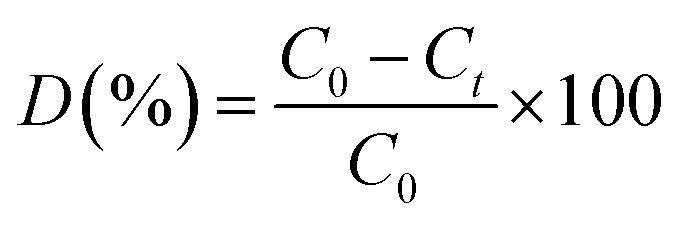DOI:
10.1039/C7RA03633G
(Paper)
RSC Adv., 2017,
7, 24754-24763
Highly efficient visible-light photocatalytic activity of MoS2–TiO2 mixtures hybrid photocatalyst and functional properties†
Received
29th March 2017
, Accepted 20th April 2017
First published on 9th May 2017
Abstract
2-D-layered molybdenum disulfide (MoS2) and MoS2/TiO2 nanocomposite were synthesized by a hydrothermal method. The effects of the concentration of TiO2 on the formation of MoS2/TiO2 composites and functional properties were investigated. X-ray diffraction patterns revealed the formation of hexagonal and anatase structure of MoS2 and TiO2, respectively. Core-level X-ray photoelectron spectroscopy confirmed the presence of Mo and Ti interaction by a significant peak shift. Morphological analysis revealed the formation of TiO2 on the surface of the MoS2 nanosheets. The photocatalytic degradation of methylene blue (MB) in an aqueous suspension was employed to evaluate the visible-light activity of the as-prepared composite photocatalyst. The MB absorption peaks completely disappeared after 12 min with 99.33% of degradation under visible-light irradiation at the TiO2 concentration of 0.005 M. It was found that hydroxyl radical (˙OH) played an important role in the degradation of MB under visible-light irradiation. The possible charge-transfer mechanism has been proposed in this study.
1. Introduction
Energy crisis and environmental remediation are the two most important issues that need to be resolved for living beings and the society. The sun irradiates 1.5 × 1018 kW h energy to earth every year, which is approximately 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times of the total annual energy consumption.1 Hence, semiconductor photocatalysis seems to be more attractive and easier to apply for clean energy production and environmental remediation.2–4 The photocatalytic activities of a semiconductive material depend on several factors such as absorption ability of photocatalysts, separation and transport rate of the photogenerated electrons and holes, and photoabsorption ability in the available light energy region. Titanium dioxide (TiO2) is one of the most explored and traditional semiconductor photocatalysts.5 It has a strong oxidizing power, high stability, low-cost, an abundant source, and is relatively non-toxic.6–10 However, TiO2 responds only to UV light, which is only 3–5% of the total sunlight.11 The band gap of TiO2 is 3.0 and 3.2 eV for the rutile and anatase phases, respectively, which is not compatible with visible-light excitation.12 Since the electron–hole recombination rate is faster, it is mainly responsible for its limited application in catalysis.13 Therefore, it is necessary to overcome this drawback by charge separation via suppressing the recombination rate of the electron and hole pairs.
000 times of the total annual energy consumption.1 Hence, semiconductor photocatalysis seems to be more attractive and easier to apply for clean energy production and environmental remediation.2–4 The photocatalytic activities of a semiconductive material depend on several factors such as absorption ability of photocatalysts, separation and transport rate of the photogenerated electrons and holes, and photoabsorption ability in the available light energy region. Titanium dioxide (TiO2) is one of the most explored and traditional semiconductor photocatalysts.5 It has a strong oxidizing power, high stability, low-cost, an abundant source, and is relatively non-toxic.6–10 However, TiO2 responds only to UV light, which is only 3–5% of the total sunlight.11 The band gap of TiO2 is 3.0 and 3.2 eV for the rutile and anatase phases, respectively, which is not compatible with visible-light excitation.12 Since the electron–hole recombination rate is faster, it is mainly responsible for its limited application in catalysis.13 Therefore, it is necessary to overcome this drawback by charge separation via suppressing the recombination rate of the electron and hole pairs.
MoS2 has drawn extensive attention because of its layered structure, similar to that of graphene. Each layer is composed of three atomic layers stacked together, in which Mo atom in the middle is strongly bonded to S atoms present above and below.14,15 This two-dimensional (2D)-layered crystal structure provides convenient electron transfer and many active sites for sunlight absorption. The band gap of MoS2 increases with the decreasing number of layers due to quantum confinement effect. The direct band gap of ∼1.8 eV makes it a visible-light active photocatalyst.16,17 The advantages of MoS2 include strong oxidizing activity, high hardness, high stability and reliability, low-cost, and non-toxicity.18–23 Researchers have focused on synthesizing heterostructure nanocomposites such as MoS2/RGO, MoS2/TiO2, MoS2/CuO, MoS2/CuS, and MoS2/MoO3.24–29 Among these, MoS2/TiO2 heterostructure is the best photocatalyst under visible-light irradiation due to the separation of charge carriers. MoS2/TiO2 nanostructures possess more surface-active sites than other nanostructures and this enhances the light absorption by the photocatalysts.30,31 Recently, Z. B. Chen and his co-workers have prepared MoO3–MoS2 core–shell nanostructures by chemical vapor deposition method and studied the photocatalytic hydrogen evolution reaction.32 B. Pourabbas et al. have synthesized MoS2 photocatalysts by hydrothermal method at 300 °C and 12 h and investigated the photooxidation of phenol.33 X. Zu and co-workers have synthesized TiO2-decorated MoS2 nanosheets by hydrothermal method at 220 °C and 24 h.34 L. Cao et al. have synthesized MoS2-hybridized TiO2 nanosheets by hydrothermal growth at 210 °C for 24 h and measured its visible light photocatalytic activity.35 R. Tang et al. have demonstrated a facile two-step hydrothermal method (200 °C for 24 h) to synthesize layered MoS2-coupled metal–organic framework (MOF)-derived dual-phase TiO2 (MDT) as photoanodes for photoelectrochemical water splitting and dye-sensitized solar cells (DSSCs).36 H. Li et al. have synthesized layered MoS2 nanosheet-coated TiO2 nanobelts by hydrothermal method at 200 °C and 24 h.37 Meng Shen et al. have prepared MoS2 nanosheet/TiO2 nanowire hybrid nanostructures by a hydrothermal method at 240 °C and 24 h and studied their visible-light photocatalytic activities.38 However, these reports are related to high-temperature synthesis; therefore, relatively low temperature (<200 °C) synthesis of MoS2/TiO2 nanocomposites is limited because of poor crystallinity and non-uniform distribution in the morphology. Therefore, a new approach has to be developed to realize the mass production of MoS2 at relatively low temperature (i.e. <200 °C). X. Zong et al. have synthesized CdS/MoS2 heterostructure photocatalyst using a hot-wire chemical vapor deposition method (HWCVD), and they found that the H2 production enhanced under visible-light irradiation.39 Weijia Zhou et al. have synthesized few layers of 2-D MoS2/TiO2 composite photocatalyst by a hydrothermal method at 200 °C and 24 h, and they found that the light absorption range for the photocatalytic H2 production was enhanced.40 As is well-known, coupling of two semiconductors with narrow and wide band gaps could extend the solar spectrum for light utilization. Therefore, it is necessary to couple two semiconductors with matched energy levels to enhance the photocatalytic activity under visible-light irradiation through interfacial charge transfer between MoS2 and TiO2 nanosheets.41
Herein, the layered MoS2 and MoS2/TiO2 nanosheets were synthesized by a hydrothermal method. The effects of TiO2 concentration on the formation of the nanosheets were investigated. The photocatalytic activity of the synthesized materials was characterized by quantifying the rate of methylene blue (MB) degradation in an aqueous suspension under visible-light irradiation. MoS2/TiO2 nanocomposite photocatalyst exhibited enhanced light absorption capacity as compared to pure MoS2 and TiO2 photocatalysts. Kinetics, electron trapping, and possible photocatalytic mechanism were studied.
2. Experimental
Sodium molybdate dihydrate [(Na2MoO4·2H2O), 99.5%], thioacetamide [(C2H5NS), 98%], citric acid [(C6H8O7), 98%], and diammonium hexafluorotitanate (DAHFT) [((NH4)2TiF6), 95%] were purchased from Wako chemicals (Japan) and used without further purification.
2.1 Synthesis of 2D-layered MoS2 nanostructures
2D-layered MoS2 nanosheets were prepared by a hydrothermal method similar to a previous report.42 In a typical synthesis, 0.04 M of sodium molybdate dihydrate, 0.08 M of thioacetamide, and 0.04 M of citric acid were dissolved in 50 mL of deionized water under vigorous stirring for 4 h. The transparent aqueous solution was transferred into a 100 mL stainless steel autoclave and maintained at 180 °C for 24 h. The final product was obtained from the autoclave through centrifuging thrice with deionized water and ethanol and drying at 80 °C for 10 h. The sample was labelled as S1.
2.2 Synthesis of layered MoS2–TiO2 mixture heterostructure
The MoS2–TiO2 mixture heterostructures were prepared by a hydrothermal method. The amount of DAHFT, as a Ti source, was changed as a parameter. In a typical reaction process, 0.04 M of sodium molybdate dihydrate, 0.08 M of thioacetamide, (0.0015, 0.0030, 0.0050, 0.010, and 0.015 M) of DAHFT, and 0.04 M of citric acid were dissolved in 50 mL deionized water under vigorous stirring for 4 h. The transparent aqueous solution was transferred into a 100 mL stainless steel autoclave and maintained at 180 °C for 24 h. The final product was obtained from the autoclave and centrifuged thrice with deionized water and ethanol and dried at 80 °C for 10 h. The samples were labelled as S2 (0.0015 M), S3 (0.0030 M), S4 (0.0050 M), S5 (0.010 M), and S6 (0.015 M).
2.3 Photocatalytic dye degradation
Photocatalytic degradation of methylene blue (MB) was carried out under visible-light irradiation. The optical absorption of MB at 664 nm was used as a monitor wavelength of photodegradation. Then, 5 mg of MB was added to 100 mL of aqueous solution and stirred for 30 min. After this, 50 mg of MoS2 and MoS2/TiO2 photocatalysts were separately added to the abovementioned solution. To establish absorption/desorption equilibrium, the mixed solution was stirred for 30 min in the dark before the photodegradation reaction. The xenon lamp as a visible light source was placed 15 cm away from the surface of the solution. The dye degradation surveys were conducted at the interval of 4 min, and the catalyst was isolated via centrifugation. The photodegradation percentage of MB was calculated using the following equation:43| |
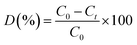 | (1) |
where C0 and Ct are the concentrations of MB at time 0 and t (s), respectively, and t is the irradiation time.
2.4 Characterization
The structure of the product was characterized by powder X-ray diffraction (XRD) using a Rigaku diffractometer (RINT ULTIMA – 2200, Japan, Cu Kα radiation). X-ray photoelectron spectroscopy (XPS) was performed via a Kratos analytical instrument (Shimadzu Corporation, ESCA 3400, Japan). Surface morphologies were characterized by field-emission scanning electron microscopy (FESEM) using a JEOL JSM 7001F microscope at an accelerating voltage of 15 kV. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM 2100F microscope at an accelerating voltage of 200 kV. UV-visible spectroscopy (JASCO V – 670) was used to detect the absorbance of the MB dye degradation.
3. Results and discussion
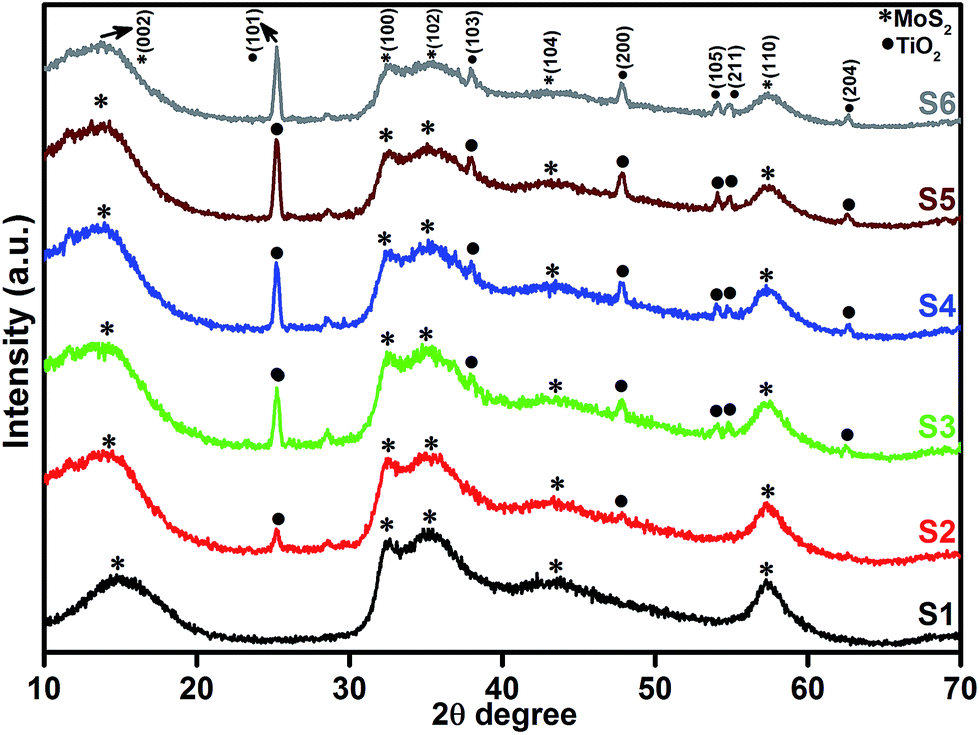
Fig. 1 shows the powder XRD patterns of pure MoS2 and MoS2/TiO2 nanocomposites. Fig. 1 (S1) shows pure MoS2 with its diffraction peaks. The peaks corresponded to the hexagonal phase of MoS2, which matched well with the standard JCPDS card number (024-0513). Peaks at 14.4°, 32.6°, 35.8°, 44.2°, and 58.2° can be indexed to the (002), (100), (102), (104), and (110) crystal planes, respectively. After the addition of TiO2, new peaks appeared at 25.2°, 37.8°, 48.0°, 53.8°, 55.0°, and 62.6°, which corresponded to the (101), (103), (200), (105), (211), and (204) crystal planes, respectively. This confirmed the presence of TiO2 in the nanocomposites. As the concentration of TiO2 increased, the peak intensity of TiO2 also increased. The structures of MoS2 and TiO2 in all the samples were indexed to the hexagonal (024-0513) and anatase structure (021-1272), respectively. No other characteristic peaks were observed.
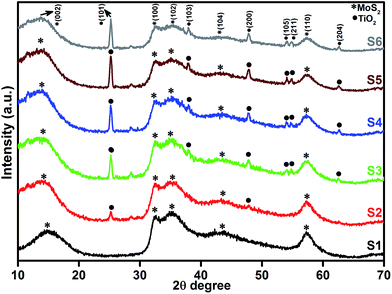 |
| | Fig. 1 XRD patterns of the layered MoS2 and TiO2 nanosheets. | |
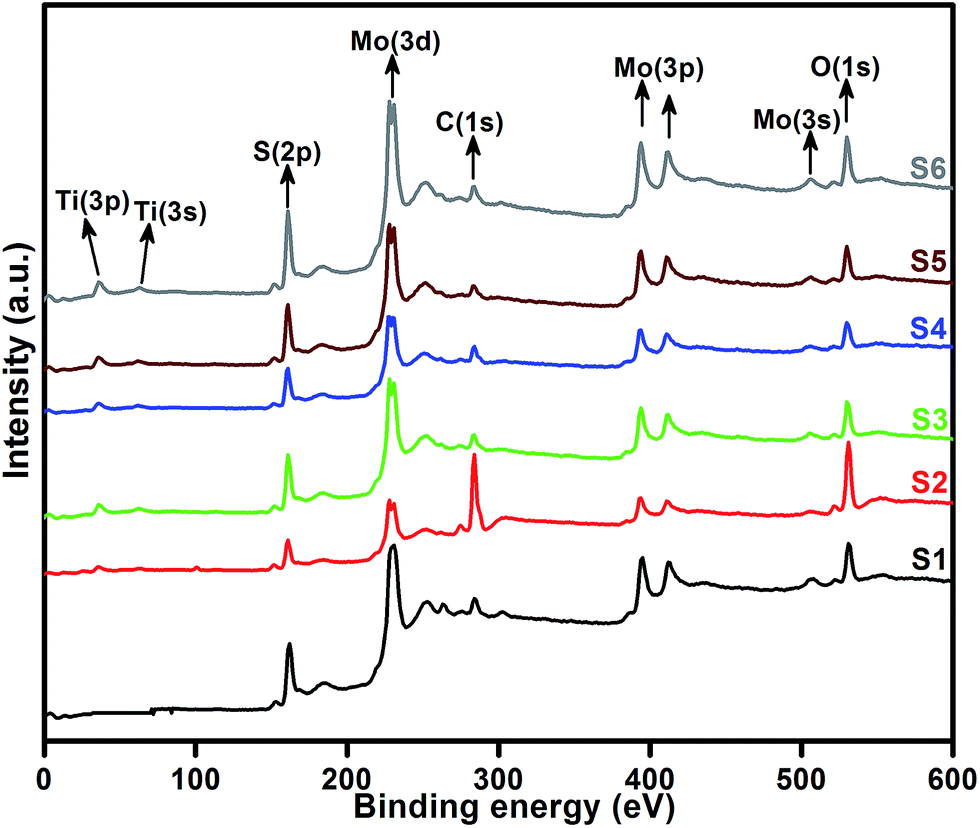
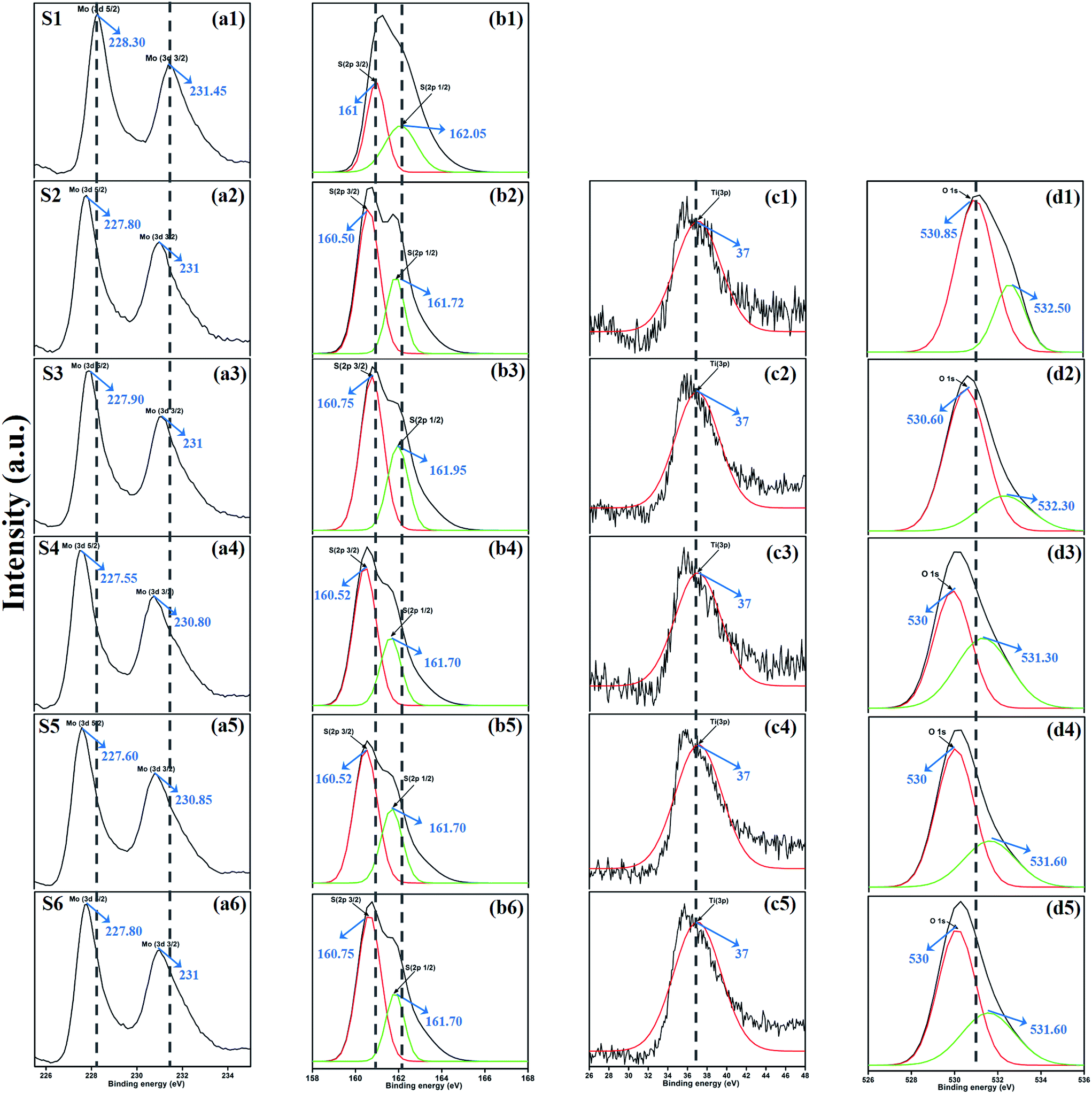
XPS measurement was carried out to observe the chemical state of the MoS2 and MoS2/TiO2 nanocomposite. Fig. 2 shows the survey spectra of MoS2/TiO2. The characteristic peaks at 36.90, 161.80, 229.40, and 529.89 eV can be attributed to Ti 3p, S 2p, Mo 3d, and O 1s, respectively. The high resolution XPS spectra of Ti 3p, S 2p, Mo 3d, and O 1s are shown in Fig. 3. Herein, two symmetric peaks appeared for Mo 3d state, as shown in Fig. 3(a1–a6). The peaks centered at 228.30 and 231.45 eV (S1) can be attributed to the Mo 3d5/2 and Mo 3d3/2 states, respectively, indicating a +4 oxidation state.44 The energy separation between the Mo 3d5/2 and Mo 3d3/2 states was 3.1 eV, which was in good agreement with the standard value.45 The binding energies of the sample S2 were shifted to 227.80 and 231 eV from 228.30 and 231.45 eV of pure MoS2, respectively. On further increasing the concentration of TiO2, the peaks shifted to 227.90 and 231 eV for the sample S3, 227.55 and 230.80 eV for the sample S4, 227.55 and 230.80 eV for the sample S5, and 227.5 and 230.80 eV for the sample S6. The samples showed significant peak shift because of strong interaction between Mo and Ti. The S 2p spectra, as shown in Fig. 3(b1–b6), can be deconvoluted into two peaks located at 161 and 162.05 eV, which can be attributed to the S 2p3/2 and S 2p1/2 orbitals of divalent sulfide ions (S2−). The energy separation between S 2p3/2 and S 2p1/2 was 1.1 eV, which was in good agreement with the reported value.46 The binding energies shifted to 160.50 and 161.72 for the sample S2, 160.75 and 161.95 for the sample S3, 160.52 and 161.70 for the sample S4, and 160.52 and 161.70 for the sample S5. Moreover, for the samples S4 (a4, b4), S5 (a5, b5), and S6 (a6, b6), binding energies of Mo 3d5/2, Mo 3d3/2, S 2p3/2, and S 2p1/2 were shifted about 0.20 eV when compared with those of the samples S2 (a2) and S3 (a3). This slight shift was attributed to the strong interaction between MoS2 and TiO2. Fig. 3(c1–c5) shows the Ti 3p state with the peak centered at 37 eV. There was no significant peak shift observed in the Ti 3p state. O 1s states of the samples are shown in Fig. 3(d1–d5). All the samples exhibited an asymmetrical shape and two symmetrical peaks centered at 530.85 eV and 532.50 eV, indicating two different types of O species in the samples. The binding energy of 530.85 eV corresponded to O2− ions surrounded by Ti in the TiO2 compound system. The shoulder peak observed at 532.50 eV corresponded to the chemisorbed oxygen, dissociated oxygen, or OH− groups on the surface. Similar shift in the peaks was observed as the concentration of TiO2 was increased, 530.60 eV for sample S3, 530 eV for sample S4, 530 eV for sample S5, and 530 eV for sample S6. O 1s binding energy of the abovementioned samples was shifted about 0.20 eV when compared with those of the samples S1 (d1) and S2 (d2). The XPS measurement confirmed the presence of TiO2 in the prepared nanocomposites.
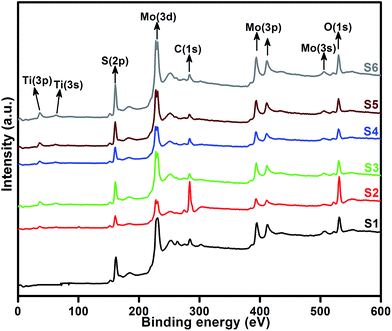 |
| | Fig. 2 High resolution XPS spectra of the layered MoS2 and TiO2 nanosheets. | |
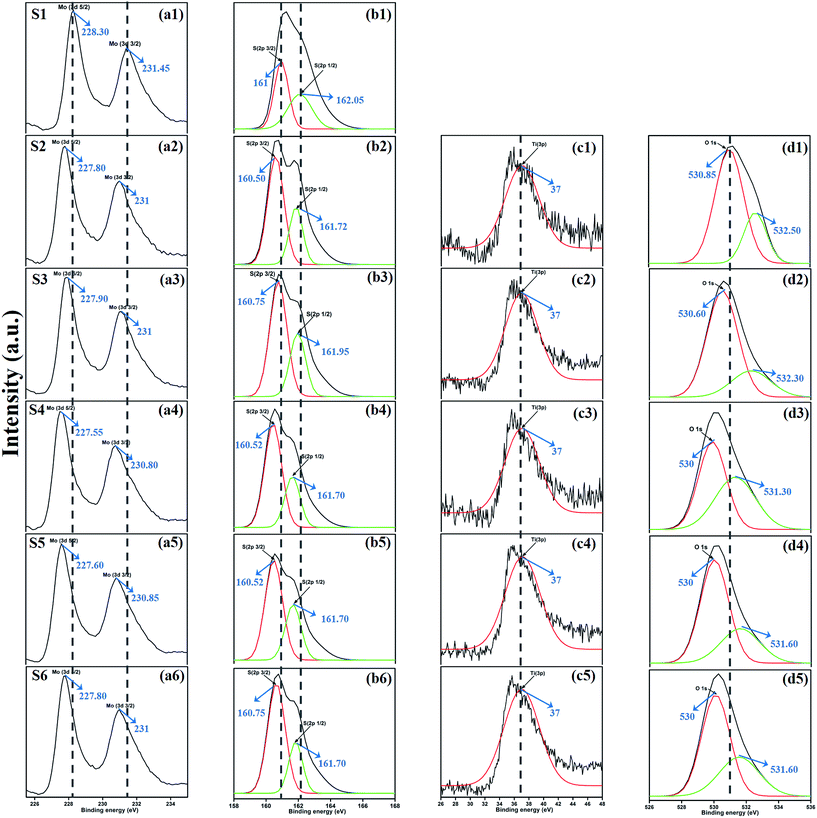 |
| | Fig. 3 XPS spectra of the Mo 3d state (a1–a6), S 2p state (b1–b6), Ti 3p state (c1–c5), and O 1s state (d1–d5) of samples S1 to S6. | |
The morphological analysis was performed using FESEM, TEM, and HRTEM, as shown in Fig. 4. Fig. 4(a) shows the monodispersed pure MoS2 (S1) nanosheets with a layered structure. The thickness of each layer was about 20–50 nm. When TiO2 was incorporated into the MoS2 nanosheets, TiO2 agglomerated on the surface of the nanosheets. This was evident from the FESEM images of S4 (j), S5 (m), and S6 (p). This agglomeration was attributed to the increased ionic strength of the TiO2 nanoparticles. The layered structure of the sample was further characterized by TEM and HRTEM, as shown in Fig. 4(b) and (c). MoS2 nanosheets had 5–9 layers and the lattice d-spacing was 0.61 nm, which corresponded to the (002) plane. Fig. 4(d)–(f) show the FESEM, TEM, and HRTEM images of the MoS2/TiO2 nanocomposites of the S2 sample, respectively. In the HRTEM images (f, i, l, o, and r), white dashed line corresponded to the MoS2-layered nanosheets and white squared dot line indicated the TiO2 nanosheets. When the concentration of TiO2 was increased from 0.0015 to 0.015 M, formation of TiO2 nanosheets was observed in the samples S2 to S6. Nanocomposites were composed of MoS2 and TiO2, as shown in Fig. 4. It can be concluded from XPS and TEM analysis that the layered MoS2 nanosheets and TiO2 were formed as composites.
 |
| | Fig. 4 (a, d, g, j, m, and p) FESEM, (b, e, h, k, n, and q) TEM and (c, f, i, l, o, and r) HRTEM images of the samples S1 to S6. | |
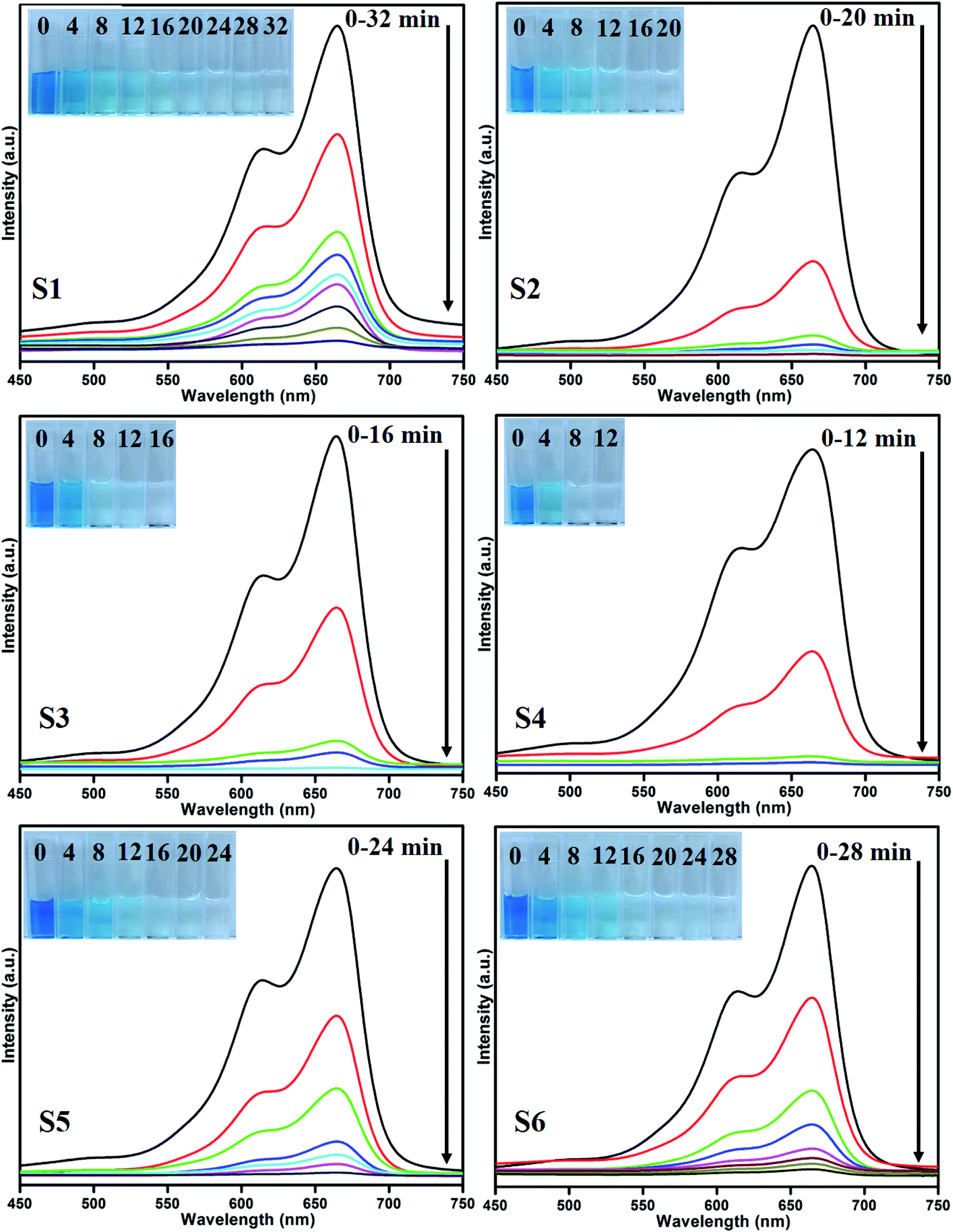
3.1 Photocatalytic activity
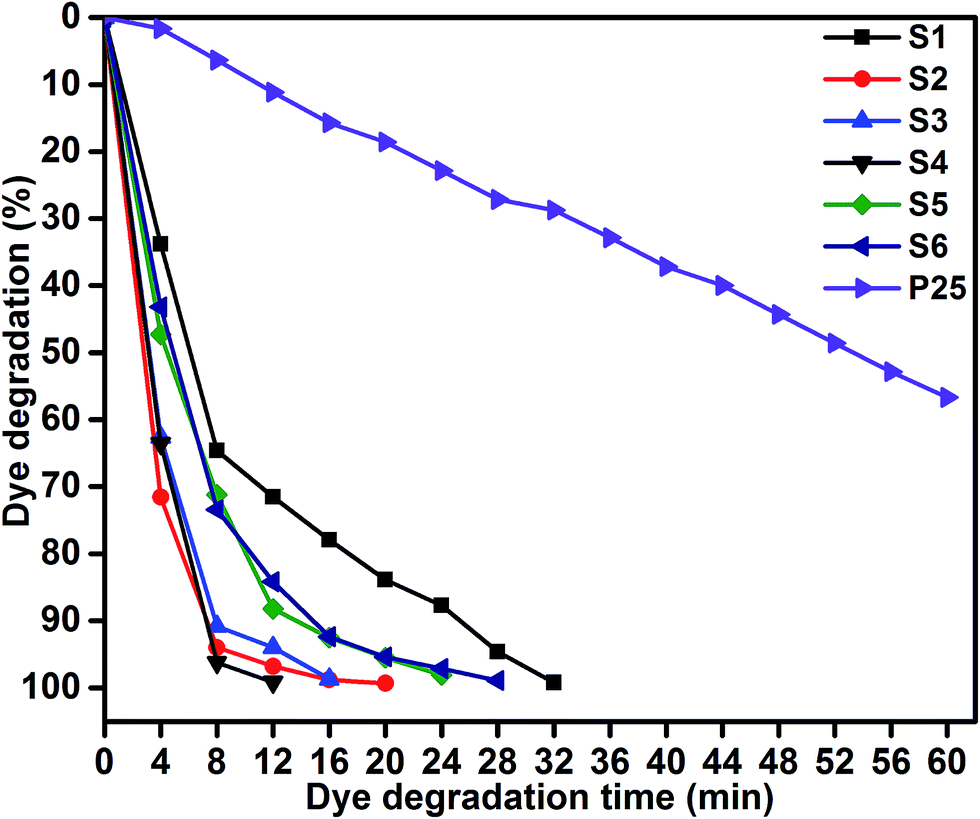
Photocatalytic performances of MoS2 and MoS2/TiO2 photocatalyst were examined by degrading MB under visible-light irradiation. The characteristic absorption peak of MB at 664 nm was chosen to monitor the photocatalytic degradation of MB. Fig. 5 shows the time-dependent absorption spectra of pure MoS2 (S1) and MoS2/TiO2 (S2–S6). The intensity of the absorption peak at 664 nm rapidly decreased with illumination time and the peak almost disappeared after 32 min for sample S1. However, for S2, S3, S4, S5, and S6 samples, irradiation time was decreased to 20, 16, 12, 24, and 28 min, respectively. Fig. 6 shows that the percentage of degradation for the samples S1, S2, S3, S4, S5, S6, and P25 (TiO2) is 99.20, 99.17, 98.68, 99.33, 98.08, 98.90, and 56.89%, respectively. Moreover, 71.50, 93.99, 96.79, 99.17, 88.25, and 84.21% MB was degraded in 12 min of irradiation for sample S1, S2, S3, S4, S5, and S6, respectively. It was evident that the photodegradation efficiency of the MoS2/TiO2 photocatalyst was significantly higher than that of pure MoS2 and TiO2 (P25). Among all the synthesized samples, S4 showed enhanced photocatalytic activity because the oxidation state of MoS2/TiO2 photocatalyst provided more surface-active sites and strong absorption ability towards organic dye molecules. Moreover, the layered nanosheets favored the photogenerated charge carrier transfer from TiO2 to MoS2. It is a key factor in determining the photocatalytic activities of layered MoS2/TiO2 nanocomposites as a photocatalyst. The formation of synergetic interaction between the MoS2 and TiO2 semiconductors provided an efficient transport platform for charge carrier transfer and enhanced the photocatalytic activities.
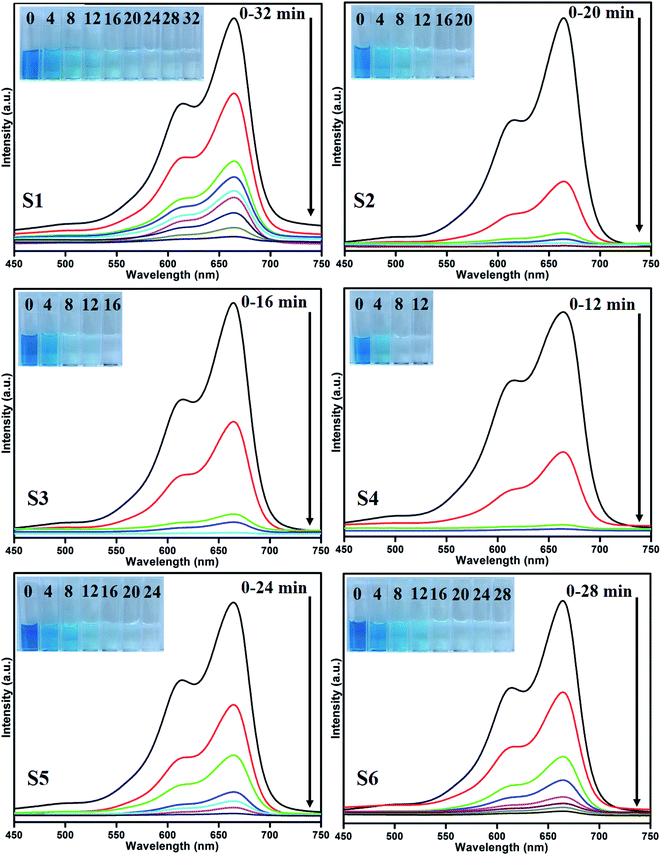 |
| | Fig. 5 UV absorbance spectra of the MB samples S1 (a), S2 (b), S3 (c), S4 (d), S5 (e), and S6 (f). | |
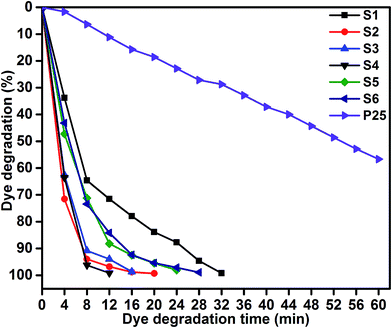 |
| | Fig. 6 MB degradation of samples S1, S2, S3, S4, S5, S6, and P25: time (min) vs. dye degradation (%). | |
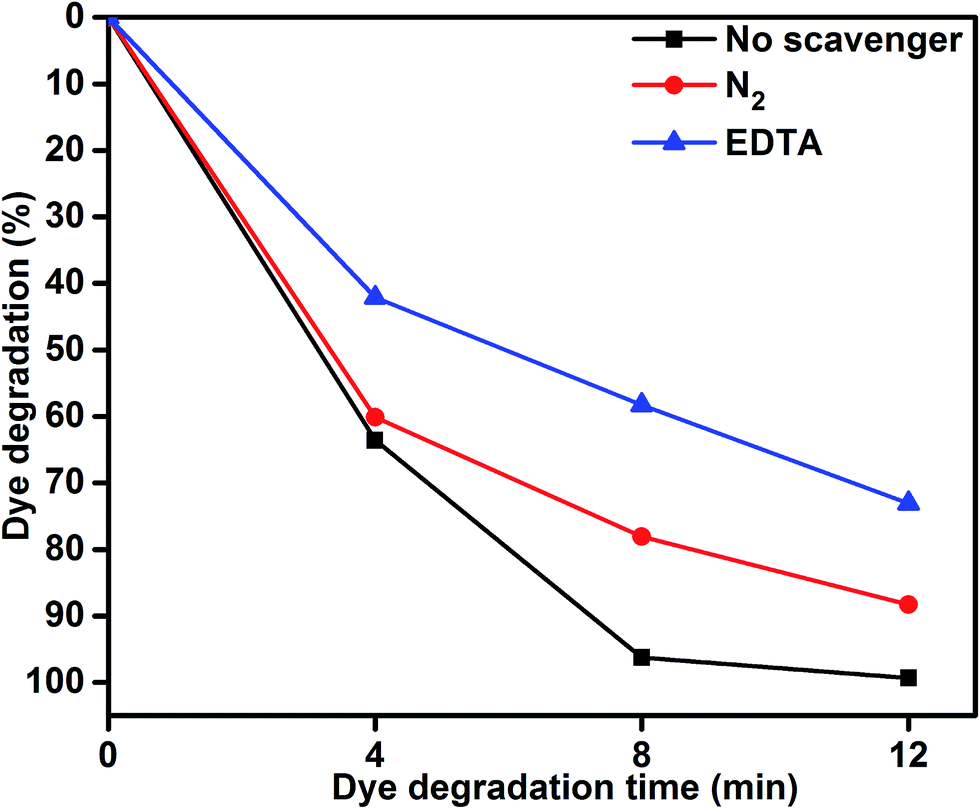
To understand the role of photocatalytic activity, free radical trapping experiments were carried out, as shown in Fig. 7. In general, during photocatalysis, hydroxyl radicals (˙OH) and superoxide anions (O2−˙) are the possible reactive species for the degradation of organic pollutants. Disodium ethylenediaminetetraacetate (EDTA-2Na) and N2 were used as a hole (h+) and electron acceptor scavenger, respectively.47,48 A high purity N2 gas was continuously purged throughout the reaction process under ambient conditions, which eliminated the dissolved oxygen from the reaction solution and thereby prevented the formation of O2−˙. The degradation percentage of MB was 88.39% after 12 min irradiation as compared 99.33% in the scavenger-free photocatalyst. This conformed that superoxide anions played a minor role in the photocatalytic degradation of organic pollutants. To further determine the degradation mechanism, EDTA was added into the solution as a ˙OH radical scavenger. The dye degradation was 72.90%, which indicated suppression of the MB degradation rate. This result confirmed that the photoinduced holes (h+) were one of the main reactive species for the degradation of MB. Therefore, these results clearly determined that MB degradation mainly depended on the photogenerated ˙OH radical. The possible reason for dye degradation by MoS2/TiO2 as a photocatalyst was that superoxide anion radical had the potential to directly react via oxidative pathways. It can also produce singlet oxygen or decompose to H2O2, and transform to hydroxyl radical during MoS2/TiO2 photocatalysis process.49–51 This process was explained in the dye degradation mechanism of MB under visible-light irradiation. These results suggested that the rate of photocatalytic degradation of MB was suppressed in the presence of EDTA as a hole (h+) scavenger. Moreover, the reactive species of superoxide anions (O2−˙) played a minor role in the degradation of MB. The obtained results indicated that the rate of photocatalytic degradation not only depended on organics but also on the nature of the catalysts.
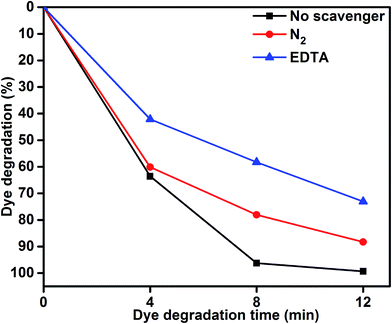 |
| | Fig. 7 MB degradation using the MoS2/TiO2 nanosheets without scavenger and with scavengers under visible-light irradiation. | |
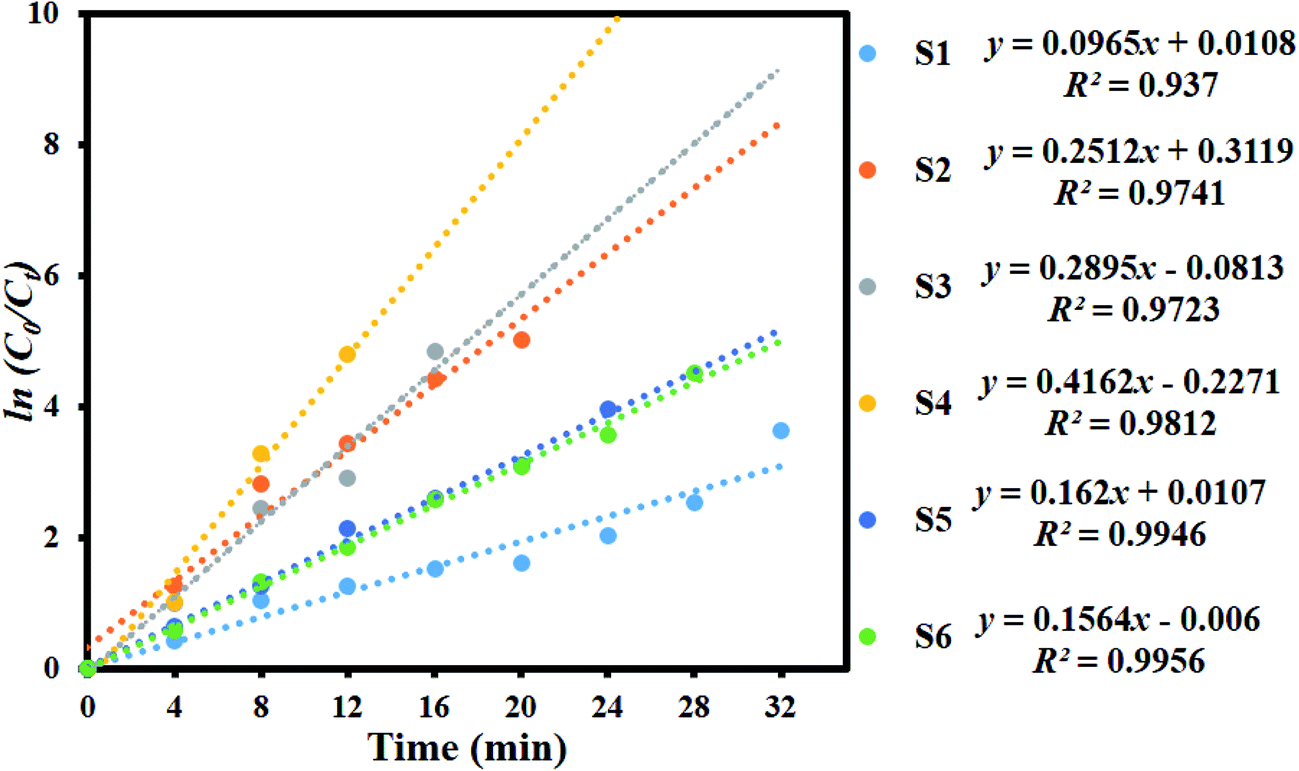
Fig. 8 shows the kinetic plot of ln(C0/Ct) versus irradiation time to investigate whether the process obeyed pseudo-first-order model. A linear correlation existed between ln(C0/Ct) versus irradiation time. The kinetic data was obtained by the pseudo-first order model. The apparent rate constants (Kapp), corresponding correlation coefficients (R2), and maximum dye degradation in presence of MoS2 and MoS2/TiO2 nanocomposites are presented in Table 1. The apparent rate constant Kapp was 0.0965, 0.2512, 0.2895, 0.4162, 0.162, and 0.1564 min−1 for S1, S2, S3, S4, S5, and S6, respectively. K value increased with the increase in the concentration of TiO2 from 0.0965 to 0.4162 min−1 for the samples S1 to S4. With further increase in the TiO2 concentration, K value decreased to 0.1564 min−1 for sample S6. Plots of ln(C0/Ct) versus irradiation time show that apparent rate constant was about two times higher for MoS2/TiO2 nanocomposites than that for pure MoS2. A comparison was carried out between the photocatalytic performance of the materials developed in this study and those of other recently reported MoS2/TiO2 composite nanostructures, as shown in Table 2.
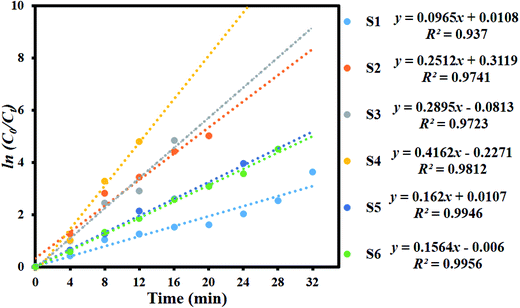 |
| | Fig. 8 Kinetic plot of ln(C0/Ct) as a function of time (min) for the degradation of MB. | |
Table 1 Observed pseudo-first-order rate constants, R2 values, maximum degradation (%), and time required for maximum degradation by the MoS2/TiO2 nanosheets
| Sample code |
Kapp (MoS2/TiO2) |
R2 |
Maximum degradation (%) |
Time taken for maximum degradation (min) |
| S1 |
0.0965 |
0.937 |
99.17 |
32 |
| S2 |
0.2512 |
0.9741 |
99.10 |
20 |
| S3 |
0.2895 |
0.9723 |
98.68 |
16 |
| S4 |
0.4162 |
0.9812 |
99.33 |
12 |
| S5 |
0.162 |
0.9946 |
98.08 |
24 |
| S6 |
0.1564 |
0.9956 |
98.90 |
28 |
Table 2 Comparison of the photocatalytic performances of the MoS2/TiO2 composites in this research with that of those reported in literatures
| Material |
Light source |
Dye |
Dye degradation (%) |
Time taken for degradation (min) |
Ref. |
| MoS2/TiO2 |
500 W xenon lamp |
RhB |
81.8 |
180 |
40 |
| MoS2/TiO2 |
30 W day lamp |
MB |
98 |
90 |
55 |
| MoS2/TiO2 |
30 W day lamp |
MO |
95.1 |
120 |
56 |
| MoS2/TiO2 |
500 W xenon lamp |
MO |
97 |
60 |
57 |
| MoS2/TiO2 |
300 W xenon lamp |
RhB |
86.9 |
20 |
58 |
| MoS2/TiO2 |
400 W Asahi spectra(xenon lamp) |
MB |
99.33 |
12 |
This work |
Based on the abovementioned results, we proposed the possible photocatalytic mechanism for the degradation of MB using MoS2/TiO2 as photocatalysts:52
| | |
MoS2/TiO2 + hv → MoS2 (h+)/TiO2 (e−)
| (1) |
| | |
˙O2− + H2O → ˙HO2 + OH−
| (4) |
| | |
˙HO2 + H2O → H2O2 + ˙OH
| (5) |
| | |
˙OH + MB → CO2 + H2O + NO−3 + NH+4 + Cl−
| (7) |
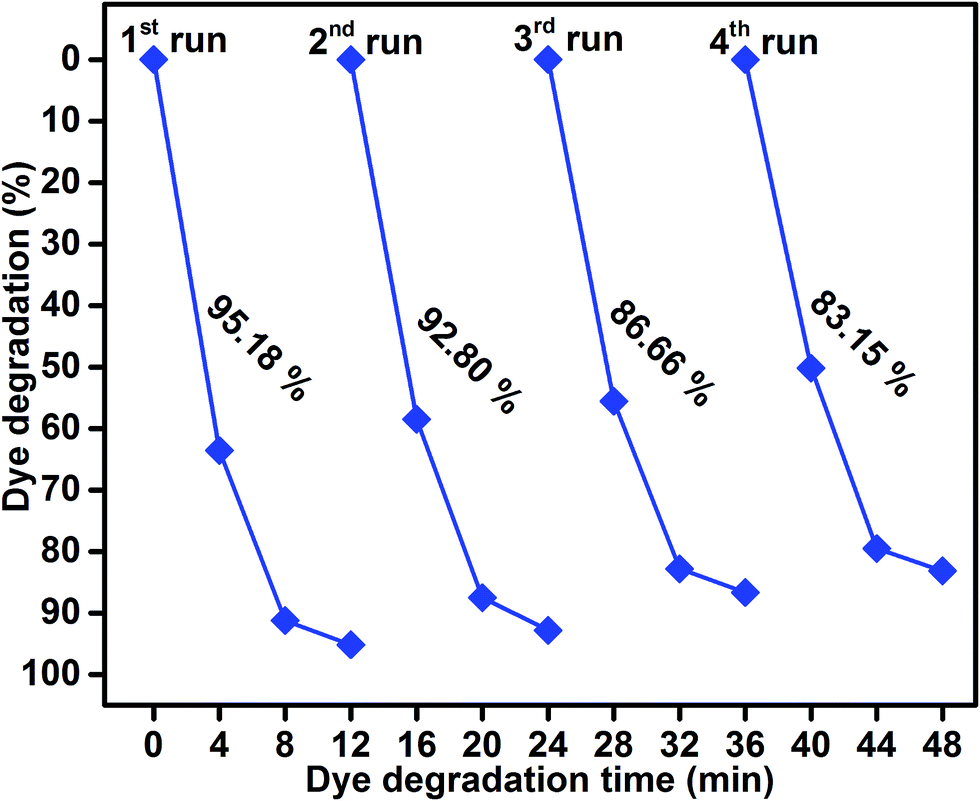
As shown in Fig. 9, the reusability of the sample S4 photocatalyst for the degradation of methylene blue was studied over four cycles under visible-light irradiation. The dye concentration was adjusted each time to its initial value. Photocatalysts were reused for four cycles and the obtained degradation values were 95.18, 92.80, 86.66, and 83.15. The efficiency of the sample S4 did not significantly decline, which suggested that the catalyst had good stability and sustainability. (Fig. S1, ESI†) shows the XRD patterns of the sample S4 after five runs. It can be clearly seen that the phase and structure of the sample S4 remained the same. In addition, the chemical state of Mo 3d, S 2p, Ti 3p, and O 1s were investigated by XPS spectra, as shown in (Fig. S2, ESI†). It suggested that the binding energies of Mo, S, Ti, and O showed no peak shift after four cycles as compared to those of the as-prepared sample.
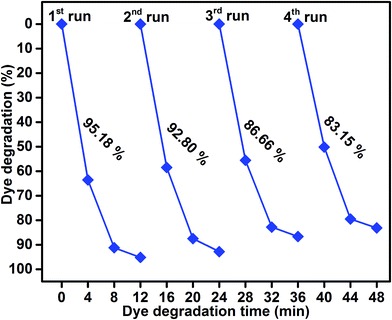 |
| | Fig. 9 Reusability of sample S4 photocatalyst under visible-light irradiation. | |
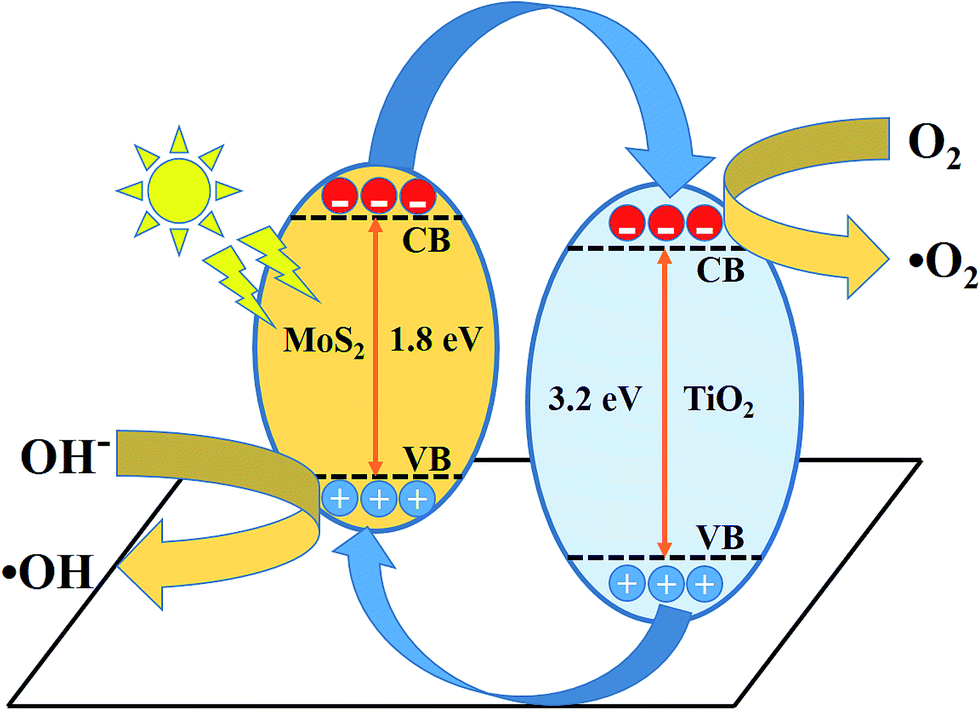
The schematic (Fig. 10) illustrates the energy band structure and charge transfer mechanism. MoS2 is a narrow band gap (1.9 eV) semiconductor with a work function of 4.52 eV, whereas anatase TiO2 is a wide band gap (3.2 eV) semiconductor with a work function of 4.5 eV.19,53,54 Under visible-light irradiation, electrons are excited from the valance band of MoS2 to the conduction band, leaving behind holes in the valence band. The photoinduced electrons in the conduction band of MoS2 were transferred to the conduction band of TiO2, which acted as a photoelectronic receiver. The photogenerated holes reacted with either water (H2O) or hydroxyl ions (OH−) adsorbed onto the catalyst surface to produce hydroxyl radicals (˙OH), and the photogenerated electrons reacted with oxygen (O2) to form superoxide radicals (˙O2−). Consequently, both ˙OH and ˙O2− radicals can decompose the organic compounds to form CO2, H2O, and other inorganic molecules as harmless compounds.
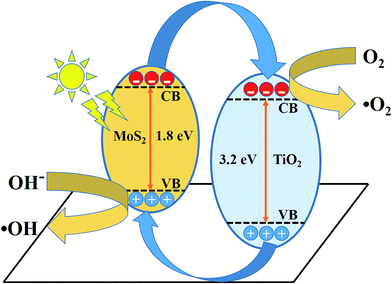 |
| | Fig. 10 Schematic of the charge transfer mechanism from MoS2 to TiO2 photocatalysis. | |
4. Conclusion
Pure layered MoS2 and MoS2–TiO2 nanocomposites were successfully synthesized by a facile hydrothermal method. The XRD patterns revealed the hexagonal MoS2 and anatase structure of TiO2. The morphological analysis confirmed the presence of layered MoS2 and TiO2 nanosheets. The obtained results confirmed that the MoS2/TiO2 nanocomposite photocatalyst had enhanced photocatalytic activity for MB degradation under visible-light irradiation. The interfacial charge carriers were transferred between MoS2 and TiO2 nanosheets and effectively suppressed the electron hole pair recombination. The maximum degradation efficiency was attained for the MoS2/TiO2 (S4) nanocomposite photocatalyst in 12 min. The degradation efficiency obtained for the nanocomposite was three times higher than that obtained for pure MoS2 and commercial P25 TiO2. The free radicals trapping experiment confirmed that photogenerated (˙OH) radicals played an important role in the degradation of the organic pollutant.
References
- C. Minero, E. Pelizzetti, S. Malato and J. Blanco, Chemosphere, 1993, 26, 2103–2119 CrossRef CAS.
- H. Liu, T. Lv, X. H. Wu and C. K. Zhu, Appl. Surf. Sci., 2014, 30, 242–246 CrossRef.
- S. Ameen, M. S. Akhtar, M. Nazim and H. S. Shin, Mater. Lett., 2013, 96, 228–232 CrossRef CAS.
- S. Harish, M. Navaneethan, J. Archana, A. Silambarasan, S. Ponnusamy, C. Muthamizhchelvan and Y. Hayakawa, Dalton Trans., 2015, 44, 10490–10498 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Archana, M. Navaneethan and Y. Hayakawa, J. Power Sources, 2013, 242, 803–810 CrossRef CAS.
- J. Archana, S. Harish, M. Sabarinathan, M. Navaneethan, S. Ponnusamy, C. Muthamizhchelvan, M. Shimomura, H. Ikeda, D. K. Aswal and Y. Hayakawa, RSC Adv., 2016, 6, 68092–68099 RSC.
- B. Banerjee, V. Amoli, A. Maurya, A. Sinha and A. Bhaumik, Nanoscale, 2015, 7, 10504–10512 RSC.
- G. H. Moon, W. Kim, D. Bokare, N. Sung and W. Choi, Energy Environ. Sci., 2014, 7, 4023–4028 CAS.
- S. Hernandez, D. Hidalgo, A. Sacco, A. Chiodoni, A. Lamberti, V. Cauda, E. Tresso and G. Saracco, Phys. Chem. Chem. Phys., 2015, 17, 7775–7786 RSC.
- C. Wang, H. Lin, Z. Liu, J. Wu, Z. Xu and C. Zhang, Part. Part. Syst. Charact., 2016, 33, 221–227 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- L. Zheng, S. Han, H. Liu, P. Yu and X. Fang, Small, 2016, 12, 1527–1536 CrossRef CAS PubMed.
- Y. G. Li, H. L. Wang, L. M. Xie, Y. Y. Liang, G. S. Hong and H. J. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- I. Song, C. Park and H. Cheul Choi, RSC Adv., 2015, 5, 7495–7514 RSC.
- T. S. Li and G. L. Galli, J. Phys. Chem. C, 2007, 111, 16192–16196 CAS.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C. Yung, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- J. R. Chen, F. X. Qiu, W. Z. Xu, S. S. Cao and H. J. Zhu, Appl. Catal., A, 2015, 495, 131–140 CrossRef CAS.
- H. Y. Liu, J. B. Joo, M. Dahl, L. S. Fu, Z. Z. Zeng and Y. D. Yin, Energy Environ. Sci., 2015, 8, 286–296 CAS.
- C. J. Liu, S. Y. Tai, S. W. Chou, Y. C. Yu, K. D. Chang, S. Wang, F. S. Chien, J. Y. Lin and T. W. Lin, J. Mater. Chem., 2012, 22, 21057–21064 RSC.
- K. H. Hu, Y. Xu, E. Z. Hu, J. H. Guo and X. G. Hu, Tribol. Int., 2016, 104, 131–139 CrossRef CAS.
- Y. Jun Yuan, Z. Jun Ye, H. Wei Lu, B. Hu, Y. Hui Li, D. Qin Chen, J. Song Zhong, Z. Tao Yu and Z. Gang Zou, ACS Catal., 2016, 6, 532–541 CrossRef.
- H. Li, K. Yu, X. Lei, B. Guo, C. Li, H. Fu and Z. Zhu, Dalton Trans., 2015, 44, 10438–10447 RSC.
- N. Meng, Y. Zhou, W. Nie, L. Song and P. Chen, J. Nanopart. Res., 2015, 17, 300–310 CrossRef.
- G. Zhou, X. Xu, J. Yu, B. Feng, Y. Zhang, J. Hu and Y. Zhou, CrystEngComm, 2014, 16, 9025–9032 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- Y. Gai, J. Li, S. S. Li, J. B. Xia and S. H. Wei, Phys. Rev. Lett., 2009, 102, 036402–036405 CrossRef PubMed.
- Z. B. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168–4175 CrossRef CAS PubMed.
- B. Pourabbas and B. Jamshidi, Chem. Eng. J., 2008, 138, 55–62 CrossRef CAS.
- X. Zu, C. Yang, F. Xiao, J. Wang and X. Su, New J. Chem., 2015, 39, 683–688 RSC.
- L. Cao, R. Wang, D. Wang, X. Li and H. Jia, Mater. Lett., 2015, 160, 286–290 CrossRef CAS.
- R. Tang, R. Yin, S. Zhou, T. Ge, Z. Yuan, L. Zhang and L. Yin, J. Mater. Chem. A, 2017, 5, 4962–4971 CAS.
- H. Li, Y. Wang, G. Chen, Y. Sang, H. Jiang, J. He, X. Li and H. Liu, Nanoscale, 2016, 8, 6101–6109 RSC.
- M. Shen, Z. Yan, L. Yang, P. Du, J. Zhang and B. Xiang, Chem. Commun., 2014, 50, 15447–15449 RSC.
- X. Zong, H. J. Yan, G. P. Wu, G. J. Ma, F. Y. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS PubMed.
- W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li and Z. H. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- M. Sabarinathan, S. Harish, J. Archana, M. Navaneethan, H. Ikeda and Y. Hayakawa, RSC Adv., 2016, 6, 109495–109505 RSC.
- S. Harish, J. Archana, M. Navaneethan, A. Silambarasan, K. D. Nisha, S. Ponnusamy, C. Muthamizhchelvan, H. Ikeda, D. K. Aswal and Y. Hayakawa, RSC Adv., 2016, 6, 89721–89731 RSC.
- Y. Yan, B. Xia, N. Li, Z. Xu, A. Fisher and X. Wang, J. Mater. Chem. A, 2015, 3, 131–135 CAS.
- F. A. Deorsola, N. Russo, G. A. Blengini and D. Fino, Chem. Eng. J., 2012, 1–6, 195–196 Search PubMed.
- J. Baltrusaitis, C. R. Usher and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 3011–3024 RSC.
- L. Peitao, L. Yonggang, Y. Weichun, M. Ji and G. Daqiang, Nanotechnology, 2016, 27, 225403–225410 CrossRef PubMed.
- G. Gong, Y. Liu, B. Mao, B. Wang, L. Tan, D. Li, Y. Liu and W. Shi, RSC Adv., 2016, 6, 99023–99033 RSC.
- S. Zheng, Y. Cai and K. E. O'Shea, J. Photochem. Photobiol., A, 2010, 210, 61–68 CrossRef CAS PubMed.
- P. Zheng, Z. Pan, H. Li, B. Bai and W. Guan, J. Mater. Sci.: Mater. Electron., 2015, 26, 6399–6410 CrossRef CAS.
- X. V. Doorslaer, P. M. Heynderickx, K. Demeestere, K. Debevere, H. V. Langenhove and J. Dewulf, Appl. Catal., B, 2012, 111–112, 150–156 CrossRef.
- W. Ho, J. C. Yu, J. Lin, J. Yu and P. Li, Langmuir, 2004, 20, 5865–5869 CrossRef CAS PubMed.
- Y. Liu, Y. X. Yu and W. D. Zhang, J. Phys. Chem. C, 2013, 117, 12949–12957 CAS.
- E. Scalise, M. Houssa, G. Pourtois, V. V. Afanas'ev and A. Stesmans, Phys. E, 2014, 56, 416–421 CrossRef CAS.
- K. Hong Hu, Y. Kui Cai and S. Li, Adv. Mater. Res., 2011, 197–198, 996–999 Search PubMed.
- K. Hong Hu, X. Guo Hu and Y. Fu Xu, J. Mater. Sci., 2010, 45, 2640–2648 CrossRef.
- W. Zhang, X. Xiao, L. Zheng and C. Wan, Can. J. Chem. Eng., 2015, 93, 1594–1602 CrossRef CAS.
- D. Yong, Z. Yifeng, N. Wangyan and C. Pengpeng, Appl. Surf. Sci., 2015, 357, 1606–1612 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03633g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
H. Ikedab and
Y. Hayakawa
*b,
H. Ikedab and
Y. Hayakawa