
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base catalysed N-functionalisation of boroxazolidones†
J. Raunioa,
J. Mannojaa,
T. Nguyenb,
N. Ahmadb,
N. M. Kemppainena,
R. G. Franzéna,
M. Kandhavelu*b and
N. R. Candeias *a
*a
aLaboratory of Chemistry and Bioengineering, Tampere University of Technology, Korkeakoulunkatu 8, 33101 Tampere, Finland. E-mail: nuno.rafaelcandeias@tut.fi
bMolecular Signalling Lab, TUT-BMT Unit, Tampere University of Technology, P.O.Box 553, 33101 Tampere, Finland. E-mail: meenakshisundaram.kandhavelu@tut.fi
First published on 10th April 2017
Abstract
A method for the condensation of boroxazolidones derived from L-valine with aromatic aldehydes, catalysed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene was developed. The preparation and isolation of a series of highly functionalised stable ketimines derived from the reaction of 2,2-diaryl-1,3,2-oxazaborolidin-5-ones with aryl aldehydes is herein described. Several unreported boroxazolidones were prepared by condensation of triethylammonium tetra-arylborates with L-valine in up to 98% yield. The newly synthesised compounds were determined to be moderately cytotoxic against colorectal adenocarcinoma cells, with the best compound in this series having an IC50 of 76 μM. A brief inspection of the effect of the same compound against human brain astrocytoma cells showed an IC50 of 268 μM.
Introduction
Synthesis of boron compounds derived from biomolecules such as sugars, amino acids, peptides and nucleic acids has been an extensive and foremost area of research in the last two decades.1 Among other subjects, much interest has been devoted to molecules containing boron–nitrogen bonds. These compounds can possess broad biological activity2 such as insecticidal, fungicidal, herbicidal and antibacterial properties.3 The ability of several boron compounds, namely those derived from α-amino acids, to interact with tumour cells has opened new venues for their use as part of boron neutron capture therapy.4 The biological properties of chelates containing N–B bonds have also been reported to include apoptotic activity in tumour cells,5 an ability to disturb calcium channel transporters,6 inhibition of human neutrophile elastase,7 and modulation of human phenylalanine hydroxylase activity.8 Boroxazolidones, a particular kind of N–B bond-containing compounds, generally obtained by reacting α-amino acids with boranes or borinates, were firstly reported in 1962.9 Glycine and L-methionine derivatives were then prepared by the reaction of these amino acids with trialkyl and triaryl boranes in refluxing xylene.9a Since then, several methods for preparation of boroxazolidones with alkyl groups, hydrogens and halogens on boron have been developed. Skoog reported the use of a diaryl alkyl borinate in the condensation with glycine, alanine and leucine10 and later Nefkens described the preparation of several α-amino acid derivatives after reaction with a slight excess of triethylborane or triphenylborane in THF.11 A different procedure, comprising the reaction of α-amino acids with sodium tetraphenylborate in the presence of hydrochloric acid in water was developed by Baum.12 This procedure was then applied to the preparation of boroxazolidones derived from glycine, alanine, phenylalanine, proline, cysteine and tyrosine in moderate to good yields. The preparation of 2,2-diphenyl-boroxazolidones is usually achieved by reaction of the α-amino acid with diphenylborinic acid under basic conditions.5b,13Boroxazolidones have also been employed in organic synthesis. The higher reactivity of the 2,2-dialkyl boroxazolidones towards solvolysis, namely with diluted HCl or refluxing methanol, when compared with the 2,2-diphenyl counterpart, allows these compounds to be used as a double protecting group of amino acids.11,14 Due to the increased solubility of the amino acids with 9-borabicyclononane (9-BBN) in organic solvents, this moiety has been particularly explored as a protecting group.15 Boroxazolidones were also explored as a derivatization procedure to ease the HPLC analysis of α-amino acids.16 The preparation of isoquinoline and isoindoline derivatives,17 asymmetric α-alkylation of α-amino acids using boron as a stereogenic centre,18 and asymmetric hydroboration reactions19 are other transformations where these compounds have been employed. Despite the many reports on preparation of boroxazolidones, and their interesting biological properties, few attempts have been made to functionalize these bench stable compounds. The discovery of new reactions of boroxazolidones would allow the formation of highly functionalized compounds derived from biological molecules, with a vast potential for further transformations (Scheme 1).
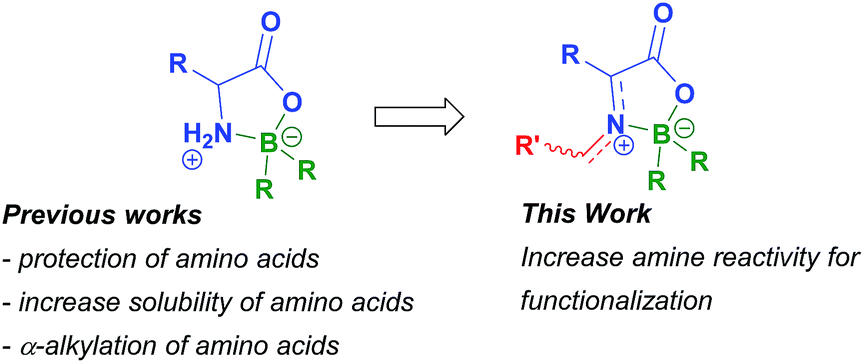 | ||
| Scheme 1 Uses of boroxazolidones in synthetic organic chemistry. | ||
Nefkens and Zwanenburg17 reported that 2,2-diethyl boroxazolidone derived from glycine could be transformed into the correspondent Schiff base in 60% yield upon reaction with benzaldehyde with azeotropic removal of water. On the other hand, boroxazolidones are known to react preferentially with nucleophiles by the boron atom, rather than the carbonyl functionality.20 Interestingly, when attempting the condensation of L-valine derived 2,2-diphenyl-1,3,2-oxazaborolidin-5-one 1a with benzaldehyde, using standard dehydrating procedures,21 we could not obtain the correspondent imine in more than 17% yield. The use of molecular sieves, trimethylorthoformate,22 pyridinium p-toluenesulfonate and magnesium sulphate23 or zinc chloride24 were some of the methods attempted. Taking these observations as a starting point we hypothesized that the nucleophilicity of the nitrogen atom of the boroxazolidone towards aldehydes could be increased upon presence of a catalytic amount of a nucleophile, as this could lead to the momentary disruption of the N–B bond of the boroxazolidone.
Results and discussion
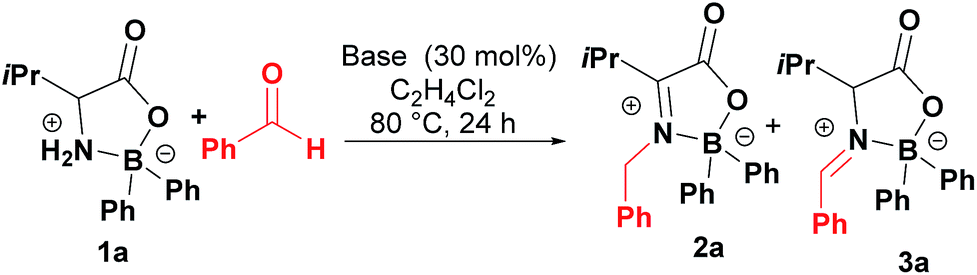
The condensation reaction of boroxazolidone 1a with benzaldehyde, using powdered molecular sieves as dehydrating agent, provided aldimine 3a in only 7% (Table 1, entry 1), which isomerized to ketimine 2a upon treatment with triethylamine. Although in low yield, condensation products 2a and 3a were obtained using DMAP as catalyst (Table 1, entry 2), which prompted us to test other catalysts. Using phosphorus derived Lewis bases led to the formation of aldimine 3a in low yields (Table 1, entries 3–5), while the use of nitrogen bases resulted in equilibration of aldimine 3a to ketimine 2a in better yields (Table 1, entries 6–9). The addition of powdered molecular sieves or increasing the amount of DBU resulted in similar or worse yields (Table 1, entries 10 and 11). Other solvents tested, such as acetonitrile, dimethoxyethane, dioxane, THF, DMF and toluene resulted in formation of ketimine 2a in lower yields. On the other hand, increasing the temperature to refluxing 1,2-dichloroethane and the use of 5 equivalents of benzaldehyde allowed formation of 2a in moderate 46% yield (Table 1, entry 12). Other Brønsted bases such as proton sponge and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) were observed to be better catalysts promoting the formation of condensation product 2a in up to 57% yield (Table 1, entries 13 and 14).
|
|||
|---|---|---|---|
| Entry | Base | Conversionb (%) | |
| 2a | 3a | ||
| a Reaction conditions: 1a (0.5 mmol), benzaldehyde (1.5 equiv.); base (0.3 equiv.), C2H4Cl2 (1.5–2.0 mL), 80 °C, 24 h.b Determined by 1H NMR of reaction mixture.c With powdered molecular sieves 4 Å, 70 °C.d Isolated yields.e With powdered molecular sieves 4 Å.f 1 equiv. of DBU.g Reflux.h 5 equiv. of benzaldehyde. | |||
| 1 | Nonec | — | 7d |
| 2 | DMAP | 6 | 12 |
| 3 | HMPA | — | 9 |
| 4 | PPh3 | — | 13 |
| 5 | DPPE | — | 14 |
| 6 | TEA | 25 | 3 |
| 7 | DABCO | 6 | 6 |
| 8 | DIPEA | 20 | 2 |
| 9 | DBU | 27d | — |
| 10 | DBUe | 25d | — |
| 11 | DBUf | 11d | |
| 12 | DBUg,h | 46d | — |
| 13 | Proton spongeg,h | 53d | — |
| 14 | TBDg,h | 57d | — |
Taking boroxazolidone 1a as starting material, the reaction conditions were applied to the condensation reaction with several other aromatic aldehydes, affording the corresponding products in reasonable yields (Table 2). Condensation of 1a with para-bromo and fluoro-substituted aldehydes afforded the corresponding ketimines 2b and 2c in moderate 60% yield. Trifluoromethyl-substituted benzaldehydes were also successful partners for condensation with 1a, leading to the formation of products 2d and 2e. The reaction seems to be sensitive to steric hindrance, as the decoration of benzaldehyde with the trifluoromethyl group in the ortho position resulted in isolation of the desired compound 2f in only 8% yield. Other functionalities prone for further modification of the products such as nitro, nitrile and esters could also be introduced by this method in up to 43% yields (2g–2i). Electron rich aldehydes such as anisaldehyde were reasonably good partners, affording product 2j in 40% yield.
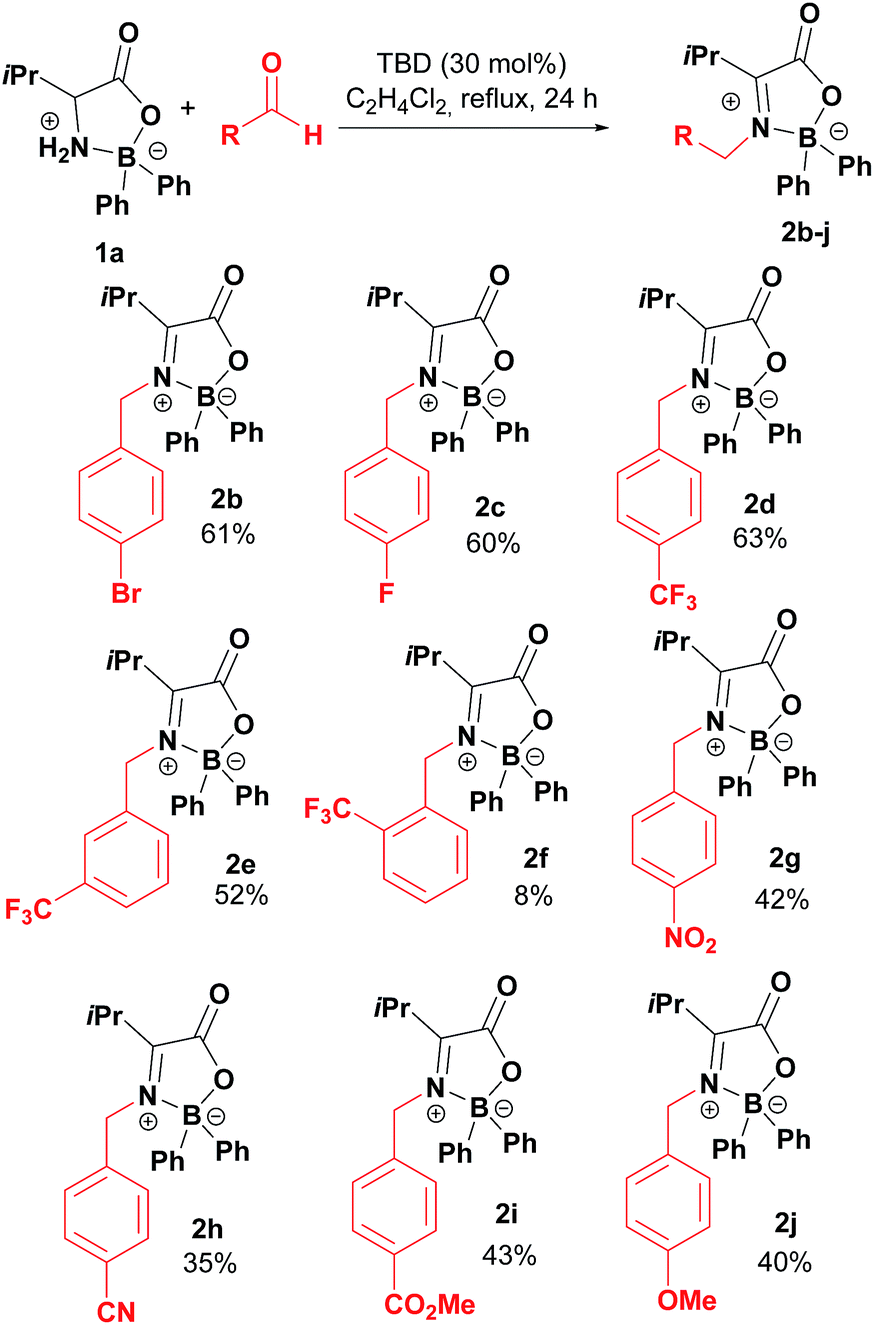 |
The method used for preparation of 1a was then extended for preparation of several L-valine derived boroxazolidones (Scheme 2) to be further coupled with benzaldehyde. Preparation of 2,2-diaryl-1,3,2-oxazaborolidin-5-ones was achieved by first preparation of the triethylammonium tetra-arylborate as previously reported,25 followed by condensation of the ammonium salt with L-valine in overnight refluxing toluene in excellent yields. Generally, the desired compounds were purified by precipitation or recrystallization, overcoming the use of chromatography.
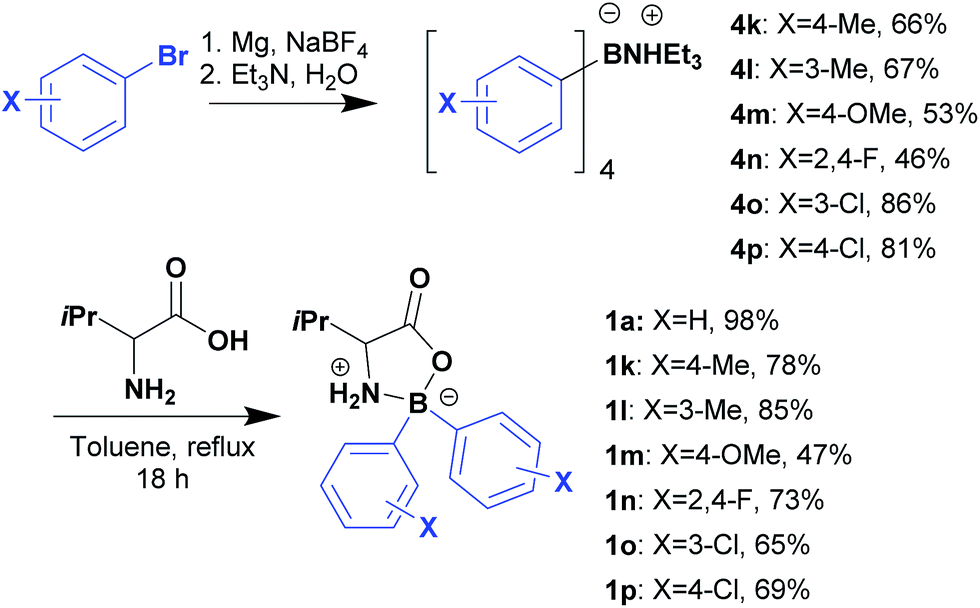 | ||
| Scheme 2 Preparation of boroxazolidones. | ||
Ketimines 2k–2p were prepared in reasonable yields upon reflux in DCE in presence of 5 equivalents of benzaldehyde for 24 h, using TBD or DBU as catalysts (Table 3). Notably, decoration of the boron aryl ring with electron rich methoxy groups led to the formation of condensation product 2m in only 29% yield, whilst halogens or methyl substituents at different positions had little impact in the condensation process.
| a 30 mol% DBU used as base. |
|---|
 |
Antitumour activity
Preliminary bioactive assays on the newly synthesised ketimines derived from boroxazolidones were performed, impelled by the recent interest of medicinal chemists on the introduction of boron for discovery of new drugs,26 and the previously reported apoptotic activity of boroxazolidones.5 The cytotoxic effect of these compounds was evaluated against human epithelial colorectal adenocarcinoma (CACO2) cell line. A smaller set of boroxazolidones was considered for the sake of comparison. The limited solubility of the compounds in DMSO hampered the extension of the biological assay to every ketimine-derived boroxazolidones. Boroxazolidones 1a, 1k, 1l–m and ketimines derived boroxazolidones 2a–d, 2g–i and 2l–n were determined to induce the mortality of CACO2 cells at some extent (Fig. 1). From all compounds tested, 2d showed a slightly higher cytotoxic effect, inducing 50% mortality of CACO2 cells upon treatment with 100 μM solutions. However, the similar profile observed for both families of compounds seem to suggest a modest class effect, as antineoplastic activity was previously reported for boroxazolidones.5 Furthermore, the slightly higher cytotoxic effect observed for boroxazolidones 1l–n (30–46%) compared with the corresponding boroxazolidones derived ketimines 2l–n (20–37%) might be due to their conversion into the starting boroxazolidone. However, this is not corroborated by the higher cytotoxic effect observed for ketimines 2b–d (45–50%) and 2h–i (40%) when compared with their synthetic precursor 1a (32%). A weaker mortality effect than the one caused by sodium orthovanadate (33%) was observed for 1l (30%), 2g (26%) and 2l (20%). | ||
| Fig. 1 Effects of ketimines derived from boroxazolidones, DMSO and sodium orthovanadate (PC) on CACO2 cells mortality (%) at 100 μM. | ||
Among the compounds tested, 2d showed the best cytotoxicity effect. This compound was selected for further dynamic test on CACO2 and 1321N1 (human brain astrocytoma) cell lines at various concentrations. Fig. 2 shows the cytotoxicity effect of 2d on both cell lines in a dose-dependent manner, from where the IC50 value against CACO2 cells was calculated as 76 ± 2.7 μM and 268 ± 4.4 μM for 1321N1.
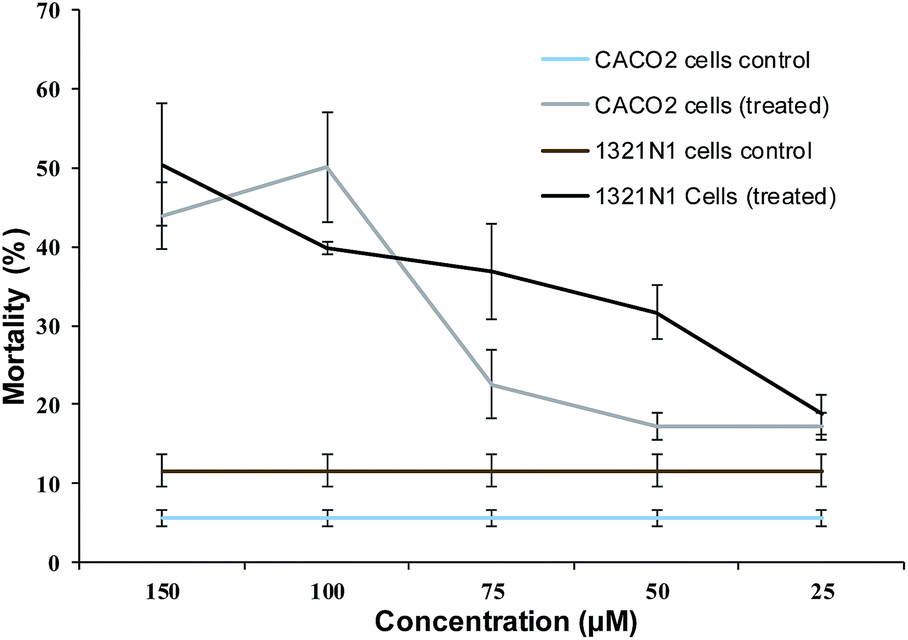 | ||
| Fig. 2 Dose-dependent effect of lead compound: the effect of compound 2d at different concentrations and DMSO on the mortality of CACO2 and 1321N1 cells. | ||
Conclusion
In this study we report the preparation of highly functionalized ketimines derived from boroxazolidones. The lack of nucleophilicity of boroxazolidones derived from L-valine can be overcome by the use of catalytic amounts of 1,5,7-triazabicyclo[4.4.0]dec-5-ene and super-stoichiometric amounts of the aryl aldehydes in refluxing 1,2-dichloroethane. Despite the known stability of the N–B bond dependency on boron's substituents, 2,2-diaryl-1,3,2-oxazaborolidin-5-ones of different electronic natures on the aryl moiety could be condensed with benzaldehyde in reasonable yields. The newly synthesized ketimines showed modest antitumour activity against colorectal adenocarcinoma cells and lower activity against human brain astrocytoma cells, making this one more entry to the growing array of biological properties of N–B bond containing compounds. The continuation of this work will focus on the expansion of the reaction scope to other amino acid derived boroxazolidones.Experimental
General information
All reagents were obtained from Sigma-Aldrich or TCI and were used without further purification. The reactions were performed under argon atmosphere and monitored by thin-layer chromatography carried out on pre-coated (Merck TLC silica gel 60 F254) aluminium plates by using UV light as visualizing agent and cerium molybdate solution or ninhydrin as developing agents. Flash column chromatography was performed on silica gel 60 (Merck, 0.040–0.063 mm). NMR spectra were recorded with Varian Mercury 300 MHz instrument using CDCl3, DMSO-d6 or acetone-d6 as solvents and calibrated using tetramethylsilane as internal standard. Chemical shifts are reported in ppm relative to TMS and coupling constants are reported in Hz. High resolution mass analysis (ES, positive or negative) was determined on a WatersSynapt G1.Preparation of triethylammonium tetra-arylborates 4
Compound 4a was obtained from commercially available sodium tetraphenylborate, while compounds 4m, 4o and 4p were obtained from addition of arylmagnesium bromide to NaBF4 as previously reported.25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:200 Na2CO3/NaOH/water solution while stirring. After organic layer separation, the water layer was saturated with NaCl and extracted with diethyl ether (4 × 60 mL). The combined organic layers were washed with brine (100 mL) and concentrated under vacuum. The resulting solid was dissolved in a 1:1 MeOH/water solution (300 mL) and filtered through celite to get a clear solution. A solution of 10 wt% aqueous triethylamine (68.1 mmol, 2.6 eq.) with MeOH (7:1) (80 mL) was slowly added to the solution, and the solution stirred for 2.5 h, yielding the triethylammonium tetra(4-methylphenyl)borate as a white precipitate that was filtered and washed with hexane and water. White solid (8.3 g, 66% yield); 1H NMR (300 MHz, acetone-d6) δ 7.20–7.25 (m, 8H), 6.74 (d, J = 9.0 Hz, 8H), 3.32 (quar, J = 7.0 Hz, 6H), 2.17 (s, 12H), 1.33 (t, J = 7.5 Hz, 9H) ppm; 13C NMR (75 MHz, acetone-d6) δ 161.2 (quar, JBC = 49.0 Hz), 136.4 (quar, JBC = 1.5 Hz), 129.4, 126.1 (quar, JBC = 2.8 Hz), 47.4, 20.6, 8.8 ppm. HRMS (ESI−): calcd for C28H28B [(M − Et3NH)−]: 375.2284; found: 375.2284.
:1:200 Na2CO3/NaOH/water solution while stirring. After organic layer separation, the water layer was saturated with NaCl and extracted with diethyl ether (4 × 60 mL). The combined organic layers were washed with brine (100 mL) and concentrated under vacuum. The resulting solid was dissolved in a 1:1 MeOH/water solution (300 mL) and filtered through celite to get a clear solution. A solution of 10 wt% aqueous triethylamine (68.1 mmol, 2.6 eq.) with MeOH (7:1) (80 mL) was slowly added to the solution, and the solution stirred for 2.5 h, yielding the triethylammonium tetra(4-methylphenyl)borate as a white precipitate that was filtered and washed with hexane and water. White solid (8.3 g, 66% yield); 1H NMR (300 MHz, acetone-d6) δ 7.20–7.25 (m, 8H), 6.74 (d, J = 9.0 Hz, 8H), 3.32 (quar, J = 7.0 Hz, 6H), 2.17 (s, 12H), 1.33 (t, J = 7.5 Hz, 9H) ppm; 13C NMR (75 MHz, acetone-d6) δ 161.2 (quar, JBC = 49.0 Hz), 136.4 (quar, JBC = 1.5 Hz), 129.4, 126.1 (quar, JBC = 2.8 Hz), 47.4, 20.6, 8.8 ppm. HRMS (ESI−): calcd for C28H28B [(M − Et3NH)−]: 375.2284; found: 375.2284.Preparation of 2,2-diaryl-1,3,2-oxazaborolidin-5-ones 1
:3). White solid (73% yield); 1H NMR (300 MHz, acetone-d6) δ 7.23–7.32 (m, 2H), 6.78–6.99 (m, 4H), 5.93 (br. s, 1H), 3.85–3.92 (m, 1H), 2.37–2.46 (m, 1H), 1.12 (d, J = 6.0 Hz, 3H), 1.08 (d, J = 6.0 Hz, 3H) ppm; 13C NMR (75 MHz, acetone-d6) δ 172.3, 166.21 (dd, JCF = 196.1, 11.6 Hz), 166.18 (dd, JCF = 195.0, 12.0 Hz), 162.99 (dd, JCF = 200.6, 11.6 Hz), 162.95 (dd, JCF = 199.1, 12.4 Hz), 135.60–136.01 (m), 110.8 (t, JCF = 3.8 Hz), 110.5 (t, JCF = 3.4 Hz), 102.68 (dd, JCF = 29.6, 24.4 Hz), 102.65 (dd, JCF = 29.3, 24.8 Hz), 60.77, 60.75, 18.2, 17.3, 17.2 ppm. HRMS (ESI+): calcd for C17H17BF4NO2 [(M + H)+]: 354.1288; found: 354.1287.Condensation of 2,2-diaryl-1,3,2-oxazaborolidin-5-ones with aryl aldehydes
Cell culture
Human colorectal adenocarcinoma (CACO2, a kind gift from Prof. Gabriella Marucci, University of Camerino, Italy) and human brain astrocytoma (1321N1, 86030402 SIGMA, sigma-Aldrich) cell lines were used for screening the activity of all compounds described in Fig. 1. CACO2 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) with the addition of 10% heat inactivated Fetal Bovine Serum (FBS), 2 mM sodium pyruvate, 0.1 mM non-essential amino acids (NEAA) solution (Sigma-Aldrich, St. Louis, MO) and antibiotic supplements: 0.1 mg mL−1 streptomycin, 100 U mL−1 penicillin and 0.025 mg mL−1 amphotericin B. 1321N1 cell line was cultured in the same culture medium as CACO2 but without the supplement of NEAA. These two cell lines were maintained in the incubator with humidified condition at 37 °C and 5% CO2.In vitro cytotoxicity assay
At first, the cytotoxicity effect of compounds reported in Fig. 1 were studied on CACO2. For this, cells were seeded on 12 well plates with the density 1 × 105 cell per well. After 24 hours the cells were treated with 100 μM concentration of the compounds of interest. Sodium orthovanadate and dimethyl sulfoxide (DMSO) were used as positive control (PC) and negative control, respectively. Treated cells were collected using trypsin followed by the centrifugation at 3000 rpm in 7 min. Cells were then ressuspended in completed culture medium and staining dye trypan blue at the ratio 1:1. To determine the number of live and dead cell, a Bürker hemocytometer (Heinz Herenz, Hamburg, Germany) was used. Biological and technical repeats were conducted to obtain the final results. The percentage of mortality for each sample was calculated according to the following equation: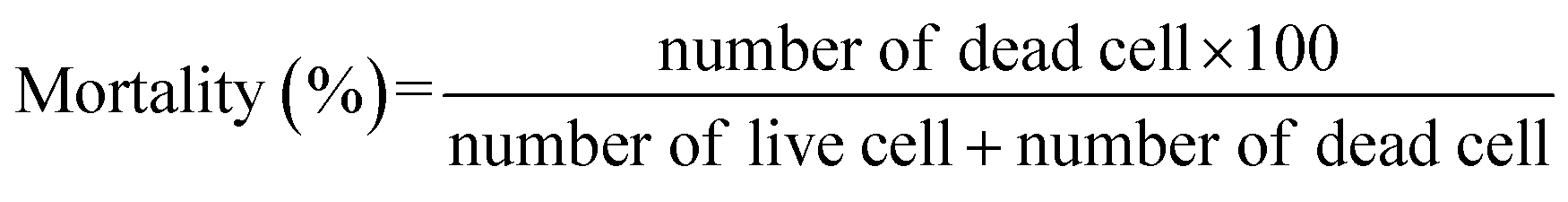
Dynamic assay
The lead compound 2d was identified from cytotoxicity assay, which showed the highest cytotoxicity effect. Further test was carried at different concentrations of 2d (150 μM, 100 μM, 75 μM, 50 μM and 25 μM) to confirm a dose-dependent effect on CACO2. To validate the activity of this compound on another tumour cell, 1321N1 was also used. The percentage of mortality was calculated as described above. The dose–response curve was plotted by using GraphPad software and the half maximal inhibitory concentration (IC50) was calculated.Acknowledgements
NRC acknowledges the Academy of Finland for the Academy Research Fellowship (Decisions No. 287954 and 294067). We acknowledge Mrs Päivi Joensuu (University of Oulu) for analysing the HRMS data.Notes and references
-
(a) P. Andres, G. Ballano, M. I. Calaza and C. Cativiela, Chem. Soc. Rev., 2016, 45, 2291–2307 RSC
; (b) S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC
- A. Jabbour, R. Smoum, K. Takrouri, E. Shalom, B. Zaks, D. Steinberg, A. Rubinstein, I. Goldberg, J. Katzhendler and M. Srebnik, Pure Appl. Chem., 2006, 78, 1425–1453 CrossRef CAS
-
(a) V. M. Dembitsky and M. Srebnik, Tetrahedron, 2003, 59, 579–593 CrossRef CAS
-
(a) W. K. George and Y. Min-Liang, Anti-Cancer Agents Med. Chem., 2006, 6, 111–125 CrossRef
-
(a) B. Velasco, J. G. Trujillo-Ferrara, L. H. F. Castillo, R. Miranda and L. E. Sánchez-Torres, Life Sci., 2007, 80, 1007–1013 CrossRef CAS PubMed
-
(a) Y. Dobrydneva, C. J. Abelt, B. Dovel, C. M. Thadigiri, R. L. Williams and P. F. Blackmore, Mol. Pharmacol., 2006, 69, 247–256 CAS
- F. Montalbano, P. M. S. D. Cal, M. A. B. R. Carvalho, L. M. Goncalves, S. D. Lucas, R. C. Guedes, L. F. Veiros, R. Moreira and P. M. P. Gois, Org. Biomol. Chem., 2013, 11, 4465–4472 CAS
- F. Montalbano, J. Leandro, G. D. V. F. Farias, P. R. Lino, R. C. Guedes, J. B. Vicente, P. Leandro and P. M. P. Gois, RSC Adv., 2014, 4, 61022–61027 RSC
-
(a) K. Lang, K. Nuetzel and F. Schubert, Germany Pat., 1130445, 1962
- I. H. Skoog, J. Org. Chem., 1964, 29, 492–493 CrossRef CAS
- G. H. L. Nefkens and B. Zwanenburg, Tetrahedron, 1983, 39, 2995–2998 CrossRef CAS
- G. Baum, J. Organomet. Chem., 1970, 22, 269–271 CrossRef CAS
-
(a) J. Trujillo, H. Höpfl, D. Castillo, R. Santillan and N. Farfán, J. Organomet. Chem., 1998, 571, 21–29 CrossRef CAS
- F. Albericio, E. Nicolás, J. Rizo, M. Ruiz-Gayo, E. Pedroso and E. Giralt, Synthesis, 1990, 119–122 CrossRef CAS
-
(a) W. H. Dent, W. R. Erickson, S. C. Fields, M. H. Parker and E. G. Tromiczak, Org. Lett., 2002, 4, 1249–1251 CrossRef CAS PubMed
-
(a) R. Flückiger, E. Henson, G. M. Hess and P. M. Gallop, Biol. Mass Spectrom., 1984, 11, 611–615 CrossRef PubMed
- G. H. L. Nefkens and B. Zwanenburg, Tetrahedron, 1985, 41, 6063–6066 CrossRef CAS
-
(a) E. Vedejs, S. C. Fields and M. R. Schrimpf, J. Am. Chem. Soc., 1993, 115, 11612–11613 CrossRef CAS
- H. C. Brown and A. K. Gupta, J. Organomet. Chem., 1988, 341, 73–81 CrossRef CAS
- B. Garrigues and M. Mulliez, J. Organomet. Chem., 1986, 314, 19–24 CrossRef CAS
-
(a) R. D. Patil and S. Adimurthy, Asian J. Org. Chem., 2013, 2, 726–744 CrossRef CAS
- G. C. Look, M. M. Murphy, D. A. Campbell and M. A. Gallop, Tetrahedron Lett., 1995, 36, 2937–2940 CrossRef CAS
- B. P. Branchaud, J. Org. Chem., 1983, 48, 3531–3538 CrossRef CAS
- J. H. Billman and K. M. Tai, J. Org. Chem., 1958, 23, 535–539 CrossRef CAS
- N. M. Kuuloja, T. M. Kylmälä, J. E. Tois, R. E. Sjöholm and R. G. Franzén, Synth. Commun., 2011, 41, 1052–1063 CrossRef CAS
-
(a) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed
- K. E. Bessler, E. B. Dourado, M. de S. Carvalho, S. S. Lemos and J. Ellena, Z. Anorg. Allg. Chem., 2005, 631, 1935–1940 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra of all new compounds. See DOI: 10.1039/c7ra03266h |
| This journal is © The Royal Society of Chemistry 2017 |