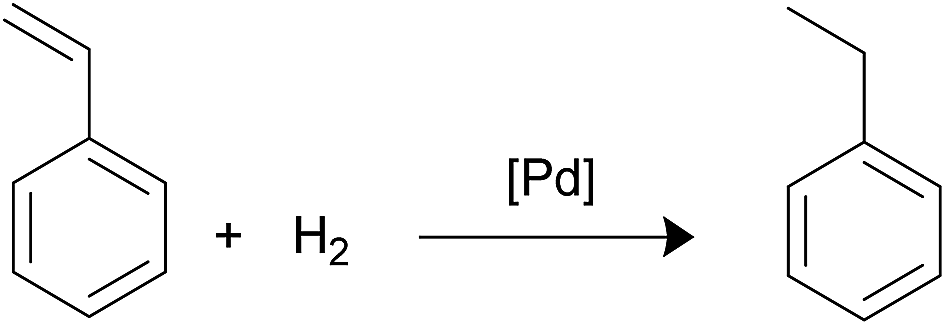DOI:
10.1039/C7RA03639F
(Paper)
RSC Adv., 2017,
7, 27872-27880
NH2- or PPh2-functionalized linkers for the immobilization of palladium on magnetite nanoparticles?
Received
29th March 2017
, Accepted 17th May 2017
First published on 25th May 2017
Abstract
Immobilization of palladium on magnetite nanoparticles has been carried out with the assistance of two differently functionalized linkers containing phosphino- or amino-terminated groups. The linkers have been anchored to the magnetite surface by means of catechol, mercapto or carboxylate groups. The nature of the resulting Pd nanoparticles deposited has been examined by HAADF-STEM images and XPS electron spectroscopy. The efficiency of the two kinds of catalysts has been checked and compared for the Suzuki–Miyaura reaction, 4-nitrophenol reduction and styrene hydrogenation. The results evidence that the nanoparticles equipped with the phosphino fragment are better catalysts than those functionalized with the amino group and, in some processes, they are among the most active catalysts reported in the literature.
Introduction
Palladium nanoparticles (NPs) are being widely used as catalysts for many processes in organic chemistry, mainly for carbon–carbon cross-coupling reactions.1 Owing to the tendency of Pd NPs for agglomeration it is convenient to deposit the metal nanoparticles on the surface of supporting materials as an effective route to preserve the integrity of the catalyst. For this purpose, the use of metal oxides has been shown to be an excellent option. In particular magnetite is one of the oxides most extensively employed nowadays since it can be easily separated from the solution with a simple external magnet. This strategy favours the reuse of the catalyst and avoids the need for filtration, centrifugation or other time-consuming workup processes.2 The simplest method for the deposition of palladium NPs on the surface of magnetic nanoparticles consists of the addition of palladium salts to a suspension of bare magnetite nanoparticles followed by reduction with hydrogen or NaBH4. However, this strategy does not provide convenient control of the morphology and size of the nanoparticles formed.3 A straightforward approach for the effective deposition of palladium involves the previous functionalization of the support material surface with linkers equipped with terminal functions (generally amino groups) able to capture metal ions that then will be reduced and deposited on the magnetic support.4 The choice of appropriate linkers is crucial for the nucleation and growth of the immobilized metal NPs.5 The number of studies addressed to investigate the influence of functional linkers on the size of Pd NPs deposited on the magnetite surface is surprisingly scarce. For example, Rossi et al. reported that amine and ethylenediamine groups grafted on the surface of silica support assisted the preparation of magnetically recoverable Pd NPs that showed very different average particle diameters, 6 and 1 nm, respectively.3b Interestingly, the use of non-functionalized surfaces gave metal aggregates. This study was completed afterwards with the use of the mercapto function.6 Yi et al. functionalized the silica-coated magnetic Fe2O3 NPs with mercapto or amine groups and found that the size of the Pd nanoclusters deposited with the assistance of the amino group was smaller than that obtained with the mercapto function (2.2 and 2.9 nm, respectively).7 The unique comparative study relative to the influence of NH2 and PPh2 functions on the palladium nanoparticles size was published by Rossi et al.8 The authors prepared samples of magnetite NPs functionalized with NH2 or iminophosphine and proceeded to immobilize the Pd NPs. The average diameters measured were 3.8 and 2.9 nm respectively. Besides, efficiency investigations of both catalysts in the coupling reaction of phenylboronic acid with 4-bromoanisole revealed that the phosphine-terminated catalyst was more active and selective than the amino-counterpart.
As commented above, linkers can determine the Pd NPs size and this factor is crucial for the final efficiency of the system. However, it is important to underline that although small Pd nanoparticles are generally better catalysts than those containing larger ones, in the nanosize range catalytic activities arise also from geometric effects (presence of low-coordinated sites in edges and corners), lattice distortions, type of facets, support interaction and others.9 In this paper, we have worked with two types of magnetite nanoparticles; in one case they were functionalized with linkers terminated with the NH2-function and in the other, with the PPh2 group. The immobilization of Pd NPs on the magnetite surface using both types of linkers permitted to compare them in terms of Pd NPs size and catalytic efficiency for the Suzuki–Miyaura coupling reaction, 4-nitrophenol reduction and styrene hydrogenation processes.
Results and discussion
Synthesis of the catalysts
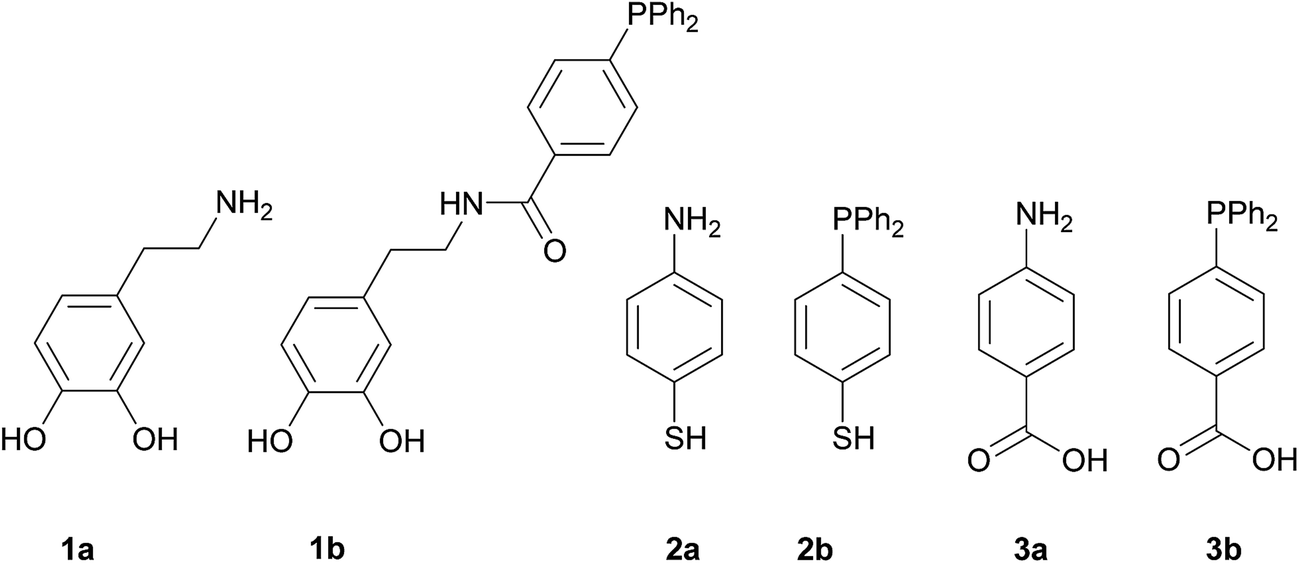
In order to carry out this work we thought that the use of the three pairs of molecules shown in Scheme 1 would permit to compare the influence of the NH2- or PPh2-functions in the formation and immobilization of the Pd NPS on the magnetite surface in terms of size and/or morphology and catalytic applications. The main difference in each pair is the terminal function (NH2 or PPh2) that capture the Pd ions. In addition, 1a and 1b display a catechol fragment that strongly coordinates to Fe ions by a chelating effect while 2a and 2b exhibit a mercapto group and 3a and 3b the COOH function for anchoring to the magnetite surface.
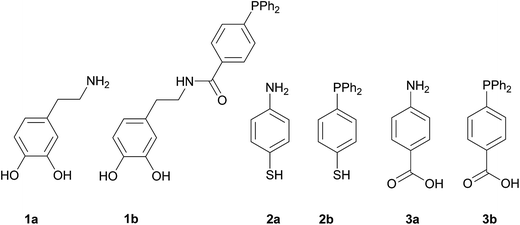 |
| | Scheme 1 Linkers used for the functionalization of the magnetite nanoparticles. | |
1a, 2a, 3a, and 3b are commercially available. 1b10 and 2b11 were synthesized according to the literature. The anchorage of these molecules onto the surface of the bare magnetite NPs was achieved by sonication in methanol for two hours. The Pd deposition was carried out by means of two reactions: (i) addition of K2[PdCl4] to a previously sonicated suspension in water of the functionalized magnetite nanoparticles, followed by isolation, washing and redispersion in water and (ii) Pd(II) reduction with NaBH4 and separation of the new catalysts with an external magnet (Scheme 2).12
 |
| | Scheme 2 Pd immobilization on the magnetite surface. | |
This method was indistinctly used for the synthesis of hybrids containing phosphino or amino functions. The organic functionalized ligands are considered to have a detrimental effect on the catalytic activity as they can block the catalyst from the action of the substrates. However, it has been suggested that they can improve the selectivity, stability, solubility and recyclability.2b In the course of our previous works we demonstrated that the immobilization of the Pd NPs using Fe3O4-L (L = 2b, and 3b) produced the loss of a fraction of L, but this fact was not detected for Fe3O4-1b due to the chelate effect of the linker. The partial loss of the capping ligand probably does not affect the size or morphology of the Pd NPs deposited, but undoubtedly will affect the catalytic activity of the system. However, the target of this paper was mainly to compare the two elements of each pair of catalysts in terms of the Pd nanoparticle size and resulting catalytic activity. This objective was possible because the ligand L content (μmol g−1 NPs) is very similar for the pair Fe3O4-2a@Pd and Fe3O4-2b@Pd and for the pair Fe3O4-3a@Pd and Fe3O4-3b@Pd so that steric effects due to the organic linkers will not interfere in this study. The comparison of catalytic efficiency of the functionalized NPs required inevitably the synthesis of catalysts with the same Pd content. We decided to work with samples containing 0.5 wt% Pd so that the amount of K2[PdCl4] for the synthesis of the catalysts was properly adjusted. Table 1 lists the linker and the palladium content of the six catalysts prepared.
Table 1 Linker (μmol g−1) and palladium (%) content of the six catalysts
| Catalyst |
Linker (μmol g−1 NPs) |
% Pd |
| Fe3O4-1a@Pd |
94.6 |
0.48 |
| Fe3O4-1b@Pd |
91.9 |
0.49 |
| Fe3O4-2a@Pd |
57.1 |
0.51 |
| Fe3O4-2b@Pd |
52.2 |
0.51 |
| Fe3O4-3a@Pd |
42.1 |
0.50 |
| Fe3O4-3b@Pd |
48.2 |
0.51 |
Characterization of the catalysts
The catalysts, including the new amino-functionalized species, have been characterized by elemental analyses and ICPoes (Inductive Coupled Plasma optical emission spectroscopy) measurements for palladium (Table 1), FT-IR spectroscopy, X-ray photoelectron spectroscopy (XPS) and scanning transmission electron microscopy (STEM).
The FT-IR spectra of the catalysts are not very informative since no obvious changes took place after the immobilization of the palladium on the magnetite surface. All of them show a sharp peak at about 594 cm−1, which is the IR signature ν(Fe–O) for ferrite nanoparticles and weak signals at 1920 and 2920 cm−1 due to aryl C–H vibrations. The ν(N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) appears at 1633 cm−1 for Fe3O4-1b@Pd.
O) appears at 1633 cm−1 for Fe3O4-1b@Pd.
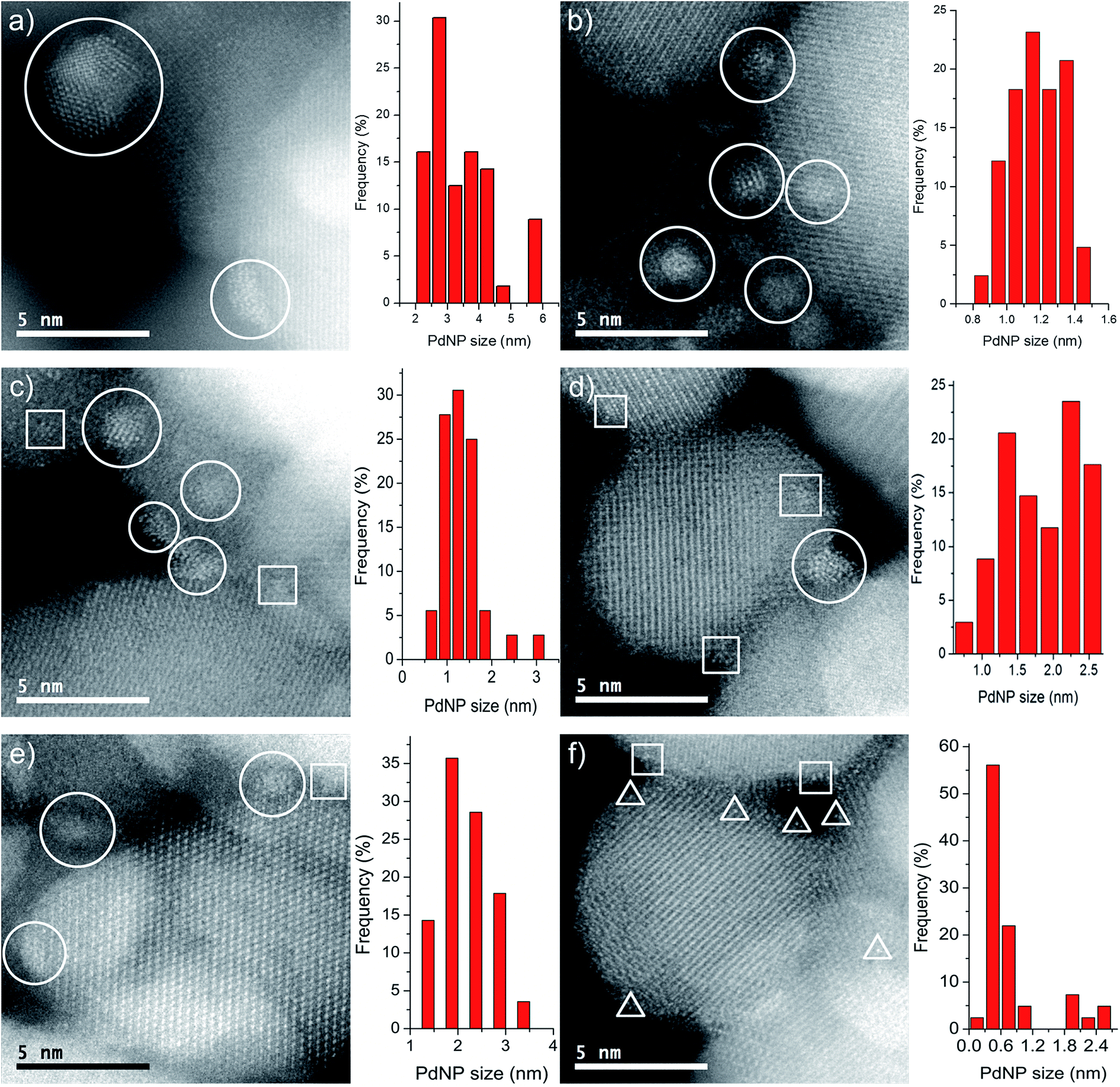
Images of the nanocatalysts were obtained by atomic-resolution high-angle annular dark-field (HAADF)-STEM. As a general tendency, we have observed that nanoparticles formed with the assistance of the PPh2 linker are more homogeneous in size than those formed by using the amino function (Fig. 1). Pd NPs immobilized on Fe3O4-1a@Pd are appreciably larger (3.4 nm) than those deposited on Fe3O4-1b@Pd (1.2 nm). The size of the Pd NPs found in Fe3O4-2a@Pd and Fe3O4-2b@Pd are very similar (about 1.5 nm), although in the first catalyst some larger nanoparticles (3.0 nm) are also present. Remarkable loss of palladium nanoparticle size homogeneity is found for the catalyst Fe3O4-3a@Pd. Thus, along with nanoparticles from 0.8 nm to 3.5 nm, unexpected Pd regions showing high wettability is clearly detected, that is, small zones where Pd atoms are spread on the magnetite surface. This situation is not observed in the Fe3O4-3b@Pd images. For this catalyst, along with small clusters and single palladium atoms, a few large nanoparticles of about 5.0 nm can be detected. The presence of wetting regions and single Pd atoms are factors that should be considered for explaining the efficiency of our catalysts. In summary, the micrographs of the samples Fe3O4-L@Pd contain only NPs (L = 1a, 1b); small NPs (clusters) and NPs (L = 2a, 2b); clusters, NPs, and wetting areas (L = 3a); single Pd atoms (SACs), clusters and NPs (3b).
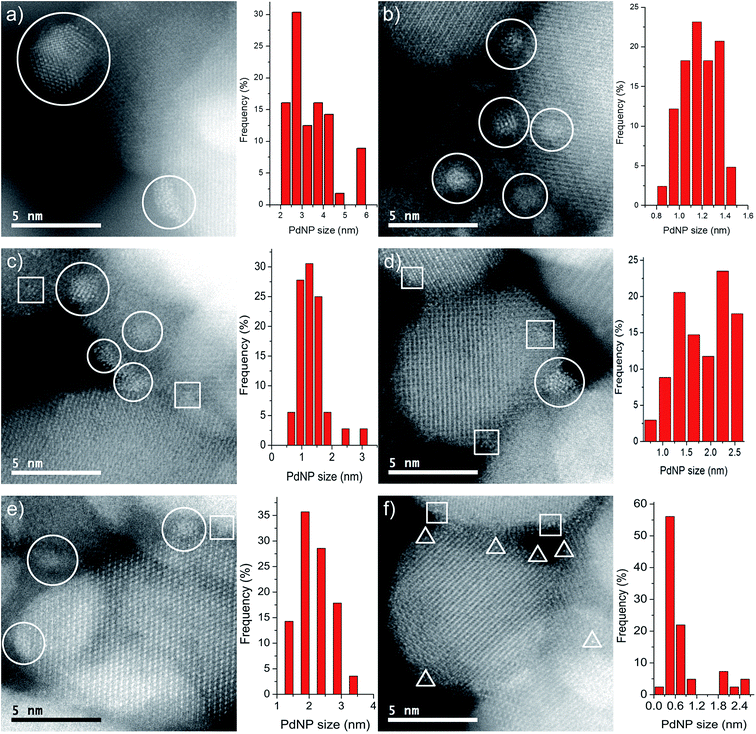 |
| | Fig. 1 Representative HAADF–STEM images and histograms of the catalysts. Pd NPs, clusters and single metal atoms are enclosed in circles, squares and triangles, respectively. (a) Fe3O4-1a@Pd (b) Fe3O4-1b@Pd (c) Fe3O4-2a@Pd (d) Fe3O4-2b@Pd (e) Fe3O4-3a@Pd (f) Fe3O4-3b@Pd. For clarity, only a few Pd species are enclosed in each panel, although many more are present. | |
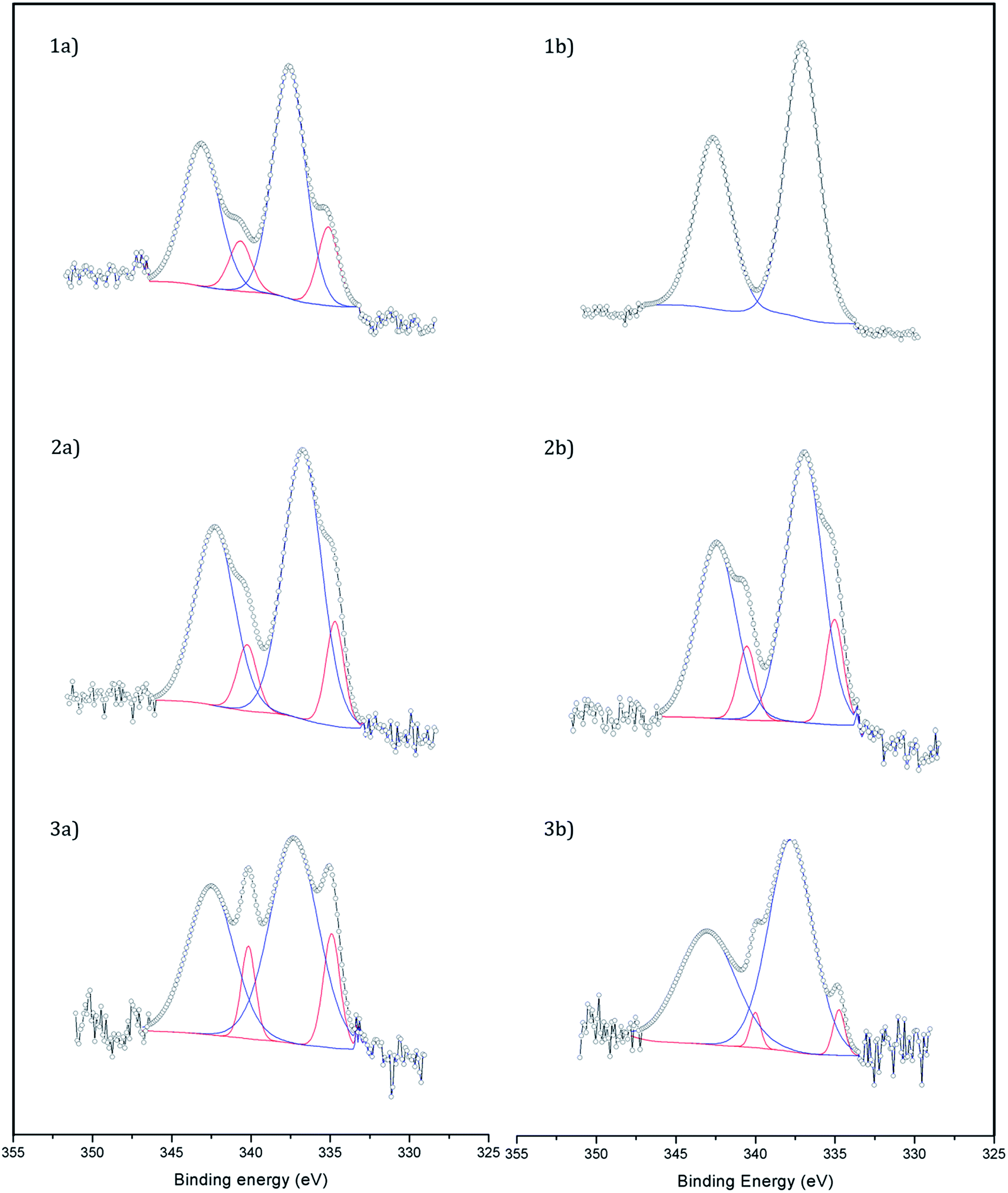
X-ray photoelectron spectroscopy (XPS) was used to study the dispersion and oxidation state of Pd (Fig. 2). The atomic ratio Pd/Fe of the samples was calculated from the core-level Pd 3d and Fe 2p photoelectrons and it was in all cases in the range 0.008–0.009 except for the catalyst Fe3O4-3b@Pd, where a Pd/Fe atomic ratio of 0.012 was recorded. Taking into account that the Pd content in the samples is virtually identical (Table 1), the larger Pd/Fe atomic ratio in Fe3O4-3b@Pd is an indication of a higher Pd dispersion in this sample, in accordance to the HAADF-STEM results, which showed mostly small Pd clusters and single atoms, as explained above. This is also in accordance with the Pd(0)/Pdox atomic ratios. The catalysts Fe3O4-1b@Pd and Fe3O4-3b@Pd containing small Pd clusters and single atoms and/or small Pd nanoparticles exhibit Pd(0)/Pdox atomic ratios of 0–0.06, which are lower than those measured over the other catalysts containing larger Pd nanoparticles, Pd(0)/Pdox = 0.18–0.22. The electronic transfer between Pd and the Fe3O4 support leading to positively charged Pd when small clusters or nanoparticles are present has been already reported in.13
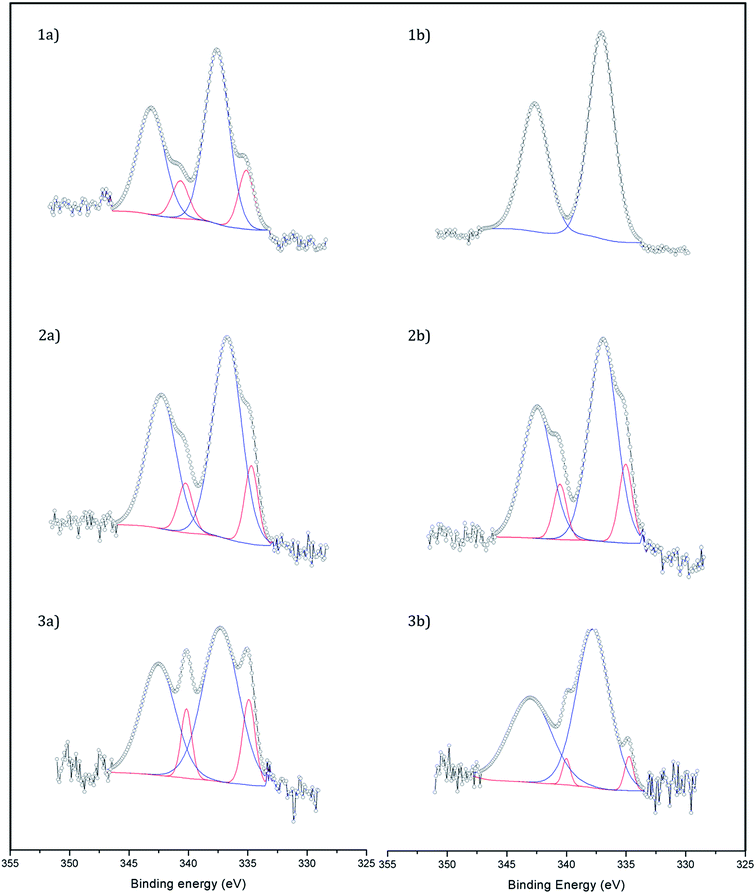 |
| | Fig. 2 XP spectra of (1a) Fe3O4-1a@Pd, (1b) Fe3O4-1b@Pd, (2a) Fe3O4-2a@Pd, (2b) Fe3O4-2b@Pd, (3a) Fe3O4-3a@Pd, and (3b) Fe3O4-3b@Pd. Pd2+ peaks are shown in blue and Pd0 ones in red. | |
Catalytic studies
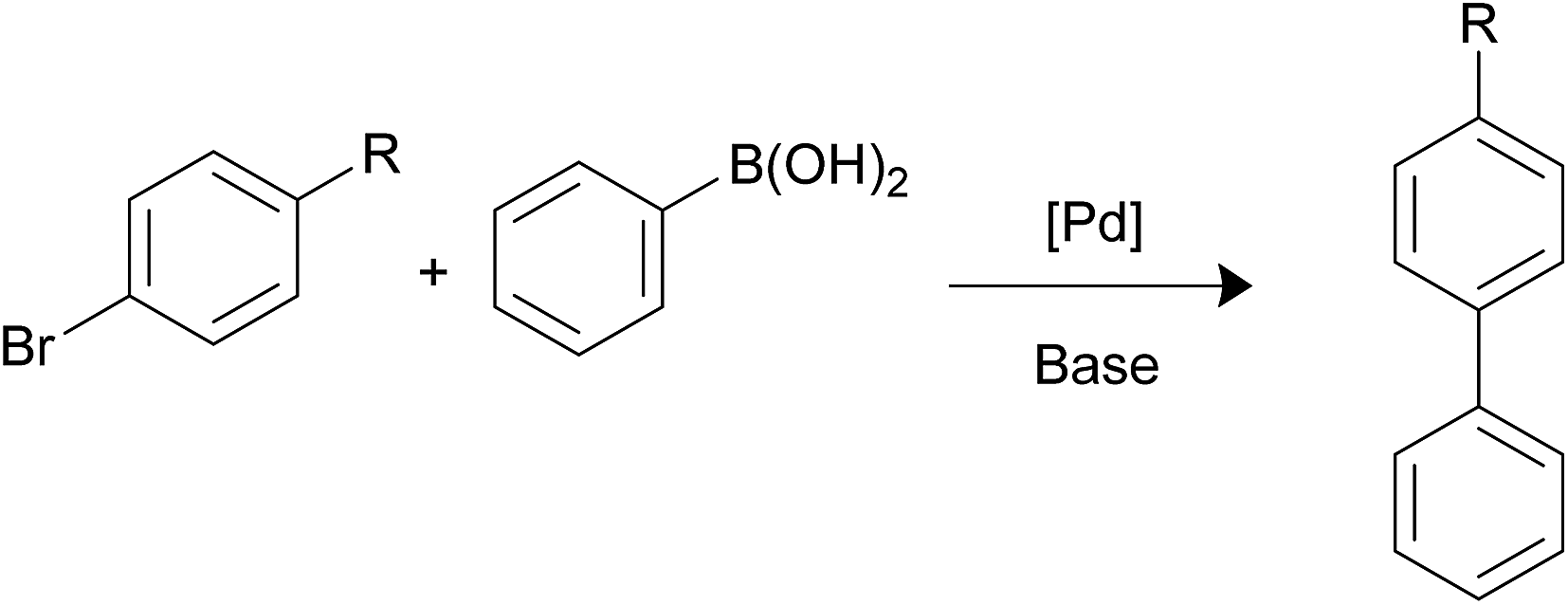
Suzuki–Miyaura reaction. Palladium-catalyzed Suzuki–Miyaura reaction is a well-known process that enables to obtain biaryl structures by reaction of aryl halides with arylboronic acids (Scheme 3). The reaction is used widely in the synthesis of agrochemicals, pharmaceuticals, natural products and other materials.14 The efficiency and rate of this reaction strongly depends on the electronegativity of the halide: chloro-aryl substrates are much more difficult to react than bromo- or iodo-aryl compounds;15 on the other hand, the reaction rate is clearly influenced by the impact of other substituents at Ar-X: electron-withdrawing groups enhance the rate, while electron-donating groups decrease it.16
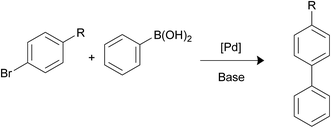 |
| | Scheme 3 Suzuki–Miyaura coupling. | |
Initially, the Suzuki–Miyaura activity of the nanocatalysts was tested for the reaction of bromobenzene and phenylboronic acid. Previous works carried out in our group permitted us to establish the best reaction conditions in terms of solvent (a mixture of ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water 1:1), base (K2CO3), temperature (65 °C) and reaction time (1 hour) and that no coupling reaction takes place using simply functionalized nanoparticles in absence of palladium. Table 2 lists the catalytic results which clearly show that the nanocatalysts containing the PPh2 terminal linkers are more efficient than those equipped with the NH2 function. This is in good accord with the smaller size Pd NPS formed with the assistance of the phosphine terminated linkers. On the other hand, the low values of the TOFs exhibited by the pair of nanohybrids containing 2a and 2b can be attributed to the poising effect of the sulphur.17 The lower catalytic efficiency of these nanocatalysts is also observed in the rest of the catalytic processes studied in this article. The scope of this reaction was expanded to 4-substituted bromoarenes bearing the electron-withdrawing nitro group or the methoxy electron-donating fragment (Table 2). The most interesting result is that the efficiency of the catalysts containing the PPh2-linker (1b-3b) is invariability higher than those obtained with the NH2-linker (1a-3a). As expected, the catalytic efficiency of the reaction increases with the presence of the 4-substituted nitro group. Notably, the turnover frequency obtained in this work using Fe3O4-L@Pd (L = 1b, 3b) for the coupling of the three bromoaryl substrates investigated in this work compares well with the best results reported in the literature (Table 3). It is well known that the activation of C–Cl bond is more difficult than that of C–Br and C–I bonds. In this paper, we have observed that neither the catalyst containing the NH2 function nor those equipped with the phosphine group are able to catalyse the coupling of chloroaryl molecules with phenylboronic acid. For example, reaction of 4-chlorotoluene with phenylboronic acid in the presence of Fe3O4-L@Pd (L = 1a, 1b) only gave the coupling product in less than 1% yield.
:water 1:1), base (K2CO3), temperature (65 °C) and reaction time (1 hour) and that no coupling reaction takes place using simply functionalized nanoparticles in absence of palladium. Table 2 lists the catalytic results which clearly show that the nanocatalysts containing the PPh2 terminal linkers are more efficient than those equipped with the NH2 function. This is in good accord with the smaller size Pd NPS formed with the assistance of the phosphine terminated linkers. On the other hand, the low values of the TOFs exhibited by the pair of nanohybrids containing 2a and 2b can be attributed to the poising effect of the sulphur.17 The lower catalytic efficiency of these nanocatalysts is also observed in the rest of the catalytic processes studied in this article. The scope of this reaction was expanded to 4-substituted bromoarenes bearing the electron-withdrawing nitro group or the methoxy electron-donating fragment (Table 2). The most interesting result is that the efficiency of the catalysts containing the PPh2-linker (1b-3b) is invariability higher than those obtained with the NH2-linker (1a-3a). As expected, the catalytic efficiency of the reaction increases with the presence of the 4-substituted nitro group. Notably, the turnover frequency obtained in this work using Fe3O4-L@Pd (L = 1b, 3b) for the coupling of the three bromoaryl substrates investigated in this work compares well with the best results reported in the literature (Table 3). It is well known that the activation of C–Cl bond is more difficult than that of C–Br and C–I bonds. In this paper, we have observed that neither the catalyst containing the NH2 function nor those equipped with the phosphine group are able to catalyse the coupling of chloroaryl molecules with phenylboronic acid. For example, reaction of 4-chlorotoluene with phenylboronic acid in the presence of Fe3O4-L@Pd (L = 1a, 1b) only gave the coupling product in less than 1% yield.
Table 2 Substrate effect on Suzuki–Miyaura coupling reaction, TOF (h−1)a
| Catalyst Fe3O4-L@Pd |
Bromoaryl substrate |
| R = H |
R = NO2 |
R = OCH3 |
| Bromoaryl (12 mmol), phenylboronic acid (14.4 mmol), K2CO3 (36 mmol), catalyst (9.4 × 10−5 mmol Pd), 65 °C, EtOH:H2O, 1 h. TOF = [mol of biphenyl/mol of Pd]/time (h). |
| L = 1a |
36979 |
47062 |
6780 |
| L = 1b |
122085 |
115568 |
86292 |
| L = 2a |
1130 |
9180 |
1796 |
| L = 2b |
1750 |
32386 |
3694 |
| L = 3a |
19820 |
29568 |
3207 |
| L = 3b |
110000 |
44795 |
6385 |
Table 3 Survey of selected palladium nanoparticles catalysts in Suzuki–Miyaura couplinga
| Catalyst |
Substrate |
Solvent |
Base |
T (°C) |
TOF (h−1) |
Ref. |
| (BB = bromobenzene, BN = 4-bromonitrobenzene, BA = 4-bromoanisole). |
| Graphene–Fe3O4@Pd |
BB |
EtOH:H2O |
K2CO3 |
80 (MW) |
111000 |
18 |
| Graphene@Pd |
BB |
EtOH:H2O |
K2CO3 |
80 (MW) |
108000 |
19 |
| Pd NPs |
BN |
EtOH:H2O |
K3PO4 |
80 |
45000 |
20 |
| Polymer@Pd |
BA |
i-propanol:H2O |
K2CO3 |
100 |
41666 |
14b |
| Polymer@Pd |
BN |
i-propanol:H2O |
K2CO3 |
100 |
41666 |
14b |
| Pd (homogenous) |
BA |
EtOH:H2O |
K3PO4 |
60 |
14050 |
21 |
| Polymer@Pd |
BA |
H2O |
K2CO3 |
100 |
8083 |
22 |
Due that reusability is a significant factor for a catalyst we have investigated the behaviour of our six catalysts for the reaction of bromobenzene with phenylboronic acid. Thus, once finished the first reaction the catalyst was recovered by using an external magnet and used in the second round by mixing them with a new substrate, base, and solvent. The third and fourth rounds were carried out similarly. Table 4 displays the results obtained. From this Table it can be observed that the decrease of the catalytic activity is remarkable in all cases and that no catalysis was detected after the fourth cycle. Palladium analysis of the resulting solution after the first reaction permitted to calculate that about the 30% of the initial palladium content in the nanoparticles was leached. Consequently, partial homogeneous catalysis is not in doubt. This was confirmed by a hot filtration test. In detail, the substrate and phenylboronic acid were reacted at 65 °C. After 10 min the solid catalyst was separated by a magnet and the filtrated was transferred to another Schlenk flask. In absence of the catalyst the reaction progressed to 70–90% conversion after 1 h.
Table 4 TOF (h−1) for the recycling tests for Suzuki–Miyaura reaction using bromobenzene as substrate
| Fe3O4-L@Pd |
Cycle 1 |
Cycle 2 |
Cycle 3 |
Cycle 4 |
| L = 1a |
36979 |
4954 |
777 |
0 |
| L = 1b |
122085 |
9404 |
1305 |
0 |
| L = 2a |
1130 |
411 |
79 |
0 |
| L = 2b |
1750 |
657 |
115 |
0 |
| L = 3a |
19820 |
8320 |
624 |
0 |
| L = 3b |
110000 |
12450 |
950 |
0 |
Styrene hydrogenation. The selective hydrogenation of styrene is of great interest for the petroleum industry because the targeted product, ethylbenzene, can be used in the gasoline pool or valued in the aromatic industry so that reaction has been used in many articles as a model31 (Scheme 5). Reaction conditions and TOFs are listed in Table 7. The catalytic results are somewhat discouraging in relation to our attempts to establish direct relationship between palladium nanoparticles size and catalytic activity. Only catalysts Fe3O4-2a@Pd and Fe3O4-2b@Pd followed the expected catalytic trend since the TOFs observed for Fe3O4-2b@Pd is higher than that found for Fe3O4-2a@Pd. It is interesting to note that Fe3O4-2b@Pd was reused until six rounds while Fe3O4-2a@Pd leached a considerable amount of palladium in the first round. Fe3O4-1a@Pd is more efficient than Fe3O4-1b@Pd but the reuse of the first catalyst is very poor; in contrast, Fe3O4-1b@Pd did not exhibit loss of catalytic activity after 5 rounds. The tendency of phosphino-functionalized linkers to assist in the stabilization of palladium nanoparticles on the magnetite surface and, in its turn, to provide high reusability to the catalysts in some processes has been previously reported.8
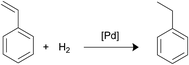 |
| | Scheme 5 Styrene hydrogenation. | |
Table 7 Catalytic results for the styrene hydrogenation reactiona
| Fe3O4-L@Pd |
TOF (h−1) |
| 3 bar H2, 5 mmol styrene, 2.3 × 10−4 mmol Pd, 25 °C, EtOH, 1 h. TOF = [mol of ethylbenzene/mol of Pd]/time (h). |
| L = 1a |
7904 |
| L = 1b |
6989 |
| L = 2a |
678 |
| L = 2b |
1090 |
| L = 3a |
16540 |
| L = 3b |
4880 |
The most exciting result is obtained by Fe3O4-3a@Pd which exhibits a TOF of 16540 h−1, the highest found in the literature up to now (13724 h−1 (ref. 32) and 8973 h−1 (ref. 13)). As discussed before a detailed analysis of the HAADF-STEM images of Fe3O4-3a@Pd showed a mixture of small and large nanoparticles and the presence of palladium wetting regions. Fig. 4 shows one of these peculiar regions with a Pd nanoparticle wetting over a relatively large region. They can be described as Pd monolayer. The high number of neighbour palladium single atoms is most probably the explanation for the unexpected high catalytic behaviour of Fe3O4-3a@Pd. To our knowledge, wetting metal areas on magnetite surfaces have not ever reported in the literature, but growth modes of metals on several surfaces have been discussed and analysed both experimentally33 and theoretically.34 For example, it has been found that palladium wetting decreases on c-ZrO2 support as the metal loading decreases.35 The drawback for using Fe3O4-3a@Pd is that this catalyst cannot be reused because the catalytic activity decreased enormously after the first round due to the leaching of palladium. A different behaviour was for observed for Fe3O4-3b@Pd since, in this case, the catalyst could be reused six rounds without evident loss of catalytic activity. Our results issue the following challenge: to synthesize immobilized Pd NPs on magnetite functionalized NPs with the high reusability provided by the PPh2 linkers, but with the exceptional catalytic behavior attained with the NH2 function. To achieve this goal might require promoting the wettability of the Pd NPs on the magnetite surfaces functionalized with phosphine-terminated linkers.
 |
| | Fig. 4 HAADF-STEM images of Pd NPs on the surface of the magnetite support. Left: wetting Pd NP spread over a large region on the magnetite surface as typically found in sample Fe3O4-3a@Pd. Right: representative image of a Pd NP showing the characteristic round morphology observed for the rest of the samples; in this particular case, it belongs to sample Fe3O4-2b@Pd. White arrows indicate the position of the Pd NPs. | |
Conclusions
Immobilized Pd NPs on the surface of magnetic NPs previously functionalized with amino (–NH2) or phosphino (–PPh2) linkers have been prepared. The HAADF-STEM images showed that the NPs obtained by means of the phosphino group are generally smaller and more homogeneous in size than those obtained by using amino functionalized linkers. In line with this finding, the catalytic efficiency of the phosphine-functionalized Pd NPs resulted to be higher than that shown by the amino-Pd-hybrids NPs for the Suzuki–Miyaura reaction and nitrophenol reduction; however, for the styrene hydrogenation the best catalytic result (and the best reported up to now) was obtained with the Fe3O4-3a@Pd catalyst, due to the unexpected presence of wetting palladium regions on the surface of this catalyst. Samples equipped with NH2-functionalized linkers showed negligible recyclability for the styrene hydrogenation whereas those decorated with PPh2-functions permitted their reuse for five or more rounds.
Experimental
All manipulations were performed under purified nitrogen using standard Schlenk Techniques. Organic Elemental Analysis (OEA) was performed in an Elemental Analyzer Thermo Scientific Flash 2000 A7 model. The Infrared Spectra (IR) were recorded with a NICOLET Impact 400 FT-IR. ICPoes (Inductively Coupled Plasma optical emission spectrometry) analysis was achieved in a Perkin Elmer 3200RL model. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was carried out using a double spherical aberration-corrected JEOL JEM-ARM200F microscope operated at 200 kV. The microscope was setup in STEM mode with a probe semiconvergence angle set to 25.3 mrad which yields a calculated probe size of about 80 pm. The annular semidetection range of the annular dark-field detector was set to collect electrons scattered between 90 and 370 mrad. Samples were gently plasma cleaned with an Ar(75%)/O2(25%) plasma for a few seconds to remove hydrocarbon contamination from the surfaces of the nanoparticles.
XPS (X-ray Photoelectron Spectroscopy) analysis was carried out in a SPECS instrument with Al X-ray source and a Phoibos 150 analyzer. The spectrum was recorded under 10−7 Pa. Binding energies were referred to the C 1s signal at 284.8 eV. All reactants were purchased from suppliers in “for synthesis” quality or higher and used without further purification. The solvents were HPLC quality and/or were dried trough pure solve instrument from Innovate Technology USA Inc. Deionized water was obtained from Milipore Helix 3 water purification system.
Ferrite nanoparticles10 and linkers 1b10 and 2b11 were synthesized according to the literature. 1a, 2a, 3a and 3b are commercially available.
Synthesis of the catalysts
400 mg of ferrite nanoparticles were sonicated in methanol (15 mL) for 30 min. Then, a solution of 1 mmol of linkers (1a, 1b, 2a, 2b, 3a and 3b) in methanol was added separately to a suspension of Fe3O4. The mixtures were sonicated for 2 h. After this time, the nanoparticles obtained Fe3O4-Lxy (x = 1–3; y = a, b) were washed with ethanol (3 × 30 mL) and acetone (30 mL), collected by magnetic separation and dried under reduced pressure. After sonication of 100 mg of the of the Fe3O4-Lxy nanoparticles in water (10 mL) for 30 min, 4.1 mg of K2[PdCl4] (for xy = 2a, 2b, 3a, 3c) or 2 mg of K2[PdCl4] (for xy = 1a, 1b) dissolved in water (2 mL) were added and stirred (1000 rpm) for 2 h. After that, the nanoparticles were washed with water (2 × 10 mL), ethanol (2 × 10 mL) and acetone (10 mL), dispersed in water (8 mL) and reacted with an aqueous solution (2.5 mL) of sodium borohydride 0.05 M. Then, the resulting nanoparticles were washed with water (3 × 30 mL) and acetone (1 × 10 mL), removed by an external magnet and dried under reduced pressure.
Suzuki–Miyaura coupling
In a typical Suzuki–Miyaura reaction 14.4 mmol of phenylboronic acid, 36 mmol of base, and 2 mg of Fe3O4-2b@Pd (9.4 × 10−5 mmol) were weighed into a Schlenk tube. Then, 60 mL of solvent (ethanol:water 1:1) was added under nitrogen. After that, the tube was brought to a preheated plate at 65 °C and 12 mmol (1 eq.) of substrate was added. The reaction was maintained for 1 hour, time where the conversion of the substrate is not complete and allows comparison of catalyst activity. The mixture was allowed to cool down and extraction with ethyl acetate was carried out. The organic phase was removed, dried over sodium sulphate and analyzed by GC. The identity of products was confirmed by GC-MS analysis.
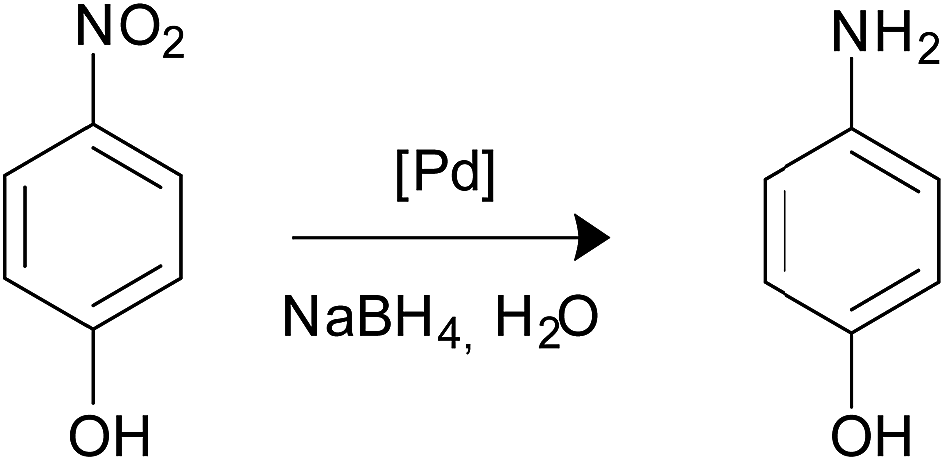
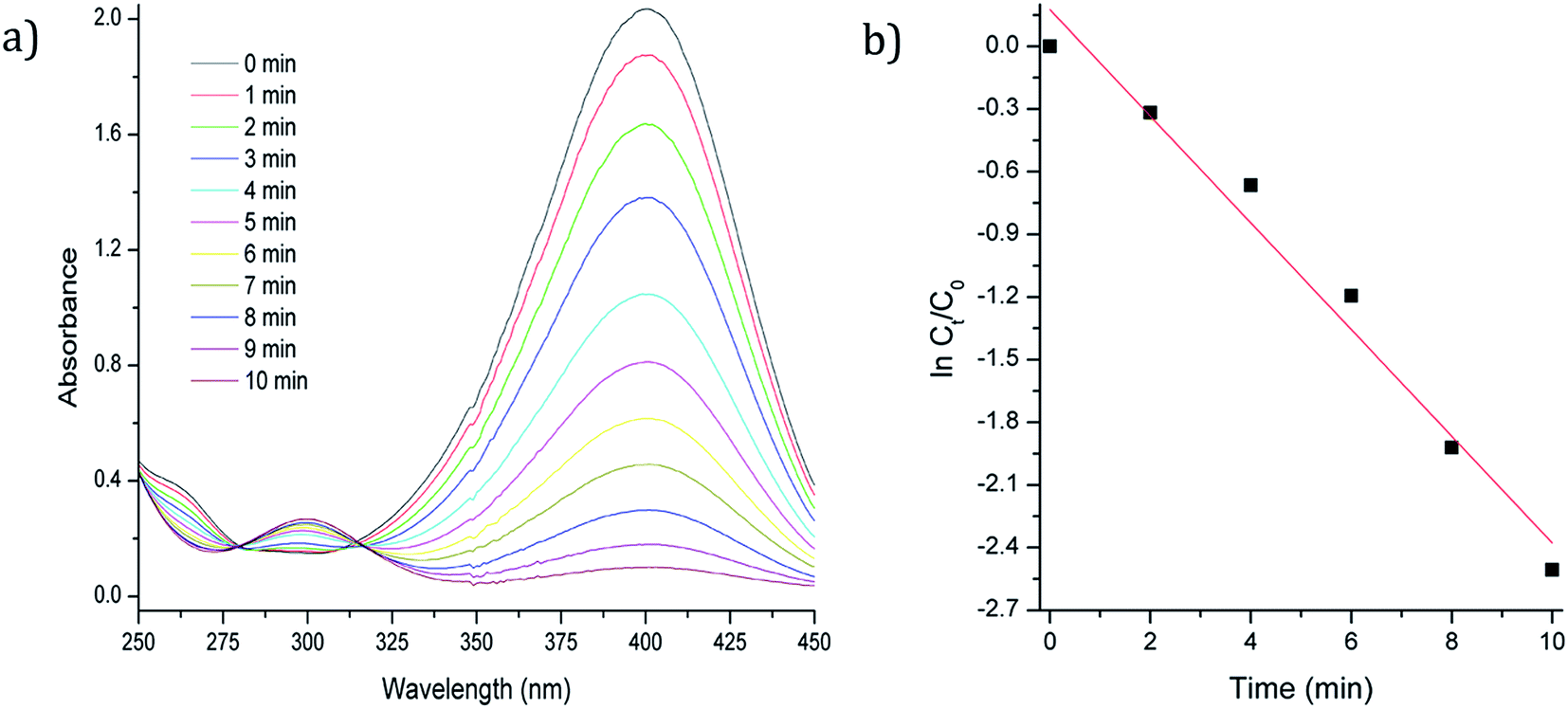
Catalytic reduction of 4-nitrophenol
30 μL of p-nitrophenol (7.4 mM) and 30 μL of NaBH4 (0.40 M) were added into a quartz cuvette containing 2 mL of water. Then, 30 μL of an aqueous solution containing catalyst nanocomposite (3 to 5 mg approximately in 5 mL of water) was injected into the cuvette to start the reaction. The intensity of the absorption peak at 400 nm in UV-Vis spectroscopy was used to monitor the process of the conversion of 4-nitrophenol to 4-aminophenol. After each cycle of reaction, another 30 μL of 4-nitrophenol and 30 μL of NaBH4 were added to the reaction to study the reuse of the catalyst. The catalytic reduction reactions were conducted at room temperature.
Hydrogenation of styrene
Hydrogenation reactions were carried out in a hydrogen atmosphere at room temperature. Typically, under nitrogen, ferrite catalyst (5 mg approximately) was dispersed in ethanol freshly distilled (20 mL), and then, styrene (5 mmol) was poured. After that, the mixture was transfer into a Fisher–Porter tube, and then filled with hydrogen (3 bar), and the mixture was stirred at constant speed for 1 hour, time where the conversion of the substrate is not complete and allows comparison of catalyst activity. The conversion was determined by GC analysis.
Acknowledgements
This work was financially supported by the MICINN (projects CTQ2012-31335 and CTQ2015-65040 P) and by the MINECO/FEDER, UE (project CTQ2016-76120 P). J. L. is a Serra Húnter Fellow and is grateful to the ICREA Academia program. Access to the TEM facilities at IBM/EMPA Master Joint Development Agreement is gratefully acknowledged. A. G. thanks the University of Tolima (Colombia) for financial support.
References
-
(a) M. Lamblin, L. Nassar-Hardy, J. C. Hierso, E. Fouquet and F. X. Felpin, Adv. Synth. Catal., 2010, 352, 33–79 CrossRef CAS
 ;
(b) A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, RSC Adv., 2016, 6, 11446–11453 RSC ;
(c) S. Paul, M. M. Islam and S. M. Islam, RSC Adv., 2015, 5, 42193–42221 RSC .
;
(b) A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, RSC Adv., 2016, 6, 11446–11453 RSC ;
(c) S. Paul, M. M. Islam and S. M. Islam, RSC Adv., 2015, 5, 42193–42221 RSC . -
(a) B. Karimi, F. Mansouri and H. M. Mirzae, ChemCatChem, 2015, 7, 1736–1789 CrossRef CAS ;
(b) D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed ;
(c) N. Baig and R. Varma, Chem. Commun., 2013, 49, 752–770 RSC ;
(d) N. J. S. Costa and L. M. Rossi, Nanoscale, 2012, 4, 5826–5834 RSC .
-
(a) L. M. Rossi, N. J. S. Costa, F. P. Silva and R. Wojcieszak, Green Chem., 2014, 16, 2906–2933 RSC ;
(b) L. M. Rossi, I. M. Nangoi and N. J. S. Costa, Inorg. Chem., 2009, 48, 4640–4642 CrossRef CAS PubMed ;
(c) M. Guerrero, N. J. S. Costa, L. L. R. Vono, L. M. Rossi, E. V. Gusevskaya and K. Philippot, J. Mater. Chem. A, 2013, 1, 1441–1449 RSC .
-
(a) N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209–2212 CrossRef CAS PubMed ;
(b) A. H. Latham and M. E. Williams, Acc. Chem. Res., 2008, 41, 411–420 CrossRef CAS PubMed ;
(c) C. W. Lim and I. S. Lee, Nano Today, 2010, 5, 412–434 CrossRef CAS ;
(d) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed ;
(e) R. B. Nasir Baig and R. J. Varma, Green Chem., 2012, 14, 625–632 RSC ;
(f) R. B. Nasir Baig and R. J. Varma, Green Chem., 2013, 15, 398–417 RSC .
- B. Karimi, F. Mansouri and H. Vali, Green Chem., 2014, 16, 2587–2596 RSC .
- F. P. S. da Silva and L. M. Rossi, Tetrahedron, 2014, 70, 3314–3318 CrossRef .
- D. K. Yi, S. S. Lee and J. Y. Ying, Chem. Mater., 2006, 18, 2459–2461 CrossRef CAS .
- N. J. S. Costa, P. K. Kiyohara, A. L. Monteiro, Y. Coppel, K. Philippot and L. M. Rossi, J. Catal., 2010, 276, 382–389 CrossRef CAS .
- J. Durand, E. Teuma and M. Gómez, Eur. J. Inorg. Chem., 2008, 23, 3577–3586 CrossRef .
- F. Gonzàlez de Rivera, I. Angurell, M. D. Rossell, R. Erni, J. Llorca, N. J. Divins, G. Muller, M. Seco and O. Rossell, Chem.–Eur. J., 2013, 19, 11963–11974 CrossRef PubMed .
- A. Guarnizo, I. Angurell, M. D. Rossell, J. Llorca, G. Muller, M. Seco and O. Rossell, RSC Adv., 2015, 5, 91340–91348 RSC .
- A. Guarnizo, I. Angurell, G. Muller, J. Llorca, M. Seco, O. Rossell and M. D. Rossell, RSC Adv., 2016, 6, 68675–68684 RSC .
- M. D. Rossell, F. J. Caparrós, I. Angurell, G. Muller, J. Llorca, M. Seco and O. Rossell, Catal. Sci. Technol., 2016, 6, 4081–4085 CAS .
-
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS ;
(b) L. Geng, Y. Li, Z. Qi, H. Faan, Z. Zhou, R. Chen, Y. Wang and J. Huang, Catal. Commun., 2016, 82, 24–28 CrossRef CAS .
-
(a) F. Lu, J. Ruiz and D. Astruc, Tetrahedron Lett., 2004, 45, 9443–9445 CrossRef CAS ;
(b) H. Shen, C. Shen, C. Chen, A. Wang and P. Zhang, Catal. Sci. Technol., 2015, 5, 2065–2071 RSC ;
(c) P. Li, L. Wang, L. Zhang and G-W Wang, Adv. Synth. Catal., 2012, 354, 1307–1318 CrossRef CAS ;
(d) Y. Huang, Z. Zheng, T. Liu, J. Lü, J. Lin, H. Li and R. Cao, Catal. Commun., 2011, 14, 27–31 CrossRef CAS .
-
(a) A. Dewan, P. Bharali, U. Bora and A. J. Thakur, RSC Adv., 2016, 6, 11758–11762 RSC ;
(b) Q. Zhang, H. Su, J. Luo and Y. Wei, Catal. Sci. Technol., 2013, 3, 235–243 RSC ;
(c) D. Rosario-Amorin, X. Wang, N. Gaboyard, R. Clérac, S. Nlate and K. Heuzé, Chem.–Eur. J., 2009, 15, 12636–12643 CrossRef CAS PubMed .
- B. P. S. Chauhan, J. S. Rathore and T. Bandoo, J. Am. Chem. Soc., 2004, 126, 8493–8500 CrossRef CAS PubMed .
- H. A. Elazab, A. R. Siamaki, S. Moussa, B. F. Gupton and M. S. El-Shall, Appl. Catal., A, 2015, 491, 58–69 CrossRef CAS .
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS .
- C. Deraedt, L. Salmon, L. Etienne, J. Ruiz and D. Astruc, Chem. Commun., 2013, 49, 8169–8171 RSC .
- Z. Guan, J. Hu, Y. Gu, H. Zhang, G. Li and T. Li, Green Chem., 2012, 14, 1964–1970 RSC .
- Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 3190–3198 CrossRef CAS PubMed .
- Y. Jang, S. Kim, S. W. Kun, B. H. Kim, S. Hwans, I. K. Song, B. M. Kim and T. Hyeon, Chem. Commun., 2011, 47, 3601–3603 RSC .
- T. Yao, H. Wang, Q. Zuo, J. Wu, X. Zhang, F. Cui and T. Cui, Chem.–Asian J., 2015, 10, 1940–1947 CrossRef CAS PubMed .
- Z. Dong, X. Le, Y. Liu, C. Dong and J. Ma, J. Mater. Chem. A, 2014, 2, 18775–18785 CAS .
- K. Jiang, H.-X. Zhang, Y.-Y. Yang, R. Mothes, H. Lang and W.-B. Cai, Chem. Commun., 2011, 47, 11924–11926 RSC .
- Y. Xue, X. Lu, X. Bian, J. Lei and C. Wang, J. Colloid Interface Sci., 2012, 379, 89–93 CrossRef CAS PubMed .
- X. Le, Z. Dong, X. Li, W. Zhang, M. Le and J. Ma, Catal. Commun., 2015, 59, 21–25 CrossRef CAS .
- J. Morère, M. J. Tenorio, M. J. Torralvo, C. Pando, J. A. R. Renuncio and A. Cabañas, J. Supercrit. Fluids, 2011, 56, 213–222 CrossRef .
- C.-H. Liu, R.-H. Liu, Q.-J. Sun, J.-B. Chang, X. Gao, Y. Liu, S.-T. Lee, Z.-H. Kang and S.-D. Wang, Nanoscale, 2015, 7, 6356–6362 RSC .
- F. Corvaisier, Y. Schuurman, A. Fecant, C. Thomazeau, P. Raybaud, H. Toulhoat and D. Farrusseng, J. Catal., 2013, 307, 352–361 CrossRef CAS .
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–801 CrossRef CAS PubMed .
- P. J. Feibelman, Phys. Rev. Lett., 1998, 81, 168–171 CrossRef CAS .
- B. D. Yu and M. Scheffler, Phys. Rev. Lett., 1996, 77, 1095–1098 CrossRef CAS PubMed .
- M. Alfredsson, C. Richard and A. Catlow, Surf. Sci., 2004, 561, 43–56 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Miquel Secoa,
Guillermo Mullera,
Jordi Llorcac and
Oriol Rossella
*a,
Miquel Secoa,
Guillermo Mullera,
Jordi Llorcac and
Oriol Rossella