
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vitro study of novel microparticle based silk fibroin scaffold with osteoblast-like cells for load-bearing osteo-regenerative applications†
Nimisha Parekh*a,
Chandni Hushyeb,
Saniya Warunkarb,
Sayam Sen Gupta c and
Anuya Nisal*a
c and
Anuya Nisal*a
aPolymer Science and Engineering Dept., National Chemical Laboratory, HomiBhabha Road, Pune – 411008, India. E-mail: n.parekh@ncl.res.in; aa.nisal@ncl.res.in
bDepartment of Zoology, Poona College, Camp, Pune – 411001, India
cDepartment of Chemical Sciences, Indian Institute of Science and Educational Research, Mohanpur – 741246, Kolkata, India
First published on 17th May 2017
Abstract
Silk Fibroin (SF) is today considered to be one of the most favorable materials for bone tissue engineering. We have prepared novel SF microparticle based 3D scaffolds, with appropriate pore size, pore interconnectivity and porosity, excellent mechanical properties and tunable bioresorption, while retaining the inherent biocompatibility of SF. These properties make them ideal candidates for osteoregenerative applications. Here, we report the in vitro cell viability, cell adhesion and proliferation with osetoblastic differentiation of MG 63 osteoblast-like cell line on these scaffolds. In addition, we have also modified the surface of these scaffolds using collagen type I and chitosan biopolymers. Our results show that although the SF scaffold does support in vitro cell attachment, proliferation and differentiation, this performance can be further enhanced using the surface coating approach. Also, the ALP activity and bone mineralization was found to be particularly superior in the chitosan modified scaffolds.
Introduction
Bone tissue engineering (BTE) has emerged as a promising method for accelerated healing of damaged bone tissues.1 BTE involves use of a three-dimensional scaffold that provides the support for cell attachment and natural tissue formation, thus mimicking the natural extracellular matrix. There are, therefore, stringent requirements on the properties of the scaffold that can be used in BTE. The ideal scaffold should have a three-dimensional architecture with optimum pore size and pore volume for efficient diffusion and/or migration of nutrients and cells. Secondly, it must have appropriate mechanical strength to provide structural rigidity and support. Thirdly, the scaffold must give the proper chemical, biological and morphological clues that will enable in the efficient functioning of the cells for formation of new tissue.2–4 The main challenge, therefore, lies in the development of a functionalizable biocompatible 3D scaffold that can promote cell adhesion by specific cell–matrix interactions.Calcium phosphate and bioresorbable bioactive glass-based micro-porous ceramic scaffolds have been extensively explored for use in BTE.5 However, they are limited by their brittleness. In contrast, variety of natural and synthetic polymeric 3D scaffolds have been extensively explored for BTE and they exhibit better biocompatibility and mechanical properties.3,4,6 However, polymer-based matrices typically do not possess the mechanical properties of ceramics and rapidly degrade in vivo. Thus they remain nondurable for load bearing applications required in BTE. Recently, a lot of interest is focused on composite materials, which attempt to combine advantages of both polymers and ceramics. However, a 3D scaffold composed of 100% biopolymer having the properties of composite materials is desirable.
In this context, from the last two decades, silk fibroin (SF) has emerged as a promising material for BTE on account of its excellent biocompatibility, tuneable bioresorption, easy processibility and thermo-mechanical stability.1,7–9 SF-scaffolds have shown to induce bone formation, stimulate osteogenic differentiation of human mesenchymal stem cells,10–13 and support tissue vascularisation in in vitro models of BTE.14–16 SF can be produced in large commercial quantities with control on quality. It allows easy chemical modification17 that is required to obtain required functionality and can also be processed into 3D materials such as hydrogels, sponges, fibers, microspheres, and electrospun fiber mats.1,18–22 The unique mechanical properties and tunable bioresorption rate of these 3D scaffolds allows attachment, proliferation and differentiation of mesenchymal stem cells along the osteogenic lineage for BTE. The efficacy of these 3D scaffolds from SF gets enhanced when they are surface functionalized with integrin binding RGD motifs, collagen, chitosan, gelatin and growth factors like transforming growth factor-β (TGF-β), VEG-F and bone morphogenic protein (BMPs).23–25 However, the main limitation of the scaffolds discussed above is that they are mostly soft in nature and do not have high load bearing capacity which is essential for BTE. Recently, high strength SF scaffold for BTE which possess high load bearing ability, has been reported by a few groups.26–28 Ak et al. have described a novel method of cryo-gelation using a cross-linker molecule.27 Also, Mandal et al. have described scaffolds prepared using salt leaching and reinforced with SF microfibers.26 We have recently developed novel processing protocols to produce unique 3D scaffolds of silk fibroin using SF micro-particles.29,30 Here, the SF micro-particles are produced by collection of drops of SF solution in a methanol bath. These micro-particles are then fused together using a dilute solution of SF in water. This novel protocol has then been used to produce 3D scaffolds with excellent mechanical properties (dry compression modulus 10–100 MPa, wet compression modulus of 25 MPa), which is one of the main prerequisite for BTE and a property that is lacking in almost all SF based scaffold. The bulk porosity in scaffolds is in the range of 40–60% and the pore sizes are of the order of 50–500 μm. It has been well documented that microparticle based scaffolds have improved pore interconnectivity and better control on pore size and porosity.31 The conformation of SF protein in the microparticle can be controlled, which implies that the bioresorption of the scaffold can be tuned. In addition to these advantages, the methodology of scaffold fabrication is flexible enough to incorporate various functionalities such as growth factors, fillers and drug molecules amongst others. These properties make this scaffold unique for BTE in comparison to other scaffolds recently reported. (ESI Fig. S1,† for a comparison of properties with other load bearing SF-scaffolds).
In this paper, we explore the in vitro cell morphology, cell viability, cell adhesion and proliferation with osteoblastic differentiation of MG 63 osteoblast-like cell line on these scaffolds containing 100% SF and having high mechanical strengths. In addition, we report a simple method to enhance the performance of this SF-scaffold for BTE by surface coating of the scaffolds with collagen type I and chitosan biopolymers. Collagen, a dominant component of the ECM in the bone tissue, is known to favour cell adhesion and provide the right chemical cues for osteogenic differentiation.32–34 Chitosan on the other hand, is known for its inherent osteogenic potential and has been a well-documented biopolymer for BTE.35–39 We show the in vitro cell viability, cell adhesion and proliferation with osetoblastic differentiation of MG 63 osteoblast-like cells in this collagen (Coll-SF)/chitosan (CS-SF) coated SF scaffolds and compare their performance to the uncoated SF-scaffolds.
Materials and methods
SF-scaffold preparation
SF-scaffold has been prepared as per the protocol mentioned in our patent applications.29,30 In brief, 4 mL syringe was filled with the SF-HFIP solution. A syringe pump with an infusion rate of 1 mL min−1 was used and the drops of SF-HFIP solution were collected in a stirred methanol coagulant bath. The particles so formed were kept in methanol bath for 3 h. Fresh methanol was then replenished after 3 h and the particles were further incubated in methanol for 12 h. The micro-particles were allowed to air dry and the 3D SF-scaffold was prepared by fusing these SF micro-particles in a cylindrical mold using aqueous SF solution as shown in Fig. 1.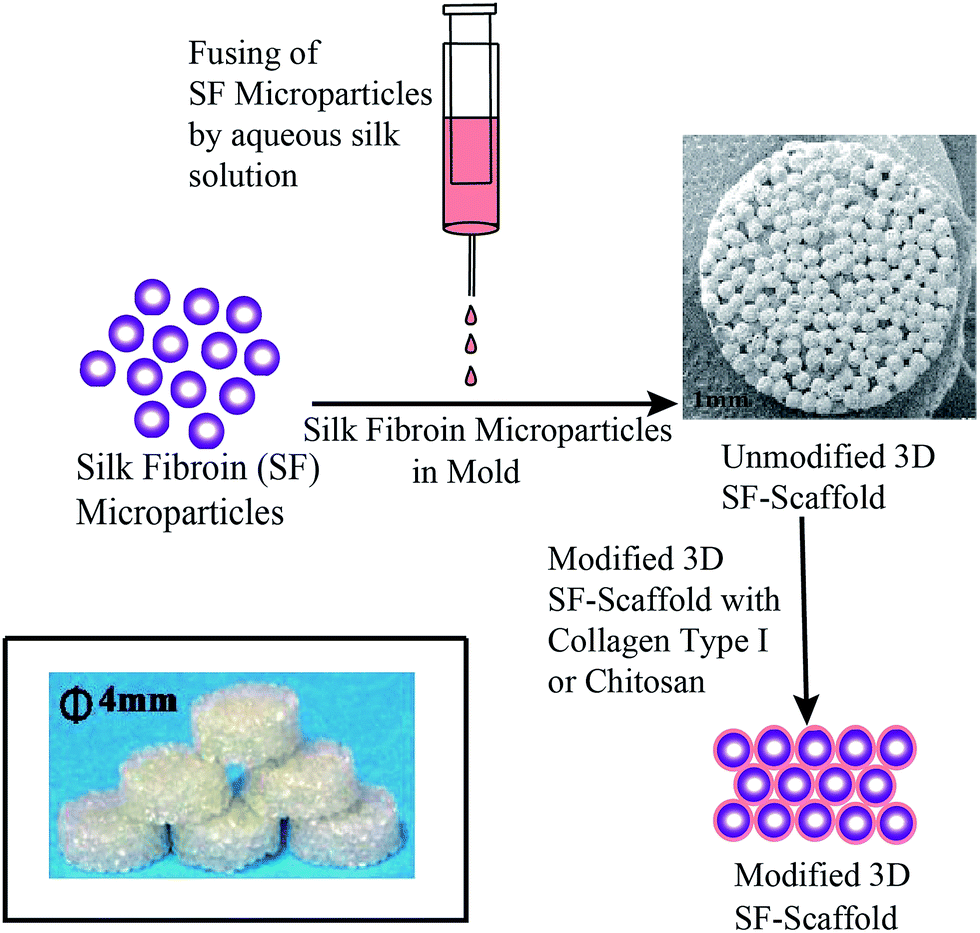 | ||
| Fig. 1 Schematic representation of SF-scaffold preparation and surface modification. | ||
Surface modification
Surface characterisation by scanning electron microscopy
The uncoated as well as coated SF scaffolds were observed for surface features using a Scanning Electron Microscope (SEM). The samples were sputter coated with 5 nm gold (Au) coating using a Polaron SC 6420 sputter coater prior to SEM imaging. Image was captured on a Quanta 200 3D SEM equipped with a tungsten filament gun, operating at WD 10.6 mm and 20 kV. Images of representative areas at suitable magnification were captured.Cell culture
MG63 human osteoblast-like cells were purchased from National Centre for Cell Science (NCCS), Pune, Maharastra, India. Cells were maintained in DMEM (Gibco) with 10% FBS (Gibco).Cell viability
Cell proliferation
Cell proliferation was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Before seeding the cells, SF, Coll-SF and CS-SF scaffolds were incubated in complete media at 37 °C with 5% CO2 for 12 h. MG 63 cells were seeded in a flat-bottomed non-adhesive 96-well plate at a density of 5 × 104 cells per scaffold in 100 μL of complete media. The plate was incubated at 37 °C with 5% CO2 for 24 h. During incubation, on the 1st, 3rd, 5th and 7th day, the media was replaced with filter sterilized MTT (0.45 mg mL−1) prepared in DMEM containing 10% FBS and further incubated for 4 h at 37 °C with 5% CO2. MTT reagent was replaced by DMSO 100 μL per well and kept at 37 °C with 5% CO2 for 20–25 minutes. Addition of DMSO dissolves the formazan crystals formed by reaction of sample with MTT and the developed color was measured at 550 nm using a microtitre plate reader (Multiskan EX, Thermo Scientific). Each absorbance was taken to be the mean of triplicate measurements.Cell morphology by actin cytoskeleton staining
MG 63 cells were seeded on SF, Coll-SF, CS-SF scaffolds at a density of 5 × 104 in DMEM containing 10% FBS. Cells were incubated at 37 °C with 5% CO2 up to 7 days. During incubation, on the 3rd, 5th and 7th day, actin cytoskeleton staining was performed using the following protocol: scaffolds were washed with PBS followed by the cell fixation with 4% paraformaldehyde for 15 minutes at room temperature. The cells were later washed with PBS two times and 0.1% Triton X-100 (Sigma-Aldrich) for 5 minutes. The cells were again subjected to PBS washing two times and incubated with 5% BSA for 20 minutes at room temperature to avoid non-specific binding. Actin filaments were stained by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution of alexafluor 488 phalloidin (Thermo Fisher Scientific) prepared in PBS and incubated for 30 minutes in dark at room temperature. Cell's nucleus was counter stained with DAPI at 300 nM concentration for 4 minutes at room temperature and washed with PBS. Images were captured by epi-fluorescence microscopy using Axio Observer Z1 Carl Zeiss Microscope.
:100 dilution of alexafluor 488 phalloidin (Thermo Fisher Scientific) prepared in PBS and incubated for 30 minutes in dark at room temperature. Cell's nucleus was counter stained with DAPI at 300 nM concentration for 4 minutes at room temperature and washed with PBS. Images were captured by epi-fluorescence microscopy using Axio Observer Z1 Carl Zeiss Microscope.
Osteogenic differentiation by alkaline phosphatase (ALP) assay
Alkaline phosphatase (ALP) activity was assayed using colorimetric ALP kit (Abcam, U.K.). Briefly, MG 63 cells were seeded on a SF, Coll-SF, CS-SF scaffolds at a density of 5 × 104 cells per scaffold in 10 μL of complete media. The cells were allowed to be settled for 5–7 minutes at 37 °C with 5% CO2. The additional 90 μL media for cells was then added and the cell culture plate was then incubated at 37 °C with 5% CO2 for 7 days. On the second day of the experiment, seeding media was replaced with osteogenic differentiation media (Invitrogen). During incubation, on the 3rd, 5th and 7th day, spent media from cell seeded scaffolds was collected and 80 μL of the spent media was incubated with 50 μL of p-nitrophenyl phosphate (5 mM) solution at room temperature for 1 h in the dark. At the end of the incubation, enzyme activity was stopped by adding 20 μL of stop solution. Simultaneously, standard curve was plotted (ESI Fig. S2†) as per manufacturer's instruction. The amount of p-nitro-phenol produced was measured by measuring absorbance at 405 nm.Extracellular mineral (Ca+2) deposition and quantification
Mineralized calcium deposition at the end of 7th day was analyzed with Alizarin Red S staining. In brief, MG 63 cells were seeded on a SF, Coll-SF, CS-SF scaffolds at a density of 5 × 104 cells per scaffold in 10 μL of complete media. The cells were allowed to be settled for 5–7 minutes at 37 °C with 5% CO2. The additional 90 μL media for cells was then added and the cell culture plate was then incubated at 37 °C with 5% CO2 for 7 days. On the day after seeding, the media was replaced with osteogenic differentiation media (Invitrogen). On 7th day, scaffolds were fixed with 4% formaldehyde at room temperature for 10 min. They were later washed with PBS and then stained with 2% Alizarin Red (Sigma) solution for 20 minutes at room temperature. After staining, the scaffolds were washed two times with DI water. Calcium deposition was observed under bright field by Axio Observer Z1 from Carl Zeiss.Ca deposition was further confirmed by quantifying the total calcium content.23,40 In brief, at the end of 7th day, scaffolds were washed with PBS and the deposited calcium was extracted with 0.5 mL of 5% trichloroacetic acid for 30 minutes. At the end of incubation, Ca was quantified using O-cresolphthalein complexone colorimetric assay (Sigma).
The developed color for scaffolds seeded with cells were measured at 575 nm and the absorbance at 575 nm for scaffolds that were not seeded with cells was subtracted to quantify the Ca content. Standard calibration curve was prepared as per manufacturer instructions (ESI Fig. S3†).
Results and discussion
3D microparticle SF-scaffold
Silk fibroin based scaffolds have been prepared using a variety of methods like lyophilisation, salt leaching, electrospinning, ice crystal templating, etc. These materials have shown promise in BTE applications. In this report, we use 3D microparticle SF-scaffold which was prepared using a methodology developed in our laboratory. Preliminary studies on the physical properties of this 3D microparticle SF-scaffold (such as mechanical integrity, pore size and pore volume and rate of bioresorption) match the requirements for a scaffold in BTE (ESI Fig. S1†).However, the hydrophobic nature of SF results in limited cell–biomaterial interaction. We have introduced surface modification of the SF-scaffold to further improve cell adhesion and proliferation. Our process involves coating the surface of this microparticle scaffold using collagen type I and chitosan – biopolymers that are known to enhance cell adhesion and proliferation. The method involved incubating the SF-scaffolds with the biopolymer and allowing them to anneal and coat the surface. The morphological features of surface coating were studied using Scanning Electron Microscopy (SEM). Fig. 2 shows the SEM images of SF scaffold (SF) and surface modified scaffolds treated with collagen (Coll-SF) and chitosan (CS-SF). The SEM images of SF scaffold (Fig. 2) display mono-disperse micro-particles packed together in hexagonal packing. The surface of SF micro-particles shows presence of dried SF solution used to fuse these particles together. The interparticle pore sizes are clearly visible and are of the order of 100 μm. The collagen modified SF scaffold shows presence of additional material domains dispersed throughout the surface (Fig. 2). These could be attributed to the collagen coating. Similar material domains were also observed in the chitosan treated scaffold (Fig. 2). It has been found that a minimum pore size of 30 microns is necessary for effective cell migration and colonization.41 As can be seen in the Fig. 2, the method used for coating here does not alter the pore size of the scaffold. This implies that the surface modification technique is not expected to inhibit transport of nutrients and waste diffusion or cell migration in the scaffold.
 | ||
| Fig. 2 SEM images of SF, Coll-SF and CS-SF and all images were at the scale bar is 50 μm. | ||
Cell viability and proliferation studies
The uncoated and coated SF scaffolds were evaluated using in vitro cell culture studies. MG-63 cells, was chosen for this study as it has a high proliferation rate. In vitro studies performed in a 7 day experiment are therefore sufficient to provide reliable and sensitive information on performance of this material as a possible scaffold for BTE.42–44 Cell viability of MG 63 is prerequisites for further application of any developed material and hence it was quantitatively assayed by using trypan blue dye exclusion method and qualitatively by performing live/dead imaging using AO and PI stains. Fig. 3 shows the viable cell count on the 1st, 3rd, 5th and 7th day after seeding the MG 63 on SF, Coll-SF and CS-SF scaffolds and it shows that SF scaffold does support attachment of the MG63 cells. | ||
| Fig. 3 Viable MG 63 cell density counted by trypan blue dye exclusion method (data are represented as the mean ± SD of two independent experiments). | ||
Also, as the experiment progresses the number of viable cells on the SF-scaffold increases. However, coating or surface modifying the scaffolds resulted in increased number of viable cells. On a given day of the experiment, the Coll-SF scaffolds and CS-scaffolds have a higher number of viable cells as compared to the uncoated SF scaffold. However, viability of the cells on the Coll-SF and CS-SF scaffold is comparable and no significant differences between these two scaffolds were observed here.
The trends observed in cell viability were further corroborated using live-dead assay. Fig. 4 shows the individual channel images of AO and PI on the 1st, 3rd, 5th and 7th day after seeding the MG 63 on SF, Coll-SF and CS-SF scaffold. For the SF-scaffold, MG 63 cells did adhere and found to be live to the micro-particle surface. Also, there were increased number of viable cells visible as the days progressed. Insignificant or no red coloured spots in these images indicate absence of dead cells on these scaffolds. Also, the number of live cells observed for modified scaffolds is always higher as compared to the uncoated SF scaffold. The living cell with elongated and spread morphology was most clearly visible on the Coll-SF scaffold as compared to the other two scaffolds used in the study (ESI Fig. S4†). Interestingly, as the experiment progressed, cells formed clumps on CS-SF scaffold.
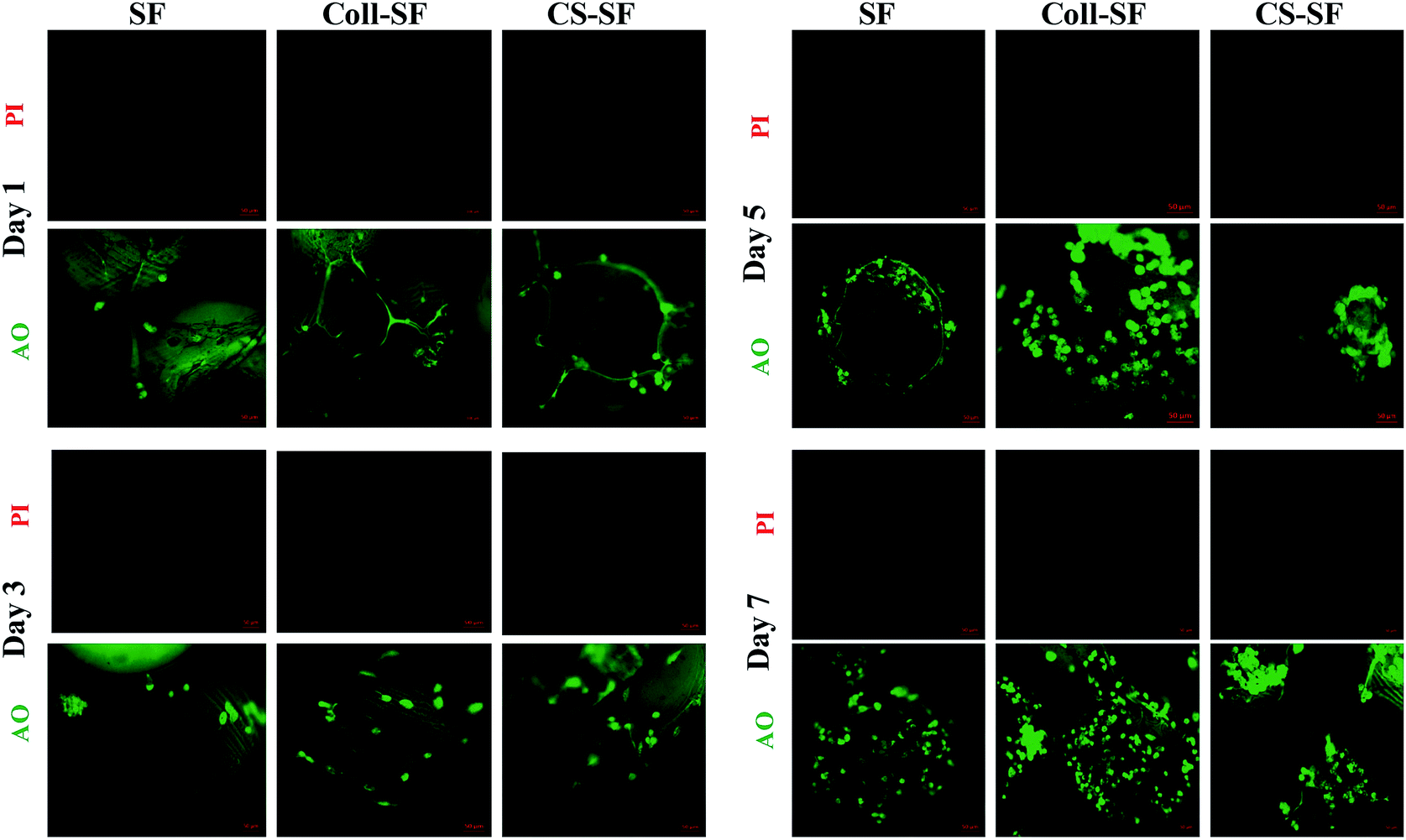 | ||
| Fig. 4 Epi-fluorescence images of live (green) and dead (red) MG 63 cells grown on SF, Coll-SF and CS-SF. All images were taken at same exposure time and scale bar is 50 μm. | ||
This clumping was not observed in the SF or Coll-SF scaffold. It has been reported that degree of acetylation of chitosan is responsible for the clustering of cells.41 In our study, 15–25% acetylated chitosan was used to modify the surface and our observations are in accordance with the literature reports.35,41
Cell proliferation was analyzed by using MTT assay. As can be seen in Fig. 5, all three scaffolds do support proliferation of the cells. Increased absorbance as a result of proliferation of viable cells were observed with both surface modified scaffolds as compare to unmodified SF scaffold. These results are in-line with those observed in trypan blue and live-dead assay experiments. The viable cell count nearly doubled on the 7th day after incubation. Similar trends in cell proliferation were also observed for the Coll-SF and CS-SF scaffolds.
 | ||
| Fig. 5 Cell proliferation assay (MTT assay) of MG 63 cultured on SF, Coll-SF and CS-SF till 7 days. Data are presented as the mean ± SD of two independent experiments. | ||
Cell morphology by actin cytoskeleton staining
Cellular morphology was evaluated by F-actin cytoskeleton staining with alexafluor 488 phalloidin. Fig. 6 shows the actin cytoskeleton of MG 63 grown on SF, Coll-SF and CS-SF scaffolds. From the images, the significant cell growth with clear morphology of cells with respect to all three scaffolds is clearly visible. More elongated cell morphology with increased number of cells was visible with Coll-SF as compare to SF and CS-SF. Also, for the CS-SF scaffolds, we did observe spreading out of some cells, but the predominant morphology was spherical (ESI Fig. S5†). This variation in morphology of the cells can be attributed to the spread in degree of acetylation in the chitosan used for experimentation. Both the results are in accordance with MTT and live/dead assay results.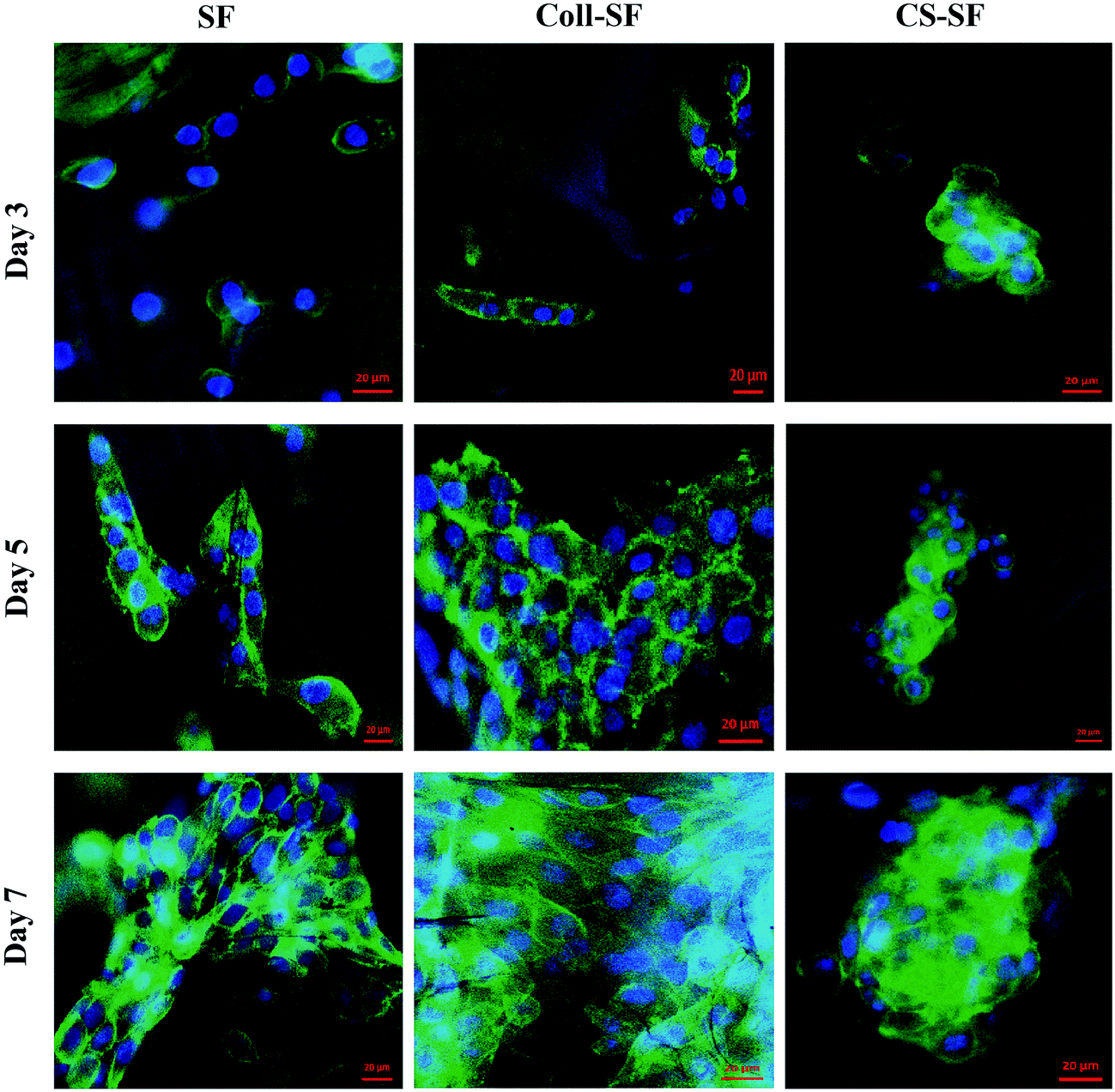 | ||
| Fig. 6 Actin cytoskeleton staining of MG63 cells with alexafluor phalloidin 488 and nucleus were counter stained with DAPI. Scale bar is 20 μm. | ||
In short, data obtained from trypan blue dye extraction method, live/dead assay, actin cytoskeleton staining and cell proliferation assayed by MTT (Fig. 3–6) showed improved performance of the biopolymer coated scaffolds towards cell viability, proliferation and adhesion with retained cell morphology. This is in line with several reports where collagen and chitosan has been shown to improve cell adhesion and proliferation. Collagen type I is the dominant component of the ECM in a bone tissue and hence provides the right chemical cues for cell recognition. The integrins present on the cell surface are known to recognize the triple helix structure of collagen. Also, it has been acknowledged that collagen plays an important role in cytoskeleton reorganization and signal transduction, which in turn regulates the cell adhesion and proliferation.46 Chitosan has been used in BTE and is shown to induce proliferation of osteoblast cells. The chemical structure of chitosan is similar to glycosaminoglycans (GAGs), which is an important component in ECM. This has been cited as one of the possible reasons for improved adhesion of cells to chitosan.39 Our observations support this literature.
Osteogenic differentiation
Bone is a mineralized connective tissue, which is continuously bioresorbed and neoformed. The formation of new bone tissue requires commitment from the cells towards the osteogenic lineage. The osteoblasts are responsible for synthesis of the bone matrix and as the cells proliferate it shows higher ALP activity and mineral deposition. Thus, in addition to cell adhesion and proliferation, understanding the functioning of cells is also of prime importance in BTE. In this study, we used ALP activity and calcium deposition to monitor osteogenic differentiation of MG 63 cells on unmodified and modified scaffolds.ALP is an initial marker for the osteogenic differentiation process and is secreted into the ECM, playing a crucial role in bone ECM mineralization.
In our study Fig. 7, we detected measurable ALP activity with all three scaffolds and within the standard calibration curve (ESI Fig. S3†). The ALP activity was evident on day 3 for the SF scaffold and was found to be significantly enhanced on day 7. For the Coll-SF scaffold too, a similar increased ALP activity was observed for day 7. On 3rd and 7th day, the ALP activity between the three types of scaffold studied were comparable, with CS-SF scaffold showing the highest ALP activity. However, on the 5th day, the ALP activity in the CS-SF scaffold was significantly higher than the other two scaffolds studied. Here, the absorbance for CS-SF scaffold was nearly double that of SF and Coll-SF scaffold.
 | ||
| Fig. 7 ALP assay- MG 63 cells cultured on SF, Coll-SF and CS-SF till 7 days. Absorbance was taken at 405 nm and data are presented as the mean ± SD of two independent experiments. | ||
Presence of ALP enzyme is the predetermining early marker as the cells started to differentiate into osteogenic lineage. Throughout the assay, we got higher ALP activity with CS-SF scaffold. The invigorating effect of chitosan on osteogenesis has been extensively reported45,46 however the exact mechanism of this osteogenic behavior is unknown. One of the possible reasons that have been discussed is the cationic charge on chitosan.47 Also, it has been suggested that high molecular weight (>10 kDa) of chitosan results in improved osteogenic behaviour of MG63 cells. The molecular weight of chitosan used here is 190–310 kDa and thus our observations are in sync with literature.46,48,49
Alizarin red S staining was used to detect in vitro extra-cellular mineral (calcium) deposition as a result of osteoblastic differentiation initiation process. After the 7th day of seeding the MG 63 cells, calcium deposited nodules were detected on all three scaffold Fig. 8. Amount of deposition was not significantly different in SF scaffold and Coll-SF scaffold.
 | ||
| Fig. 8 Calcium depositions by alizarin red S staining. Arrow indicates deposited calcium nodules. All bright field images were taken at same exposure time and scale bar is 50 μm. | ||
This deposition has been considerably increased as a part of differentiation process with the CS-SF scaffold as is evident in the images. For the Coll-SF scaffolds, Ca deposition is slightly higher than the SF and this improvement can been attributed to the GER sequence in collagen, which acts as an integrin binding site.1,50 Also, our ALP activity assay shows moderately increased ALP activity with Coll-SF as compare to SF on 7th day but considerable difference has been noticed with CS-SF. Further, the total amount of Ca deposition was quantified by using O-cresolphthalein complexone colorimetric assay (ESI Fig. S6†). Higher Ca content was observed with CS-SF scaffolds as compared to SF and Coll-SF scaffolds. These results are in accordance with the alizarin red S staining studies.
Conclusions
We report here in vitro cell behavior of osteoblast-like cells on novel SF microparticle scaffolds for applications in load-bearing osteo-regenerative applications. Additionally, we have modified the surface of these scaffolds using natural biopolymers – collagen type I and chitosan. Our results suggest that SF micro-particle scaffold does support cell attachment, proliferation and differentiation of cells. This behavior can be further enhanced by coating the surface of these scaffolds with collagen type I and chitosan biopolymers. Although the cell attachment, viability and morphology were found to be superior in collagen modified scaffolds, the osteogenic differentiation is better in chitosan-modified scaffolds. The efficacy of these scaffolds for load bearing bone tissue engineering application in vivo is currently underway.Acknowledgements
The authors acknowledge BiolMed Innovations Pvt. Ltd. for providing SF micro-particles and SF scaffolds as per the Materials Transfer Agreement signed between BiolMed Innovations and CSIR-National Chemical Laboratory, Pune. We also thank Mr Abhijit Shete for providing help in sample preparation for Scanning Electron Microscopy. We acknowledge Dr MujeebShaikh from Poona College for supporting the project. Also, this work was funded through the CSIR 12th Five Year Plan Project CSC0134.References
- J. Melke, S. Midha, S. Ghosh, K. Ito and S. Hofmann, Acta Biomater., 2015, 31, 1–16 CrossRef PubMed.
- X. Le, G. E. J. Poinern, N. Ali, C. M. Berry and D. Fawcett, Int. J. Biomater., 2013, 2013, 1–16 CrossRef PubMed.
- X. Liu and P. X. Ma, Ann. Biomed. Eng., 2004, 32, 477–486 CrossRef PubMed.
- D. W. Hutmacher, Biomaterials, 2000, 21, 2529–2543 CrossRef CAS PubMed.
- S.-J. Ding, C.-K. Wei and M.-H. Lai, J. Mater. Chem., 2011, 21, 12793–12802 RSC.
- E. Sachlos and J. T. Czernuszka, Eur. Cells Mater., 2003, 5, 29–40 CrossRef CAS PubMed.
- N. Kasoju and U. Bora, Adv. Healthcare Mater., 2012, 1, 393–412 CrossRef CAS PubMed.
- L. D. Koh, Y. Cheng, C. P. Teng, Y. W. Khin, X. J. Loh, S. Y. Tee, M. Low, E. Ye, H. D. Yu, Y. W. Zhang and M. Y. Han, Prog. Polym. Sci., 2015, 46, 86–110 CrossRef CAS.
- A. E. Thurber, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2015, 71, 145–157 CrossRef CAS PubMed.
- L. Meinel, O. Betz, R. Fajardo, S. Hofmann, A. Nazarian, E. Cory, M. Hilbe, J. McCool, R. Langer, G. Vunjak-Novakovic, H. P. Merkle, B. Rechenberg, D. L. Kaplan and C. Kirker-Head, Bone, 2006, 39, 922–931 CrossRef CAS PubMed.
- S. Saha, B. Kundu, J. Kirkham, D. Wood, S. C. Kundu and X. B. Yang, PLoS One, 2013, 8, 1–10 Search PubMed.
- B. Kundu, R. Rajkhowa, S. C. Kundu and X. Wang, Adv. Drug Delivery Rev., 2013, 65, 457–470 CrossRef CAS PubMed.
- P. Gupta, M. Adhikary, M. Christakiran, M. Kumar, N. Bhardwaj and B. B. Mandal, ACS Appl. Mater. Interfaces, 2016, 8, 30797–30810 CAS.
- S. Ghanaati, R. E. Unger, M. J. Webber, M. Barbeck, C. Orth, J. A. Kirkpatrick, P. Booms, A. Motta, C. Migliaresi, R. A. Sader and C. J. Kirkpatrick, Biomaterials, 2011, 32, 8150–8160 CrossRef CAS PubMed.
- R. E. Unger, A. Sartoris, K. Peters, A. Motta, C. Migliaresi, M. Kunkel, U. Bulnheim, J. Rychly and C. James Kirkpatrick, Biomaterials, 2007, 28, 3965–3976 CrossRef CAS PubMed.
- S. Fuchs, X. Jiang, H. Schmidt, E. Dohle, S. Ghanaati, C. Orth, A. Hofmann, A. Motta, C. Migliaresi and C. J. Kirkpatrick, Biomaterials, 2009, 30, 1329–1338 CrossRef CAS PubMed.
- S. Das, D. Pati, N. Tiwari, A. Nisal and S. Sen Gupta, Biomacromolecules, 2012, 13, 3695–3702 CrossRef CAS PubMed.
- D. L. Kaplan and C. Vepari, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef PubMed.
- A. Nisal, M. Dhanalakshmi, A. Lele, B. L. V Prasad and J. Bellare, Proc. 70th Annu. Tech. Conf. Exhib., Soc. Plast. Eng., Mumbai, India, 6-7 December 2012, pp. 270–275 Search PubMed.
- A. Nisal, C. Kalelkar, J. Bellare and A. Lele, Rheol. Acta, 2013, 52, 833–840 CrossRef CAS.
- P. Dubey, L. Nawale, D. Sarkar, A. Nisal and A. Prabhune, RSC Adv., 2015, 5, 33955–33962 RSC.
- P. Dubey, S. Kumar, V. K. Aswal, S. Ravindranathan, P. R. Rajamohanan, A. Prabhune and A. Nisal, Biomacromolecules, 2016, 17, 3318–3327 CrossRef CAS PubMed.
- V. Karageorgiou, M. Tomkins, R. Fajardo, L. Meinel, B. Snyder, K. Wade, J. Chen, V. Vunjak-Novakovic and D. L. Kaplan, J. Biomed. Mater. Res., Part A, 2006, 33, 324–334 CrossRef PubMed.
- H. J. Chung and T. G. Park, Adv. Drug Delivery Rev., 2007, 59, 249–262 CrossRef CAS PubMed.
- W. Zhang, C. Zhu, D. Ye, L. Xu, X. Zhang, Q. Wu, X. Zhang, D. L. Kaplan and X. Jiang, PLoS One, 2014, 9, 1–9 Search PubMed.
- B. B. Mandal, A. Grinberg, E. Seok, B. Panilaitis and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7699–7704 CrossRef CAS PubMed.
- F. Ak, Z. Oztoprak, I. Karakutuk and O. Okay, Biomacromolecules, 2013, 14, 719–727 CrossRef CAS PubMed.
- A. M. Collins, N. J. V Skaer, T. Gheysens, D. Knight, C. Bertram, H. I. Roach, R. O. C. Oreffo, S. Von-Aulock, T. Baris, J. Skinner and S. Mann, Adv. Mater., 2009, 21, 75–78 CrossRef CAS.
- A. Nisal, P. Venugopalan and N. Sinha, CSIR-National Chemical Laboratory, WO2014125505 A1, 2014, pp. 1–10.
- A. Nisal, P. Venugopalan and B. Khude, CSIR-National Chemical Laboratory, WO2016110873 A1, 2016, pp. 1–9.
- B. Dhandayuthapani, Y. Yoshida, T. Maekawa and D. S. Kumar, Int. J. Polym. Sci., 2011, 290602 Search PubMed.
- M. Vandrovcová, T. Douglas, D. Hauk, B. Grössner-Schreiber, J. Wiltfang, L. Bačáková and P. H. Warnke, Physiological Research, 2011, 60, 797–813 Search PubMed.
- A. M. Ferreira, P. Gentile, V. Chiono and G. Ciardelli, Acta Biomater., 2012, 8, 3191–3200 CrossRef CAS PubMed.
- M. H. Lee, C. You and K. H. Kim, Materials, 2015, 8, 1150–1161 CrossRef.
- J. Y. Lee, S. H. Nam, S. Y. Im, Y. J. Park, Y. M. Lee, Y. J. Seol, C. P. Chung and S. J. Lee, J. Controlled Release, 2002, 78, 187–197 CrossRef CAS PubMed.
- J. Venkatesan and S. K. Kim, Mar. Drugs, 2010, 8, 2252–2266 CrossRef CAS PubMed.
- A. R. Costa-Pinto, R. L. Reis and N. M. Neves, Tissue Eng., Part B, 2011, 17, 331–347 CrossRef CAS PubMed.
- S. Zeng, L. Liu, Y. Shi, J. Qiu, W. Fang, M. Rong, Z. Guo and W. Gao, PLoS One, 2015, 10, e0128658 Search PubMed.
- R. LogithKumar, A. KeshavNarayan, S. Dhivya, A. Chawla, S. Saravanan and N. Selvamurugan, Carbohydr. Polym., 2016, 151, 172–188 CrossRef CAS PubMed.
- C. Li, C. Vepari, H.-J. Jin, H. J. Kim and D. L. Kaplan, Biomaterials, 2006, 27, 3115–3124 CrossRef CAS PubMed.
- I. F. Amaral, P. Sampaio and M. A. Barbosa, J. Biomed. Mater. Res., Part A, 2006, 76, 335–346 CrossRef CAS PubMed.
- B. Liu, S. Wu, L. Han and C. Zhang, Int. J. Mol. Med., 2015, 1572–1582 CAS.
- M. Yang, Y. Shuai, W. He, S. Min and L. Zhu, Int. J. Mol. Sci., 2012, 13, 7762–7775 CrossRef CAS PubMed.
- C. Pautke, M. Schieker, T. Tischer, A. Kolk, P. Neth, W. Mutschler and S. Milz, Anticancer Res., 2004, 24, 3743–3748 CAS.
- Y. Zhang, M. Ni, M. Zhang and B. Ratner, Tissue Eng., 2003, 9, 337–345 CrossRef CAS PubMed.
- A. Fakhry, G. B. Schneider, R. Zaharias and S. Şenel, Biomaterials, 2004, 25, 2075–2079 CrossRef CAS PubMed.
- X. Yang, X. Chen and H. Wang, Biomacromolecules, 2009, 10, 2772–2778 CrossRef CAS PubMed.
- K. Lee, G. Jin, C. H. Jang, W.-K. Jung and G. Kim, J. Mater. Chem. B, 2013, 1, 5831 RSC.
- S. Sundelacruz and D. L. D. L. Kaplan, Semin. Cell Dev. Biol., 2009, 20, 646–655 CrossRef CAS PubMed.
- C. G. Knight, L. F. Morton, D. J. Onley, A. R. Peachey, J. Messent, P. A. Smethurst, S. Danny, R. W. Farndale, J. Michael, C. G. Knight, L. F. Morton, D. J. Onley, A. R. Peachey, A. J. Messent, P. A. Smethurst, D. S. Tuckwell, R. W. Farndale and M. J. Barnes, J. Biol. Chem., 1998, 273, 33287–33294 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparative data of scaffolds and comparative image of cell morphology. See DOI: 10.1039/c7ra03288a |
| This journal is © The Royal Society of Chemistry 2017 |