
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical properties of PX-phase PbTiO3 electrode for sodium ion batteries†
Yixiang Zhanga,
Muwei Jiabc,
Ziyi Liua,
Yiqing Hea,
Yao Hua,
Qinghua Yanga,
Bo Lia and
Jin Wang *a
*a
aAdvanced Materials Institute, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, P. R. China. E-mail: Wang.jin@sz.tsinghua.edu.cn
bInstitute of Chemistry, Chinese Academy of Science, Beijing, 100190, P. R. China
cCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, 518061, P. R. China
First published on 12th May 2017
Abstract
PX-phase PbTiO3 (PT) nanowires have been investigated for the first time as an anode material for sodium ion batteries (NIBs). Their sodium storage properties are presented by employing sodium-carboxyl-methyl-cellulose (CMC) and styrene-butadiene-rubber (SBR) as binders. Electrochemical results show that the PT/CMC–SBR electrode delivers a specific capacity of 84.2 mA h g−1 after 100 cycles. The PT nanowires are found to undergo an irreversible conversion reaction to transform into a composite structure composed of 5–8 nm Pb nanoparticles uniformly dispersed in the amorphous matrixes in the initial discharge process. In the subsequent cycling process, the discharge capacity is primarily attributed to alloying reaction between Pb and Na and conversion reaction between Pb and PbO2. Furthermore, we use polyvinylidene fluoride (PVDF) instead of CMC–SBR as binder to study the influence of binders on the electrochemical performance of PX-phase PT anode. It is found that PT/CMC–SBR electrode exhibits better electrochemical properties than PT/PVDF electrode in both cyclic performance and rate capability. Detailed analysis is conducted to understand the underlying mechanism for the distinct NIBs anode performance of PT nanowires caused by use of different binders. It is found that compared to PT/CMC–SBR electrode, the conversion reaction between Pb and PbO2 is missing in the PT/PVDF electrode, and that Pb active material has diffused out from the wires and reprecipitated as microsized hexagonal particles during the discharge/charge process, leading to the inferior electrochemical performance of PT/PVDF electrodes. This work signifies the importance of binders to optimize the electrochemical performance for alloy anode materials in NIBs.
Introduction
In recent years, sodium-ion batteries (NIBs) have attracted lots of attention due to the abundance, low cost and environmental friendliness of sodium resources.1,2 A number of cathode materials are proposed for NIBs, including NaxCoO2,3 NaxMnO2,4 NaFeO2,5 NaV6O15,6 NaFePO4,7 and Na3V2(PO4)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 et al. A wide category of candidate materials for NIBs anode materials have also been intensively investigated. It turns out sodium ions could not intercalate into graphite sheets which is the sate-of-the-art lithium-ion anode material.9 On the contrary, hard carbon,10 petroleum coke11 and carbon microspheres12 show good sodium storage capability. TiO2,13,14 sodium titanates15,16 and alloying materials such as Sn,17 Sb,18,19 SnSb20 and SnO221,22 are also reported as promising sodium-ion anode materials. However, a key issue for alloying anode materials is significant volume change and agglomeration of nanoparticles during charge/discharge process, which results in the material pulverization, capacity reduction and unsatisfactory cyclic performance.23 Therefore, delicate composite systems such as core/shell structures24 or introducing secondary carbon phases20 are explored to solve the problem. Another important factor in the stabilization of the cycling capacity of alloying anode is binders, which can maintain the structural integrity during the volume change. Polyvinylidene fluoride (PVDF) is a most conventional binder in NIBs. Recently, alloy anodes with some other kinds of binders such as sodium-carboxyl-methyl-cellulose (CMC), polyacrylic acid (PAA), sodium polyacrylate (PANa), and styrene-butadiene-rubber (SBR) have been reported to exhibit better performance than that with PVDF in NIBs.25–27
8 et al. A wide category of candidate materials for NIBs anode materials have also been intensively investigated. It turns out sodium ions could not intercalate into graphite sheets which is the sate-of-the-art lithium-ion anode material.9 On the contrary, hard carbon,10 petroleum coke11 and carbon microspheres12 show good sodium storage capability. TiO2,13,14 sodium titanates15,16 and alloying materials such as Sn,17 Sb,18,19 SnSb20 and SnO221,22 are also reported as promising sodium-ion anode materials. However, a key issue for alloying anode materials is significant volume change and agglomeration of nanoparticles during charge/discharge process, which results in the material pulverization, capacity reduction and unsatisfactory cyclic performance.23 Therefore, delicate composite systems such as core/shell structures24 or introducing secondary carbon phases20 are explored to solve the problem. Another important factor in the stabilization of the cycling capacity of alloying anode is binders, which can maintain the structural integrity during the volume change. Polyvinylidene fluoride (PVDF) is a most conventional binder in NIBs. Recently, alloy anodes with some other kinds of binders such as sodium-carboxyl-methyl-cellulose (CMC), polyacrylic acid (PAA), sodium polyacrylate (PANa), and styrene-butadiene-rubber (SBR) have been reported to exhibit better performance than that with PVDF in NIBs.25–27
Sodium has been shown to electrochemically insert into Pb at room temperature. The full sodiation lead of Na15Pb4 phase has a capacity of 485 mA h g−1, corresponding to a volume expansion of 365%. Both Jow et al.28 and Komaba et al.29 suggested the formation of NaPb3, NaPb, Na5Pb2 and Na15Pb4 in order during the sodiation of lead, but without any evidence of their formation. However, it was demonstrated by Ellis et al.30 that NaPb3, NaPb, Na9Pb4 and Na15Pb4 were formed sequentially instead of NaPb3, NaPb, Na5Pb2 and Na15Pb4 during the sodiation, when they studied the sodiation process of lead film by in situ XRD. Recently, Darwiche et al.31 utilized in situ XRD to study the phase transformation of the bulk lead during sodiation process and reported phase formation process of Na–Pb alloy consistent with that found by Ellis et al. In addition, the highly loaded Pb in this system showed sustainable capacity retention of 464 mA h g−1 after 50 cycles at 13 mA g−1. Pb(NO3)2 was also investigated as anode materials for sodium ion batteries with a specific capacity of 81.5 mA h g−1 after 50 cycles.32 Different from the case of bulk Pb, the sodiation process of Pb(NO3)2 does not show four distinct potential plateaus.
In our previous studies, it was found that PX-phase PbTiO3 (PT) presents high reversible specific capacity, excellent cycling property and rate capacity in the lithium-ion batteries because it can combine the high specific capacity of Pb and good rate capability of TiO2-based systems. In this work, PX-phase PbTiO3 nanowires are studied as novel anode material for NIBs. Electrochemical results show that the PT/CMC–SBR electrode delivers a specific capacity of 84.2 mA h g−1 after 100 cycles. The PT nanowires are found to undergo an irreversible conversion reaction to transform into a composite structure composed of 5–8 nm Pb nanoparticles uniformly dispersed in the amorphous matrixes in the initial discharge process. In the subsequent cycling process, the discharge capacity was primarily attributed to alloying reaction between Pb and Na and conversion reaction between Pb and PbO2. Furthermore, we use PVDF instead of CMC–SBR as binder to study the influence of binders on the electrochemical performance of PX-phase PT anode. It is found that PT/CMC–SBR electrode exhibited better electrochemical properties than PT/PVDF electrode in both cyclic performance and rate capability. Detailed analysis is conducted to understand the underlying mechanism for the distinct NIBs anode performance of PT nanowires caused by use of different binders. This work signifies the importance of binders to optimize the electrochemical performance of alloy anode materials in NIBs.
Experiments
Materials preparation
PX-phase PT nanowires are prepared by a reported hydrothermal method.33 4 mmol of Ti(OC4H9)4 (Sigma-Aldrich) is first dissolved in 8 mL of ethanol and the obtained transparent solution is hydrolyzed in 8 mL of deionized H2O. Subsequently, 20 mmol of KOH (Sigma-Aldrich), 5.2 mmol of Pb(NO3)2 (Alfa Aesar), and 0.05 g of polyvinyl alcohol (PVA) (Alfa Aesar) are added and mixed homogeneously under stirring. The volume of the mixture is then adjusted to 40 mL with deionized H2O, and transfered into a 50 mL Teflon-lined autoclave. Hydrothermal treatment is carried out at 200 °C for 3.5 h, followed by washing with deionized H2O until neutral pH is achieved. The Pb oxide remnant is removed with 10 wt% CH3COOH aqueous solution. The final pure PX-phase sample is obtained by another cycle of washing with deionized H2O and dried at 60 °C in air.Material characterization
X-ray diffraction (XRD) is conducted using a Rint-2000V/PC diffractometer (Rigaku, Japan) with Cu/Kα radiation (λ = 1.5406 Å). The scanning electron microscope (SEM, ZEISS SUPRA® 55), which is equipped with an EDS detector (Oxford X-Max 20), is used for the morphologies and EDS analysis of materials. Transmission electron microscopy (TEM) analysis is conducted with Tecnai G2 F30. X-ray photoelectron spectroscopy (XPS) is performed using PHI-5000 Versaprobe II (Ulvac-Phi, Japan).Electrochemical measurements
Electrochemical tests of the anode materials are carried out using coin cell CR2032. For preparing the PT/PVDF electrodes, a slurry of 80% active materials (PX-phase PT), 10% carbon black (Super P) and 10% PVDF in NMP is coated onto a copper foil and then dried at 110 °C over night in a vacuum oven. The procedure for preparing PT/CMC–SBR electrodes is the same except that a mixture of 5 wt% CMC (1.5 wt% aqueous solution) and 5 wt% SBR (50 wt% aqueous solution) is used instead of PVDF. The sodium cells are assembled in an argon-filled glove box (mbraum) with metallic sodium wafer as the counter electrode, glass fibers as the separator, and 1 M NaClO4 in 1:1 ethylene carbonate (EC)/dimethylcarbonate (DMC) as the electrolyte. All the cells are aged for 12 h before conducting the electrochemical measurements. The galvanostatic discharge–charge tests are conducted in the voltage window of 0.005–3 V at room temperature using LanHe BT2000 multichannel testing equipment. Cyclic voltammograms (CVs) measurement is conducted using a Bio-Logic VMP2 multichannel potentiostat. The scan rate and potential interval in the CV tests are set at 0.1 mV s−1 and 0.005–3 V respectively.
Result and discussion
As shown in Fig. 1(a), all the diffraction peaks of as-prepared PX-phase PT nanowires match well with the standard XRD pattern (JCPDS #48-0105). The PX-phase PT material has a body-centered tetragonal unit cell and contains ∼5.5 Å wide pores in the a–b plane (inset in Fig. 1(a)). Fig. 1(b) presents a typical SEM images of the as-prepared PX-phase PT, showing fibrous morphology with the diameter in the range of 10 nm to more than 300 nm and the length in the range of hundreds of nanometers to over 30 μm.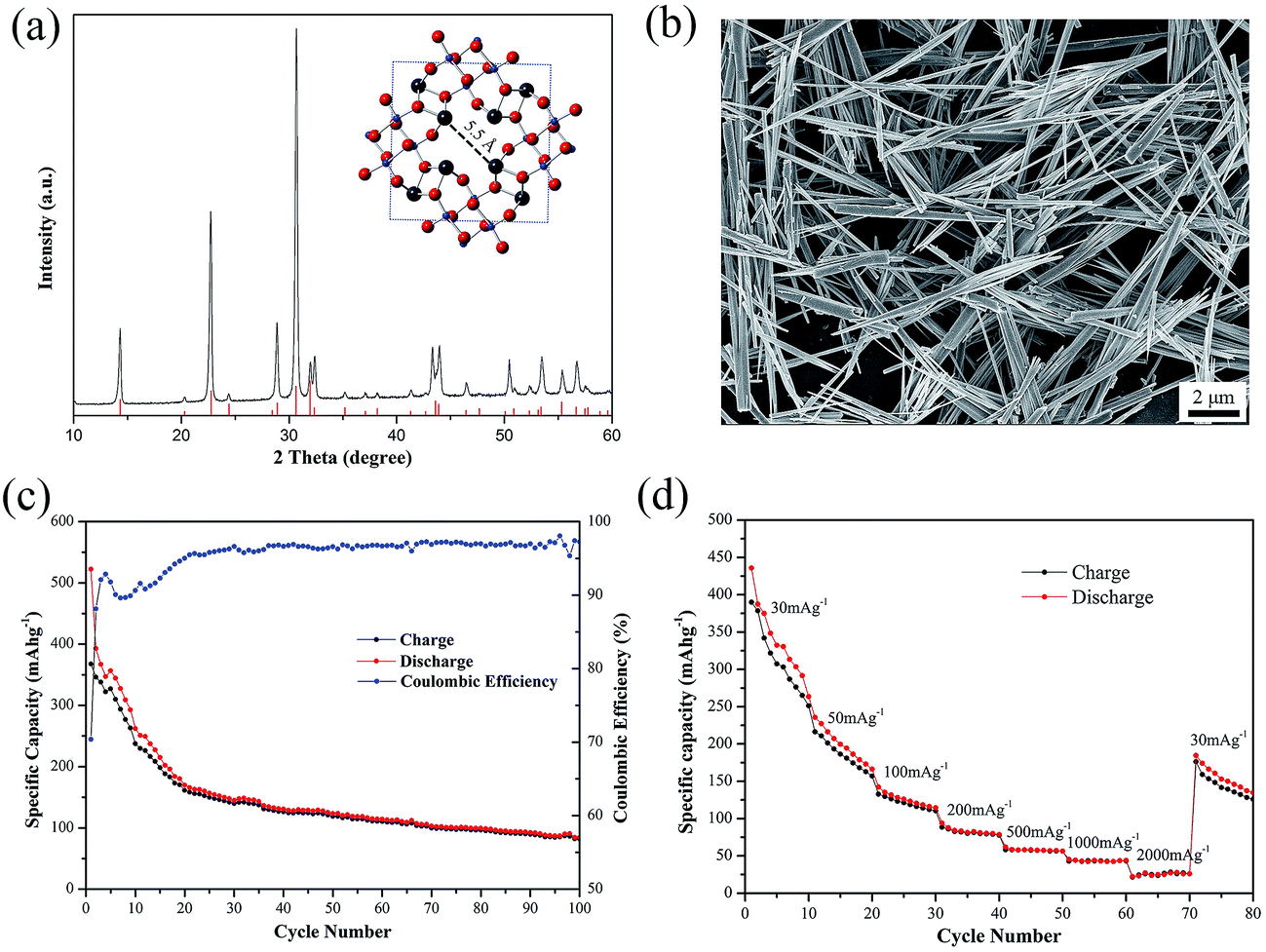 | ||
| Fig. 1 (a) XRD pattern, and (b) SEM image of pristine PX-phase PT nanowires; (c) cycling capacity and coulombic efficiency of PT/CMC–SBR electrode; (d) rate capability of PT/CMC–SBR electrode. The inset in (a) shows the atomic structure of PX-phase PT within a unit cell, in which the black, blue and red balls represent Pb, Ti, O atoms, respectively. | ||
The according electrochemical performance of PT as a NIB anode material with CMC and SBR as binders is evaluated by charge–discharge tests at the current intensity of 30 mA g−1 with the voltage ranging from 0.005 V to 3 V. As depicted in Fig. 1(c), the initial discharge/charge capacity of PT/CMC–SBR electrode is 522.3/367.6 mA h g−1. An obvious gradual fading similar to those normally observed in the PX-phase PT as LIB anodes is seen in first several cycles. After 100 cycles, the capacity still remains 84.2 mA h g−1. Fig. 1(d) shows the rate capability of PT/CM–SBR electrodes. The specific current is increased stepwise from 30 mA g−1 to 2000 mA g−1 every 10 cycles, and finally reverts to the initial value of 30 mA g−1. It is seen that PT/CM–SBR electrode delivers discharge capacity of 263.5 mA h g−1 at 30 mA g−1 and then reduces to 166.3, 114.5 and 78.6 mA h g−1 at a specific current of 50, 100 and 200 mA g−1, respectively. A discharge capacity of 30 mA h g−1 is delivered at the highest tested specific current of 2000 mA g−1 and 134.7 mA h g−1 is remained when the specific current is set back to 30 mA g−1.
Cyclic voltammograms (CVs) analysis has been conducted to illuminate the Na-ions insertion/extraction mechanism of PT/CMC–SBR electrode. As presented in Fig. 2(a), the first discharge curve shows three reduction peaks, located at 0.43, 0.18 and 0.01 V respectively, whereas the peak at 0.43 V disappears in the subsequent cycles. The reduction peak at 0.43 V corresponded to a multiphase transition PbTiO3 + 2Na+ + e− → Pb + TiO2 + Na2O. The peaks at 0.18 and 0.01 V are corresponding to the alloying process of Pb to NaxPb as well as sodium ion inserting into the super P carbon.16,32 In the subsequent charge process, three oxidation peaks at 0.28, 0.45 and 0.58 V are corresponding to the progressive dealloying reactions from NaxPb to Pb.32 There are two broad oxidation peaks ranging from 1.2 V to 1.5 V and two broad reduction peaks ranging from 1.4 V to 0.7 V in the CV curves, which can be attributed to the conversion reaction between Pb and PbO2.21 Based on the CV profiles and reaction mechanisms of PX-phase PT as anode materials of LIBs, the reactions that are involved in PX-phase PT anode as NIBs can be expressed as follows:
| PbTiO3 + 2Na+ + 2e− → Pb + TiO2 + Na2O | (1) |
| Pb + xNa+ +xe− ↔ NaxPb | (2) |
| TiO2 + yNa+ + ye− ↔ NayTiO2 | (3) |
| Pb + Na2O− ↔ PbO2 + 2Na+ + 2e− | (4) |
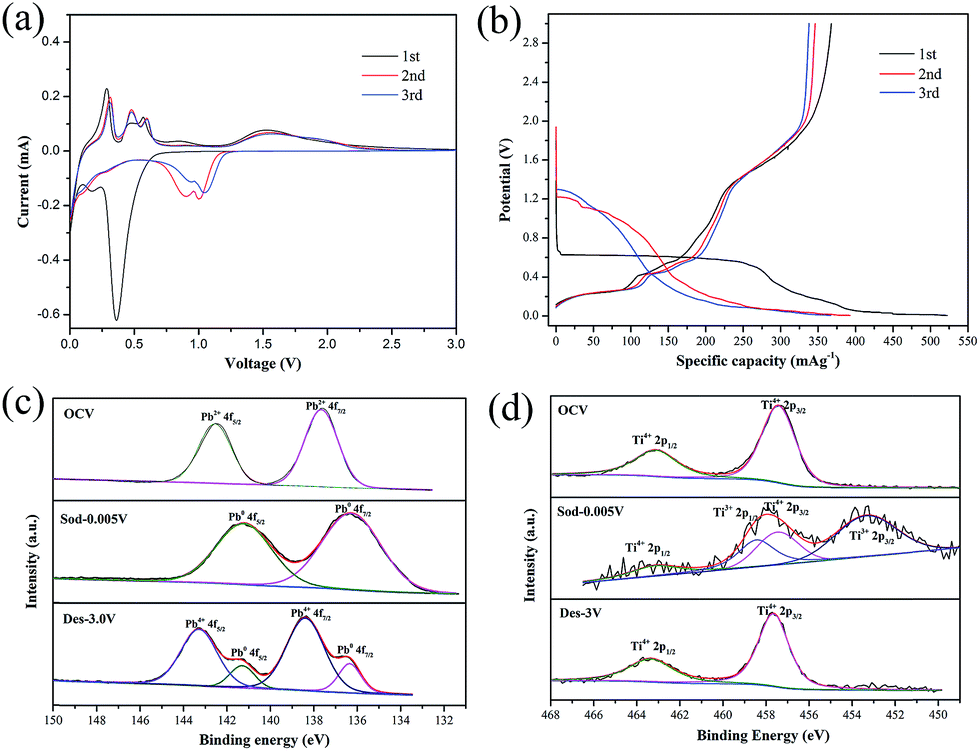 | ||
| Fig. 2 (a) Cyclic voltammogram curves of PT/CMC–SBR electrode; (b) voltage profiles of selected cycles for PT/CMC–SBR electrode; XPS spectra of (c) Pb and (d) TiO2 in the PT/CMC–SBR electrode at various cut-off voltages: open circuit voltage, first discharged to 0.005 V, first charged to 3.0 V. | ||
The voltage profiles (vs. capacity) for first three cycles are shown in Fig. 2(b). Two long potential plateaus at 0.6 V and 0.01 V and one short potential plateaus at 0.2 V appear during the initial discharge process. The potential plateaus at 0.6 V disappears in the subsequent cycles. This potential plateau corresponds to the reaction described by eqn (1), which was the main reason leading to low initial coulombic efficiency (70.38%) of PT/CMC–SBR electrode. The slope (1.2–1.8 V) that delivers a specific capacity of 100 mA h g−1 was corresponding to the reaction described by eqn (4). Different from the case for PX-phase PT LIBs, the conversion reaction between Pb and PbO2 only proceeds in the PX-phase PT NIBs. It is first time to find this reaction in the PT/CMC–SBR electrode. Darwiche et al.31 have suggested that the four distinct plateaus observed in the discharge process of bulk lead, corresponding to the formation of NaPb3, NaPb, Na9Pb4 and Na15Pb4 crystalline phases sequentially, and phase transformation in the charge process is just the opposite. However, for PT/CMC–SBR electrode, the four plateaus cannot be clearly observed, especially for the second and third cycles, with only two plateaus at 1.2 and 0.01 V in the discharge process and three plateaus at 0.2, 0.4 and 0.56 V in the charge process. This indicates that the sodiation process of Pb produced by the decomposition of PX-phase PbTiO3 is different from that of bulk Pb.
XPS analysis of cycled electrode is performed to further track the chemical state of Pb and Ti elements in the PT/CMC–SBR electrode (Fig. 2(c) and (d)). At open circuit voltage (OCV), the Pb 4f peaks located at 137.65 eV and 142.55 eV are consistent with the Pb2+ state in PX-phase PbTiO3. After discharged to 0.005 V, the Pb 4f peaks shift to 136.27 eV and 141.22 eV, indicating the occurrence of reduction of Pb2+ to Pb0 in the first discharge process as shown in reaction (1) and (2). Upon the completion of the subsequent charging, the peaks for Pb4+ (138.39 eV and 143.29 eV) appear as the dominant ones, indicating that a large portion of metallic Pb are oxidized and thus conforming the presence of reaction (4) in the PT/CMC–SBR electrode. Fig. 2(d) shows the reversible valance state change of Ti from Ti4+ to Ti3+ in good accord with reaction (3). It is interesting to note that no appreciable reversible peaks that are expected for the insertion/de-insertion process of Na+ in crystalline TiO2 have been observed in the CV measurements. This is due to the amorphous nature of the formed TiO2, similar to the case of PX-phase PT-fiber electrode in LIBs.34
As indicated by reaction (1), PX-phase PT has undergone a decomposition process during the first discharge. It is thus interesting to investigate their structural change induced by the reaction. Fig. 3(a) shows the microstructural image of PT/CMC–SBR electrode after first discharged to 0.005 V. It is seen that the active material has retained their integrity as wires. Nevertheless, a close inspection of the magnified TEM image (inset in Fig. 3(a)) reveals a substantial change in microstructure, leading to formation of a composite structure with 5–8 nm ultrafine nanoparticles uniformly dispersed in an amorphous matrix. The exposed crystalline fringes of the nanoparticles observed in the HRTEM images (Fig. 3(b)) as well as the SAED pattern (inset in Fig. 3(b)) indicate that those crystalline particles are fcc Pb nanoparticles. Fig. 3(c) shows the HRTEM image and SAED pattern of PT/CMC–SBR electrode after first charged to 3.0 V. The results show that the nanoparticles in the matrix are Pb, but their content is relatively reduced compared with Fig. 3(b). Fig. 3(d) shows the XRD of PT/CMC–SBR electrode at various cut-off voltages. Upon first discharged to 0.25 V new peaks ascribed to Pb appeared instead of PX-phase PbTiO3. This change indicates the occurrence of reaction (1). The TiO2·Na2O phase had been reported to be amorphous which explains the lack of associated Bragg peaks. The XRD of fully discharged state shows only Pb phase. While that of fully charged state also shows only Pb phase, but with much weaken intensity. And this indicates that Pb has been transformed, consisting with the result of XPS analysis.
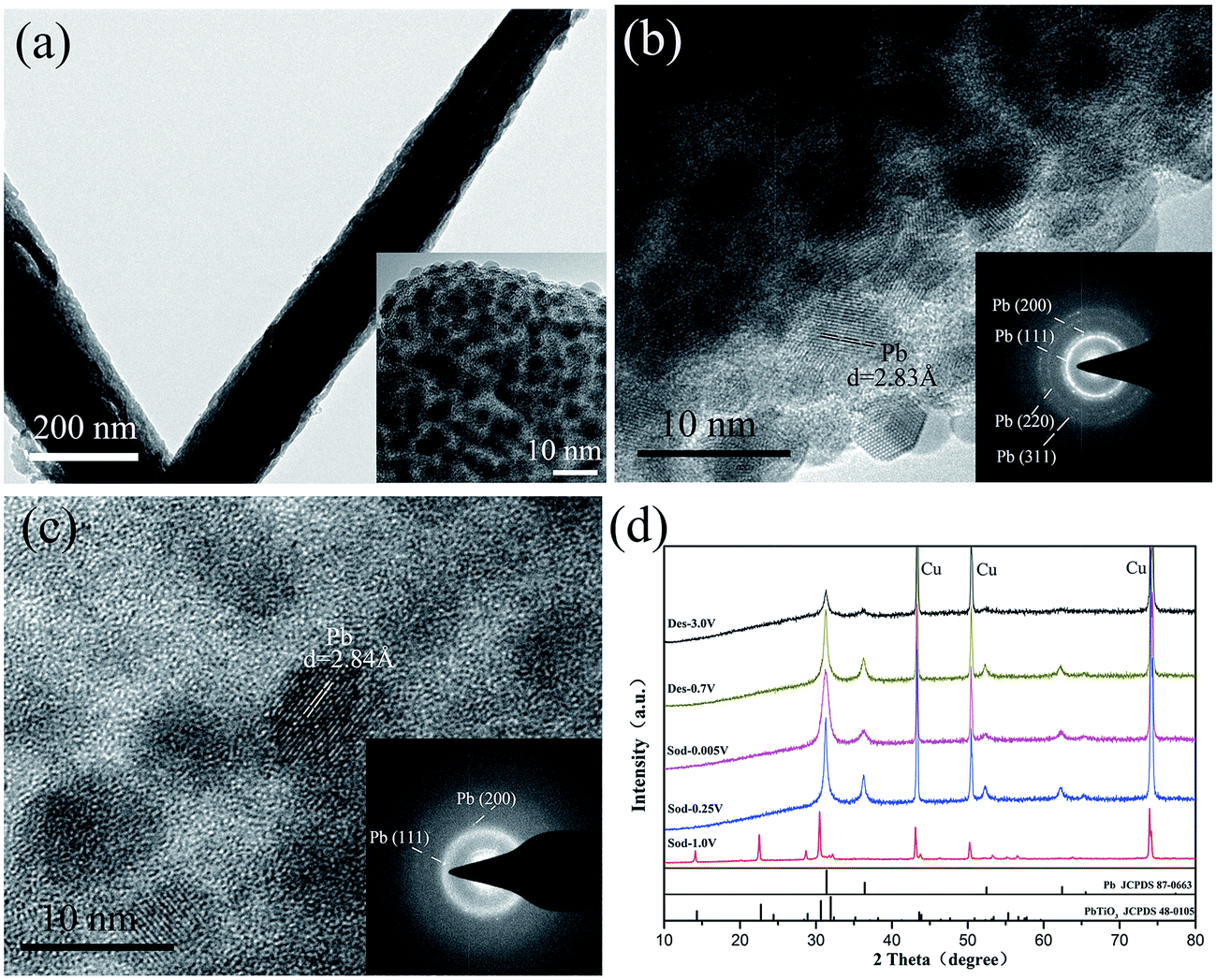 | ||
| Fig. 3 (a) TEM image and (b) HRTEM image of the PT/CMC–SBR electrode first discharged to 0.005 V; (c) HRTEM image of the PT/CMC–SBR electrode first charged to 3.0 V; (d) XRD patterns of PT/CMC–SBR electrode at various cut-off voltages: first discharged to 1.0 V, first discharge to 0.25 V, first discharged to 0.005 V, first charged to 0.7 V and first charged to 3.0 V (“Cu” represents the peaks contributed from the Cu current collector substrate). The inset in (a) presents a magnified view of the wires. The inset in (b) and (c) show the corresponding selected-area electron diffraction patterns. | ||
Although the CV curves show the alloying reaction (Pb + Na+ + e− ↔ NaxPb) can proceed, the results of XRD and repeated TEM characterizations do not show the presence of crystalline NaxPb alloy phases. Therefore we conclude that the NaxPb alloy phases after discharge process are amorphous and alloying reaction is not fully completed. This is significantly different from the case of bulk Pb.31 The fact that a substantial portion of metallic Pb that is reduced from pristine PT does not further participate in the sodiation reaction largely accounts for the lower capacity of PX-phase PT relative to bulk Pb. The possible reason might be ascribed to the existence of amorphous TiO2·Na2O matrix formed after the PT decomposition, which makes it difficult for Na+ to diffuse through the amorphous surrounding to reach Pb and completely alloy with it. This is similar to the case of SnO2 anode materials in NIBs.21
As indicated by Zhang et al.,27 the choice of binders could have a significant influence on the anode performance in NIBs. To have a deeper view of the role of CMC/SBR binder we used, a comparative study is conducted on PX-phase PT anode with PVDF as binder. As shown in Fig. S1(a),† the specific capacity of PT/PVDF electrode is down to 26.8 mA h g−1 after 100 cycles, which is only one third of PT/CMC–SBR electrode (84.2 mA h g−1). Fig. S1(b)† shows the rate capability of PT/PVDF electrode. The capacity of PT/PVDF electrode is dropped to 0 mA h g−1 when the current density is larger than 200 mA g−1, while that of PT/CMC–SBR is still 100 mA h g−1 at 200 mA h g−1 and 30 mA h g−1 at 2000 mA g−1. Therefore, it is clearly seen that CMC/SBR is superior to PVDF as binder in the NIBs anode with PT nanowires as active material. To elucidate the detailed mechanism, further analysis has been performed. As shown in Fig. S1(c),† the CV curves of PT/PVDF electrode nearly coincide with that of PT/CMC–SBR electrode in the low potential range (<0.7 V). However, in the potential range higher than 0.7 V, the two broad oxidation peaks ranging from 1.2 V to 1.5 V and two broad reduction peaks ranging 1.4 V to 0.7 V in the CV curves of PT/CMC–SBR electrode are not present in PT/PVDF electrode. This pair of oxidation/reduction peaks is corresponding to the reaction described by eqn (4) and delivers a specific capacity of 100 mA h g−1 in the initial cycle. The missing reaction (4) in the PT/PVDF electrode is in good accord with its lower specific capacity compared to PT/CMC–SBR electrode.
SEM analysis of the cycled electrodes at different discharge/charge states is carried out to check the structural change during discharge and charge processes (Fig. 4). Upon the initial discharge, the pristine PX-phase PT wires with smooth surface (Fig. 4(a) and (e)) undertake a decomposition into Pb and TiO2 and subsequent alloying process of Pb with Na, which lead to a huge volume expansion and roughened wire surface (Fig. 4(b) and (f)). In the charge process, the surface of the nanowires become smooth again accompanied with a reduction in the thickness due to deintercalation of Na ions from the matrix. However, in the case of PT/PVDF electrode, hexagonal particles appear on the surface of the electrode at 1.5 V (Fig. 4(c)), and continues growing in size and number upon further charge to 3.0 V (Fig. 4(d)) while no presence of hexagonal particles is detected in the PT/CMC–SBR electrode during the whole charge/discharge process (Fig. 4(g) and (h)). The energy dispersive analysis (EDS) result shows that the main elements in the hexagonal particles are Pb, Na and O, which excludes that the phase is PT (ESI Fig. S2†). Thus it can be inferred that instead of reaction (4) which take place within the similar potential range in the PT/CMC–SBR electrode, the Pb species have diffused out from the wires and reprecipitated as hexagonal particles in combination with Na and O when PVDF is employed as binder. The formed microsized hexagonal particles lead to a bigger volume change and further aggravate electrode's disintegration, resulting the inferior cyclic and rate capability of PT/PVDF electrode in NIBs.
 | ||
| Fig. 4 (a–d) SEM images of the PT/PVDF electrodes at various cut-off voltages: (a) a pristine electrode, (b) first discharged to 0.005 V, (c) first charged to 1.5 V, (d) first charged to 3.0 V. The inset in (d) presented a magnified view of hexagonal particles. (e–h) SEM images of the PT/CMC–SBR electrodes at various cut-off voltages: (e) a pristine electrode, (f) first discharged to 0.005 V, (g) first charged to 1.5 V, (h) first charged to 3.0 V. | ||
Conclusions
In this paper, PX-phase PbTiO3 nanowires is investigated as anode materials for sodium-ion batteries. PT nanowires is found to undergo an irreversible conversion reaction to transform into a composite structure composed of 5–8 nm Pb nanoparticles uniformly dispersed in the amorphous matrixes in the first discharge process. In the subsequent process, the capacity is primarily attributed to alloying reaction between Pb and Na which formed amorphous NaxPb alloys. With different binders (CMC–SBR vs. PVDF), PT nanowires exhibit distinct electrochemical performance, PT/CMC–SBR electrode delivers a specific capacity of 84.2 mA h g−1 at 30 mA g−1 after 100 cycles, which is three times as the PT/PVDF electrode (26.8 mA h g−1). The initial coulombic efficiency of PT/CMC–SBR electrode (70.38%) is also obviously higher than that of PT/PVDF (55.21%). Meanwhile, PT/CMC–SBR electrode exhibits better rate capability. It is found that compared to PT/CMC–SBR electrode, the conversion reaction between Pb and PbO2 is missing in the PT/PVDF electrode, and that Pb active material has diffused out from the wires and reprecipitated as microsized hexagonal particles during the discharge/charge process, leading to the inferior electrochemical performance of PT/PVDF electrodes. This work manifests that binders have an important influence on the reaction mechanism between PX-phase PT and sodium ion and signifies the importance of binders to optimize the electrochemical performance for alloy anode materials in NIBs.Acknowledgements
This work is supported by Guangdong Natural Science Fund for Distinguished Young Scholars (2016A030306020), Guangdong Youth Talent Plan (2015TQ01C536), Shenzhen Special Fund for the Development of Emerging Industries (JCYJ20140417115840233), and Shenzhen Peacock Plan (Grant No. KQCX20140521161756228).Notes and references
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N. S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067–2081 CAS.
- R. C. Massé, E. Uchaker and G. Cao, Sci. China Mater., 2015, 58, 715–766 CrossRef.
- R. Berthelot, D. Carlier and C. Delmas, Nat. Mater., 2011, 10, 74–80 CrossRef CAS PubMed.
- Y. Cao, L. Xiao, W. Wang, D. Choi, Z. Nie, J. Yu, L. V. Saraf, Z. Yang and J. Liu, Adv. Mater., 2011, 23, 3155–3160 CrossRef CAS PubMed.
- E. Lee, D. E. Brown, E. E. Alp, Y. Ren, J. Lu, J. J. Woo and C. S. Johnson, Chem. Mater., 2015, 27, 6755–6764 CrossRef CAS.
- H. M. Liu, H. S. Zhou, L. P. Chen, Z. F. Tang and W. S. Yang, J. Power Sources, 2011, 196, 814–819 CrossRef CAS.
- W. Tang, X. Song, Y. Du, C. Peng, M. Lin, S. Xi, B. Tian, J. Zheng, Y. Wu, F. Pan and K. P. Loh, J. Mater. Chem. A, 2016, 4, 4882–4892 CAS.
- W. Shen, H. Li, Z. Y. Guo, Z. H. Li, Q. J. Xu, H. M. Liu and Y. G. Wang, RSC Adv., 2016, 6, 71581–71588 RSC.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- M. M. Doeff, Y. P. Ma, S. J. Visco and L. C. Dejonghe, J. Electrochem. Soc., 1993, 140, L169–L170 CrossRef CAS.
- R. AlcáNtara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222–A225 CrossRef.
- L. Wu, D. Bresser, D. Buchholz, G. A. Giffin, C. R. Castro, A. Ochel and S. Passerini, Adv. Energy Mater., 2015, 5, 1401142 CrossRef.
- Y. Xu, E. M. Lotfabad, H. Wang, B. Farbod, Z. Xu, A. Kohandehghan and D. Mitlin, Chem. Commun., 2013, 49, 8973–8975 RSC.
- A. Rudola, K. Saravanan, S. Devaraj, H. Gong and P. Balaya, Chem. Commun., 2013, 49, 7451–7453 RSC.
- A. Rudola, K. Saravanan, C. W. Mason and P. Balaya, J. Mater. Chem. A, 2013, 1, 2653–2662 CAS.
- Y. H. Xu, Y. J. Zhu, Y. H. Liu and C. S. Wang, Adv. Energy Mater., 2013, 3, 128–133 CrossRef CAS.
- N. Zhang, Y. C. Liu, Y. Y. Lu, X. P. Han, F. Y. Cheng and J. Chen, Nano Res., 2015, 8, 3384–3393 CrossRef CAS.
- S. Liao, G. Z. Yang and C. X. Wang, RSC Adv., 2016, 6, 114790–114799 RSC.
- L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321–3323 RSC.
- J. Ding, Z. Li, H. Wang, K. Cui, A. Kohandehghan, X. Tan, D. Karpuzov and D. Mitlin, J. Mater. Chem. A, 2015, 3, 7100–7111 CAS.
- M. Gu, A. Kushima, Y. Shao, J. G. Zhang, J. Liu, N. D. Browning, J. Li and C. Wang, Nano Lett., 2013, 13, 5203–5211 CrossRef CAS PubMed.
- W. J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- M. Sawicki and L. L. Shaw, RSC Adv., 2015, 5, 53129–53154 RSC.
- Y. Yui, M. Hayashi, K. Hayashi and J. Nakamura, Solid State Ionics, 2016, 288, 219–223 CrossRef CAS.
- W. J. Zhang, M. Dahbi and S. Komaba, Curr. Opin. Chem. Eng., 2016, 13, 36–44 CrossRef.
- T. R. Jow, L. Y. Shacklette, M. Maxfield and D. Vernick, J. Electrochem. Soc., 1987, 134, 1730–1733 CrossRef CAS.
- S. Komaba, Y. Matsuura, T. Ishikawa, N. Yabuuchi, W. Murata and S. Kuze, Electrochem. Commun., 2012, 21, 65–68 CrossRef CAS.
- L. D. Ellis, B. N. Wilkes, T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2014, 161, A416–A421 CrossRef CAS.
- A. Darwiche, R. Dugas, B. Fraisse and L. Monconduit, J. Power Sources, 2016, 304, 1–8 CrossRef CAS.
- X. T. Lin, P. Li, L. Y. Shao, X. Zheng, M. Shui, N. B. Long, D. J. Wang and J. Shu, Electrochim. Acta, 2015, 169, 382–394 CrossRef CAS.
- J. Wang, K. Schenk, A. Carvalho, B. Wylie-van Eerd, J. Trodahl, C. S. Sandu, M. Bonin, I. Gregora, Z. B. He, T. Yamada, H. Funakubo, P. R. Briddon and N. Setter, Chem. Mater., 2011, 23, 2529–2535 CrossRef CAS.
- Z. Shi, J. Wang, W. Wang, Y. Zhang, B. Li, Z. Lu and Y. Li, Nano Res., 2015, 9, 353–362 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03622a |
| This journal is © The Royal Society of Chemistry 2017 |