
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridoids and bis-iridoids from Patrinia scabiosaefolia†
Zhen-Hua Liuab,
Bo Houab,
Liu Yanga,
Rui-Jing Maab,
Jin-Yu Liab,
Jiang-Miao Hu*a and
Jun Zhou*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650204, People's Republic of China. E-mail: yxcheng@mail.kib.ac.cn; Fax: +86-871-65223048; Tel: +86-871-65223048
bUniversity of Chinese Academy of Sciences, Yuquan Road 19, Beijing 100049, People's Republic of China
First published on 10th May 2017
Abstract
Ten new iridoids, patriscabioins A–J (1–10), and three unique bis-iridoids, patriscabiobisins A–C (11–13), together with seven known analogues, have been identified from whole plants of Patrinia scabiosaefolia. Compounds 1 to 8 are a series of 5,6-dihydrovaltrate hydrins with unique substituent groups in the Valerianaceae family such as isovaleryl and 3-methylcrotonyl. Furthermore, compounds 11 and 12 are the first reported bis-iridoids with two units connected by a 1,3-dioxane group, whereas compound 13 is linked by an ether bond between two units. The structures of all the compounds were established on the basis of extensive spectroscopic analysis as well as experimental and calculated ECD spectra. Compounds 1 and 3 showed moderate inhibitory activities on AChE with IC50 values of 37.6 and 10.5 µM, respectively. Moreover, compounds 1, 3, and 5 also showed moderate cytotoxic activity against HL-60, with IC50 values ranging from 1.2 to 27.6 µM.
1. Introduction
Iridoids, which derive their name from iridomyrmecin, iridolactone and iridodial, usually contain a bicyclic H-5/H-9β, β-cis-fused cyclopentan pyran ring. Iridoids are found in a large number of folk medicinal plants. Cleaving the cyclopentan or pyran ring results in derivatives called secoiridoids. Iridoids and secoiridoids have remarkable biological activities such as antiallergic, antiarthritic, anti-inflammatory, antispasmodic, antibacterial, antifungal, antiviral, antiprotozoal, anticancer, anticoagulant, antioxidant, neuroprotective, and nerve growth factor-potentiating; thus, they are currently attracting increasing attention.1–4 Furthermore, iridoids are regarded as the bioactive compounds in some plants used in traditional medicines and are also considered to be chemotaxonomic markers in some cases such as the revision of Asteridae in 1959.5Previous phytochemical investigations on genus Patrinia (Valerianaceae) showed that these plants contain many iridoids and their saponins.6 Patrinia scabiosaefolia is a perennial herb that is distributed widely across China, except for the provinces of Ningxia, Qinghai, Xinjiang, Tibet and Hainan Island; it is used in sedation, antibacterial, and antiviral applications.7 In order to enrich our knowledge of iridoids and further explore their bioactivities, the ethyl acetate extracts of whole plants of P. scabiosaefolia were investigated. This resulted in the isolation of ten new iridoids (1–10) and three unique bis-iridoids (11–13) along with seven known iridoids, confirmed to be stenopterin A (14),8 jatamanvaltrate P (15),9 (1S,3R,5S,7S,8S,9S)-3,8-epoxy-7-hydroxy-1-butoxy-4,11-dihyronepetane (16),10 (1S,3R,5S,7S,8S,9S)-3,8-epoxy-7-hydroxy-1-methoxy-4,11-dihyronepetane (17),10 jatamanin A (18),11 6-hydroxy-7-(hydroxymethyl)-4-methylenehexahydro-cyclopenta[c]pyran-1 (3H)-one (19),12 and villosol (20).13 In addition, all the new compounds were evaluated for their inhibitory activities on acetylcholine esterase (AChE). Meanwhile, considering the cytotoxicity of iridoids,14 we also tested the cytotoxicities of the new compounds against four human tumor cell lines (HL-60, SMMC-7721, MCF-7, and SW-480). Herein, we describe the isolation, structure elucidation, and biological evaluation of these new iridoids and bis-iridoids (Fig. 1).
 | ||
| Fig. 1 The chemical structures of compounds 1–13. | ||
2. Results and discussion
2.1. Structure elucidation
The EtOH extract of P. scabiosaefolia was suspended in H2O and partitioned with EtOAc. The EtOAc extract was repeatedly chromatographed to yield 20 iridoids, including ten new iridoids, patriscabioins A–J (1–10), and three new bis-iridoids, patriscabiobisins A–C (11–13).Compound 1 was obtained as a light yellow oil, which was analyzed and determined to have the molecular formula C25H38O8 based on HRESIMS at m/z 489.2454 [M + Na]+ (calcd 489.2459) and its 13C NMR spectrum. The 1H and 13C spectroscopic data (Tables 1 and 3) showed a hemiketal methine at δH 5.86 (1H, d, J = 5.6 Hz, H-1) and δC 91.5 (d, C-1); a trisubstituted olefinic bond at δH 6.38 (1H, s, H-3), δC 140.2 (d, C-3), and δC 113.7 (s, C-4); two oxygenated methylenes at δC 62.5 (t, C-10) and δC 63.6 (t, C-11); and an oxymethine at δC 71.1 (C-7). The above data clearly suggest 1 to be a 7,10,11-trihydroxy-3-ene iridoid with ten carbons in its skeleton.8 Careful analysis of the 13C NMR and 2D NMR spectra led to the discovery of two isovaleryl substituents at δC 174.1 (s), δC 43.4 (t), δC 25.7 (d), δC 22.4 (q), δC 22.4 (q) as well as δC 173.0 (s), δC 43.3 (t), δC 25.7 (d), δC 22.3 (q), δC 22.3 (q). In addition, a 3-methylcrotonyl group was discovered at δC 164.8 (s), δC 160.0 (s), δC 115.1 (d), δC 27.6 (q), δC 20.5 (q).14 The 3-methylcrotonyl group should be attached to C-1 on the basis of HMBC correlation from H-1 (δH 5.86) to the ester carbonyl carbon (δC 164.8). The correlations from H-10 (δH 4.10, 4.52) to one isovaleryl substituent at δC 174.1 (s) and from H-11 (δH 4.57, 4.40) to the other isovaleryl at δC 173.0 (s) in HMBC suggested that the two isovaleroxy substituents were positioned at C-10 and C-11, respectively (Fig. 2).
| No. | 1a | 2b | 3b | 4b | 5b |
|---|---|---|---|---|---|
| a 1H NMR data recorded at 400 MHz.b 1H NMR data recorded at 500 MHz. | |||||
| 1 | 5.86 (d, 5.6) | 5.86 (d, 4.8) | 5.84 (d, 5.5) | 5.86 (d, 5.6) | 5.86 (d, 5.8) |
| 3 | 6.38 (s) | 6.40 (s) | 6.37 (s) | 6.39 (s) | 6.39 (s) |
| 5 | 2.99 (dd, 8.2) | 3.00 (dd, 7.1) | 2.98 (dd, 8.0) | 2.92 (dd, 7.5) | 2.99 (dd, 8.0) |
| 6a | 2.13 (m) | 2.14 (m) | 2.15 (m) | 2.02 (m) | 2.13 (m) |
| 6b | 1.67 (m) | 1.87 (m) | 1.68 (m) | 1.81 (m) | 1.68 (m) |
| 7 | 4.15 (t, 3.5) | 5.34 (br s) | 4.16 (t, 3.4) | 4.45 (m) | 4.19 (t, 4.3) |
| 8 | 2.06 (m) | 2.07 (m) | 2.04 (m) | 2.01 (m) | 2.08 (m) |
| 9 | 2.18 (m) | 2.19 (m) | 2.18 (m) | 2.40 (m) | 2.20 (m) |
| 10a | 4.10 (dd, 11.4, 4.8) | 4.10 (dd, 11.4, 4.6) | 4.10 (dd, 11.4, 4.6) | 3.97 (dd, 11.2, 3.5) | 4.13 (dd, 11.4, 4.9) |
| 10b | 4.52 (dd, 11.4, 9.9) | 4.54 (d, 10.7) | 4.51 (dd, 11.4, 10.0) | 3.81 (dd, 11.2, 6.8) | 4.49 (dd, 11.4, 9.8) |
| 11a | 4.57 (d, 12.3) | 4.58 (d, 12.2) | 4.57 (d, 12.3) | 4.60 (d, 12.2) | 4.57 (d, 12.3) |
| 11b | 4.40 (d, 12.3) | 4.42 (d, 12.2) | 4.41 (d, 12.3) | 4.38 (d, 12.2) | 4.41 (d, 12.3) |
| R1-2′ | 5.70 (s) | 5.71 (s) | 2.19 (m) | 5.68 (s) | 5.70 (s) |
| 3′ | 2.09 (m) | ||||
| 4′ | 1.92 (s) | 1.94 (s) | 0.94 (d, 2.6) | 1.91 (s) | 1.93 (s) |
| 5′ | 2.18 (s) | 2.20 (s) | 0.94 (d, 2.6) | 2.17 (s) | 2.19 (s) |
| R3-2″ | 2.21 (d, 7.2) | 2.23 (d, 7.4) | 2.23 (m) | 2.09 (s) | |
| 3″ | 2.08 (m) | 2.08 (m) | 2.09 (m) | ||
| 4″ | 0.95 (d, 5.6) | 0.96 (d, 5.5) | 0.96 (d, 3.1) | ||
| 5″ | 0.95 (d, 5.6) | 0.96 (d, 5.5) | 0.96 (d, 3.1) | ||
| R4-2‴ | 2.17 (d, 8.9) | 2.19 (d, 9.4) | 2.22 (m) | 2.16 (d, 8.3) | 2.18 (d 8.3) |
| 3‴ | 2.08 (m) | 2.08 (m) | 2.09 (m) | 2.07 (m) | 2.10 (m) |
| 4‴ | 0.94 (d, 5.6) | 0.96 (d, 5.5) | 0.94 (d, 2.6) | 0.95 (d, 6.7) | 0.95 (d, 6.6) |
| 5‴ | 0.94 (d, 5.6) | 0.96 (d, 5.5) | 0.94 (d, 2.6) | 0.95 (d, 6.7) | 0.95 (d, 6.6) |
| No. | 6c | 7b | 8c | 9b | 10a |
|---|---|---|---|---|---|
| a 1H NMR data recorded at 400 MHz.b 1H NMR data recorded at 500 MHz.c 1H NMR data recorded at 600 MHz. | |||||
| 1 | 5.87 (d, 5.5) | 5.87 (d, 5.5) | 5.88 (d, 5.6) | 4.65 (d, 6.0) | 4.80 (d, 2.0) |
| 3a | 6.40 (s) | 6.29 (s) | 6.41 (s) | 7.21 (br s) | 4.09 (d, 9.2) |
| 3b | 3.92 (d, 9.2) | ||||
| 4 | 2.05 (m) | ||||
| 5 | 3.00 (q, 7.8) | 2.95 (q, 7.4) | 2.99 (q, 6.8) | 3.16 (q, 8.2) | 1.73 (br s) |
| 6a | 2.16 (m) | 2.03 (m) | 2.13 (m) | 2.30 (m) | 1.90 (m) |
| 6b | 1.68 (m) | 1.54 (m) | 1.73 (m) | 1.65 (m) | 1.86 (m) |
| 7 | 4.16 (br s) | 4.45 (q, 5.5) | 4.24 (t, 5.0) | 4.44 (m) | 4.19 (t, 3.3) |
| 8 | 2.07 (m) | 2.02 (m) | 2.12 (m) | 1.97 (m) | 2.67 (m) |
| 9 | 2.20 (m) | 2.40 (m) | 2.21 (m) | 2.32 (m) | 2.16 (m) |
| 10a | 4.53 (d, 10.9) | 3.97 (dd, 11.2, 8.3) | 4.52 (dd, 11.2, 9.8) | 3.96 (dd, 11.2, 4.2) | 4.15 (dd, 8.3, 2.6) |
| 10b | 4.11 (dd, 11.2, 4.0) | 3.82 (dd, 11.2, 3.7) | 4.41 (dd, 11.2, 4.9) | 3.82 (dd, 11.2, 6.5) | 3.87 (dd, 8.3, 3.5) |
| 11a | 4.58 (d, 12.2) | 3.95 (d, 11.8) | 4.59 (d, 12.3) | 9.29 (s) | 1.05 (d, 6.9) |
| 11b | 4.41 (d, 12.2) | 3.74 (d, 11.8) | 4.41 (d, 12.3) | ||
| R1-2′ | 5.70 (s) | 5.69 (s) | 5.71 (s) | 3.56 (s) | |
| 4′ | 1.94 (s) | 1.92 (s) | 1.94 (s) | ||
| 5′ | 2.20 (s) | 2.17 (s) | 2.20 (s) | ||
| R3-2″ | 2.19 (d, 10.7) | 2.53 (s) | |||
| 3″ | 2.09 (m) | ||||
| 4″ | 0.96 (d, 6.5) | 1.25 (s) | |||
| 5″ | 0.96 (d, 6.5) | 1.31 (s) | |||
| R4-2‴ | 2.33 (m) | 3.37 (m) | 2.19 (d, 9.3) | ||
| 3.30 (m) | |||||
| 3‴ | 1.58 (m) | 1.53 (m) | 2.10 (m) | ||
| 4‴ | 1.25 (m) | 1.35 (m) | 0.96 (d, 6.6) | ||
| 5‴ | 1.25 (m) | 0.91 (t, 7.4) | 0.96 (d, 6.6) | ||
| No. | 1a | 2b | 3b | 4b | 5b | 6c | 7b | 8c | 9b | 10b |
|---|---|---|---|---|---|---|---|---|---|---|
| a 13C NMR data recorded at 100 MHz.b 13C NMR data recorded at 125 MHz.c 13C NMR data recorded at 150 MHz. | ||||||||||
| 1 | 91.5 | 91.3 | 91.8 | 91.2 | 91.5 | 91.5 | 91.4 | 91.3 | 103.5 | 91.8 |
| 3 | 140.2 | 141.0 | 140.1 | 140.8 | 140.4 | 140.3 | 138.9 | 140.4 | 161.3 | 63.0 |
| 4 | 113.7 | 112.8 | 114.0 | 112.9 | 113.7 | 113.8 | 114.6 | 113.4 | 123.6 | 41.1 |
| 5 | 33.0 | 33.4 | 32.9 | 33.1 | 33.1 | 33.0 | 32.9 | 33.0 | 30.3 | 30.1 |
| 6 | 39.5 | 37.7 | 39.5 | 40.4 | 39.7 | 39.5 | 40.4 | 39.8 | 41.4 | 36.3 |
| 7 | 71.1 | 75.4 | 71.1 | 73.9 | 71.2 | 71.1 | 74.2 | 71.3 | 74.3 | 76.9 |
| 8 | 46.7 | 47.8 | 46.8 | 46.4 | 46.5 | 46.8 | 46.4 | 45.9 | 47.3 | 36.1 |
| 9 | 40.7 | 40.7 | 40.7 | 39.9 | 40.8 | 40.6 | 40.1 | 40.9 | 40.4 | 49.4 |
| 10 | 62.5 | 60.9 | 62.4 | 61.8 | 62.9 | 62.6 | 61.9 | 63.1 | 62.0 | 69.0 |
| 11 | 63.6 | 63.7 | 63.6 | 63.7 | 63.7 | 63.7 | 70.0 | 63.6 | 190.7 | 12.9 |
| R1-1′ | 164.8 | 164.7 | 171.8 | 165.1 | 164.9 | 164.9 | 165.2 | 164.9 | 57.3 | |
| 2′ | 115.1 | 115.0 | 43.3 | 115.2 | 115.1 | 115.1 | 115.2 | 115.0 | ||
| 3′ | 160.0 | 160.4 | 25.7 | 160.1 | 160.2 | 160.2 | 159.9 | 160.3 | ||
| 4′ | 27.6 | 27.7 | 22.4 | 27.7 | 27.7 | 27.7 | 27.7 | 27.7 | ||
| 5′ | 20.5 | 20.6 | 22.4 | 20.6 | 20.6 | 20.6 | 20.5 | 20.6 | ||
| R3-1″ | 174.1 | 173.6 | 174.2 | 172.0 | 173.0 | 173.2 | ||||
| 2″ | 43.4 | 43.5 | 43.5 | 21.0 | 43.5 | 46.6 | ||||
| 3″ | 25.7 | 25.8 | 25.7 | 25.7 | 69.3 | |||||
| 4″ | 22.4 | 22.4 | 22.5 | 22.4 | 29.4 | |||||
| 5″ | 22.4 | 22.4 | 22.4 | 22.4 | 29.3 | |||||
| R4-1‴ | 173.0 | 173.0 | 173.0 | 173.2 | 173.1 | 175.0 | 69.2 | 173.0 | ||
| 2‴ | 43.3 | 43.5 | 43.4 | 43.5 | 43.5 | 34.3 | 31.8 | 43.5 | ||
| 3‴ | 25.7 | 25.8 | 25.7 | 25.7 | 25.7 | 25.0 | 19.4 | 25.7 | ||
| 4‴ | 22.3 | 22.4 | 22.4 | 22.4 | 22.5 | 29.7 | 14.0 | 22.4 | ||
| 5‴ | 22.3 | 22.4 | 22.4 | 22.4 | 22.4 | 29.7 | 22.4 | |||
| Palmitoyl of 6: δC 29.7–29.1 (t, C-6-13), 31.9 (t, C-14), 22.7 (t, C-15), 14.2 (q, C-16) | ||||||||||
 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of compound 1. | ||
The absolute configurations of C-1 and C-9 were both S, as found in all naturally occurring valepotriates.15 Thus, the α-orientations of H-7 and H-8 were determined by the ROESY correlations of H-1 with H-7 and H-8. Additionally, the relative configuration of H-5 was β-orientation, as deduced by the correlations from H-9 to H-5 in the ROESY experiments. Thus, the structure of 1 was elucidated as (1S,5S,7S,8S,9S)-1-O-(3-methylcrotonyl)-7-hydroxy-10,11-diisovaleroxy-5,6-dihydrovaltrate hydrin, named patriscabioin A (Fig. 3).
 | ||
| Fig. 3 Key ROESY correlations of compound 1. | ||
Compound 2 possessed the same molecular formula as compound 1, C25H38O8, based on HRESIMS at m/z 489.2462 [M + Na]+ (calcd 489.2459). The 13C and 1H NMR spectroscopic features were consistent with 1 but with a difference in the oxymethine signal at δC 75.4 (d, C-7), which was higher than that in compound 1 (δC 71.1, d, C-7). Meanwhile, there are some differences between compounds 1 and 2 at C-6, C-8, and C-10 in their 13C NMR spectra. Because the positions of the two isovaleryl substituents and the 3-methylcrotonyl group were the same, it can be speculated that the configuration of C-7 is α-orientation; this was proved by the correlations of H-7 with H-6β and H-6β with H-5 and H-9 in the ROESY experiments. Hence, the structure of 2 was determined as (1S,5S,7R,8S,9S)-1-O-(3-methylcrotonyl)-7-hydroxy-10,11-diisovaleroxy-5,6-dihydrovaltrate hydrin; this compound was named patriscabioin B.
Compound 3 gave a molecular formula of C25H40O8, deduced from HRESIMS at m/z 491.2607 [M + Na]+ (calcd 491.2602), which is two mass units higher than that of 1. The NMR data were very similar to those of compound 1 except for the appearance of an isovaleryl substituent [δC 171.8.0 (s), δC 43.3 (t), δC 25.7 (d), δC 22.4 (q), δC 22.4 (q)] instead of the 3-methylcrotonyl group in compound 1. Therefore, compound 3 has three isovaleryl substituents. These three isovaleryl substituents were assigned separately at C-1, C-10, and C-11 by means of HMBC spectra. Finally, the structure of 3 was characterized as (1S,5S,7S,8S,9S)-7-hydroxy-1,10,11-triisovaleroxy-5,6-dihydrovaltrate hydrin, named patriscabioin C.
Compound 4 had a molecular formula of C20H30O7 as established from HRESIMS at m/z 405.1889 [M + Na]+ (calcd 405.1884) and 13C NMR spectroscopic data. Compared with compound 1, compound 4 has one isovaleryl substituent and a 3-methylcrotonyl group according to careful analysis of the NMR data. The 3-methylcrotonyl group and isovaleryl group were attached at C-1 and C-11, respectively, according to correlations from H-1 to the ester carbonyl carbon at δC 165.1 (s) and from H-11 to the ester carbonyl carbon at δC 173.2 (s) in the HMBC spectra. Accordingly, the structure of compound 4 was characterized as (1S,5S,7S,8S,9S)-1-O-(3-methylcrotonyl)-7,10-dihydroxy-11-isovaleroxy-5,6-dihydrovaltrate hydrin, named patriscabioin D.
Compound 5 was analyzed to have a molecular formula of C22H32O8 by a combination of HRESIMS at m/z 447.1983 [M + Na]+ (calcd 447.1989) and 13C NMR data. Compared with the 1H NMR and 13C NMR spectroscopic data of compound 4, it could be determined that 5 has a similar structure to 4 except for the appearance of an acetyl group at δC 172.0 (s), δC 21.0 (q) in the 13C NMR spectrum and δH 2.09 (s) in the 1H NMR spectrum. This hinted that compound 5 is an acetyl derivative of 4 at C-10 on account of the correlation from H-10 to the acetyl carbonyl carbon at δC 172.0 (s) in the HMBC spectrum. Consequently, compound 5 was elucidated as (1S,5S,7S,8S,9S)-1-O-(3-methylcrotonyl)-7-dihydroxy-10-acetoxy-11-isovaleroxy-5,6-dihydrovaltrate hydrin, named patriscabioin E.
Compound 6 was formulated as C36H60O8 from HRESIMS at m/z 659.3924 [M + K]+ (calcd 659.3920). Comparison of the NMR data showed signals similar to those of 1 except for the presence of 14 methylenes and a methyl group in the high field region, which implied that a palmitoyl group replaced the original isovaleryl substituent. This finding agrees with the molecular weight exactly. Ultimately, the structure of 6, interpreted from 2D NMR spectra, was confirmed as (1S,5S,7S,8R,9S)-1-O-(3-methylcrotonyl)-7-hydroxy-10-isovaleroxy-11-palmitoyl-oxy-5,6-dihydrovaltrate hydrin, named patriscabioin F.
The molecular formula of compound 7 was inferred to be C19H30O6 by HRESIMS at m/z 377.1934 [M + Na]+ (calcd 377.1935). The 1H and 13C NMR data revealed that compound 7 is an analogue of 4; the only difference is the C-11 substituent group, which was defined as an n-butoxy unit by the 1H and 13C NMR data. By analysis of 2D NMR data, compound 7 was further identified as (1S,5S,7S,8S,9S)-1-O-(3-methylcrotonyl)-7-hydroxy-10-isovaleroxy-11-n-butoxy-5,6-dihydrovaltrate hydrin, named patriscabioin G.
Compound 8 possessed a molecular formula of C25H38O9, deduced from HRESIMS at m/z 505.2407 [M + Na]+ (calcd 505.2408). The 1H and 13C NMR spectroscopic data were almost identical to those of 1; however, a methine (δC 25.7) was absent and an oxygenated tetrahedral carbon (δC 69.3) was present along with two methyl groups downfield at δC 29.3 (q), δH 1.31 (s); δC 29.4 (q), δH 1.25 (s). These spectroscopic differences suggested the presence of a 3-hydroxylisovaleryl group in 8 at C-10, in accordance with the HMBC correlation from H-10 to the carbonyl (δC 173.2) of the 3-hydroxylisovaleryl group. After assignment of the 1D and 2D NMR data of 8, the structure was characterized as (1S,5S,7S,8S,9S)-1-O-(3-methylcrotonyl)-7-hydroxy-10-(3-hydroxylisovaleroxy)-11-isovaleroxy-5,6-dihydrovaltrate hydrin, named patriscabioin H.
Compound 9 was assigned a molecular formula of C11H16O5 from HRESIMS at m/z 251.0885 [M + Na]+ (calcd 251.0890) and 13C NMR data. In addition to a methoxy group at δC 57.3, we observed a typical acetal group at δC 103.5 (d), δH 4.65 (d, J = 6.0 Hz); a conjugated aldehyde group at δC 190.7 (d), δH 9.29 (s); and an adjacent double bond at δC 161.3 (d), δC 123.6 (s). All these spectroscopic features are close to those of the known compound 1-O-methyl cachinol,16 except for the absence of a methyl and the presence of an oxymethylene, suggesting that the methyl group at C-10 was oxygenated to an O-methylene group. After further confirmation of the structure by 1H–1H COSY, HSQC, HMBC, and ROESY data, it was assigned as (1R,5S,7S,8S,9S)-1-methoxy-7,10-dihydroxy-11-aldehyde-5,6-dihydrovaltrate hydrin, named patriscabioin I.
Compound 10 was found to have a molecular formula of C10H16O3 by HRESIMS at m/z 207.0991 [M + Na]+ (calcd 207.0992), with 3 degrees of unsaturation. The NMR spectra of 10 revealed a hemiketal methine at δC 91.8 (d, C-1) and δH 4.80 (d, J = 2.0 Hz); two oxygenated methylenes at δC 63.0 (t) and δC 69.0 (t); an oxymethine at δC 76.9 (t); and a methyl at δC 12.9 (q) and δH 1.05 (d, J = 6.9 Hz). These data suggested that the compound was a 7-hydroxy-3,4,5,6-tetrahydrovaltrate hydrin in which a methyl was changed to an oxymethylene. However, the cyclization of C-10 with OH-1 was confirmed by the correlations from H-1 to C-3 (δC 63.0, t) and C-10 (δC 69.0, t) in the HMBC spectrum. The β-configurations of H-1, Me-11, and H-8 and the α-configuration of H-7 were determined by the correlations of H-1/H-9, Me-11/H-1, H-8/H-9, and H-7/H-4 in the ROESY spectrum. Hence, the structure of compound 10 was established as (1R,4R,5R,7S,8R,9S)-1,10-epoxy-7-hydroxy-3,4,5,6-tetrahydrovaltrate hydrin, named patriscabioin J.
Compounds 11 and 12 possessed the same molecular formula, C30H40O9, according to HRESIMS, with 11 degrees of unsaturation. The IR spectrum of compound 11 showed absorption bands of hydroxy groups (3435 cm−1), a conjugated group consisting of an aldehyde function (1731 cm−1) and a double bond (1644 cm−1), and two ethers (1126 and 1089 cm−1). Furthermore, compound 12 showed similar absorptions to 11 in its IR spectrum. Careful analysis of the NMR data indicated that two compounds exhibited similar NMR spectroscopic features except for their data in the low magnetic field. Furthermore, their NMR spectroscopic data showed two distinct regions, indicating a dimer of two iridoid units. One was easily assigned to compound 4; another was identified as 8,9-didehydro-7-hydroxydolichodial,17 except for the absence of an aldehyde group and the presence of an acetal. This suggested that two units were linked through the aldehyde group. Also, the difference between compounds 11 and 12 was the position of the aldehyde group of 8,9-didehydro-7-hydroxydolichodial. In 11, this group was linked at C-1′ based on the correlations of H-1′/C-10, C-7 in the HMBC spectrum, while compound 12 was placed at C-3′ by detailed analysis of the HMBC correlations from H-3′/C-10, C-7 (Fig. 5). In addition, detailed analysis of the ROESY data showed that the configurations of the two iridoid units are the same as in compound 4 and 8,9-didehydro-7-hydroxydolichodial. The α-configuration of H-1′ in 11 and the α-configuration of H-3′ in 12 were assigned by the correlations of H-1′/H-7 and H-3′/H-7 in the ROESY experiments, respectively (Fig. 6). The good agreement between the experimental and calculated ECD spectra of compounds 11 and 12 (Fig. 4) further proved the α-configurations of H-1′ of compound 11 and H-3′ of compound 12. Hence, the structures of patriscabiobisin A (11) and patriscabiobisin B (12) were characterized as shown.
 | ||
| Fig. 4 Calculated and experimental ECD spectra of compounds 11 and 12 at the TDDFT/B3LYP/6-31G(d) level. | ||
 | ||
| Fig. 5 Key 1H–1H COSY and HMBC correlations of compound 11. | ||
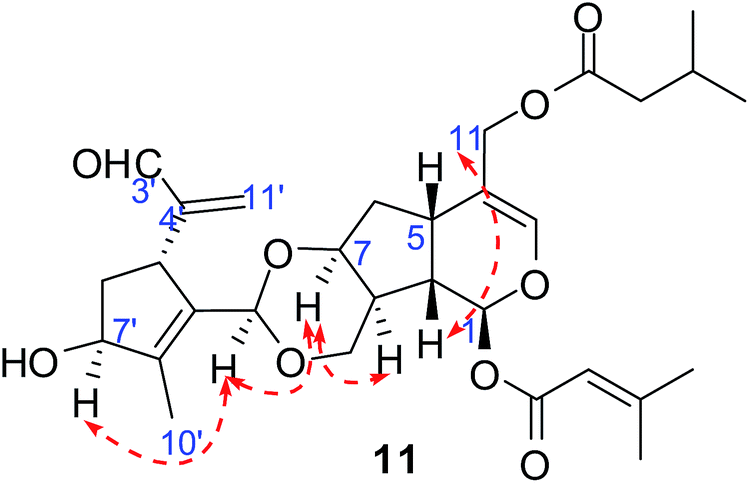 | ||
| Fig. 6 Key ROESY correlations of compound 11. | ||
Compound 13 was isolated as a light yellow oil with a molecular formula of C21H28O8 based on HRESIMS at m/z 447.1418 [M + K]+ (calcd 447.1416). Its UV spectrum displayed absorptions of conjugated aldehyde and olefinic groups at 247 nm and 202 nm. The IR spectrum showed broad absorptions for a hydroxy group (3431 cm−1), an α, β-unsaturated aldehyde group (1713, 1662 cm−1), and an ether (1076 cm−1). Detailed analysis of the 1H and 13C NMR spectroscopic data of 13 revealed that it exhibited two sets of C10-iridoid signals, of which one unit was determined to be patriscabioin I (9) and the other was judged to be jatamanin D.11 The difference was that the two sets of data shifted to a lower field [δC 62.0 (t) → δC 68.7 (t, C-10); δC 91.1 (d) → δC 97.5 (d, C-1′)]. This suggested that those two units were connected through C-10—O—C-1′, which was further verified by correlations from H-1′ (δH 5.15) to C-10 (δC 68.7) in the HMBC spectrum. The configurations of the two iridoid units were the same as in compound 9 and jatamanin D. Therefore, patriscabiobisin C (13) was characterized as shown.
Compounds 1 to 8 are a series of 5,6-dihydrovaltrate hydrins with substituent groups unique to the Valerianaceae family such as isovaleryl, 3-methylcrotonyl, 3-hydroxylisovaleryl, and palmitoyl groups. Compound 9 contains a conjugated aldehyde group with a double bond, and compound 10 contains a 6/5/5 ring system. Furthermore, compounds 11 to 13 are the first reported bis-iridoids; among these, compounds 11 and 12 are connected with a 1,3-dioxane group between two units, while compound 13 is linked by an ether bond.
2.2. Biological evaluation
P. scabiosaefolia has been used for sedation,7 in which acetylcholine esterase inhibitors may be responsible for the sedation effects;18 therefore, the inhibitory activities on AChE of all the new compounds were tested. The screening results (Table 5) showed that at concentrations of 50 µM, compounds 1 and 3 inhibited acetylcholine esterase activity over 60%, and compounds 2, 6, 9, and 11 to 13 inhibited acetylcholine esterase activity from 20% to 50%; however, compounds 4, 5, 7, 8, and 10 basically had no inhibitory activities. Then, the IC50 values of compounds 1 and 3 were examined. Compared with the reference compound tacrine (IC50 = 0.4 µM), these compounds showed moderate inhibitory activities on AChE, with IC50 values of 37.6 and 10.5 µM, respectively. These results suggested that the presence of 7β-OH and 10-Iv may be essential to the inhibitory activities of the compounds on acetylcholine esterase. Of course, the structure-activity relationships of the inhibitory activities of these compounds on acetylcholine esterase remain to be further explored.| No. | 11 | 12 | 13 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 5.89 (d, 5.2) | 91.4 | 5.94 (d, 4.9) | 91.1 | 5.06 (d, 3.8) | 104.2 |
| 3 | 6.36 (s) | 139.9 | 6.36 (s) | 139.8 | 7.39 (s) | 163.7 |
| 4 | 114.3 | 114.3 | 126.2 | |||
| 5 | 2.93 (q, 8.2) | 33.2 | 3.04 (q, 8.0) | 33.1 | 3.02 (q, 7.6) | 30.3 |
| 6a | 2.06 (m) | 38.3 | 2.17 (m) | 38.2 | 2.17 (m) | 41.4 |
| 6b | 1.58 (m) | 77.8 | 1.68 (m) | 78.0 | 1.74 (m) | 73.5 |
| 7 | 4.15 (t, 3.7) | 4.23 (t, 3.8) | 4.21 (m) | |||
| 8 | 1.65 (m) | 40.7 | 1.70 (m) | 40.6 | 2.03 (m) | 47.0 |
| 9 | 2.58 (m) | 39.9 | 2.67 (m) | 39.9 | 2.34 (m) | 44.5 |
| 10a | 4.07 (d, 12.1) | 66.1 | 4.15 (d, 12.1) | 66.2 | 3.91 (dd, 9.4, 7.6) | 68.7 |
| 10b | 3.98 (dd, 11.9, 2.8) | 4.06 (dd, 12.1, 3.1) | 3.74 (dd, 9.4, 6.3) | |||
| 11a | 4.55 (d, 12.3) | 63.7 | 4.59 (d, 12.3) | 63.7 | 9.21 (s) | 193.5 |
| 11b | 4.41 (d, 12.3) | 4.41 (d, 12.3) | ||||
| OMe | 3.53 (s) | 57.2 | ||||
| Cr-1 | 165.0 | 165.0 | ||||
| 2 | 5.70 (s) | 115.2 | 5.69 (s) | 115.2 | ||
| 3 | 160.1 | 160.0 | ||||
| 4 | 1.94 (s) | 27.7 | 1.94 (s) | 27.7 | ||
| 5 | 2.20 (s) | 20.6 | 2.20 (s) | 20.6 | ||
| Iv-1 | 173.0 | 173.1 | ||||
| 2 | 2.19 (d, 5.9) | 43.5 | 2.19 (d, 4.4) | 43.5 | ||
| 3 | 2.07 (m) | 25.7 | 2.10 (m) | 25.8 | ||
| 4 | 0.96 (d, 6.7) | 22.5 | 0.96 (d, 6.6) | 22.4 | ||
| 5 | 0.96 (d, 6.7) | 22.4 | 0.96 (d, 6.6) | 22.4 | ||
| 1′ | 5.11 (s) | 97.2 | 9.99 (s) | 188.6 | 5.15 (d, 3.0) | 97.5 |
| 3′ | 9.52 (s) | 194.0 | 4.95 (s) | 101.6 | 5.04 (s) | 95.0 |
| 4′ | 152.8 | 147.3 | 151.7 | |||
| 5′ | 3.98 (m) | 41.8 | 3.84 (m) | 42.5 | 3.12 (t, 5.9) | 43.6 |
| 6a′ | 2.09 (m) | 41.0 | 2.43 (m) | 40.7 | 2.09 (m) | |
| 6b′ | 1.96 (m) | 41.0 | 1.90 (m) | 40.7 | 1.87 (m) | |
| 7′ | 4.74 (br s) | 80.1 | 4.88 (br s) | 80.0 | 3.81 (dd, 7.6, 3.1) | 79.9 |
| 8′ | 143.2 | 161.5 | 83.6 | |||
| 9′ | 135.2 | 139.1 | 2.34 (m) | 44.5 | ||
| 10′ | 1.88 (s) | 11.7 | 2.23 (s) | 11.5 | 1.38 (s) | 19.4 |
| 11a′ | 6.18 (s) | 133.2 | 5.18 (s) | 112.8 | 4.92 (s) | 107.4 |
| 11b′ | 5.90 (s) | 4.76 (s) | 4.82 (s) | |||
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Inhibition (%) | 68.53 | 29.73 | 75.23 | −5.30 | 7.19 | 40.49 | −42.42 |
| SD | 0.90 | 2.87 | 0.42 | 2.47 | 4.45 | 2.05 | 1.81 |
| Inhibitiory activity | +++ | ++ | +++ | − | − | ++ | − |
| Compound | 8 | 9 | 10 | 11 | 12 | 13 | TA |
| Inhibition (%) | 17.78 | 46.97 | −14.54 | 36.03 | 21.91 | 37.87 | 51.01 |
| SD | 2.58 | 4.43 | 2.76 | 4.31 | 2.79 | 6.33 | 1.96 |
| Inhibitiory activity | + | ++ | − | ++ | ++ | ++ |
The final concentration of tacrine (TA) was 0.333 µM, and the final concentrations of the compounds were 50 µM. “−”: Inhibition (%) < 10%; “+”: Inhibition (%) from 10% to 20%; “++”: Inhibition (%) from 20% to 60%; “+++”: Inhibition (%) > 60%.
Considering the cytotoxicity of iridoids,14 all new compounds were evaluated for their cytotoxicities in vitro against four human cancer cell lines (HL-60, SMMC-7721, MCF-7, and SW-480) by MTT assay,19 using cisplatin (DDP) and paclitaxel as positive controls (Table 6). As a result, compound 1 showed moderate cytotoxic activity, with IC50 values of 1.4, 7.2, and 7.1 µM against HL-60, SMMC-7721, and SW480, respectively, which is comparable to cisplatin (DDP). Meanwhile, compound 5 showed cytotoxicity against HL-60, with an IC50 value of 1.2 µM. Consequently, 7β-OH and the substituent at C-10 may be responsible for the cytotoxic activity of these iridoids. Furthermore, the Cr-group may improve the activity. Moreover, in order to know their selectivity, compounds 1–3, 5 and 11 were tested for their cytotoxicities towards human normal epithelium cells (BEAS-2B). The results (Table 7) showed that human normal epithelium cells showed viable, even the concentration of these compounds was increased to 40 µM (Table 7).
| Compound | HL-60 | SMMC-7721 | MCF-7 | SW-480 |
|---|---|---|---|---|
| a “—”: inactive for cell lines. Cisplatin and paclitaxel: positive controls. | ||||
| 1 | 1.4 ± 0.02 | 7.2 ± 0.29 | 27.6 ± 1.68 | 7.1 ± 0.35 |
| 2 | — | — | — | 24.3 ± 2.39 |
| 3 | 9.9 ± 1.52 | 13.8 ± 0.17 | 17.8 ± 0.54 | 10.0 ± 0.28 |
| 5 | 1.2 ± 0.05 | 7.1 ± 0.38 | — | 18.2 ± 0.19 |
| 11 | 17.9 ± 0.73 | 19.7 ± 0.62 | 23.9 ± 1.85 | 17.6 ± 0.26 |
| Cisplatin | 2.8 ± 0.12 | 5.9 ± 0.17 | 20.4 ± 1.07 | 7.6 ± 0.54 |
| Paclitaxel | <0.008 | <0.008 | <0.008 | <0.008 |
| Concentration (µM) | Cell viability (%) | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 5 | 11 | Cisplatin | Paclitaxel | |
| 0.064 | 95.9 | 96.3 | 97.9 | 97.9 | 97.7 | 93.0 | 93.1 |
| 0.32 | 98.8 | 98.0 | 99.1 | 98.1 | 97.6 | 94.2 | 93.5 |
| 1.6 | 98.9 | 98.8 | 97.4 | 97.1 | 94.9 | 92.1 | 89.1 |
| 8 | 99.1 | 99.5 | 96.9 | 97.6 | 95.8 | 82.9 | 83.3 |
| 40 | 97.5 | 97.2 | 92.9 | 94.4 | 86.9 | 61.3 | 53.9 |
3. Experimental section
3.1. General procedure
Optical rotations were obtained on a JASCO P-1020 digital polarimeter (Horiba, Tokyo, Japan). UV spectra were measured using a Shimadzu UV-2401 PC spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were obtained on a Bruker Tensor 27 infrared spectrophotometer (Bruker Optics GmbH, Ettlingen, Germany) with KBr pellets. Mass spectra were performed on an API QSTAR time-of-flight spectrometer (MDS Sciqaszex, Concord, Ontario, Canada) and an LCMS-IT-TOF (Shimadzu, Kyoto, Japan) spectrometer. NMR spectra were recorded on Bruker AM-400, DRX-500 and Av III-600 instruments with TMS as the internal standard (Bruker, Bremerhaven, Germany). The chemical shifts were given in δ (ppm) with reference to the solvent signal. Column chromatography was performed on silica gel (200–300 and 300–400 mesh, Qingdao Marine Chemical Inc., Qingdao, China), Lichroprep Rp-18 gel (40 to 63 µm, Merck, Darmstadt, Germany), MCI gel CHP–20P (75 to 150 µm, Mitsubishi Chemical Corp., Tokyo, Japan), Sephadex LH-20 (20 to 150 µm, Amersham Biosciences, Uppsala, Sweden), and YMC*GEL ODS-A-HG (50 µm, YMC Co. Ltd. Japan). The fractions were monitored by TLC, and the spots were visualized by UV light and sprayed with 10% H2SO4 in EtOH, followed by heating.3.2. Plant material
Whole plants of P. scabiosaefolia were collected in October 2001 from Shucheng County, Anhui Province, People's Republic of China; the plants were stored in a cool and dry place at room temperature. The material was identified by Prof. Shou-Jin Liu at the Anhui University of Chinese Medicine, and a voucher specimen (Wan1295) was deposited at the Anhui University of Chinese Medicine.P. scabiosaefolia plants are abundant in local resources, and collection was permitted. Also, we ensured that the local population of P. scabiosaefolia was not destroyed by collecting specimens at different locations.
3.3. Extraction and isolation
The air-dried and powdered whole plants (29 kg) of P. scabiosaefolia were extracted with 95% ethanol (3 × 75 L) under room temperature and concentrated under reduced pressure. Then, the residue (3 kg) was dissolved in water and partitioned successively with EtOAc to yield EtOAc extract (0.85 kg) after concentration. The EtOAc extract was subjected to silica gel column chromatography eluted with a gradient of petroleum ether-ethyl acetate (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 0:1, v/v) to obtain six fractions (1 to 6) by TLC plate analysis. Fraction 2 (64 g) was separated by silica gel column chromatography eluted with a gradient of petroleum ether-acetone (5:1 → 1:1, v/v) to afford 4 subfractions (Fr.2-1 to Fr.2-4). Fr.2-1 (5.9 g) was separated by Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v), and Rp-18 column chromatography (MeOH–H2O, 50:50 → 100:0, v/v) and was further purified by semi-prep. HPLC (MeOH–H2O, 75:25, v/v) to afford 1 (14.1 mg, tR = 30.9 min) and 16 (15.0 mg, tR = 23.2 min). Fr.2-2 (1.6 g) was chromatographed on a Sephadex LH-20 column (MeOH–H2O, 90:10, v/v) and on a silica gel column with petroleum ether-ethyl acetate (10:1, v/v) to acquire 3 (5.7 mg). Fraction 3 (62 g) was separated by silica gel column chromatography eluted with a gradient of petroleum ether-acetone (5:1 → 1:1, v/v) to afford 6 subfractions (Fr.3-1 to Fr.3-6). Fr.3-1 (507.2 mg) was separated by Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v) and silica gel column chromatography eluted with petroleum ether-ethyl acetate (6:1, v/v) and purified by semi-prep. HPLC (MeOH–H2O, 95:5, v/v) to afford 6 (2.2 mg, tR = 29.8 min). Fr.3-5 (10 g) and Fr.3-6 (5.7 g) were separated by Rp-18 column chromatography (MeOH–H2O, 50:50 → 100:0, v/v), Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v), and silica gel column chromatography eluted with petroleum ether-ethyl acetate (5:1, v/v) to obtain 2 (1.8 mg), 15 (2.0 mg), and 17 (23.0 mg). Fraction 4 (69 g) was separated by Rp-18 column chromatography (MeOH–H2O, 50:50 → 100:0, v/v) to afford 7 subfractions (Fr.4-1 to Fr.4-6). Fr.4-1 (2.2 g) and Fr.4-2 (1.0 g) were all separated by Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v) and silica gel column chromatography (CHCl3–MeOH, 30:1, v/v) to obtain 10 (6.0 mg), 4 (8.0 mg), and 7 (3.5 mg). Fr.4-6 (1.7 g) was separated by successive silica gel column chromatography (CHCl3–MeOH, 30:1, v/v and petroleum ether-ethyl acetate 1:1, v/v) to yield 5 (9.0 mg), 14 (20.0 mg), and 18 (5.0 mg); meanwhile, purification by semi-prep. HPLC (MeOH–H2O, 72:28, v/v) afforded 8 (2.20 mg, tR = 29.8 min). Fraction 6 (5.2 g) was isolated by the same method as Fr.4-6, and compounds 9 (3.0 mg), 19 (6.0 mg) and 20 (22.0 mg) were obtianed; in addition, compounds 11 (1.0 mg, tR = 10.0 min) and 12 (1.0 mg, tR = 12.5 min) were purified by semi-prep. HPLC (MeOH–H2O, 50:50, v/v), and compound 13 (1.0 mg, tR = 19.8 min) was obtained by semi-prep. HPLC (MeOH–H2O, 35:65, v/v).
:1 → 0:1, v/v) to obtain six fractions (1 to 6) by TLC plate analysis. Fraction 2 (64 g) was separated by silica gel column chromatography eluted with a gradient of petroleum ether-acetone (5:1 → 1:1, v/v) to afford 4 subfractions (Fr.2-1 to Fr.2-4). Fr.2-1 (5.9 g) was separated by Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v), and Rp-18 column chromatography (MeOH–H2O, 50:50 → 100:0, v/v) and was further purified by semi-prep. HPLC (MeOH–H2O, 75:25, v/v) to afford 1 (14.1 mg, tR = 30.9 min) and 16 (15.0 mg, tR = 23.2 min). Fr.2-2 (1.6 g) was chromatographed on a Sephadex LH-20 column (MeOH–H2O, 90:10, v/v) and on a silica gel column with petroleum ether-ethyl acetate (10:1, v/v) to acquire 3 (5.7 mg). Fraction 3 (62 g) was separated by silica gel column chromatography eluted with a gradient of petroleum ether-acetone (5:1 → 1:1, v/v) to afford 6 subfractions (Fr.3-1 to Fr.3-6). Fr.3-1 (507.2 mg) was separated by Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v) and silica gel column chromatography eluted with petroleum ether-ethyl acetate (6:1, v/v) and purified by semi-prep. HPLC (MeOH–H2O, 95:5, v/v) to afford 6 (2.2 mg, tR = 29.8 min). Fr.3-5 (10 g) and Fr.3-6 (5.7 g) were separated by Rp-18 column chromatography (MeOH–H2O, 50:50 → 100:0, v/v), Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v), and silica gel column chromatography eluted with petroleum ether-ethyl acetate (5:1, v/v) to obtain 2 (1.8 mg), 15 (2.0 mg), and 17 (23.0 mg). Fraction 4 (69 g) was separated by Rp-18 column chromatography (MeOH–H2O, 50:50 → 100:0, v/v) to afford 7 subfractions (Fr.4-1 to Fr.4-6). Fr.4-1 (2.2 g) and Fr.4-2 (1.0 g) were all separated by Sephadex LH-20 column chromatography (MeOH–H2O, 90:10, v/v) and silica gel column chromatography (CHCl3–MeOH, 30:1, v/v) to obtain 10 (6.0 mg), 4 (8.0 mg), and 7 (3.5 mg). Fr.4-6 (1.7 g) was separated by successive silica gel column chromatography (CHCl3–MeOH, 30:1, v/v and petroleum ether-ethyl acetate 1:1, v/v) to yield 5 (9.0 mg), 14 (20.0 mg), and 18 (5.0 mg); meanwhile, purification by semi-prep. HPLC (MeOH–H2O, 72:28, v/v) afforded 8 (2.20 mg, tR = 29.8 min). Fraction 6 (5.2 g) was isolated by the same method as Fr.4-6, and compounds 9 (3.0 mg), 19 (6.0 mg) and 20 (22.0 mg) were obtianed; in addition, compounds 11 (1.0 mg, tR = 10.0 min) and 12 (1.0 mg, tR = 12.5 min) were purified by semi-prep. HPLC (MeOH–H2O, 50:50, v/v), and compound 13 (1.0 mg, tR = 19.8 min) was obtained by semi-prep. HPLC (MeOH–H2O, 35:65, v/v).
3.4. Spectral data of the new compounds
ε): 217 (3.94) nm; IR (KBr) νmax 3432, 2960, 2930, 1731, 1645, 1126 cm−1; positive ESIMS m/z 489 [M + Na]+, HREIMS m/z 489.2454 [M + Na]+ (calcd for C25H38O8Na, 489.2459); 1H and 13C NMR data, see Tables 1 and 3.ε): 218 (3.87) nm; IR (KBr) νmax 3453, 2960, 2930, 1732, 1646, 1125 cm−1; positive ESIMS m/z 489 [M + Na]+, HREIMS m/z 489.2462 [M + Na]+ (calcd for C25H38O8Na, 489.2459); 1H and 13C NMR data, see Tables 1 and 3.ε): 207 (3.41) nm; IR (KBr) νmax 3428, 2961, 2931, 1734, 1665, 1191 cm−1; positive ESIMS m/z 491 [M + Na]+, HREIMS m/z 491.2607 [M + Na]+ (calcd for C25H40O8Na, 491.2602); 1H and 13C NMR data, see Tables 1 and 3.ε): 217 (4.05) nm; IR (KBr) νmax 3432, 2958, 2932, 1731, 1648, 1408, 1127 cm−1; positive ESIMS m/z 405 [M + Na]+, HREIMS m/z 405.1889 [M + Na]+ (calcd for C20H30O7Na, 405.1884); 1H and 13C NMR data, see Tables 1 and 3.ε): 218 (4.11) nm; IR (KBr) νmax 3436, 2961, 2930, 1734, 1647, 1381, 1251, 1127 cm−1; positive ESIMS m/z 447 [M + Na]+, HREIMS m/z 447.1983 [M + Na]+ (calcd for C22H32O8Na, 447.1989); 1H and 13C NMR data, see Tables 1 and 3.ε): 223 (2.91) nm; IR (KBr) νmax 3430, 2925, 2856, 1630, 1384, 1031 cm−1; positive ESIMS m/z 620 [M + K]+, HREIMS m/z 659.3924 [M + K]+ (calcd for C36H60O8K, 659.3920); 1H and 13C NMR data, see Tables 2 and 3.ε): 220 (3.64) nm; IR (KBr) νmax 3427, 2930, 1726, 1638, 1384, 1128, 1085 cm−1; positive ESIMS m/z 377 [M + Na]+, HREIMS m/z 377.1934 [M + Na]+ (calcd for C19H30O6Na, 377.1935); 1H and 13C NMR data, see Tables 2 and 3.ε): 217 (3.41) nm; IR (KBr) νmax 3431, 2963, 2929, 1729, 1633, 1382, 1125 cm−1; positive ESIMS m/z 505 [M + Na]+, HREIMS m/z 505.2407 [M + Na]+ (calcd for C25H38O9Na, 505.2408); 1H and 13C NMR data, see Tables 2 and 3.ε): 248 (3.56) nm; IR (KBr) νmax 3421, 2928, 1670, 1627, 1385, 1188 cm−1; positive ESIMS m/z 251 [M + Na]+, HREIMS m/z 251.0885 [M + Na]+ (calcd for C11H16O5Na, 251.0890); 1H and 13C NMR data, see Tables 2 and 3.ε): 216 (2.24) nm; IR (KBr) νmax 3417, 2928, 1735, 1629, 1384, 1121 cm−1; positive ESIMS m/z 207 [M + Na]+, HREIMS m/z 207.0991 [M + Na]+ (calcd for C10H16O3Na, 207.0992); 1H and 13C NMR data, see Tables 2 and 3.ε): 217 (4.24) nm; IR (KBr) νmax 3435, 2960, 2928, 1731, 1644, 1127, 1090 cm−1; positive ESIMS m/z 567 [M + Na]+, HREIMS m/z 567.2567 [M + Na]+ (calcd for C30H40O9Na, 567.2565); 1H and 13C NMR data, see Table 4.ε): 219 (3.69) nm; IR (KBr) νmax 3422, 2928, 1730, 1643, 1383, 1125, 1093 cm−1; positive ESIMS m/z 567 [M + Na]+, HREIMS m/z 567.2562 [M + Na]+ (calcd for C30H40O9Na, 567.2565); 1H and 13C NMR data, see Table 4.ε): 247 (3.82), 202 (3.66) nm; IR (KBr) νmax 3431, 2929, 1625, 1384, 1121, 1077 cm−1; positive ESIMS m/z 447 [M + K]+, HREIMS m/z 447.1418 [M + K]+ (calcd for C21H28O8K, 447.1416); 1H and 13C NMR data, see Table 4.3.5. Computational studies
CHARMM force field and DFT/TDDFT calculations were performed with Discovery Studio 4.0 and the Gaussian 09 program package, respectively.20 A conflex conformational search generated low-energy conformers within a 20 kcal mol−1 energy window that were subjected to geometry optimization using DFT without imposing any symmetry constraints at the B3LYP/6-31G(d) level. Frequency calculations were carried out at the same level to verify that the molecular structures were true minima. The calculated ECD spectra were generated by the program SpecDis2 using a Gaussian band shape with an exponential half-width of 0.3 eV from the dipole-length dipolar and rotational strengths.213.6. Acetylcholinesterase inhibitory activity
The acetylcholinesterase (AChE) inhibitory activities of the isolated compounds were assayed by the spectrophotometric method developed by Ellman et al.22 with slight modifications. S-Acetylthiocholine iodide, S-butyrylthiocholine iodide, 5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB, Ellman's reagent), and acetylcholinesterase derived from human erythrocytes were purchased from Sigma Chemical. The compounds were dissolved in DMSO. The reaction mixture (totally 200 µL) containing phosphate buffer (pH 8.0), test compound (50 µM), and acetyl cholinesterase (0.02 U mL−1), was incubated for 20 min (37 °C). Then, the reaction was initiated by the addition of 40 µL of solution containing DTNB (0.625 mM) and acetylthiocholine iodide (0.625 mM) for the AChE inhibitory activity assay, respectively. The hydrolysis of acetylthiocholine was monitored at 405 nm every 30 seconds for one hour. Tacrine was used as a positive control with a final concentration of 0.333 µM. All these actions were performed in triplicate. The percentage inhibition was calculated as follows: % inhibition = (E − S)/E × 100 (E is the activity of the enzyme without test compound and S is the activity of enzyme with test compound).3.7. Cytotoxicity assays
The following human tumor cell lines were used: HL-60, SMMC-7721, A-549, MCF-7, and SW-480. The cells were obtained from ATCC (Manassas, VA, USA). All the cells were cultured in RPMI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone) at 37 °C in a humidified atmosphere with 5% CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan formed in living cells based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-5 (3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (Sigma, St. Louis, MO, USA).23 Briefly, 100 µL of adherent cells were seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1 × 105 cells per mL in 100 µL medium. Each tumor cell line was exposed to the test compound at various concentrations in triplicate for 48 h, with cisplatin and paclitaxel (Sigma) as positive controls. After the incubation, MTS (100 µg) was added to each well, and the incubation continued for 4 h at 37 °C. The cells were lysed with 100 µL of 20% SDS-50% DMF after removal of 100 µL medium. The optical density of the lysate was measured at 490 nm in a 96-well microtiter plate reader (Bio-Rad 680). The IC50 value of each compound was calculated by Reed and Muench's method.244. Conclusions
In conclusion, we have isolated and characterized 13 new and 7 known iridoids from the EtOAc extract of P. scabiosaefolia. Among these, compounds 11 to 13 are the first reported bis-iridoids. Several of the new compounds exhibited moderate inhibitory activities on AChE and moderate cytotoxic activities. This contribution enriches our knowledge of iridoids and their bioactivities.Acknowledgements
The authors are grateful to the staff of the analytical group of the State Key Laboratory of Phytochemistry and Plant Resources in West China, the Kunming Institute of Botany, Chinese Academy of Sciences, for measurements of all spectra. This work was funded by the Natural Science Foundation of Yunnan Province, China (no. 2015FB168, 2015HB093 and the provincial academician free exploration project).Notes and references
- B. Dinda, S. Debnath and Y. Harigaya, Chem. Pharm. Bull., 2007, 55, 159 CrossRef CAS PubMed.
- B. Dinda, S. Debnath and Y. Harigaya, Chem. Pharm. Bull., 2007, 55, 689 CrossRef CAS PubMed.
- B. Dinda, D. R. Chowdhury and B. C. Mohanta, Chem. Pharm. Bull., 2009, 57, 765 CrossRef CAS PubMed.
- B. Dinda, S. Debnath and R. Banik, Chem. Pharm. Bull., 2011, 59, 803 CrossRef CAS PubMed.
- S. R. Jensen, B. J. Nielsen and R. Dahlgren, Bot. Not., 1975, 128, 148 CAS.
- J. S. Kim and S. S. Kang, Nat. Prod. Sci., 2013, 19, 77 CAS.
- X. M. Gao, D. Q. Zhang and J. J. Zhang, Applied Illustrated Compendium of Materia Medica, Foreign Languages Press, Beijing, 2006, vol. 1, p. 164 Search PubMed.
- F. W. Dong, Z. K. Wu, L. Yang, C. T. Zi, D. Yang, R. J. Ma, Z. H. Liu, H. R. Luo, J. Zhou and J. M. Hu, Phytochemistry, 2015, 118, 51 CrossRef CAS PubMed.
- S. J. Wang, X. Q. Qiu, J. Y. Zhu, X. Q. Ma, B. Lin, C. J. Zheng and L. P. Qin, Helv. Chim. Acta, 2014, 97, 722 CrossRef CAS.
- P. W. Thies, Tetrahedron Lett., 1970, 35, 3087 CrossRef.
- S. Lin, T. Chen, X. H. Liu, Y. H. Shen, H. L. Li, L. Shan, R. H. Liu, X. K. Xu, W. D. Zhang and H. Wang, J. Nat. Prod., 2010, 73, 632 CrossRef CAS PubMed.
- Y. Zhang, Y. Lu, L. Zhang, Q. T. Zheng, L. Z. Xu and S. L. Yang, J. Nat. Prod., 2005, 68, 1131 CrossRef CAS PubMed.
- I. Kouno, I. Yasuda, H. Mizoshiri, T. Tanaka, N. Marubayashi and D. M. Yang, Phytochemistry, 1994, 37, 467 CrossRef CAS PubMed.
- Y. M. Xu, S. P. McLaughlin and A. A. L. Gunatilaka, J. Nat. Prod., 2007, 70, 2045 CrossRef CAS PubMed.
- S. Popov, N. Handjieva and N. Marekov, Phytochemistry, 1974, 13, 2815 CrossRef CAS.
- J. L. Jin, S. Lee, Y. Y. Lee, J. E. Heo, J. M. Kim and H. S. Yun-Choi, Planta Med., 2005, 71, 578 CrossRef CAS PubMed.
- S. Georg and V. Joerg, Arch. Pharm., 1985, 318, 515 CrossRef.
- N. E. Slatkin and M. Rhiner, J. Supportive Oncol., 2003, 1, 53 CAS.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise and A. Vaigro-Wolff, J. Natl. Cancer Inst., 1991, 83, 757 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Y. Nakajima, O. Honda, H. Kitao, T. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. J. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88 CrossRef CAS PubMed.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise and A. Vaigro-Wolff, J. Natl. Cancer Inst., 1991, 83, 757 CrossRef CAS PubMed.
- L. J. Reed and H. Muench, Am. J. Hyg., 1938, 27, 493 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D, 2DNMR, and MS spectra, crystallographic data, MTT assay. See DOI: 10.1039/c7ra03345a |
| This journal is © The Royal Society of Chemistry 2017 |