2 and UV/PDS processes in the presence of CO32−/HCO3− and Cl− (mM range).33 In addition, previous studies have been reported that NOM as a photosensitizer can lead to the photochemical formation of various reactive intermediates such as singlet oxygen (1O2), peroxy radical (ROO˙), and 3DOM* in direct UV photolysis, and these species are considerably reactive toward organic contaminants such as atenolol, carbamazepine, and glimepiride.37 In our previous work, the influence of water constituents (i.e., HCO32−/CO32−, Cl−, and NOM) on ATZ degradation kinetics have been investigated, and these factors on the relative contributions of HO˙ and SO4˙− in UV/PDS and UV/H2O2 processes have been evaluated.16 However, the effects of various water constituents on the formation of primary products (i.e., DEPs and DIA) from ATZ are still unknown.
The main objective of this study was to investigate the formation of primary dealkylation products (i.e., DEPs and DIA) along with the degradation of ATZ in UV/PDS and UV/H2O2 processes in the absence vs. presence of various water constituents, including HCO32−/CO32−, Cl−, and NOM. Then the characteristics of secondary reactive radicals were evaluated by monitoring the changes of the molar ratio of DEPs to DIA. Furthermore, the formation of DEPs and DIA was also examined in three natural waters.
Materials and methods
Reagents
ATZ, DEA, DIA, potassium peroxodisulfate, nitrobenzene (NB), sodium carbonate, hypochlorite (HClO), and sodium chloride of analytical grade were purchased from Sigma-Aldrich Chemical Co. Ltd. (USA). Suwannee River Natural Organic Matter (SRNOM) was purchased from the International Humic Substances Society. H2O2 solution (35% w/w), sodium hypochlorite, phosphate monobasic monohydrate, sodium phosphate dibasic, sodium thiosulfate were of analytical-reagent grade and purchased from Sinopharm Chemical Reagent Co., Ltd., China. Other chemicals were of analytical grade or better and used without further purification. Three real water samples were collected from Songhua River (SW) (China), drinking water plants (EW), and groundwater (GW), respectively. All of the three samples were filtered through 0.45 μm glass fiber membranes (Whatman) prior to using. The water quality parameters of these samples were shown in Table 1. ATZ was spiked to the real water samples to prepare ATZ-containing water.| pH | TOC (mg L−1) | Alkalinity (mM) | Cl− (mM) | |
|---|---|---|---|---|
| SW | 7.6 ± 0.02 | 3.12 | 0.4 | 0.15 |
| GW | 7.9 ± 0.02 | 1.62 | 2.3 | 3.50 |
| EW | 6.8 ± 0.02 | 1.53 | 0.3 | 0.22 |
Analytical methods
ATZ, DEA, and DIA were analyzed using high performance liquid chromatography (HPLC) with Hitachi 5110 pump and 5420 UV detector (Hitachi Chromaster), and the separation was carried out with a symmetry C18 column (4.6 × 150 mm, 5 μm particle size, Waters). For the mobile phase, various isocratic mixtures and gradients of 1‰ acetic acid and methanol were used. The flow rate was set at 1.0 mL min−1, and the sample injection volume was 100 μL. The wavelengths of UV detection were set at 230 nm (ATZ), 260 nm (DEA), and 237 nm (DIA), respectively.23 PDS and H2O2 concentrations were measured by the iodometric method.38 The stock solutions of residual chlorine and active chlorine were standardized by using the triiodide spectrophotometric method.39 NOM (mg C L−1) was measured with a Multi 3100 N/C TOC analyzer (Shimadzu).Experimental procedures
The bench-scale UV irradiator equipped with four Low Pressure Hg UV lamps (254 nm, Heraeus, GPH212T5 L/4). 100 mL of solution with water depth of 4.0 cm was placed directly beneath the collimated tube for UV irradiation. The surface of the solution was approximately 30 cm from the UV lamp (Fig. 1). The irradiance of the UV irradiator was measured by a UV detector UVC radiometer (Photoelectric Instrument Factory of Beijing Normal University, Beijing, China) and calibrated following the suggestion of Bolton and Linden,40 which was estimate to be 0.16 mW cm−2. All experiments were conducted at room temperature (20 °C). Phosphate buffer (5 mM) was used to adjust pH value at 7.0. The samples were withdrawn at predetermined time intervals and immediately quenched using excess sodium sulfite. Then, they were kept at room temperature for 72 h in order to achieve complete conversion of Atra-imine and simazine amide to DEA and DIA, respectively. Thus, the concentrations of initially formed deethyl products (DEPs) and deisopropyl products (DIPs) could be represented by the concentrations of DEA and DIA, respectively.1 All kinetic experiments were replicated independently at least three times, and the error bars represented the standard deviations. | ||
| Fig. 1 The experimental device used for the UV photolysis experiments. | ||
Results and discussion
Degradation of ATZ and formation of DIA and DEPs
Fig. 2a showed the time courses of DEPs and DIA formation during ATZ oxidation in direct UV, UV/H2O2 and UV/PDS processes. As could be seen, the degradation rates of ATZ in UV/PDS and UV/H2O2 processes were much higher than that in direct UV process. DEPs and DIA were generated along with ATZ degradation in UV/PDS and UV/H2O2 processes, and then were completely depleted within the investigated time scales in the UV/PDS process. Comparatively, there were still residuals of the products after 60 min of reaction in the UV/H2O2 process. For direct UV process, neither DEPs nor DIA was detected, which was consistent with the finding that hydroxylated atrazine rather than dealkylation products was the primary product of direct ATZ photolysis in previous study.41 | ||
| Fig. 2 (a) Degradation of ATZ and formation of DEPs and DIA as a function of time in direct UV, UV/H2O2, and UV/PDS processes. (b) Yields of DEPs and DIA as a function of time in UV/H2O2 and UV/PDS processes. The insert showed the DEPs to DIA ratio. Experiment conditions: [ATZ] = 10 μM, [H2O2] = [PDS] = 1.5 mM, and pH = 7. | ||
The second-order rate constants of parent ATZ and its primary oxidation products (e.g. Atra-imine, DEA, DIA, terbuthylazine, and propazine) with HO˙ and SO4˙− were comparable.12 Nevertheless, the higher molar extinction coefficient at 254 nm and the larger quantum efficiency of PDS (21.1 M−1 cm−1 and 1.4 mol einstein−1) than those of H2O2 (18 M−1 cm−1 and 1.0 mol einstein−1) would result in higher steady-state concentrations of SO4˙− than HO˙16. For instance, Lutze et al. reported that the concentration of SO4˙− in UV/PDS process was 1.5–1.7 fold higher than that of HO˙ in UV/H2O2 process.42 These factors lead to the faster degradation of ATZ as well as DEPs and DIA in the UV/PDS process.
In initial stage of ATZ reaction with SO4˙− (i.e., UV/PDS), the formation of DIA and DEPs accounted for approximate 7% and 75% of ATZ consumption, respectively, as shown in Fig. 2b. As for ATZ degradation by HO˙, the initial yields of DIA and DEPs were 20% and 51%, respectively. Accordingly, the DEPs to DIA ratio was calculated to be >10 in UV/PDS process, while it was ≈2.5 in UV/H2O2 process. This finding was consistent with previous reports,12,27,43 which could be attributed to that SO4˙− displayed a more distinctive preference to the ethyl function group than HO˙.
Effect of CO32−/HCO3−
The effect of CO32−/HCO3− on ATZ degradation as well as the formation of predominant products (i.e., DEPs and DIA) was investigated. It was observed that CO32−/HCO3− (5 mM) had a significant inhibitory effect on ATZ removal in both processes of UV/PDS and UV/H2O2, and the efficiency of ATZ degradation in UV/PDS was still much higher than that in UV/H2O2 (Fig. 3a). The negative effect of CO32−/HCO3− on these two systems might result from the fact that significant amounts of HO˙ and SO4˙− could react with CO32−/HCO3− to form the inorganic radical CO3˙−.30 Previous studies have reported that CO3˙− presumably attacked on the site of the nitrogen atom of ATZ by electron transfer or hydrogen transfer, but it was much less reactive than SO4˙− or HO˙, with a relatively low second order rate constant of 4 × 106 M−1 s−1 (vs. kSO4˙−,ATZ = 3 × 109 M−1 s−1).34 | ||
| Fig. 3 Effect of CO32−/HCO3− on the degradation of ATZ (a), the formation of DIA (b), the formation of DEPs (c), and the DEPs to DIA ratio (d) as a function of time in UV/H2O2 and UV/PDS processes. The inserts in (b) and (c) showed the effect of CO32−/HCO3− on the yield of DEPs and DIA, respectively. Experiment conditions: [ATZ] = 10 μM, [H2O2] = [PDS] = 1.5 mM, and pH = 7. | ||
Further, results exhibited that the impact of CO32−/HCO3− on the formation of products (i.e., the concentrations of DEPs and DIA as well as the DEPs to DIA ratio) was relatively slight (Fig. 3b–d). Lutze et al. suggested that the reactivity of the side groups of ATZ (i.e., ethylamine and isopropylamine group) was comparable to ethanol and 2-propanol.12 The second order rates of CO3˙− plus 2-propanol and ethanol were <4.0 × 104 M−1 s−1 and 2.2 × 104 M−1 s−1, respectively.44,45 This suggested the low reactive CO3˙− toward the side groups of ATZ was unable to impact the yields of DEPs and DIA.
Effect of NOM
As shown in Fig. 4a, the degradation of ATZ by UV/PDS or UV/H2O2 was inhibited in the presence of NOM (1 mg C L−1), and the former was still more efficient. Meanwhile, the degradation of generated DEPs and DIA was also significantly suppressed (Fig. 4b and c). Both yields of these two products as well as the DEPs to DIA ratio were not affected by NOM (Fig. 4b and c inserts and Fig. 4d), similar to the case of CO32−/HCO3−. The significant inhibition of NOM on ATZ and these two products degradation could be explained as following: (i) NOM usually acted as radical scavenger via competing for HO˙ and SO4˙− (kHO˙,NOM = 1.4 × 104 L mg C−1, kSO4˙−,NOM = 6.8 × 103 L mg C−1);12 (ii) NOM could exert an inner filter effect for the photolysis of oxidants.46 Previous studies reported that the reaction of HO˙ or SO4˙− with organic matters (e.g., aromatic, aliphatic, ester, and ketone etc.) could yield organic radicals. Howbeit, the redox potentials of varied organic radicals (i.e., −1.09 V to +0.40 V) have been determined in previous studies,47,48 which are much lower than those of HO˙ and SO4˙−. Thus, the yield of oxidation products could not be impacted by these organic radicals similar to CO3˙−. | ||
| Fig. 4 Effect of NOM on the degradation of ATZ (a), the formation of DIA (b), the formation of DEPs (c), and the DEPs to DIA ratio (d) as a function of time in UV/H2O2 and UV/PDS processes. The inserts in (b) and (c) showed the effect of NOM on the yield of DEPs and DIA, respectively. Experiment conditions: [ATZ] = 10 μM, [H2O2] = [PDS] = 1.5 mM, NOM = 1 mg C L−1, and pH = 7. | ||
In addition, singlet oxygen (1O2), peroxy radical (ROO˙), and 3DOM* could be produced by direct photolysis of NOM as above mentioned. Minero et al. reported that the rate of ATZ photolysis was increased and the formation of dealkylation products was enhanced in the presence of humic material, suggesting that NOM promoted the formation of various reactive intermediates in direct UV process.49 However, the result in this work indicated that the degradation of ATZ by direct photolysis was slightly inhibited in the presence of NOM, and no DEPs or DIA was detected (Fig. 5), consistent with Torrents' study.41 This discrepancy could be explained by the fact that different functional groups present in NOM could influence the photochemical reactions.37
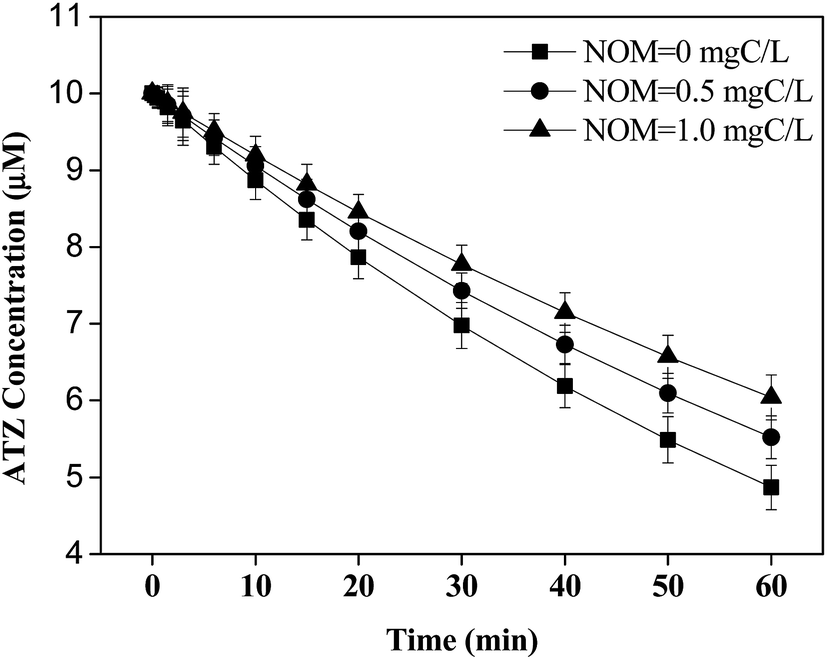 | ||
| Fig. 5 Effect of NOM on the degradation of ATZ in UV direct process. Experiment conditions: [ATZ] = 10 μM, pH = 7. | ||
Effect of Cl−
Experiments were carried out in the presence of Cl− (5 mM) to investigate its effect on ATZ degradation as well as the formation of DEPs and DIA. As shown in Fig. 6, Cl− had a negligible effect on the transformation of ATZ in UV/H2O2 process, including the kinetics and products formation. However, the presence of Cl− inhibited ATZ degradation and significantly influenced the formation trend of DEPs and DIA in UV/PDS process.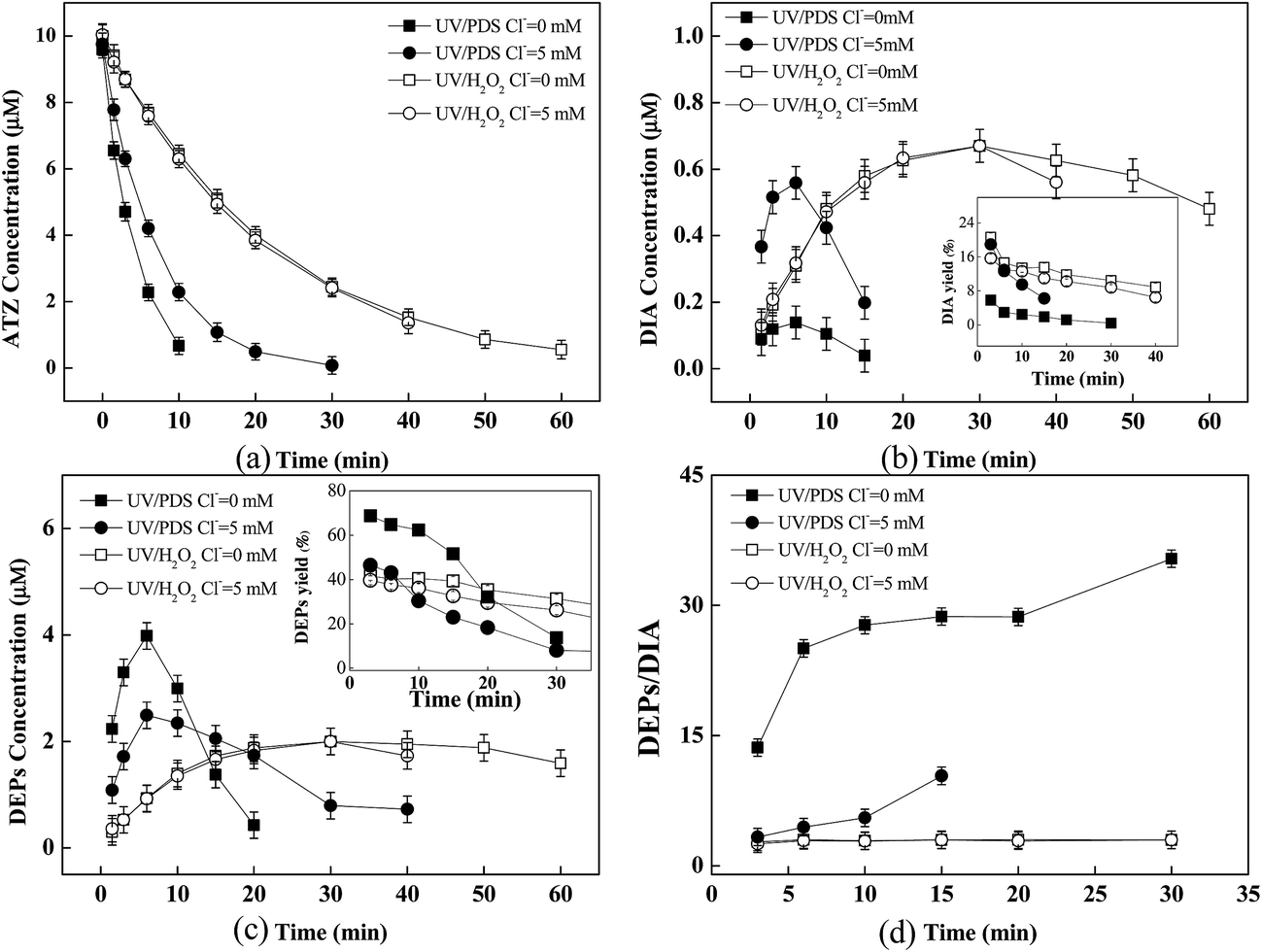 | ||
| Fig. 6 Effect of Cl− on the degradation of ATZ (a), the formation of DIA (b), the formation of DEPs (c), and the DEPs to DIA ratio (d) as a function of time in UV/H2O2 and UV/PDS processes. The inserts in (b) and (c) showed the effect of Cl− on the yield of DEPs and DIA, respectively. Experiment conditions: [ATZ] = 10 μM, [H2O2] = [PDS] = 1.5 mM, Cl− = 5 mM, and pH = 7. | ||
It was observed that the initial yield of DIA increased from approximate 6.8–18.9% when 5 mM Cl− was present, while the initial yield of DEPs decreased from approximate 75–46.4% (Fig. 6b and c insets). Correspondingly, the initial ratio of DEPs to DIA decreased from 13.6 to 3.3. In UV/H2O2 process, Cl− scavenging for HO˙ was apparently weak considering the fast backward reaction process (i.e., 6.1 × 109 s−1) in the equilibrium reaction of HO˙ with Cl− to form ClOH−˙,50,51 which explained no impact of Cl− on ATZ degradation. In contrast, the significant inhibitory effect of Cl− on ATZ oxidation by UV/PDS could be attributed to the much higher second-order rate constant of Cl− reacting with SO4˙− (i.e., 3.0 × 108 M−1 s−1) than that of HO˙ (i.e., 1.8 × 105 M−1 s−1) at neutral pH.52
It was well-known that Cl− could be oxidized by SO4−˙ to form reactive RCs such as Cl˙, ClO˙ and Cl2˙−.53 Recent studies reported that RCs were responsible for the degradation of certain organic pollutants (e.g., N,N-diethyl-3-methylbenzamide, caffeine, trimethoprim, and acid orange 7).50,54 In order to clarify the pattern of DEPs and DIA formation during the reaction of RCs with ATZ, UV/HClO, a typical process of generating RCs, was employed for oxidation of ATZ. NB was used herein as an effective quencher for HO˙ (kNB, HO˙ = 6.0 × 108 M−1 s−1), but not for RCs (<106 M−1 s−1) (e.g., Cl˙, ClO˙ and Cl2˙−).53,55–57 As shown in Fig. 7, ATZ was effectively oxidized in UV/HClO process, and the initial yields of DIA and DEPs were approximately 12% and 45%, respectively. Hence, the DEPs to DIA ratio could be calculated to be ≈4 (Fig. 7b). The co-existence of NB significantly inhibited the degradation of ATZ, as expected. It was noteworthy that the yield of DEPs was about 45% but no DIA was detected in the presence of NB, suggesting that RCs had a great selectivity on attacking the ethylamine group of ATZ to form DEPs. Accordingly, it seemed likely that the promoted yield of DIA in the UV/PDS process in the presence of Cl− could not be attributed to the generation of RCs.
 | ||
| Fig. 7 (a) Degradation of ATZ and formation of DEPs and DIA as a function of time in UV/HClO process. (b) Yields of DEPs and DIA as a function of time in UV/HClO process. HClO = 1.5 mM, ATZ = 10 μM, NB = 100 μM, and pH = 7. | ||
Actually, in the literature one could find an important conclusion on the influence of Cl− on SO4˙− based oxidation, Cl− could turn SO4˙− into HO˙ at pH ≥ 7. Zhang et al. investigated the effect of Cl− on pharmaceuticals degradation in UV/PDS process, and obtained that the steady-state concentration of HO˙ increased from 1.75 × 10−13 M to 2.78 × 10−11 M due to the addition of 0.1 M Cl− by kinetic modeling.58 Lutze et al. suggested that the ratio of degradation rates of 4-nitrobenzoic acid (pNBA) vs. 4-chlorobenzonic acid (pCBA) could be used as an indicator for the transformation of SO4˙− to HO˙ in the UV/PDS with Cl−.42 It was found that the slope (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C/C0 (pNBA) vs. lnC/C0 (pCBA)) for UV/PDS in presence of Cl− was equal to the UV/H2O2 without Cl− present, indicating HO˙ were the major reactive species in both processes. In this study, the initial ratio of DEPs to DIA in the presence of Cl− in UV/PDS process (i.e., 3) was very close to that in UV/H2O2 process (i.e., 2.5), which provided further evidence for the facilitated effect of Cl− on the transformation of SO4˙− to HO˙.
C/C0 (pNBA) vs. lnC/C0 (pCBA)) for UV/PDS in presence of Cl− was equal to the UV/H2O2 without Cl− present, indicating HO˙ were the major reactive species in both processes. In this study, the initial ratio of DEPs to DIA in the presence of Cl− in UV/PDS process (i.e., 3) was very close to that in UV/H2O2 process (i.e., 2.5), which provided further evidence for the facilitated effect of Cl− on the transformation of SO4˙− to HO˙.
Degradation of atrazine under natural water background
The degradation of ATZ as well as the formation of DEPs and DIA in three natural waters were investigated. As could be seen in Fig. 8a, UV/PDS system was more effective than UV/H2O2 system under the identical conditions. This was ascribed to that the quantum efficiency for photolysis and the molar extinction coefficient for S2O82− were higher than H2O2, as described above. The yields of DIA and DEPs in these three natural waters were fairly close in UV/H2O2 system. Distinguishingly, the yield of DIA in UV/PDS system in the effluent from GW was much higher than those in effluents from SW and EW, and meanwhile the yield of DEPs showed the reverse change trend. As shown in Fig. 8b and c, the initial yields of DIA and DEPs were about 15% and 50% in GW during UV/PDS process, respectively, while the initial yields of DIA and DEPs were approximately 5% and 65% in SW or EW, respectively. This could be attributed to the higher concentration of Cl− in GW than that in SW and EW (Table 1), and this further confirmed that Cl− played an important role in the transformation of reactive oxidative species as well as the yields of ATZ oxidation products. | ||
| Fig. 8 The degradation of ATZ (a), the formation of DIA (b), the formation of DEPs (c), and the DEPs to DIA ratio (d) as a function of time in UV/H2O2 and UV/PDS processes in actual waters. Experiment conditions: [ATZ] = 10 μM, [H2O2] = [PDS] = 1.5 mM, and pH = 7. | ||
Conclusion
The formation of DEPs and DIA was investigated in UV/H2O2 and UV/PDS processes under various conditions. For all studied processes the following conclusions were obtained:(i) UV/PDS system was more effective than UV/H2O2 system for the degradation of ATZ and its oxidation products under the same conditions.
(ii) SO4˙− displayed a more distinctive preference to the ethyl function group than HO˙, leading to the higher ratio of DEPs/DIA in UV/PDS system than UV/H2O2 system in pure water.
(iii) CO32−/HCO3− and NOM had a certain inhibitory effect on ATZ degradation in both UV/PDS and UV/H2O2 processes, due to their competition on SO4˙− and/or HO˙. The generated CO3˙− and organic radicals had very low reactivity toward ATZ as well as its side groups, and thus they showed a negligible effect on the yields of DEPs and DIA.
(iv) Cl− suppressed the degradation of ATZ in UV/PDS, but had a slight effect on that in UV/H2O2. The decrease of DEPs concentration and the increase of DIA concentration in UV/PDS process were observed when a certain concentration of Cl− was present, attributed to that Cl− enhanced the transformation of SO4˙− into HO˙. In addition, RCs (i.e., Cl˙, ClO˙ and Cl2˙−) were found to be more selective on attacking the ethylamine group of ATZ.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51578203 and 51408107), the Funds of the State Key Laboratory of Urban Water Resource and Environment (HIT, 2016DX13), the Foundation for the Author of National Excellent Doctoral Dissertation of China (201346), the National Key Research and Development Program (2016YFC0401107), and the Heilongjiang Province Postdoctoral Science Foundation (LBH-Q15057).References
- J. L. Acero, K. Stemmler and U. von Gunten, Environ. Sci. Technol., 2000, 34, 591–597 CrossRef CAS.
- E. Pelizzrtti, V. Maurino, C. Minero, V. Carlin, E. Pramauro, O. Zerbinati and M. L. Tosato, Environ. Sci. Technol., 1990, 24, 1559–1565 CrossRef.
- W. Hua, E. R. Bennett and R. J. Letcher, Water Res., 2006, 40, 2259–2266 CrossRef CAS PubMed.
- H. R. Buser, Environ. Sci. Technol., 1990, 24, 1049–1058 CrossRef CAS.
- C. Sulmon, F. Ramel, G. Gouesbet and I. Couée, Environ. Sci. Technol., 2014, 48, 6055–6056 CrossRef CAS PubMed.
- N. C. Meakins, J. M. Bubb and J. N. Lester, Chemosphere, 1994, 28, 1611–1622 CrossRef CAS.
- S. R. D. Solla and P. A. Martin, Chemosphere, 2011, 85, 820–825 CrossRef PubMed.
- X. Kong, J. Jiang, J. Ma, Y. Yang, W. Liu and Y. Liu, Water Res., 2015, 90, 15–23 CrossRef PubMed.
- S. Komtchou, A. Dirany, P. Drogui, N. Delegan, M. A. E. Khakani, D. Robert and P. Lafrance, Chemosphere, 2016, 157, 79–88 CrossRef CAS PubMed.
- L. Xu, H. Zang, Q. Zhang, Y. Chen, Y. Wei, J. Yan and Y. Zhao, Chem. Eng. J., 2013, 232, 174–182 CrossRef CAS.
- Y. Yang, J. Jiang, X. Lu, J. Ma and Y. Liu, Environ. Sci. Technol., 2015, 49, 7330–7339 CrossRef CAS PubMed.
- H. V. Lutze, S. Bircher, I. Rapp, N. Kerlin, R. Bakkour, M. Geisler, C. von Sonntag and T. C. Schmidt, Environ. Sci. Technol., 2015, 49, 1673–1680 CrossRef CAS PubMed.
- M. Cheng, G. Zeng, D. Huang, C. Lai, P. Xu, C. Zhang, Y. Liu, J. Wan, X. Gong and Y. Zhu, J. Hazard. Mater., 2016, 312, 184–191 CrossRef CAS PubMed.
- J. A. Khan, X. He, H. M. Khan, N. S. Shah and D. D. Dionysiou, Chem. Eng. J., 2013, 218, 376–383 CrossRef CAS.
- Y. H. Guan, J. Ma, Y. M. Ren, Y. Z. Liu, J. Xiao, L. Lin and C. Zhang, Water Res., 2013, 47, 5431–5438 CrossRef CAS PubMed.
- C. W. Luo, J. Ma, J. Jiang, Y. Z. Liu, Y. Song, Y. Yang, Y. H. Guan and D. J. Wu, Water Res., 2015, 80, 99–108 CrossRef CAS PubMed.
- J. A. Khan, X. He, H. M. Khan, N. S. Shah and D. D. Dionysiou, Chem. Eng. J., 2013, 218, 376–383 CrossRef CAS.
- S. M. Arnold, W. J. Hickey and R. F. Harris, Environ. Sci. Technol., 1995, 29, 2083–2089 CrossRef CAS PubMed.
- F. J. Beltrán, G. Ovejero and B. Acedo, Water Res., 1993, 27, 1013–1021 CrossRef.
- S. K. Papiernik and R. F. Spalding, J. Agric. Food Chem., 1998, 46, 749–754 CrossRef CAS PubMed.
- E. M. Thurman, M. T. Meyer, M. S. Mills, L. R. Zimmerman, C. A. Perry and D. A. Goolsby, Environ. Sci. Technol., 1994, 28, 2267–2277 CrossRef CAS PubMed.
- F. J. Beltran, M. Gonzalez, F. J. Rivas and P. Alvarez, Environ. Toxicol. Chem., 1996, 15, 868–872 CrossRef CAS.
- B. D. Amaral, J. A. de Araujo, P. G. Peralta-Zamora and N. Nagata, Microchem. J., 2014, 117, 262–267 CrossRef CAS.
- Y. Ji, C. Dong, D. Kong and J. Lu, J. Hazard. Mater., 2015, 285, 491–500 CrossRef CAS PubMed.
- Y. Ji, C. Dong, D. Kong, J. Lu and Q. Zhou, Chem. Eng. J., 2015, 263, 45–54 CrossRef CAS.
- C. L. Bianchi, C. Pirola, V. Ragaini and E. Selli, Appl. Catal., B, 2006, 64, 131–138 CrossRef CAS.
- A. Tauber and C. V. Sonntag, Acta Hydrochim. Hydrobiol., 2000, 28, 15–23 CrossRef CAS.
- M. Alipour, Chem. Phys., 2014, 434, 11–14 CrossRef CAS.
- Y. Xu, Z. Lin and H. Zhang, Chem. Eng. J., 2016, 285, 392–401 CrossRef CAS.
- R. Zhang, P. Sun, T. H. Boyer, L. Zhao and C. Huang, Environ. Sci. Technol., 2015, 49, 3056–3066 CrossRef CAS PubMed.
- A. A. Presto, K. E. Huff Hartz and N. M. Donahue, Environ. Sci. Technol., 2005, 39, 7036–7045 CrossRef CAS PubMed.
- S. Canonica, T. Kohn, M. Mac, F. J. Real, J. Wirz and U. von Gunten, Environ. Sci. Technol., 2005, 39, 9182–9188 CrossRef CAS PubMed.
- J. P. Huang and S. A. Mabury, Environ. Toxicol. Chem., 2000, 19, 2181–2188 CrossRef CAS.
- J. Huang and S. A. Mabury, Environ. Toxicol. Chem., 2000, 19, 1501–1507 CrossRef CAS.
- J. Fang, Y. Fu and C. Shang, Environ. Sci. Technol., 2014, 48, 1859–1868 CrossRef CAS PubMed.
- Z. Wu, J. Fang, Y. Xiang, C. Shang, X. Li, F. Meng and X. Yang, Water Res., 2016, 104, 272–282 CrossRef CAS PubMed.
- E. Lee, H. K. Shon and J. Cho, J. Hazard. Mater., 2014, 276, 1–9 CrossRef CAS PubMed.
- N. A. Frigerio, Anal. Chem., 1963, 35, 412–413 CrossRef CAS.
- W. Li, T. Jain, K. Ishida and H. Liu, Environ. Sci.: Water Res. Technol., 2017, 3, 128–138 CAS.
- J. R. Bolton and K. G. Linden, J. Environ. Eng., 2003, 129, 209–215 CrossRef CAS.
- A. Torrents, B. G. Anderson, S. Bilboulian, W. E. Johnson and C. J. Hapeman, Environ. Sci. Technol., 1997, 31, 1476–1482 CrossRef CAS.
- H. V. Lutze, N. Kerlin and T. C. Schmidt, Water Res., 2015, 72, 349–360 CrossRef CAS PubMed.
- A. Tauber and C. von Sonntag, Acta Hydrochim. Hydrobiol., 2000, 28, 15–23 CrossRef CAS.
- Y. Liu, X. He, X. Duan, Y. Fu and D. D. Dionysiou, Chem. Eng. J., 2015, 276, 113–121 CrossRef CAS.
- S. Canonica, T. Kohn, M. Mac, F. J. Real, J. Wirz and U. von Gunten, Environ. Sci. Technol., 2005, 39, 9182–9188 CrossRef CAS PubMed.
- J. Brame, M. Long, Q. Li and P. Alvarez, Water Res., 2014, 60, 259–266 CrossRef CAS PubMed.
- P. S. Rao and E. Hayon, J. Am. Chem. Soc., 1974, 96, 1287–1294 CrossRef CAS.
- S. Bank and D. A. Juckett, J. Am. Chem. Soc., 1976, 24, 7742–7746 CrossRef.
- C. Minero, E. Pramauro, E. Pelizzetti, M. Dolci and A. Marchesini, Chemosphere, 1992, 24, 1597–1606 CrossRef CAS.
- P. Sun, W. Lee, R. Zhang and C. Huang, Environ. Sci. Technol., 2016, 50(24), 13265–13273 CrossRef CAS PubMed.
- X. Lou, D. Xiao, C. Fang, Z. Wang, J. Liu, Y. Guo and S. Lu, Environ. Sci. Pollut. Res., 2016, 23, 4778–4785 CrossRef CAS PubMed.
- T. N. Das, J. Phys. Chem. A, 2001, 105, 9142–9155 CrossRef CAS.
- Z. Wu, J. Fang, Y. Xiang, C. Shang, X. Li, F. Meng and X. Yang, Water Res., 2016, 104, 272–282 CrossRef CAS PubMed.
- R. Yuan, Z. Wang, Y. Hu, B. Wang and S. Gao, Chemosphere, 2014, 109, 106–112 CrossRef CAS PubMed.
- Y. Xiang, J. Fang and C. Shang, Water Res., 2016, 90, 301–308 CrossRef CAS PubMed.
- M. J. Watts and K. G. Linden, Water Res., 2007, 41, 2871–2878 CrossRef CAS PubMed.
- J. Fang, Y. Fu and C. Shang, Environ. Sci. Technol., 2014, 48, 1859–1868 CrossRef CAS PubMed.
- R. Zhang, P. Sun, T. H. Boyer, L. Zhao and C. Huang, Environ. Sci. Technol., 2015, 49, 3056–3066 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |