DOI:
10.1039/C7RA03411C
(Paper)
RSC Adv., 2017,
7, 21883-21889
Ervaoffines E–G, three iboga-type alkaloids featuring ring C cleavage and rearrangement from Ervatamia officinalis†
Received
23rd March 2017
, Accepted 12th April 2017
First published on 19th April 2017
Abstract
Ervaoffine E (1), a new pseudoindoxyl alkaloid possessing a unique rearranged 1,4-diazacycloheptane skeleton, ervaoffine F (2), the first 5,6-seco-6-nor iboga-type alkaloid featuring ring C cleavage, and ervaoffine G (3), bearing an unusual contracted valerolactam ring, together with six known alkaloids were isolated from Ervatamia officinalis. Their structures and absolute configurations were determined by extensive spectroscopic analysis, single-crystal X-ray diffraction and quantum chemical ECD calculations. Plausible biogenetic pathways of these new alkaloids were also proposed and provided new insights into the structural plasticity of ring C in iboga-type alkaloids. Compound 3 exhibited a significant neuroprotective effect against oxygen-glucose deprivation (OGD)-induced damage of cultured cortical neurons, an in vitro model of ischemic stroke.
Introduction
Iboga-type alkaloids are a special class of monoterpenoid indole alkaloids that generally display a characteristic indole fused with an isoquinuclidine unit by a seven-membered azepane ring (ring C).1 Iboga-type alkaloids also exhibit a wide range of biological activities, such as anti-addiction, anti-acetylcholinesterase and antitumor activities.2 The complex scaffolds, multiple stereogenic centers and promising biological effects of the iboga-type alkaloids make them challenging targets for configurational assignment and total synthesis.3 The plant Ervatamia officinalis (Apocynaceae), a rich source of iboga-type alkaloids, is distributed in the Guangdong and Hainan provinces of China and has been used as a folk medicine for the treatment of stomachaches, snakebites, and hypertension.4 Our previous chemical investigations on the genus Ervatamia has led to the isolation of a number of iboga-type alkaloids, some of which possessed novel structures and showed neuroprotective activity or in vitro cytotoxicity.5 Through our continuing investigation, three novel alkaloids, ervaoffines E–G (1–3), and six putative biosynthetic relative iboga-type alkaloids are herein reported from the twigs and leaves of E. officinalis (Fig. 1). Structurally, compound 1 features a unique rearranged 1,4-diazacycloheptane ring C that fuses the pseudoindoxyl and isoquinuclidine units, forming an unprecedented 6/5/7/6/6 pentacyclic ring system with seven chiral centers. Compound 2 is the first example of 5,6-seco-6-nor iboga-type alkaloid with ring C cleavage, and 3 presents an unusual contracted valerolactam ring C. Intriguingly, these special structural features, highlighted by ring C cleavage and reconstructions, reveal the high structural plasticity of iboga-type alkaloids. Furthermore, 3 shows significant neuroprotective effects against oxygen-glucose deprivation (OGD)-induced damage of cultured cortical neurons, an in vitro model of ischemic stroke.6 Herein, the isolation, structural elucidation, and neuroprotective activity evaluation of 1–9 are described. In addition, the hypothetical biogenetic pathways of the new molecules are presented.
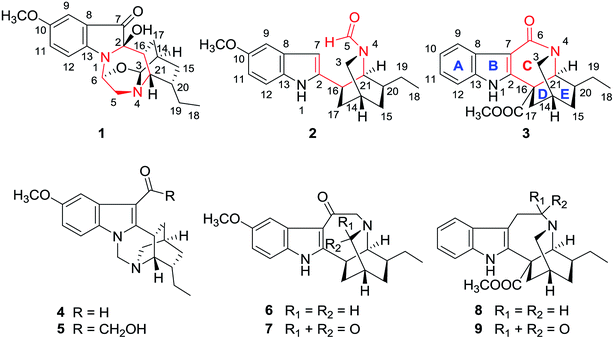 |
| | Fig. 1 Chemical structures of compounds 1–9. | |
Results and discussion
Compound 1 was isolated as bright yellow oil. The molecular formula of 1 was determined by HRESIMS analysis to be C20H24N2O4 by observing a quasi-molecular ion at m/z 357.1809 [M + H]+ (calcd for C20H25N2O4: 357.1809). The UV spectrum of 1 displayed absorption maxima at λmax 232 and 426 nm. The IR spectrum revealed the presence of hydroxyl (3421 cm−1) and carbonyl (1706 cm−1) groups and an aromatic ring (1493 and 1455 cm−1). The 1H NMR spectrum of 1 exhibited signals for a 1,2,4-trisubstituted benzene ring [δH 7.26 (1H, dd, J = 8.9, 2.7 Hz, H-11), 7.17 (1H, d, J = 8.9 Hz, H-12), and 7.02 (1H, d, J = 2.7 Hz, H-9)] and a methoxyl group [δH 3.78 (3H, s)] as well as signals for an ethyl group [δH 1.59 (1H, m, H-19a), 1.47 (1H, overlapped, H-19b), and 0.96 (3H, t, J = 7.3 Hz, H-18)]. A combination of the 13C NMR and DEPT spectra of 1 revealed the presence of twenty carbon signals, including one carbonyl, six olefinic carbons, one quaternary carbon, six methines, four methylenes, and two methyls. The above data implied that 1 was a pseudoindoxyl alkaloid. Detailed analysis of the 1H–1H COSY, HSQC, HMBC and NOESY spectra resulted in the unambiguous assignment of all the 1H and 13C NMR signals of 1, as shown in Table 1.
Table 1 1H and 13C NMR data of 1–3a
| No. |
1 |
2 |
3 |
| |
δH |
δC |
|
δH |
δC |
|
δH |
δC |
| Measured at 600 (1H) and 150 (13C) MHz in CD3OD. δ in ppm, J in Hz. Overlapped signals are reported without designating multiplicity. |
| 2 |
|
|
92.4 |
|
|
143.4 |
|
|
157.3 |
| 3 |
|
4.81 d (4.0) |
96.3 |
a |
3.49 d (12.4) |
48.4 |
a |
3.35 dt (12.0, 3.0) |
53.2 |
| |
|
|
|
b |
3.26 d (12.4) |
|
b |
3.32 m |
|
| 5 |
α |
3.19 dd (12.7, 5.5) |
54.5 |
|
7.55 s |
164.8 |
|
|
|
| β |
3.92 d (12.7) |
| 6 |
|
5.83 d (5.5) |
81.6 |
|
|
|
|
|
179.0 |
| 7 |
|
|
203.4 |
|
6.15 s |
99.9 |
|
|
105.6 |
| 8 |
|
|
120.9 |
|
|
133.2 |
|
|
126.3 |
| 9 |
|
7.02 d (2.7) |
105.8 |
|
6.96 d (2.4) |
103.0 |
|
7.85 dd (8.0, 2.2) |
121.3 |
| 10 |
|
|
155.0 |
|
|
155.2 |
|
7.19 |
123.2 |
| 11 |
|
7.26 dd (8.9, 2.7) |
129.1 |
|
6.69 dd (8.7, 2.4) |
111.9 |
|
7.17 |
124.0 |
| 12 |
|
7.17 d (8.9) |
113.0 |
|
7.16 d (8.7) |
112.2 |
|
7.36 dd (8.0, 2.2) |
113.0 |
| 13 |
|
|
157.1 |
|
|
130.2 |
|
|
137.9 |
| 14 |
|
1.79 m |
31.4 |
|
2.18 |
27.1 |
|
2.22 m |
32.8 |
| 15 |
α |
1.06 m |
30.8 |
α |
2.15 |
32.9 |
α |
1.91 td (13.0, 11.0) |
30.7 |
| β |
1.90 m |
β |
1.30 |
β |
1.40 m |
| 16 |
|
2.18 m |
43.3 |
|
3.31 |
39.6 |
|
|
49.1 |
| 17 |
α |
1.06 |
19.9 |
α |
2.20 m |
31.1 |
α |
1.79 dt (14.0, 2.0) |
34.5 |
| β |
1.14 m |
β |
2.02 m |
β |
2.78 m |
| 18 |
|
0.96 t (7.3) |
12.1 |
|
0.99 t (7.4) |
11.7 |
|
0.98 t (7.4) |
12.3 |
| 19 |
a |
1.59 m |
28.6 |
|
1.36 m |
29.4 |
a |
1.70 m |
29.3 |
| b |
1.47 |
|
|
1.31 |
|
b |
1.56 m |
|
| 20 |
|
1.48 |
40.5 |
|
1.95 m |
40.7 |
|
1.75 m |
36.4 |
| 21 |
|
3.30 |
55.3 |
|
3.58 t (1.8) |
57.4 |
|
4.17 d (2.6) |
59.7 |
| 22 (OCH3) |
|
3.78 s |
56.3 |
|
3.78 s |
56.3 |
|
|
|
| COOCH3 |
|
174.0 |
| COOCH3 |
3.85 s |
53.5 |
The 1H–1H COSY spectrum of 1 established the presence of three spin coupling systems shown in bold in Fig. 2, indicating the presence of an ethyl-substituted isoquinuclidine unit. The HMBC correlations between H-6 and C-2/C-13, between H-16 and C-7, and between H2-17 and C-2 as well as the correlation between H-21 and C-3/C-5/C-17/C-19 revealed that the pseudoindoxyl moiety and the isoquinuclidine unit were fused by a 1,4-diazacycloheptane framework. Moreover, C-3 and C-6 were connected by an oxygen atom to form a 1,3-oxazolidine moiety, which was deduced from the HMBC correlations between H-3 and C-5/C-6 as well as from the molecular formula of 1. To provide direct confirmation for the position and configuration of the hydroxyl group in 1, the 1D and 2D NMR spectra of 1 were also recorded in DMSO-d6. The HMBC correlations (in DMSO-d6) between the hydroxyl group [δH 6.39 (1H, br s)] and C-7/C-16 verified that the hydroxyl was attached to the C-2 position. Further interpretation of the HMBC correlations allowed the establishment of the planar structure of 1 (Fig. 2). In the NOESY spectrum (in DMSO-d6), correlations between H-20 and H-15β/H-16 suggested that these protons were cofacial and assigned as being β-orientated. The NOE correlations between H-3 and H-5α/H-15α and between H-6 and H-12 indicated that these protons were α-oriented. The β-orientation of 2-OH was supported by the NOE correlations between 2-OH and H-5β/H-16/H-21 (Fig. 3). The absolute configuration of 1 was established by the comparison of the calculated electronic circular dichroism (ECD) spectra of its two enantiomers with the experimental spectra. The good agreement of the calculated ECD curve of (2R,3S,6S,14R,16R,20S,21S)-1 with the experimental ECD spectrum of 1 unequivocally proved the absolute configuration of 1 (Fig. 4). Thus, the structure of 1 was elucidated, and this compound was named ervaoffine E.
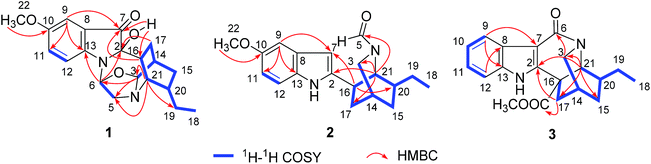 |
| | Fig. 2 1H–1H COSY and key HMBC correlations of 1–3. | |
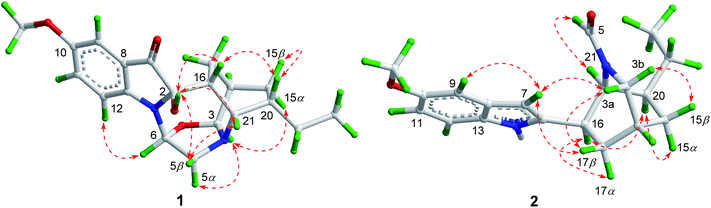 |
| | Fig. 3 Key NOESY correlations of 1–2. | |
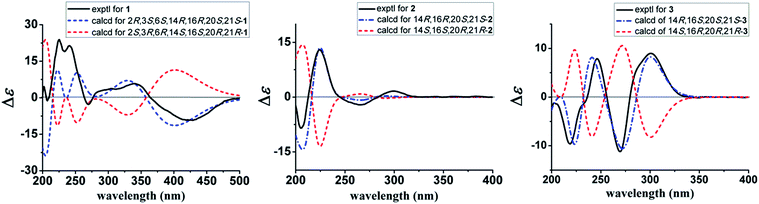 |
| | Fig. 4 Calculated and experimental ECD spectra of 1–3. | |
The molecular formula of C19H24N2O2 for 2 was established by its HRESIMS data (m/z 335.1733 [M + Na]+, calcd for C19H24N2O2Na: 335.1730). The UV spectrum of 2 revealed absorption maxima at 204 and 271 nm. The IR spectrum displayed absorption bands for a carbonyl group (1651 cm−1) and an aromatic ring (1541, 1488 cm−1). Comparison of the NMR data of 2 with those of 6-oxo-ibogaine (6) suggested their similarity, except for the existence of signals for a formyl group [δH 7.55 (1H, s); δC 164.8] and an olefinic methine [δH 6.15 (1H, s); δC 99.9] as well as the absence of resonances for a keto group, a quaternary olefinic carbon and a methylene (Table 1). The 1H–1H COSY correlations of 2 further confirmed the presence of an ethyl-substituted isoquinuclidine unit (Fig. 2). In the HMBC spectrum, cross peaks between H-21 and C-2/C-3/C-17 and between H-16 and C-7/C-20 indicated that the isoquinuclidine unit and the indole ring were connected via a C-2–C-16 bond. Furthermore, the HMBC correlations between H-21 and C-5 confirmed that the aldehyde group was attached to the N-4 position. The relative configuration of 2 could be assigned through its NOESY spectrum, the NOE correlations between H-7 and H-3a/H-9/H-17β suggested that these protons were cofacial, while the correlations between H-16 and H-17α/H-20, and between H-20 and H-15α indicated that H-15α, H-16, H-17α and H-20 were on the opposite face (Fig. 3). The absolute configuration of 2 was proved by a single-crystal X-ray diffraction experiment using CuKα radiation at 100 K [Flack parameter: 0.00(7)] (Fig. 5). Additionally, the calculated ECD curve of 14R,16R,20S,21S-2 matched well with its experimental ECD spectrum (Fig. 4), which was consistent with the X-ray crystallographic analysis of 2.
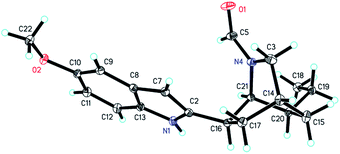 |
| | Fig. 5 X-ray ORTEP drawing of 2. | |
The molecular formula of 3 was deduced by HRESIMS analysis as C20H22N2O3 by the quasi-molecular ion at m/z 339.1703 [M + H]+ (calcd for C20H23N2O3: 339.1702). The IR spectrum of 3 showed the presence of carbonyl groups (1739, 1635 cm−1) and an aromatic ring (1583 and 1457 cm−1). The NMR data of 3 were in good agreement with those of 9, except for an upfield shift of a quaternary olefinic carbon (C-2) and a methyne (C-21) as well as the absence of resonances for a methylene in 3, suggesting that 3 was an unusual iboga-type alkaloid with one less carbon. Detailed interpretation of the 1H–1H COSY, HSQC, and HMBC data for 3 allowed the confirmation of its planar structure (Fig. 2). The valerolactam ring of 3 was verified by the key HMBC correlations between H-9 and C-7 and between H-21 and C-2/C-3/C-15/C-17 as well as the correlation between H-3 and C-6. The NOESY spectrum of 3 displayed significant NOE cross peaks between H-3a and H-17β, and between H-3b and H-15β, indicating that H-15β and H-17β were on the same face. Nevertheless, H-20 was on the opposite side, which was deduced by the NOE cross peaks between H-15α and H-17α/H-20 (Fig. S12†). Finally, the absolute configuration of 3 was elucidated to be 14R,16S,20S,21S based on the comparison between the experimental and calculated ECD data (Fig. 4).
Six known iboga-type alkaloids lirofoline A (4),7 lirofoline B (5),7 6-oxo-ibogaine (6),8 8-oxo-ibogaine lactam (7),9 coronaridine (8)10 and 5-oxo-coronaridine (9)11 were identified by the comparison of their physical and spectroscopic data with those reported in the literature. It is worth mentioning that the absolute configuration of 4 was determined for the first time by X-ray diffraction analysis with a suitable Flack parameter of −0.03(7) using CuKα radiation (Fig. S10†).
Plausible biosynthetic pathways of compounds 1–3 were shown in Scheme 1. Ibogaine (10)12 and coronaridine (8)10 are common constituents of the plant E. officinalis that could be hydroxylated and then protonated to yield intermediates A1 and B1, respectively. A1 could be further dehydrated to afford C1. Subsequently, oxidation of C1 resulted in cleavage of the C-6–C-7 bond to afford C2, which was followed by an intramolecular nucleophilic addition between the amino group (N-1) and aldehyde group (C-6) to form the N-1–C-6 bond and then by a Polonovski-type reaction13 to generate 1. The dehydration reactions of A1 and B1 coupled with electron transfer could give A2 and 5,6-dehydrocoronaridine (11),14 respectively. Similarly, oxidative cleavage of the C-5–C-6 bonds in A2 and 11 gave A3 and B3, respectively. Loss of the C-6 aldehyde group in A3 led to 2 through a decarboxylation reaction mechanism, whereas the loss of C-5 in B3 produced 3 after oxidation and amidation reactions.
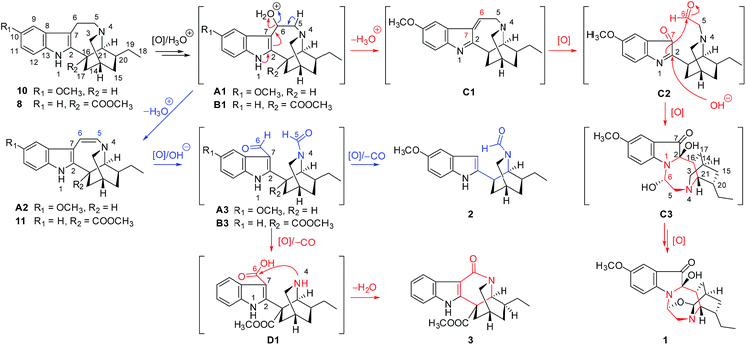 |
| | Scheme 1 Plausible biogenetic pathways of 1–3. | |
All the isolated alkaloids were tested for their neuroprotective effects against OGD-induced damage of cultured cortical neurons, an in vitro model of ischemic stroke. The result showed that compound 3 had a significant protective effect (Fig. 6), which indicates that 3 may be potentially beneficial for treating ischemic stroke.
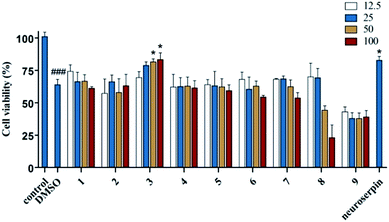 |
| | Fig. 6 Effects of compounds 1–9 on cell viability of cultured cortical neurons. The cortical neurons were treated with different alkaloids at 12.5, 25, 50 and 100 μM, and cell viability was determined by MTT assay. Neuroserpin (20 ng mL−1) served as a positive control. Data are shown as the mean ± SEM of three experiments. ###P < 0.001, control vs. DMSO, *P < 0.05, compounds 1–9 or neuroserpin vs. DMSO. One-way ANOVA followed by Bonferroni's multiple comparison test. | |
Conclusions
To sum up, three new iboga-type alkaloids (1–3) and six known ones were isolated from E. officinalis. Among them, 1 possessed an unprecedented 6/5/7/6/6 pentacyclic ring system. 2 was the first 5,6-seco-6-nor iboga-type alkaloid and 3 featured an unusual contracted valerolactam ring. In particular, plausible biogenetic pathways of these novel alkaloids have expanded the structural plasticity of iboga-type alkaloids featuring cleavage and rearrangement of ring C. Furthermore, compound 3 showed a significant protective effect against OGD-induced damage of cultured cortical neurons, which may provide a new potential candidate for treating ischemic stroke.
Experimental
General experimental procedures
Melting points were measured using an X-5 micro melting point apparatus (uncorrected). UV data were recorded using a JASCO V-550 UV/Vis spectrophotometer. Optical rotations were determined using a JASCO P-1020 digital polarimeter at 25 °C. ECD data were measured with a JASCO J-810 spectrometer. IR spectra were obtained by a JASCO FT/IR-480 Plus infrared spectrometer using KBr pellets. HRESIMS was performed on an Agilent 6210 ESI-TOF mass spectrometer. ESIMS data were measured on a Thermo Finnigan LCQ Advantage MAX mass spectrometer (Thermo Fisher Scientific Inc, USA). NMR spectra were acquired using Bruker AV-600 and AV-400 spectrometers. X-ray crystallographic data were obtained using an Agilent Gemini S Ultra diffractometer and Cu Kα radiation. Column chromatography (CC) was performed utilizing Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden), silica gel (200–300 mesh; Qingdao Marine Chemical Inc., Qingdao, P. R. China), and ODS (YMC, Kyoto, Japan). Preparative HPLC (pHPLC) was carried out on an Agilent 1260 system (equipped with a G1310B Iso pump and G1365D MWD VL detector) with a Waters Xbridge™ C18 OBD reversed-phase column (19 × 250 mm, 5 μm, USA).
Plant material
Twigs and leaves of E. officinalis were collected on Diaoluo Mountain, Hainan Province, P. R. China in September of 2014 and identified by Dr Shi-Man Huang, Hainan University, Haikou, P. R. China. The voucher specimen (no. 2014092010) was deposited at the herbarium of the College of Pharmacy, Jinan University, Guangzhou, P. R. China.
Extraction and isolation
The powdered twigs and leaves of E. officinalis (18.0 kg) were percolated at room temperature with 95% EtOH (24 h × 6) to afford a residue (0.8 kg) after evaporation, that was treated with 0.5% hydrochloric acid to adjust the pH to 3 and then extracted with CHCl3. The acidic aqueous phase was basified with aqueous ammonia to pH 9–10 and partitioned with CHCl3 to obtain a total alkaloid fraction (65.3 g), which was subjected to silica gel CC and eluted with CHCl3–MeOH (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:10, v/v) to obtain twelve fractions (Frs 1–12). Fr. 1 was re-chromatographed by silica gel CC and eluted with cyclohexane–acetone (10:0 to 0:10, v/v) to afford 5 subfractions (A–E). Fr. 1C was subjected to pHPLC using MeCN–H2O–Et2NH (7:3:0.001, v/v) as the mobile phase to yield compounds 3 (2.8 mg, tR = 28.4 min) and 9 (6.2 mg, tR = 24.0 min). Fr. 2 was successively subjected to Sephadex LH-20 using CHCl3–MeOH (1:1, v/v) as the eluent and purified by pHPLC using MeOH–H2O–Et2NH (7.3:2.7:0.001, v/v) to yield compounds 4 (15.0 mg, tR = 32.9 min), 5 (6.0 mg, tR = 29.4 min) and 8 (30.0 mg, tR = 64.4 min). Fr. 3 was chromatographed on an ODS column using MeOH–H2O (3:7 to 10:0, v/v) as the eluent, followed by pHPLC using MeOH–H2O–Et2NH (7:3:0.001, v/v) as the mobile phase to afford compound 1 (7.0 mg, tR = 36.3 min). Fr. 5 was successively separated on Sephadex LH-20 CC using CHCl3–MeOH (1:2, v/v) as the eluent and further purified by pHPLC (MeOH–H2O–Et2NH, 6.5:3.5:0.001, v/v) to afford compound 2 (4.0 mg, tR = 44.4 min). Fr. 6 was separated by repeated Sephadex LH-20 CC (MeOH) and purified by pHPLC (MeCN–H2O–Et2NH, 5:5:0.001, v/v) to obtain compounds 6 (2.3 mg, tR = 40.2 min) and 7 (3.8 mg, tR = 26.1 min).
:0 to 0:10, v/v) to obtain twelve fractions (Frs 1–12). Fr. 1 was re-chromatographed by silica gel CC and eluted with cyclohexane–acetone (10:0 to 0:10, v/v) to afford 5 subfractions (A–E). Fr. 1C was subjected to pHPLC using MeCN–H2O–Et2NH (7:3:0.001, v/v) as the mobile phase to yield compounds 3 (2.8 mg, tR = 28.4 min) and 9 (6.2 mg, tR = 24.0 min). Fr. 2 was successively subjected to Sephadex LH-20 using CHCl3–MeOH (1:1, v/v) as the eluent and purified by pHPLC using MeOH–H2O–Et2NH (7.3:2.7:0.001, v/v) to yield compounds 4 (15.0 mg, tR = 32.9 min), 5 (6.0 mg, tR = 29.4 min) and 8 (30.0 mg, tR = 64.4 min). Fr. 3 was chromatographed on an ODS column using MeOH–H2O (3:7 to 10:0, v/v) as the eluent, followed by pHPLC using MeOH–H2O–Et2NH (7:3:0.001, v/v) as the mobile phase to afford compound 1 (7.0 mg, tR = 36.3 min). Fr. 5 was successively separated on Sephadex LH-20 CC using CHCl3–MeOH (1:2, v/v) as the eluent and further purified by pHPLC (MeOH–H2O–Et2NH, 6.5:3.5:0.001, v/v) to afford compound 2 (4.0 mg, tR = 44.4 min). Fr. 6 was separated by repeated Sephadex LH-20 CC (MeOH) and purified by pHPLC (MeCN–H2O–Et2NH, 5:5:0.001, v/v) to obtain compounds 6 (2.3 mg, tR = 40.2 min) and 7 (3.8 mg, tR = 26.1 min).
Structure characterization
Ervaoffine E (1). Bright yellow oil; [α]25D +23.1 (c 0.77, MeOH); UV (MeOH) λmax (logε) 232 (4.88), 426 (4.01) nm; ECD (MeOH) λmax (Δε) 428.6 (−6.9), 339.1 (+4.3), 269.6 (−2.0), 240.9 (+15.8), 225.4 (+17.6) nm; IR (KBr) νmax 3421, 2954, 2931, 2873, 1706, 1623, 1493, 1455, 1336, 1285, 1232, 1117, 1067, 1031, 978, 889, 821 cm−1; HRESIMS m/z 357.1809 [M + H]+ (calcd for C20H25N2O4, 357.1809); 1H and 13C NMR data, see Table 1.
Ervaoffine F (2). Colorless blocks (CHCl3/MeCN); mp 166–167 °C; [α]25D +46.3 (c 0.74, MeOH); UV (MeOH) λmax (logε) 204 (4.27), 271 (3.60) nm; ECD (MeOH) λmax (Δε) 300.0 (+1.6), 265.0 (−2.1), 223.8 (+12.9), 205.2 (−8.5) nm; IR (KBr) νmax 3274, 2932, 1651, 1541, 1488, 1456, 1384, 1220, 1169, 1033, 798 cm−1; HRESIMS m/z 335.1733 [M + Na]+ (calcd for C19H24N2O2Na, 335.1730); 1H and 13C NMR data, see Table 1.
Ervaoffine G (3). Yellow oil; [α]25D +16.4 (c 0.79, CHCl3); UV (CHCl3) λmax (logε) 243 (4.26), 264 (4.09) nm; ECD (MeCN) λmax (Δε) 220 (−7.6), 246 (+5.7), 270 (−8.2), 302 (+6.2) nm; IR (KBr) νmax 3331, 2974, 2931, 2866, 1739, 1635, 1583, 1457, 1395, 1315, 1261, 1198, 947, 871, 781, 745 cm−1; HRESIMS m/z: 339.1703 [M + H]+ (calcd for C20H23N2O3, 339.1702); 1H and 13C NMR data, see Table 1.
X-ray crystallographic analysis
The structures were solved by direct methods and refined by full-matrix least-squares on F2 using SHELXL-97 package software.15 CCDC 1528868 and 1524395 ESI† for 2 and 4, respectively.
Crystal data of 2. Colorless blocks; C19H24N2O2 (fw = 312.40); orthorhombic, space group P212121; a = 7.00418(8) Å, b = 15.07922(14) Å, c = 15.68796(17) Å, α = 90°, β = 90°, γ = 90°; V = 1656.93(3) Å3, T = 100.01(10) K, Z = 4, Dc = 1.252 g cm−3, F(000) = 672. A total of 15731 reflections were collected in the range 4.07 ≤ θ ≤ 73.68, of which 3249 unique reflections with I > 2σ(I) were collected for the analysis. Final R = 0.0302 and Rw = 0.0769, and the goodness of fit on F2 was equal to 1.044; Flack parameter = −0.00(7).
Crystal data of 4. Colorless blocks; C20H24N2O2 (fw = 324.41); orthorhombic, space group P212121; a = 8.75070(10) Å, b = 14.3787(2) Å, c = 27.3576(4) Å, α = 90°, β = 90°, γ = 90°; V = 3442.23(8) Å3, T = 173(2) K, Z = 8, Dc = 1.252 g cm−3, F(000) = 1392. A total of 27736 reflections were collected in the range 3.47 ≤ θ ≤ 62.68, of which 5506 unique reflections with I > 2σ(I) were collected for the analysis. Final R = 0.0291 and Rw = 0.0737, and the goodness of fit on F2 was equal to 1.092; Flack parameter = −0.03(7).
Neuronal culture and measurement of cell viability
The cortical neurons were collected from Sprague-Dawley rats within embryo.16 Briefly, the cortical neurons were dissociated by incubation for 15 min at 37 °C in 4 mL of 0.125% trypsin. Dissociated neurons were fed with Neurobasal medium (Life Technologies) supplemented with 2% B27 (Life Technologies). Neuronal cells were seeded on 96-well dishes (1 × 105 per well), which was precoated with poly-L-lysine (0.1 mg mL−1, Sigma) for 24 h at 37 °C. The cell medium was changed every 72 h. The cells were cultured for 7 d before experiments. The isolated alkaloids were dissolved in DMSO (Sigma) and were prepared from a 50 mM L−1 stock solution immediately before use. All the compounds were then diluted in culture medium to the indicated final concentrations. The compounds were added to the cortical neurons for 4 h as pretreatment. The medium was replaced with glucose-free DMEM (Life Technologies), and the cultures were incubated in a hypoxia-chamber under a saturated atmosphere of 95% N2 and 5% CO2 for 4 h, followed by reoxygenation under normal culture conditions for 24 h. Cell survival was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) assay.17 Briefly, MTT solution (final concentration of 5 mg mL−1) was added to each well for 4 h at 37 °C. Subsequently, the dark blue formazan crystals formed in intact cells were solubilized with 100 μL DMSO. After shaking at room temperature for 10 min, absorbance at 595 nm was measured with a DTX880 multimode detector (Beckman Coulter, Brea, CA, USA). Cell viability was expressed as a percentage of the non-treated control. GraphPad Prism 5.0 software was used to perform statistical analysis. All experimental procedures involving the use of animals were approved by the Ethics Committee on Animal Experiments at Jinan University, China, and were strictly performed according to the guidelines of the Care and Use of Laboratory Animals. All efforts were made to minimize the suffering and the number of animals used.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 81373935, 81630095), the Science and Technology Planning Project of Guangdong Province (No. 2013A022100028, 2016B030301004), and the Science and Technology Development Fund of Macau (No. 042-2014-A1).
Notes and references
- R. J. Sundberg and S. Q. Smith, Alkaloids, 2002, 59, 281–386 CAS.
-
(a) B. Vastag, Science, 2005, 308, 345–346 CrossRef CAS PubMed;
(b) D. Y. He, N.-N. H. McGough, A. Ravindranathan, J. Jeanblanc, M. L. Logrip, K. Phamluong, P. H. Janak and D. Ron, J. Neurosci., 2005, 25, 619–628 CrossRef CAS PubMed;
(c) K. Ma, J. S. Wang, J. Luo, M. H. Yang and L. Y. Kong, J. Nat. Prod., 2014, 77, 1156–1163 CrossRef CAS PubMed;
(d) W. Chaiyanaa, J. Schripsemab, K. Ingkaninanc and S. Okonogia, Phytomedicine, 2013, 20, 543–548 CrossRef PubMed.
-
(a) K. Zaima, T. Hirata, T. Hosoya, Y. Hirasawa, K. Koyama, A. Rahman, I. Kusumawati, N. C. Zaini, M. Shiro and H. Morita, J. Nat. Prod., 2009, 72, 1686–1690 CrossRef CAS PubMed;
(b) G. Y. Zhao, X. G. Xie, H. Y. Sun, Z. Y. Yuan, Z. L. Zhong, S. C. Tang and X. G. She, Org. Lett., 2016, 18, 2447–2450 CrossRef CAS PubMed;
(c) A. C. Kruegel, S. Rakshit, X. G. Li and D. Sames, J. Org. Chem., 2015, 80, 2062–2071 CrossRef CAS PubMed;
(d) Y. Zhang, Y. B. Xue, G. Li, H. S. Yuan and T. P. Luo, Chem. Sci., 2016, 7, 5530–5536 RSC;
(e) G. K. Jana, S. Paul and S. Sinha, Org. Prep. Proced. Int., 2011, 43, 541–573 CrossRef CAS.
- Guangdong Nonglin College, in Chinese Flora (Zhongguo ZhiwuZhi), Science Press, Beijing, 1977, vol. 63, pp. 112–114 Search PubMed.
-
(a) Z. W. Liu, T. T. Yang, W. J. Wang, G. Q. Li, B. Q. Tang, Q. W. Zhang, C. L. Fan, D. M. Zhang, X. Q. Zhang and W. C. Ye, Tetrahedron Lett., 2013, 54, 6498–6500 CrossRef CAS;
(b) B. Q. Tang, W. J. Wang, X. J. Huang, G. Q. Li, L. Wang, R. W. Jiang, T. T. Yang, L. Shi, X. Q. Zhang and W. C. Ye, J. Nat. Prod., 2014, 77, 1839–1846 CrossRef CAS PubMed;
(c) Z. W. Liu, X. J. Huang, H. L. Xiao, G. Liu, J. Zhang, L. Shi, R. W. Jiang, X. Q. Zhang and W. C. Ye, RSC Adv., 2016, 6, 30277–30284 RSC.
- M. Papadakis, G. Hadley, M. Xilouri, L. C. Hoyte, S. Nagel, M. M. McMenamin, G. Tsaknakis, S. M. Watt, C. W. Drakesmith, R. Chen, M. J. A. Wood, Z. H. Zhao, B. Kessler, K. Vekrellis and A. M. Buchan, Nat. Med., 2013, 19, 351–357 CrossRef CAS PubMed.
- Y. Y. Low, K. H. Lim, Y. M. Choo, H. S. Pang, T. Etoh, M. Hayashi, K. Komiyama and T. S. Kam, Tetrahedron Lett., 2010, 51, 269–272 CrossRef CAS.
- K. H. Lim, V. J. Raja, T. D. Bradshaw, S. H. Lim, Y. Y. Low and T. S. Kam, J. Nat. Prod., 2015, 78, 1129–1138 CrossRef CAS PubMed.
- M. F. Bartlett, D. F. Dickel, R. C. Maxfield, L. E. Paszek and A. F. Smith, J. Am. Chem. Soc., 1959, 81, 1932–1935 CrossRef.
- T. T. Yang, B. Q. Tang, C. L. Fan, J. Zhang, X. Q. Zhang and W. C. Ye, Zhongcaoyao, 2013, 44, 1082–1085 CAS.
- J. J. De Souza, L. Mathias, R. Braz-Filho and I. J. C. Vieira, Helv. Chim. Acta, 2010, 93, 422–429 CrossRef CAS.
- P. Clivio, B. Richard, J. R. Deverre, T. Sevenet, M. Zeches and L. Le Men-Oliver, Phytochemistry, 1991, 30, 3785–3792 CrossRef CAS.
- D. Grierson, Org. React., 1990, 39, 85–295 CrossRef CAS.
- H. B. Nielsen, A. Hazell, R. Hazell, F. Ghia and K. B. G. Torssell, Phytochemistry, 1994, 37, 1729–1735 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- Y. G. Xiao, Y. H. Peng, J. Wan, G. Y. Tang, Y. W. Chen, J. Tang, W. C. Ye, N. Y. Ip and L. Shi, J. Biol. Chem., 2013, 288, 20034–20045 CrossRef CAS PubMed.
- G. Y. Tang, X. Dong, X. J. Huang, X. J. Huang, H. Liu, Y. Wang, W. C. Ye and L. Shi, Neurosci., 2015, 303, 389–401 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: UV, IR, NMR and MS of compounds 1–9. Calculation details, ECD spectra and NMR data for 1–3. CCDC 1528868 and 1524395. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra03411c |
| ‡ Z. W. L. and B. Q. T. contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Wen-Cai Ye*a
*a and
Wen-Cai Ye*a






