
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic approaches and in vitro cytotoxic evaluation of 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinones
Jaime A. Valderrama *ab,
Mónica Cabrerab,
Julio Benites*ab,
David Ríosb,
Ricardo Inostroza-Riverab,
Giulio G. Mucciolic and
Pedro Buc Calderonbd
*ab,
Mónica Cabrerab,
Julio Benites*ab,
David Ríosb,
Ricardo Inostroza-Riverab,
Giulio G. Mucciolic and
Pedro Buc Calderonbd
aInstituto de Ciencias Exactas y Naturales (ICEN), Universidad Arturo Prat, Casilla 121, Iquique 1100000, Chile. E-mail: jaimeadolfov@gmail.com; juliob@unap.cl
bFacultad de Ciencias de la Salud, Universidad Arturo Prat, Casilla 121, Iquique 1100000, Chile
cBioanalysis and Pharmacology of Bioactive Lipids Laboratory, Louvain Drug Research Institute, Université catholique de Louvain, 72 Avenue E. Mounier, BPBL 7201, 1200 Brussels, Belgium
dMetabolism and Nutrition Research Group, Louvain Drug Research Institute, Université catholique de Louvain, 73 Avenue E. Mounier, GTOX 7309, 1200 Brussels, Belgium
First published on 10th May 2017
Abstract
2-Acyl-1,4-naphthoquinones react with 3,4,5-trimethoxyaniline, under aerobic conditions, to give benzophenanthridinequinone, benzocarbazole and 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinone derivatives. The formation of the heterocyclic compounds is discussed in terms of the ring closure of C–C Michael type adduct intermediates through two alternative N–C-bond formations. The propensity of the substrates to undergo preferential C–C instead of C–N bond formation and the further heterocyclization of the C–C Michael type adduct intermediates is rationalized by using product stability parameters assessed by DFT calculations. Preliminary results are reported on a convenient access towards 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinones from 2-acylnaphthoquinones and their cytotoxic activities on cancer cells.
Introduction
Acylated 1,4-quinones are useful precursors of a variety of natural1–4 and synthetic compounds endowed with a range of biological properties.5–13 The 2-acyl-1,4-benzo- and 1,4-naphthoquinones exhibit remarkable features in terms of their reactivity with nucleophiles due to the fact that the electrophilic centers at the quinone nucleus and acyl substituent are suitably located, thus enabling reactions with compounds such as arylamines14–16 azaenamines17 enaminones18,19 and 2-aminobenzothiazoles20 to give rise to a broad variety of quinonoid compounds such as those depicted in Fig. 1.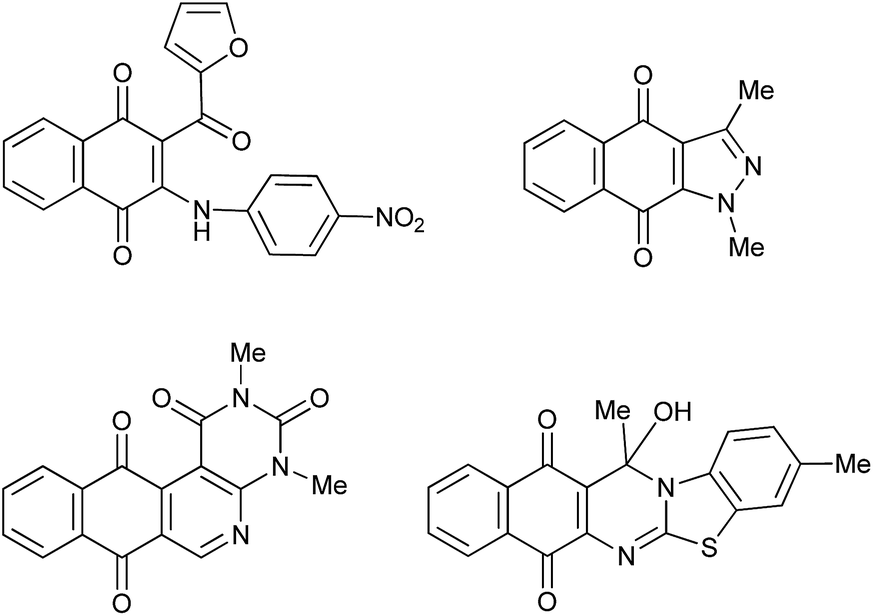 | ||
| Fig. 1 Quinones prepared from acylquinones and nucleophiles. | ||
We have previously reported that a series of 2-acyl-3-anilino-1,4-naphthoquinones possess biological properties such as inhibitors of Hsp90 chaperoning function,16 cytotoxic actions on cancer cells21 and DNA-intercalants.22 These compounds, prepared by amination reaction of 2-acyl-1,4-naphthoquinone with diverse p-substituted anilines under aerobic conditions,21 were originally designed as inhibitors of Hsp90 chaperone and their structures are based on that of the Hsp90 inhibitor, 2-acetyl-3-phenyl-1,4-naphthoquinone (HTS1).23,24
As a continuation of our work on biological active 2-acyl-3-arylamino-1,4-naphthoquinones, we are interested to expand the series to new members containing di- and tri-substituted anilino fragments in order to further structure–activity relationships studies. Based on previous results on the synthesis of arylaminoquinones15,16 we envisaged achieve new members by reaction of 2-acylnaphthoquinones with di- and tri-substituted anilines. As far as we know the only report in the literature regarding the reactivity of 2-acyl-1,4-naphthoquinones with disubstituted anilines was made by Pardo et al.,15 by using 2-acetyl-1,4-naphthoquinone 2a. According to these authors, the reaction of 2a with disubstituted anilines take place to give a variety of products derived from 2-acetyl-3-anilino-1,4-naphthoquinone, 2-acetyl-3-aminophenyl-1,4-naphthoquinone and benzophenanthridine-7,12-quinone. Based on these precedents, it seems to us that the access to 2-acyl-3-anilino-1,4-naphthoquinone derivatives by amination of 2-acyl-1,4-naphthoquinones with di- and tri-substituted anilino groups could have some limitations. To evaluate the feasibility to synthesize 2-acyl-3-anilino-1,4-naphthoquinones, substituted at the anilino group, we examined the behavior of the highly and symmetrically substituted 3,4,5-trimethoxyaniline (TMA) with a representative series of 2-acyl-1,4-naphthoquinones, as reaction models. Herein we report the results of our study, which reveals that TMA exhibits ambident nucleophilic character, acting either as nitrogen or carbon nucleophile with 2-acyl-1,4-naphthoquinones, to give hetero-annulation compounds as the main products, together with the respective amination product named 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinones. We also report preliminary results on a convenient access to 2-acyl-3-(3,4,5-trimetoxyanilino)-1,4-naphthoquinones from 2-acyl-3-anilino- and 2-acyl-3-(4-nitroanilino)-1,4-naphthoquinones and the in vitro cytotoxic activities of the new 2,3-disubstituted-1,4-naphthoquinones on normal fibroblasts and on a panel of three cancer cell lines.
Results and discussion
The reactivity of TMA with 2-acetyl-1,4-naphthoquinones 2a and 2d were firstly examined. Acylquinone 2a, prepared by oxidation of acylhydroquinone 1a with silver(I) oxide, was reacted with TMA in methanol at room temperature for 24 h. Column chromatography of the crude provided benzophenanthridinequinone 3a in 80% yield together with minor amounts of aminoquinone 4a (11%; Scheme 1). Interestingly, the reaction of TMA with 2-benzoyl-1,4-naphthoquinone 2d, prepared from 1d and conducted under the above mentioned conditions, yielded aminoquinone 4d (34%) accompanied by a yellow solid substance.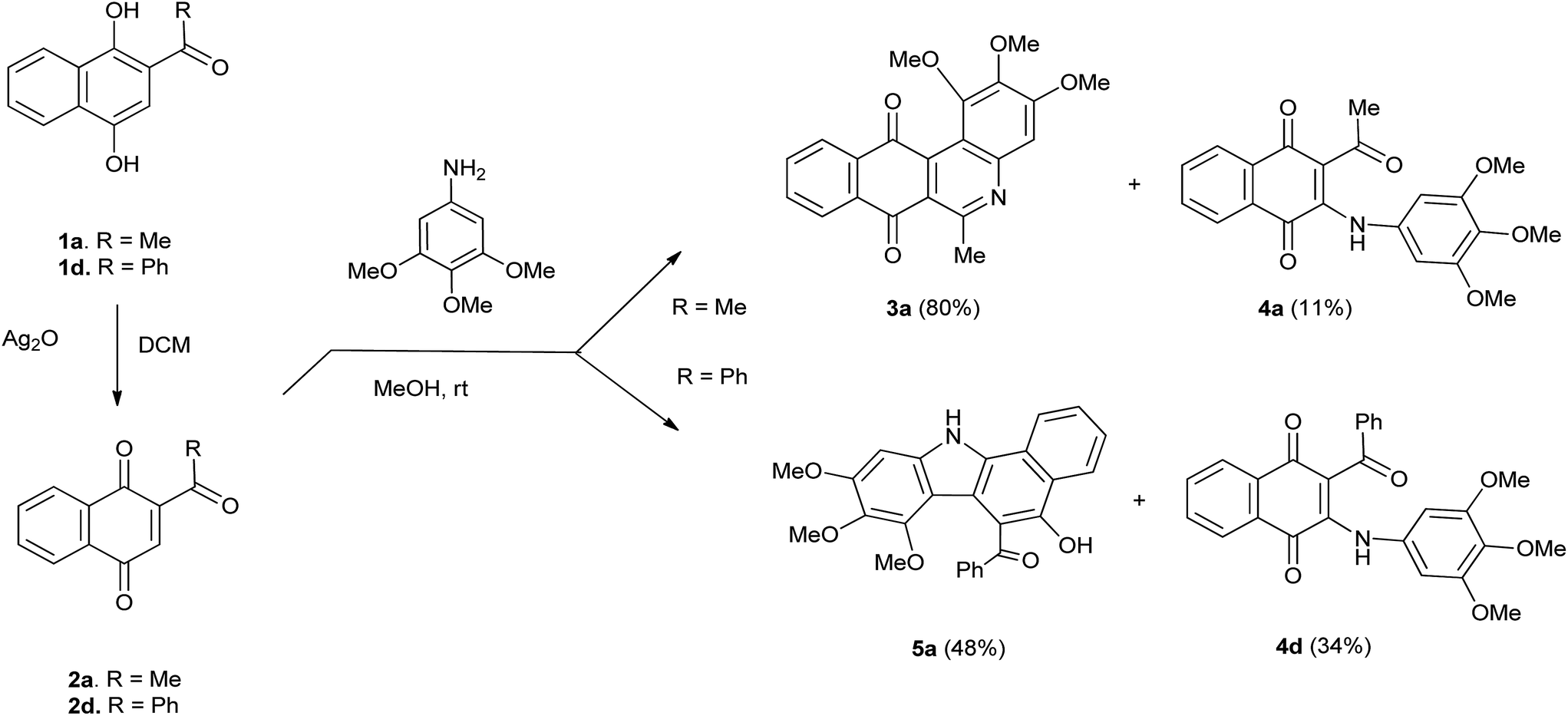 | ||
| Scheme 1 Reaction of trimethoxyaniline with acylquinones 2a and 2d. | ||
The IR spectrum of this product reveals the presence of O–H, N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands at νmáx 3382, 2935 and 1673 cm−1. The 1H NMR spectrum shows hydroxyl and amino proton signals at δ 9.66 (s) and 9.38 (br.s). In the aromatic region it was observed the aromatic pattern signals of two phenyl fragments and a proton signal at δ 6.71 (s). The 13C NMR spectrum displays the characteristic signal of a carbonyl group at δ 198.1 and the mass spectrum shows the molecular ion [M+] peak at m/z = 428.14814. These data are in agreement with the structure of benzocarbazole 5a for the new product, isolated in 48% yield (Scheme 1).
O bands at νmáx 3382, 2935 and 1673 cm−1. The 1H NMR spectrum shows hydroxyl and amino proton signals at δ 9.66 (s) and 9.38 (br.s). In the aromatic region it was observed the aromatic pattern signals of two phenyl fragments and a proton signal at δ 6.71 (s). The 13C NMR spectrum displays the characteristic signal of a carbonyl group at δ 198.1 and the mass spectrum shows the molecular ion [M+] peak at m/z = 428.14814. These data are in agreement with the structure of benzocarbazole 5a for the new product, isolated in 48% yield (Scheme 1).
Based on evidences reported in literature on the use of Lewis acids to promote the oxidative amination reaction of quinones with arylamines,24 we examined their influence on the reaction of TMA with quinones 2a and 2d, seeking to facilitate the formation of aminoquinones 4a and 4d with respect to the respective heterocycles 3a and 5a. The assays, performed in the presence of CeCl3·7H2O and InBr3, demonstrate that these Lewis acids do not show significant changes in the products ratio relative to those obtained in their absence.
To get more information about these preliminary reactivity assays, we examined the scope of the reaction of TMA with 2-alkanoyl- and 2-aroyl-1,4-naphthoquinone 2b, 2c, 2e–g. These quinones, prepared from their corresponding acylhydroquinones 1b, 1c, 1e–g, were reacted with TMA under the standard conditions and the results of the assays are summarized in Table 1.
|
||||
|---|---|---|---|---|
| R | Quinone | Product 3a (yield %) | Product 4a (yield %) | Product 5a (yield %) |
| a Isolated by column chromatography. | ||||
| Methyl | 2a | 3a (80) | 4a (11) | — |
| Propyl | 2b | 3b (47) | 4b (40) | — |
| Heptyl | 2c | 3c (59) | 4c (23) | — |
| Phenyl | 2d | — | 4d (34) | 5a (48) |
| Fur-2-yl | 2e | — | 4e (45) | 5b (53) |
| Thien-2-yl | 2f | — | 4f (45) | 5c (51) |
| Thien-3-yl | 2g | — | 4g (27) | 5d (58) |
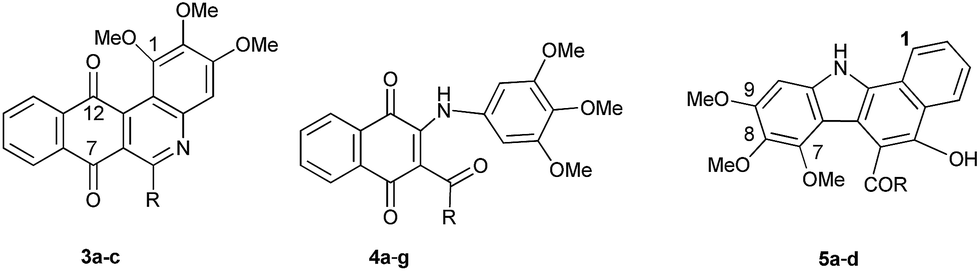
The data in Table 1 suggest that the reactions of TMA with 2-acylnaphthoquinones take place by a stepwise process that involves the formation of carbon–carbon (C–C) and carbon–nitrogen (C–N) bonds to produce benzophenanthridines 3, 2-acyl-3-trimethoxyanilinonaphthoquinones 4 and benzocarbazoles 5. Our results are in agreement with those reported by Pardo et al.,15 on the reactions of acetylnaphthoquinone 2a with disubstituted anilines in terms of the formation of compounds 3 and 4. However, the formation of benzocarbazoles 5 constitutes a new and interesting precedent on the reaction of acylnaphthoquinones with substituted anilines. The formation of benzocarbazoles 5 is a new example of the Nenitzescu reaction of 1,4-naphthoquinones with aromatic amines to produce complex 5-hydroxyindole derivatives.25–27 From these assays, it can also be deduced that the alkyl or aryl nature of the quinone acyl group determines the reaction pathways to the formation of the corresponding benzophenanthridine or benzocarbazole derivatives. The above experiments reveals that TMA behaves both as a carbon and nitrogen nucleophile toward the highly electrophilic acylquinones.
Taking into the account the above results, a reasonable mechanism for the formation of compounds 3, 4 and 5 from TMA and acylquinones 2a–g is proposed and illustrated in Scheme 2. Initially, it seems plausible that the attack of the amine to the acylquinones gives rise to C–C and C–N Michael adduct intermediates I1 and I2, through two parallel reactions. The aza-Michael intermediate I1 will evolve to the corresponding amination products 4a–g by enolization followed by oxidation. Regarding the heterocycles 3a–c and 5a–d, it seems plausible that they arise from I2 (R = alkyl, aroyl), through two alternative cyclodehydration reactions involving amino and carbonyl groups.
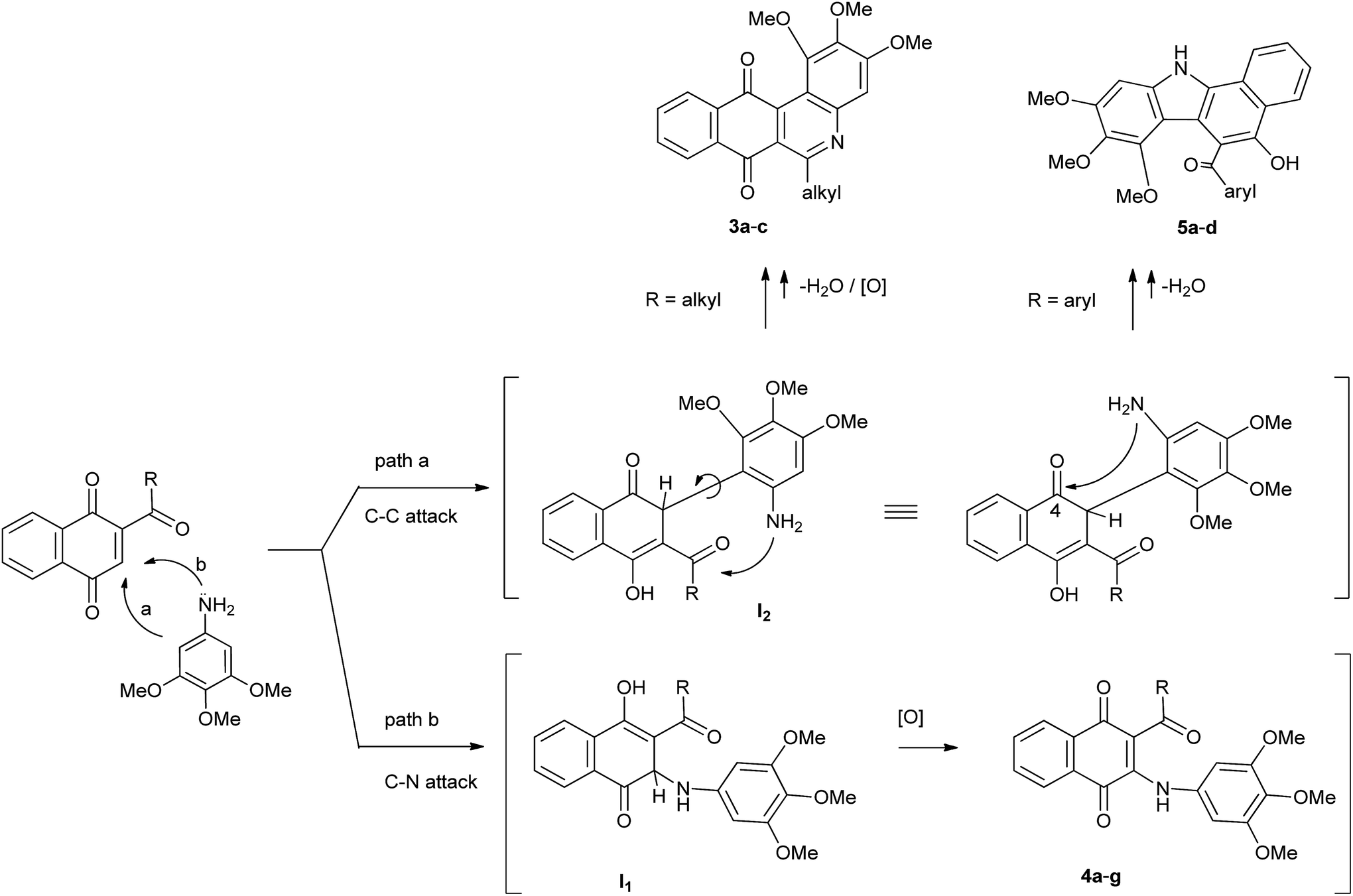 | ||
| Scheme 2 Plausible mechanism for the reaction of TMA with acylquinones 2a–g. | ||
Based on the proposed stepwise mechanism we decided to get theoretical information to rationalize the experimental results. To this end, the energies of the hypothetical intermediates I1 and I2, relative to that of 2a,d + TMA, and the HOMO–LUMO gaps of the products 3, 4 and 5 were evaluated by using Density Functional Theory (DFT) calculations.28,29 The analysis of the relative energies of intermediates I1 and I2 (R = Me, Ph) indicates that formation of the latter is more favorable than I1 (Fig. 2). As a consequence, there is a significant selectivity for the nucleophilic attack of TMA, through C-2, to the acylquinones 2a and 2d in this stepwise mechanism, in agreement with the experimental outcome.
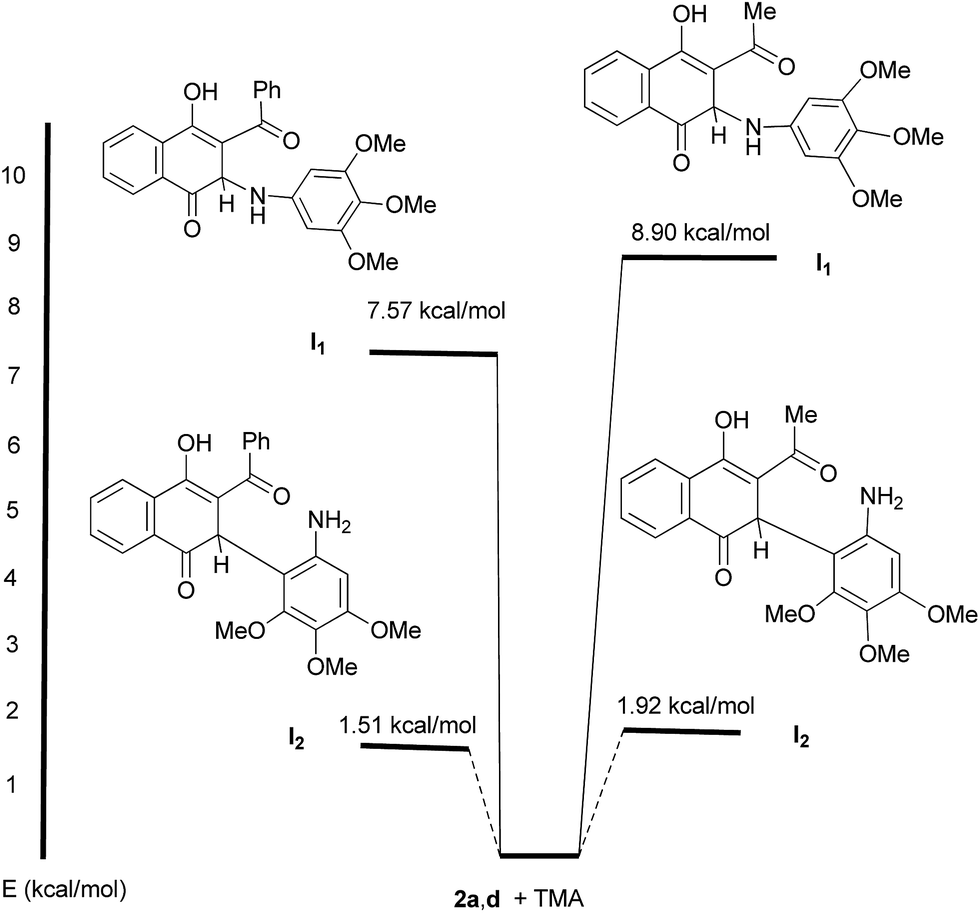 | ||
| Fig. 2 Energies of intermediates I1 and I2 related to those of 2a,d + TMA. | ||
Additionally, calculations of the HOMO–LUMO gaps of products 3a, 4a, 4d and 5a indicate that formation of 3a (3.16 eV) and 5a (3.27 eV) are energetically more favorable than that of the corresponding amination products 4a (3.06 eV) and 4d (2.91 eV). These theoretical results are in good agreement with the experimental evidences in terms that the C–C bond formation appears as a more favorable process than C–N bond formation.
Regarding the second step of the proposed mechanism for the reaction of TMA with acylquinones 2a–g, this involves the 5- and 6-endo-trig closures of intermediates I2 (R = alkyl or aryl) to give the respective heterocycles 3 and 5 (Scheme 2). The behavior of intermediates I2 (R = alkyl or aryl) to undergo these two alternative cyclization processes probably is controlled by the electrophilic capacity of carbonyl groups of the acyl group and that located at the 4-position.
Although the direct amination of TMA with acylnaphthoquinones does not offer a convenient synthetic approach to 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinones 4, it would appear that these compounds may be accessed, albeit more circuitously, by transamination reaction of TMA with 2-acyl-3-anilino-1,4-naphthoquinones. The possibility to apply this strategy to the synthesis of quinones 4 is based on literature precedents on transamination reaction involved in the synthesis of 2,l-benzisoxazolequinones from 2,5-dianilino-3-acetyl-1,4-benzoquinone and hydroxylamine.30 To check the viability of the designed strategy (Scheme in Table 2), the reactions of TMA with 2-acetyl-3-anilino-, 2-butyroyl-3-anilino- and 2-benzoyl-3-anilino-1,4-naphthoquinone 6a–c were examined. Precursors 6a–c were prepared from 2-acyl-1,4-naphthoquinone 2a, 2b, 2d and aniline. The transamination assays, performed in refluxing methanol, produced the expected substitution products 4a, 4b and 4d (Table 2).
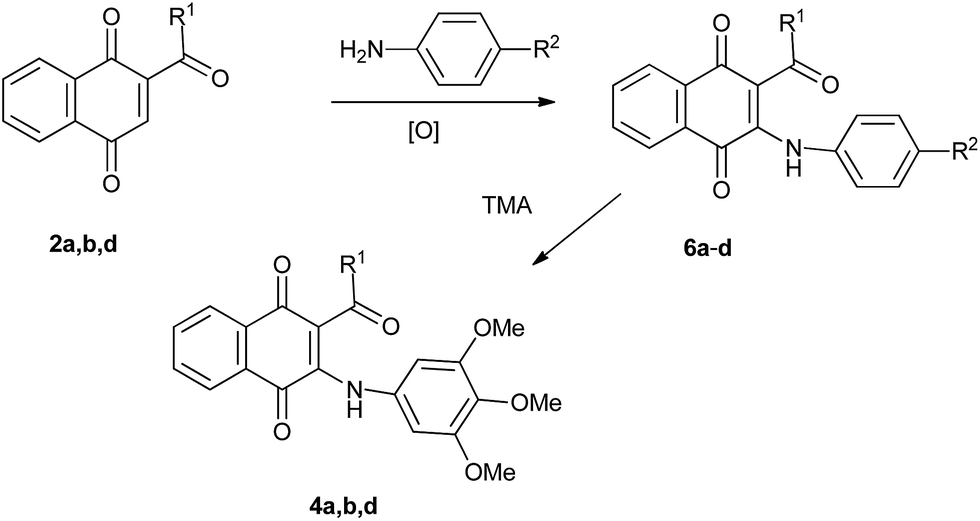
Trying to improve the access to compound 4d, we envisaged that the replacement of the aniline by nitroaniline group in 6c, as in 6d, would facilitate the transamination reaction due to the greater ability of the nitroaniline than aniline as leaving group. The required precursor 6d was prepared in 80% yield from 2-benzoylnaphthoquinone 2d and p-nitroaniline. Further reaction of TMA with 6d, in refluxing methanol, yielded the expected product 4d (24 h) albeit in 42% yield. Optimal results in terms of the preparation of compound 4d were achieved by performing the transamination reaction of TMA with 6c and 6d in refluxing ethanol that provided, after short reaction times, compound 4d in 78 and 80% yield, respectively (Table 2).
The successful results achieved on these preliminary transamination assays are of great interest to developed a new and selective access to 2-acyl-3-anilino-1,4-naphthoquinone derivatives, by transamination of 2-acyl-3-anilino-1,4-naphthoquinones with highly substituted anilines.
Quinones 4a–g were evaluated for their in vitro cytotoxic activity on non-tumor fibroblasts and on a panel of three human-derived tumor cell lines, using the conventional MTT (microculture tetrazolium reduction) assay.31 The data in Table 3 show that most of the quinones have similar cytotoxicity against the three cancer cell lines, with IC50 values around 10 μM. Compound 4a shows a rather low activity whereas quinone 4c should be considered as devoid of any cytotoxicity. It is noteworthy that doxorubicin is more potent compared to most active naphthoquinone derivatives, but doxorubicin was also active against non-tumor cells, indicating a lack of selective effect. Table 3 also shows that compounds 4d, 4e and 4g did not show a selective effect, killing both normal and cancer cells. Interestingly, compounds 4a, 4b and 4f, because of their extremely low toxicity against non-tumor fibroblasts, show a high selectivity upon cancer cells.
| IC50 ± SEMa (μM) | ||||
|---|---|---|---|---|
| Nº | T24 | DU-145 | MCF-7 | NIH 3T3 |
| a Data represent mean average values ± SEM for three separate experiments.b D = doxorubicin. | ||||
| 4a | 39.99 ± 2.44 | 27.01 ± 1.36 | 15.50 ± 0.73 | >100 |
| 4b | 18.03 ± 0.24 | 12.53 ± 0.44 | 10.16 ± 0.78 | >100 |
| 4c | >100 | 34.86 ± 1.65 | 45.26 ± 2.77 | >100 |
| 4d | 11.64 ± 0.88 | 8.12 ± 0.65 | 7.53 ± 0.54 | 14.14 ± 1.85 |
| 4e | 6.53 ± 0.95 | 9.09 ± 1.99 | 11.12 ± 1.81 | 8.89 ± 1.51 |
| 4f | 9.94 ± 1.14 | 11.36 ± 1.04 | 12.95 ± 1.45 | 114.7 ± 17.4 |
| 4g | 9.42 ± 1.41 | 11.04 ± 0.90 | 11.62 ± 1.66 | 7.00 ± 2.04 |
| Db | 0.65 ± 0.07 | 0.42 ± 0.03 | 0.33 ± 0.05 | 0.50 ± 0.02 |
Experimental
1.1. General remarks
All solvents and reagents were purchased from different companies such as Aldrich and Merck and were used as supplied. Melting points were determined on a Stuart Scientific SMP3 apparatus and are uncorrected. The IR spectra were recorded on an FT IR Bruker spectrophotometer, model vector 22, using KBr disks, and the wave numbers are given in cm−1. 1H NMR spectra were recorded on Bruker Avance-400 instrument and Bruker Ultrashield-300 in deuterochloroform (CDCl3). 13C NMR spectra were obtained in CDCl3 at 100 and 75 MHz. Bidimensional NMR techniques and DEPT were used for signal assignment. Chemical shifts are expressed in ppm downfield relative to tetramethylsilane and the coupling constants (J) are reported in Hertz. The HRMS spectra were obtained on a Thermo Finnigan spectrometer, model MAT 95XP and LTQ-Orbitrap mass spectrometer (Thermo-Fisher Scientific) with the analysis performed using an APCI source operated in positive mode. Silica gel Merck 60 (70–230 mesh) was used for preparative column chromatography and TLC aluminum foil 60F254 for analytical TL. The acylhydroquinones 1a–g, used as precursors of quinones 2a–d, were prepared by solar photoacylation of 1,4-naphthoquinone with the respective aldehydes, according to a previously reported procedure.321.2. Reaction of TMA with acylnaphthoquinones 2a,g. General procedure
Suspension of acylnaphthohydroquinone 1a,g (1 equiv.), Ag2O (2.6 equiv.), anhydrous MgSO4 (500 mg), and dichloromethane (15 mL) were left with stirring at rt for 30 min. The mixtures were filtered and the solvents were removed under reduced pressure. The residues were dissolved in methanol, 3,4,5-trimethoxyaniline (2 equiv.) was added with stirring and, the resulting solutions were left at rt for 24 h. The solvents were evaporated under reduced pressure and the residues were column chromatographied over silica gel (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 petroleum ether/EtOAc) to yield the corresponding heterocycles 3a–c, 5a–d and acylaminoquinones 4a–g.
O), 1240 (C–O–C). 1H NMR (400 MHz, CDCl3): δ 8.19 (m, 1H, 8-H), 8.04 (m, 1H, 11-H), 7.77 (m, 2H, 9-H + 10-H), 7.24 (s, 1H, 4-H), 4.07 (s, 3H, OMe), 4.06 (s, 3H, OMe), 3.97 (s, 3H, OMe), 3.13 (s, 3H, Me); 13C NMR (100 MHz, CDCl3): δ 187.2, 183.4, 158.8, 158.4, 149.0, 148.2, 144.4, 142.9, 135.4, 133.7, 133.6, 133.5, 126.8, 125.4, 123.3, 113.3, 103.8, 61.3, 61.1, 56.4, 27.1. HRMS (APCI): [M + H]+ calcd for C21H17NO5: 364.11850; found 364.11729. Compound 4a (42 mg, 0.11 mmol, 11%), red solid, mp: 188–188.5 °C. IR (KBr) νmáx cm−1: 3093 (NH), 1633 (CO), 1593 (CO), 1124 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 12.40 (s, 1H, NH), 8.18 (d, 1H, J = 7.6 Hz, 5-H), 7.93 (d, 1H, J = 7.6 Hz, 8-H), 7.80 (t, 1H, J = 7.2 Hz, 7-H), 7.66 (t, 1H, J = 7.2 Hz, 6 H), 6.36 (s, 2H, 2′-H + 6′-H), 3.88 (s, 3H, OMe), 3.83 (s, 6H, OMe), 2.67 (s, 3H, Me); 13C NMR (100 MHz, CDCl3): δ 202.9, 182.5, 181.8, 153.9 (2 × C), 137.4, 135.7 (2 × C), 135.3, 133.8, 133.1 (2 × C), 131.5, 127.1, 126.4, 102.5 (2 × C), 61.4, 56.6 (2 × C), 33.5. HRMS (APCI): [M + H]+ calcd for C21H19NO6: 382.12907; found 382.12827.O), 1251 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.19 (m, 1H, 8-H), 8.03 (m, 1H, 11-H), 7.77 (m, 2H, 9-H + 10-H), 7.26 (s, 1H, 4-H), 4.07 (s, 6H, 2 × OMe), 3.96 (s, 3H, OMe), 3.49 (dd, 2H, J = 7.8, 7.9 Hz, 1′-H), 1.82 (m, 2H, 2′-H), 1.13 (t, 3H, J = 7.3 Hz, 3′-H); 13C NMR (100 MHz, CDCl3): δ 187.5, 183.2, 162.1, 158.8, 148.9, 148.2, 144.9, 142.8, 135.3, 133.7, 133.6, 133.5, 126.8, 125.2, 123.0, 113.2, 103.9, 61.3, 61.0, 56.4, 40.7, 22.9, 14.4. HRMS (APCI): [M + H]+ calcd for C23H21NO5: 392.14980 found 392.14866. Compound 4b (142 mg, 0.35 mmol, 40%), red solid, mp: 173–174 °C. IR (KBr) νmáx cm−1: 3071 (N–H), 2962 (C–H Ar), 1633 (CO), 1122 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 11.35 (bs, 1H, NH), 8.16 (d, 1H, J = 7.7 Hz, 5-H), 7.96 (d, 1H, J = 7.6 Hz, 8-H), 7.79 (dd, 1H, J = 7.6, 7.5 Hz, 7-H), 7.66 (dd, 1H, J = 7.6, 7.5 Hz, 6-H), 6.34 (s, 2H, 2′-H + 6′-H), 3.86 (s, 3H, OMe), 3.82 (s, 6H, 2 × OMe), 2.93 (t, 2H, J = 7.3 Hz, CH3–CH2–CH2–CO), 1.56 (m, 2H, CH3–CH2–CH2–CO), 0.91 (t, 3H, J = 7.4 Hz, CH3–CH2–CH2–CO); 13C NMR (100 MHz, CDCl3): δ 204.6, 182.3, 181.4, 153.6 (2 × C), 136.9, 135.3, 134.5, 133.3, 132.6, 130.8, 126.6, 126.0, 114.6, 113.2, 102.2 (2 × C), 61.0, 56.2 (2 × C), 46.8, 17.4, 13.8. HRMS (APCI): [M + H]+ calcd for C23H23NO6: 410.16037; found 410.15896.O), 1247 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.19 (m, 1H, 8-H), 8.02 (m, 1H, 11-H), 7.76 (m, 2H, H-9 + 10-H), 7.24 (s, 1H, 4-H), 4.06 (s, 6H, 2 × OMe), 3.95 (s, 3H, OMe), 3.49 (dd, 2H, J = 7.9, 7.9 Hz, 1′-H), 1.77 (m, 2H, 2′-H), 1.53 (m, 2H, 3′-H), 1.38 (m, 2H, 4′-H), 1.31 (m, 4H, 5′-H + 6′-H), 0.88 (t, 3H, J = 6.6 Hz, 7′-H); 13C NMR (100 MHz, CDCl3): δ 187.6, 183.2, 162.4, 158.8, 148.9, 148.3, 144.9, 142.8, 135.3, 133.8, 133.6, 133.5, 126.9, 125.2, 123.0, 113.2, 103.9, 61.3, 61.0, 56.4, 38.9, 31.9, 30.0, 29.7, 29.3, 22.7, 14.1. HRMS (APCI): [M + H]+ calcd for C27H29NO5: 448.21240; found 448.21101. Compound 4c (75 mg, 0.16 mmol, 23%), red solid, mp: 103–104 °C. IR (KBr) νmáx cm−1: 3070 (NH), 2927 (C–H Ar), 1633 (CO), 1122 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 11.34 (bs, 1H, NH), 8.16 (d, 1H, J = 7.7 Hz, 5-H), 7.97 (d, 1H, J = 7.6 Hz, 8-H), 7.80 (dd, 1H, J = 8.0, 7.6 Hz, 7-H), 7.66 (dd, 1H, J = 8.0, 7.7 Hz, 6-H), 6.34 (s, 2H, 2′-H + 6′-H), 3.86 (s, 3H, OMe), 3.82 (s, 6H, 2 × OMe), 2.94 (t, 2H, J = 7.5 Hz, CH3–(CH2)5–CH2–CO), 1.50 (m, 2H, CH3–(CH2)4–CH2–CH2–CO), 1.31 (m, 8H, CH3–CH2–CH2–CH2–CH2–(CH2)2–CO), 0.91 (t, 3H, J = 7.5 Hz, CH3–(CH2)6–CO). 13C NMR (100 MHz, CDCl3): δ 204.8, 182.3, 181.4, 167.7, 153.5, 135.3, 134.5, 133.3, 132.6, 132.4, 130.8, 130.7, 128.7, 126.7, 126.0, 102.1 (2 × C), 61.0, 56.1 (2 × C), 44.9, 31.7, 29.2, 24.1, 23.0, 22.6, 10.9. HRMS (APCI): [M + H]+ calcd for C27H31NO6: 466.22297; found 466.22138.O), 1122 (C–O–C). 1H NMR (400 MHz, CDCl3) δ: 9.66 (s, 1H, OH), 9.38 (bs, 1H, NH), 8.49 (d, 1H, J = 8.3 Hz, 1-H), 8.08 (d, 1H, J = 8.2 Hz, 4-H), 7.68 (d, 2H, J = 7.6 Hz, 2′-H), 7.67 (dd, 1H, J = 8.3, 7.3 Hz, 3-H), 7.53 (dd, 1H, J = 8.3, 7.3 Hz, 2-H), 7.31 (dd, 1H, J = 7.6, 7.3 Hz, 4′-H), 7.19 (dd, 2H, J = 7.6, 7.6 Hz, 3′-H), 6.71 (s, 1H, 10-H), 3.88 (s, 3H, OMe), 3.59 (s, 3H, OMe), 3.48 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3): δ 198.1, 153.0, 149.6, 146.8, 139.6, 135.6, 135.4, 131.8 (2 × C), 129.4 (2 × C), 128.3 (2 × C), 127.7 (2 × C), 125.1, 124.8, 123.5, 123.4, 120.1, 113.9, 111.3, 88.9, 61.2, 59.5, 56.1. HRMS (APCI): [M + H]+ calcd for C26H21NO5: 428.14980; found 428.14814. Compound 4d (134 mg, 0.30 mmol, 34%), red solid, mp: 213–214 °C. IR (KBr) νmáx cm−1: 3259 (N–H), 2929 (C–H Ar), 1682 (CO), 1130 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.18 (d, 1H, J = 8.1 Hz, 5-H), 8.15 (d, 1H, J = 8.1 Hz, 8-H), 7.82 (t, 1H, J = 7.8 Hz, 3′- or 5′-H), 7.73 (t, 2H, J = 7.7 Hz, 6-H + 7-H), 7.60 (d, 2H, J = 7.7 Hz, 2′-H + 6′-H), 7.45 (t, 1H, J = 7.7 Hz, 5′- or 3′-H), 7.31 (m, 2H, –NH + 4′-H), 6.02 (s, 2H, 2′′-H + 6′′-H), 3.76 (s, 3H, OMe), 3.53 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 193.7, 182.7, 182.3, 153.4 (2 × C), 143.6, 137.8, 137.2, 135.9, 133.4, 133.3, 133.1, 132.7, 130.2, 129.2 (2 × C), 128.7 (2 × C), 127.2, 126.9, 113.6, 104.2 (2 × C), 61.2, 56.1 (2 × C). HRMS (APCI): [M + H]+ calcd for C26H21NO6: 444.14472; found 444.14310.O), 1247 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 10.25 (s, 1H, OH), 8.63 (bs, 1H, NH), 8.52 (d, 1H, J = 8.4 Hz, 1-H), 8.00 (d, 1H, J = 8.2 Hz, 4-H), 7.70 (dd, 1H, J = 8.2, 7.4 Hz, 3-H), 7.55 (dd, 1H, J = 8.4, 7.4 Hz, 2-H), 7.51 (s, 1H, 4′-H), 6.77 (s, 1H, 10-H), 6.58 (bs, 1H, 2′-H), 6.28 (m, 1H, 3′-H), 3.94 (s, 3H, OMe), 3.73 (s, 3H, OMe), 3.67 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3): δ 191.6, 154.1, 153.2, 151.9, 147.1, 145.9, 136.0, 135.5, 130.9, 128.9, 128.6, 125.5, 124.9, 123.9, 123.2, 119.9, 118.5, 111.9, 111.7, 110.1, 89.2, 61.2, 60.3, 56.1. HRMS (APCI): [M + H]+ calcd for C24H19NO6: 418.12907; found 418.12785. Compound 4e (153 mg, 0.35 mmol, 45%), red solid, mp: 204–206 °C. IR (KBr) νmáx cm−1: 3384 (NH), 2935 (C–H Ar), 1623 (CO), 1124 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 8.14 (dd, 2H, J = 6.7 Hz, 6.7 Hz, 5-H + 8-H), 7.81 (t, 1H, J = 7.6 Hz, 7-H), 7.76 (m, 2H, 3′-H + NH), 7.71 (t, 1H, J = 7.6 Hz, 6-H), 7.14 (s, 2H, 2′-H+ 4′-H), 6.09 (s, 2H, 2′′-H + 6′′-H), 3.77 (s, 3H, OMe), 3.62 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 188.7, 182.4, 181.7, 153.0 (2 × C), 143.4, 142.9, 136.6, 135.6, 133.4, 132.9, 132.8, 132.5, 129.7, 126.9, 126.8, 126.6, 125.8, 113.9, 103.4 (2 × C), 60.9, 55.8 (2 × C). HRMS (APCI): [M + H]+ calcd for C24H19NO7: 434.12398; found 434.12301.O), 1257 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 9.69 (s, 1H, OH), 8.62 (bs, 1H, NH), 8.51 (d, 1H, J = 8.5 Hz, 1-H), 8.02 (d, 1H, J = 8.0 Hz, 4-H), 7.70 (dd, 1H, J = 8.0, 7.2 Hz, 3-H), 7.55 (dd, 1H, J = 8.5, 7.3 Hz, 2-H), 7.51 (d, 1H, J = 4.8 Hz, 4′-H), 7.12 (d, 1H, J = 4.0 Hz, 2′-H), 6.78 (dd, 1H, J = 4.8, 4.0 Hz, 3′-H), 6.77 (s, 1H, 10-H), 3.93 (s, 3H, OMe), 3.71 (s, 3H, OMe), 3.61 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3): δ 189.7, 153.3, 150.3, 147.1, 146.8, 136.0, 135.5, 133.3, 132.9, 128.7, 128.6, 127.4, 125.4, 125.0, 123.6, 123.3, 119.9, 113.5, 111.5, 111.4, 89.0, 61.2, 59.9, 56.1. HRMS (APCI): [M + H]+ calcd for C24H19NO5S: 434.10622; found 434.10483. Compound 4f (150 mg, 0.33 mmol, 45%), red solid, mp: 172–173 °C. IR (KBr) νmáx cm−1: 3437 (O–H), 3294 (N–H), 2937 (C–H Ar), 1639 (CO), 1130 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.15 (dd, 2H, J = 7.7, 7.7 Hz, 5-H + 8-H), 7.81 (m, 2H, 7-H + 3′-H), 7.71 (t, 1H, J = 7.5 Hz, 6-H), 7.46 (bs, 1H, NH), 6.90 (m, 1H, 2′-H or 4′-H), 6.44 (m, 1H, 4′-H or 2′-H) 6.19 (s, 2H, 2′′-H + 6′′-H), 3.77 (s, 3H, OMe), 3.65 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 182.2, 181.4, 180.9, 153.5, 153.2 (2 × C), 145.7, 143.5, 136.9, 135.6, 132.9, 132.7, 132.5, 129.7, 126.7, 126.6, 117.3, 112.8, 112.6, 102.8 (2 × C), 60.9, 55.9 (2 × C). HRMS (APCI): [M + H]+ calcd for C24H19NO6S: 450.10114; found 450.09986.O), 1126 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 10.05 (s, 1H, OH), 8.69 (bs, 1H, NH), 8.52 (d, 1H, J = 8.5 Hz, 1-H), 8.02 (d, 1H, J = 8.0 Hz, 4-H), 7.69 (dd, 1H, J = 8.0, 7.5 Hz, 3-H), 7.58 (d, 1H, J = 5.2 Hz, 4′-H), 7.55 (dd, 1H, J = 8.0, 7.5 Hz, 2-H), 7.46 (m, 1H, 1′-H), 7.14 (dd, 1H, J = 5.2, 3.0 Hz, 3′-H), 6.75 (s, 1H, 10-H), 3.91 (s, 3H, OMe), 3.66 (s, 3H, OMe), 3.60 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3) δ: 191.4, 153.3, 150.8, 147.0, 144.0, 135.8, 135.5, 132.8, 128.7, 128.5, 127.7, 125.5, 124.9, 124.8, 123.7, 123.3, 119.9, 113.7, 111.7, 111.5, 89.0, 61.2, 59.9, 56.1. HRMS (APCI): [M + H]+ calcd for C24H19NO5S: 434.10622; found 434.10451. Compound 4g (90 mg, 0.20 mmol, 27%), red solid, mp: 102–103.5 °C. IR (KBr) νmáx cm−1: 3099 (N–H), 2939 (C–H Ar), 1662 (CO), 1128 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 8.15 (dd, 2H, J = 6.5, 6.5 Hz, 5-H + 8-H), 7.81 (m, 2H, 7-H + NH), 7.72 (t, 1H, J = 7.5 Hz, 6-H), 7.55 (d, 1H, J = 4.7 Hz, 2′-H or 3′-H), 7.42 (d, 1H, J = 4.7 Hz, 3′-H or 2′-H), 6.99 (dd, 1H, J = 4.3, 4.0 Hz, 4′-H), 6.12 (s, 2H, 2′′-H + 6′′-H), 3.79 (s, 3H, OMe), 3.60 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 185.1, 182.2, 181.5, 153.1 (2 × C), 145.5, 143.1, 136.8, 135.6, 134.5, 133.4, 132.9, 132.8, 132.2, 129.7, 127.6, 126.8, 126.6, 113.3, 103.7 (2 × C), 60.9, 55.9 (2 × C). HRMS (APCI): [M + H]+ calcd for C24H19NO6S: 450.10114; found 450.09948.
:2 petroleum ether/EtOAc) to yield the corresponding heterocycles 3a–c, 5a–d and acylaminoquinones 4a–g.
O), 1240 (C–O–C). 1H NMR (400 MHz, CDCl3): δ 8.19 (m, 1H, 8-H), 8.04 (m, 1H, 11-H), 7.77 (m, 2H, 9-H + 10-H), 7.24 (s, 1H, 4-H), 4.07 (s, 3H, OMe), 4.06 (s, 3H, OMe), 3.97 (s, 3H, OMe), 3.13 (s, 3H, Me); 13C NMR (100 MHz, CDCl3): δ 187.2, 183.4, 158.8, 158.4, 149.0, 148.2, 144.4, 142.9, 135.4, 133.7, 133.6, 133.5, 126.8, 125.4, 123.3, 113.3, 103.8, 61.3, 61.1, 56.4, 27.1. HRMS (APCI): [M + H]+ calcd for C21H17NO5: 364.11850; found 364.11729. Compound 4a (42 mg, 0.11 mmol, 11%), red solid, mp: 188–188.5 °C. IR (KBr) νmáx cm−1: 3093 (NH), 1633 (CO), 1593 (CO), 1124 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 12.40 (s, 1H, NH), 8.18 (d, 1H, J = 7.6 Hz, 5-H), 7.93 (d, 1H, J = 7.6 Hz, 8-H), 7.80 (t, 1H, J = 7.2 Hz, 7-H), 7.66 (t, 1H, J = 7.2 Hz, 6 H), 6.36 (s, 2H, 2′-H + 6′-H), 3.88 (s, 3H, OMe), 3.83 (s, 6H, OMe), 2.67 (s, 3H, Me); 13C NMR (100 MHz, CDCl3): δ 202.9, 182.5, 181.8, 153.9 (2 × C), 137.4, 135.7 (2 × C), 135.3, 133.8, 133.1 (2 × C), 131.5, 127.1, 126.4, 102.5 (2 × C), 61.4, 56.6 (2 × C), 33.5. HRMS (APCI): [M + H]+ calcd for C21H19NO6: 382.12907; found 382.12827.O), 1251 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.19 (m, 1H, 8-H), 8.03 (m, 1H, 11-H), 7.77 (m, 2H, 9-H + 10-H), 7.26 (s, 1H, 4-H), 4.07 (s, 6H, 2 × OMe), 3.96 (s, 3H, OMe), 3.49 (dd, 2H, J = 7.8, 7.9 Hz, 1′-H), 1.82 (m, 2H, 2′-H), 1.13 (t, 3H, J = 7.3 Hz, 3′-H); 13C NMR (100 MHz, CDCl3): δ 187.5, 183.2, 162.1, 158.8, 148.9, 148.2, 144.9, 142.8, 135.3, 133.7, 133.6, 133.5, 126.8, 125.2, 123.0, 113.2, 103.9, 61.3, 61.0, 56.4, 40.7, 22.9, 14.4. HRMS (APCI): [M + H]+ calcd for C23H21NO5: 392.14980 found 392.14866. Compound 4b (142 mg, 0.35 mmol, 40%), red solid, mp: 173–174 °C. IR (KBr) νmáx cm−1: 3071 (N–H), 2962 (C–H Ar), 1633 (CO), 1122 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 11.35 (bs, 1H, NH), 8.16 (d, 1H, J = 7.7 Hz, 5-H), 7.96 (d, 1H, J = 7.6 Hz, 8-H), 7.79 (dd, 1H, J = 7.6, 7.5 Hz, 7-H), 7.66 (dd, 1H, J = 7.6, 7.5 Hz, 6-H), 6.34 (s, 2H, 2′-H + 6′-H), 3.86 (s, 3H, OMe), 3.82 (s, 6H, 2 × OMe), 2.93 (t, 2H, J = 7.3 Hz, CH3–CH2–CH2–CO), 1.56 (m, 2H, CH3–CH2–CH2–CO), 0.91 (t, 3H, J = 7.4 Hz, CH3–CH2–CH2–CO); 13C NMR (100 MHz, CDCl3): δ 204.6, 182.3, 181.4, 153.6 (2 × C), 136.9, 135.3, 134.5, 133.3, 132.6, 130.8, 126.6, 126.0, 114.6, 113.2, 102.2 (2 × C), 61.0, 56.2 (2 × C), 46.8, 17.4, 13.8. HRMS (APCI): [M + H]+ calcd for C23H23NO6: 410.16037; found 410.15896.O), 1247 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.19 (m, 1H, 8-H), 8.02 (m, 1H, 11-H), 7.76 (m, 2H, H-9 + 10-H), 7.24 (s, 1H, 4-H), 4.06 (s, 6H, 2 × OMe), 3.95 (s, 3H, OMe), 3.49 (dd, 2H, J = 7.9, 7.9 Hz, 1′-H), 1.77 (m, 2H, 2′-H), 1.53 (m, 2H, 3′-H), 1.38 (m, 2H, 4′-H), 1.31 (m, 4H, 5′-H + 6′-H), 0.88 (t, 3H, J = 6.6 Hz, 7′-H); 13C NMR (100 MHz, CDCl3): δ 187.6, 183.2, 162.4, 158.8, 148.9, 148.3, 144.9, 142.8, 135.3, 133.8, 133.6, 133.5, 126.9, 125.2, 123.0, 113.2, 103.9, 61.3, 61.0, 56.4, 38.9, 31.9, 30.0, 29.7, 29.3, 22.7, 14.1. HRMS (APCI): [M + H]+ calcd for C27H29NO5: 448.21240; found 448.21101. Compound 4c (75 mg, 0.16 mmol, 23%), red solid, mp: 103–104 °C. IR (KBr) νmáx cm−1: 3070 (NH), 2927 (C–H Ar), 1633 (CO), 1122 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 11.34 (bs, 1H, NH), 8.16 (d, 1H, J = 7.7 Hz, 5-H), 7.97 (d, 1H, J = 7.6 Hz, 8-H), 7.80 (dd, 1H, J = 8.0, 7.6 Hz, 7-H), 7.66 (dd, 1H, J = 8.0, 7.7 Hz, 6-H), 6.34 (s, 2H, 2′-H + 6′-H), 3.86 (s, 3H, OMe), 3.82 (s, 6H, 2 × OMe), 2.94 (t, 2H, J = 7.5 Hz, CH3–(CH2)5–CH2–CO), 1.50 (m, 2H, CH3–(CH2)4–CH2–CH2–CO), 1.31 (m, 8H, CH3–CH2–CH2–CH2–CH2–(CH2)2–CO), 0.91 (t, 3H, J = 7.5 Hz, CH3–(CH2)6–CO). 13C NMR (100 MHz, CDCl3): δ 204.8, 182.3, 181.4, 167.7, 153.5, 135.3, 134.5, 133.3, 132.6, 132.4, 130.8, 130.7, 128.7, 126.7, 126.0, 102.1 (2 × C), 61.0, 56.1 (2 × C), 44.9, 31.7, 29.2, 24.1, 23.0, 22.6, 10.9. HRMS (APCI): [M + H]+ calcd for C27H31NO6: 466.22297; found 466.22138.O), 1122 (C–O–C). 1H NMR (400 MHz, CDCl3) δ: 9.66 (s, 1H, OH), 9.38 (bs, 1H, NH), 8.49 (d, 1H, J = 8.3 Hz, 1-H), 8.08 (d, 1H, J = 8.2 Hz, 4-H), 7.68 (d, 2H, J = 7.6 Hz, 2′-H), 7.67 (dd, 1H, J = 8.3, 7.3 Hz, 3-H), 7.53 (dd, 1H, J = 8.3, 7.3 Hz, 2-H), 7.31 (dd, 1H, J = 7.6, 7.3 Hz, 4′-H), 7.19 (dd, 2H, J = 7.6, 7.6 Hz, 3′-H), 6.71 (s, 1H, 10-H), 3.88 (s, 3H, OMe), 3.59 (s, 3H, OMe), 3.48 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3): δ 198.1, 153.0, 149.6, 146.8, 139.6, 135.6, 135.4, 131.8 (2 × C), 129.4 (2 × C), 128.3 (2 × C), 127.7 (2 × C), 125.1, 124.8, 123.5, 123.4, 120.1, 113.9, 111.3, 88.9, 61.2, 59.5, 56.1. HRMS (APCI): [M + H]+ calcd for C26H21NO5: 428.14980; found 428.14814. Compound 4d (134 mg, 0.30 mmol, 34%), red solid, mp: 213–214 °C. IR (KBr) νmáx cm−1: 3259 (N–H), 2929 (C–H Ar), 1682 (CO), 1130 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.18 (d, 1H, J = 8.1 Hz, 5-H), 8.15 (d, 1H, J = 8.1 Hz, 8-H), 7.82 (t, 1H, J = 7.8 Hz, 3′- or 5′-H), 7.73 (t, 2H, J = 7.7 Hz, 6-H + 7-H), 7.60 (d, 2H, J = 7.7 Hz, 2′-H + 6′-H), 7.45 (t, 1H, J = 7.7 Hz, 5′- or 3′-H), 7.31 (m, 2H, –NH + 4′-H), 6.02 (s, 2H, 2′′-H + 6′′-H), 3.76 (s, 3H, OMe), 3.53 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 193.7, 182.7, 182.3, 153.4 (2 × C), 143.6, 137.8, 137.2, 135.9, 133.4, 133.3, 133.1, 132.7, 130.2, 129.2 (2 × C), 128.7 (2 × C), 127.2, 126.9, 113.6, 104.2 (2 × C), 61.2, 56.1 (2 × C). HRMS (APCI): [M + H]+ calcd for C26H21NO6: 444.14472; found 444.14310.O), 1247 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 10.25 (s, 1H, OH), 8.63 (bs, 1H, NH), 8.52 (d, 1H, J = 8.4 Hz, 1-H), 8.00 (d, 1H, J = 8.2 Hz, 4-H), 7.70 (dd, 1H, J = 8.2, 7.4 Hz, 3-H), 7.55 (dd, 1H, J = 8.4, 7.4 Hz, 2-H), 7.51 (s, 1H, 4′-H), 6.77 (s, 1H, 10-H), 6.58 (bs, 1H, 2′-H), 6.28 (m, 1H, 3′-H), 3.94 (s, 3H, OMe), 3.73 (s, 3H, OMe), 3.67 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3): δ 191.6, 154.1, 153.2, 151.9, 147.1, 145.9, 136.0, 135.5, 130.9, 128.9, 128.6, 125.5, 124.9, 123.9, 123.2, 119.9, 118.5, 111.9, 111.7, 110.1, 89.2, 61.2, 60.3, 56.1. HRMS (APCI): [M + H]+ calcd for C24H19NO6: 418.12907; found 418.12785. Compound 4e (153 mg, 0.35 mmol, 45%), red solid, mp: 204–206 °C. IR (KBr) νmáx cm−1: 3384 (NH), 2935 (C–H Ar), 1623 (CO), 1124 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 8.14 (dd, 2H, J = 6.7 Hz, 6.7 Hz, 5-H + 8-H), 7.81 (t, 1H, J = 7.6 Hz, 7-H), 7.76 (m, 2H, 3′-H + NH), 7.71 (t, 1H, J = 7.6 Hz, 6-H), 7.14 (s, 2H, 2′-H+ 4′-H), 6.09 (s, 2H, 2′′-H + 6′′-H), 3.77 (s, 3H, OMe), 3.62 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 188.7, 182.4, 181.7, 153.0 (2 × C), 143.4, 142.9, 136.6, 135.6, 133.4, 132.9, 132.8, 132.5, 129.7, 126.9, 126.8, 126.6, 125.8, 113.9, 103.4 (2 × C), 60.9, 55.8 (2 × C). HRMS (APCI): [M + H]+ calcd for C24H19NO7: 434.12398; found 434.12301.O), 1257 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 9.69 (s, 1H, OH), 8.62 (bs, 1H, NH), 8.51 (d, 1H, J = 8.5 Hz, 1-H), 8.02 (d, 1H, J = 8.0 Hz, 4-H), 7.70 (dd, 1H, J = 8.0, 7.2 Hz, 3-H), 7.55 (dd, 1H, J = 8.5, 7.3 Hz, 2-H), 7.51 (d, 1H, J = 4.8 Hz, 4′-H), 7.12 (d, 1H, J = 4.0 Hz, 2′-H), 6.78 (dd, 1H, J = 4.8, 4.0 Hz, 3′-H), 6.77 (s, 1H, 10-H), 3.93 (s, 3H, OMe), 3.71 (s, 3H, OMe), 3.61 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3): δ 189.7, 153.3, 150.3, 147.1, 146.8, 136.0, 135.5, 133.3, 132.9, 128.7, 128.6, 127.4, 125.4, 125.0, 123.6, 123.3, 119.9, 113.5, 111.5, 111.4, 89.0, 61.2, 59.9, 56.1. HRMS (APCI): [M + H]+ calcd for C24H19NO5S: 434.10622; found 434.10483. Compound 4f (150 mg, 0.33 mmol, 45%), red solid, mp: 172–173 °C. IR (KBr) νmáx cm−1: 3437 (O–H), 3294 (N–H), 2937 (C–H Ar), 1639 (CO), 1130 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 8.15 (dd, 2H, J = 7.7, 7.7 Hz, 5-H + 8-H), 7.81 (m, 2H, 7-H + 3′-H), 7.71 (t, 1H, J = 7.5 Hz, 6-H), 7.46 (bs, 1H, NH), 6.90 (m, 1H, 2′-H or 4′-H), 6.44 (m, 1H, 4′-H or 2′-H) 6.19 (s, 2H, 2′′-H + 6′′-H), 3.77 (s, 3H, OMe), 3.65 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 182.2, 181.4, 180.9, 153.5, 153.2 (2 × C), 145.7, 143.5, 136.9, 135.6, 132.9, 132.7, 132.5, 129.7, 126.7, 126.6, 117.3, 112.8, 112.6, 102.8 (2 × C), 60.9, 55.9 (2 × C). HRMS (APCI): [M + H]+ calcd for C24H19NO6S: 450.10114; found 450.09986.O), 1126 (C–O–C); 1H NMR (400 MHz, CDCl3): δ 10.05 (s, 1H, OH), 8.69 (bs, 1H, NH), 8.52 (d, 1H, J = 8.5 Hz, 1-H), 8.02 (d, 1H, J = 8.0 Hz, 4-H), 7.69 (dd, 1H, J = 8.0, 7.5 Hz, 3-H), 7.58 (d, 1H, J = 5.2 Hz, 4′-H), 7.55 (dd, 1H, J = 8.0, 7.5 Hz, 2-H), 7.46 (m, 1H, 1′-H), 7.14 (dd, 1H, J = 5.2, 3.0 Hz, 3′-H), 6.75 (s, 1H, 10-H), 3.91 (s, 3H, OMe), 3.66 (s, 3H, OMe), 3.60 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3) δ: 191.4, 153.3, 150.8, 147.0, 144.0, 135.8, 135.5, 132.8, 128.7, 128.5, 127.7, 125.5, 124.9, 124.8, 123.7, 123.3, 119.9, 113.7, 111.7, 111.5, 89.0, 61.2, 59.9, 56.1. HRMS (APCI): [M + H]+ calcd for C24H19NO5S: 434.10622; found 434.10451. Compound 4g (90 mg, 0.20 mmol, 27%), red solid, mp: 102–103.5 °C. IR (KBr) νmáx cm−1: 3099 (N–H), 2939 (C–H Ar), 1662 (CO), 1128 (C–O–C); 1H NMR (400 MHz, CDCl3) δ: 8.15 (dd, 2H, J = 6.5, 6.5 Hz, 5-H + 8-H), 7.81 (m, 2H, 7-H + NH), 7.72 (t, 1H, J = 7.5 Hz, 6-H), 7.55 (d, 1H, J = 4.7 Hz, 2′-H or 3′-H), 7.42 (d, 1H, J = 4.7 Hz, 3′-H or 2′-H), 6.99 (dd, 1H, J = 4.3, 4.0 Hz, 4′-H), 6.12 (s, 2H, 2′′-H + 6′′-H), 3.79 (s, 3H, OMe), 3.60 (s, 6H, OMe); 13C NMR (100 MHz, CDCl3): δ 185.1, 182.2, 181.5, 153.1 (2 × C), 145.5, 143.1, 136.8, 135.6, 134.5, 133.4, 132.9, 132.8, 132.2, 129.7, 127.6, 126.8, 126.6, 113.3, 103.7 (2 × C), 60.9, 55.9 (2 × C). HRMS (APCI): [M + H]+ calcd for C24H19NO6S: 450.10114; found 450.09948.1.3. Synthesis of 3-acyl-2-arylamino-1,4-naphthoquinones 6a–d. General procedure21
Suspension of acylhydroquinones 1a, 1b, 1d (1 equiv.), Ag2O (2.6 equiv.), anhydrous MgSO4 (500 mg), and dichloromethane (15 mL) were left with stirring at rt after completion of the reaction as indicated by TLC. The mixtures were filtered, the solvent removed under reduced pressure and the residues dissolved in methanol. The required anilines (1.1 equiv.) were added with stirring, and the resulting solutions were left at room temperature after completion of the reaction as indicated by TLC. The solvent was evaporated under reduced pressure and the residues were column chromatographied over silica gel (8:2 petroleum ether/EtOAc) to yield the corresponding acylaminoquinones 6a–d.
O), 1507 (NO2). 1H NMR (300 MHz, CDCl3): δ 8.21 (dd, 1H, J = 7.6, 1.1 Hz, 5-H), 8.16 (dd, 1H, J = 7.6, 1.1 Hz, 8-H), 8.02 (bs, 1H, NH), 7.87 (m, 3H, 3′-, 5′- and 7-H), 7.78 (dt, 1H, J = 7.5, 1.4 Hz, 6-H), 7.59 (dd, 2H, J = 8.3, 1.2 Hz, benzoyl), 7.54 (t, 1H, J = 7.4 Hz, benzoyl), 7.35 (t, 2H, J = 7.7 Hz, benzoyl), 6.97 (d, 2H, J = 8.9 Hz, 2′-and 6′-H). 13C NMR (75 MHz, CDCl3): δ 193.5, 182.1, 181.8, 145.1, 143.1, 142.5, 136.9, 135.8, 133.9, 133.3, 132.5, 129.8, 128.8 (2 × C), 128.6 (2 × C), 126.9, 126.8, 124.8 (2 × C), 124.5 (2 × C), 115.6. HRMS (ESI): [M − H]− calcd for C23H14N2O5: 398.09027; found: 398.08264.1.4. Synthesis of 3-acyl-2-(3,4,5-trimethoxyphenyl)amino-1,4-naphthoquinones 4a, 4b and 4d. General procedure
Solutions of 6a–d (1 equiv.), 3,4,5-trimethoxyaniline (1.1 equiv.) and methanol or ethanol (15 mL) were refluxed after completion of the reaction as indicated by TLC (0:13–61 h). The solvents were removed under vacuum and the corresponding products 4a, 4b and 4d were isolated by column chromatography over silica gel (35:5 petroleum ether/EtOAc). Table 2 summarizes the results of these experiments.
1.5. Computational details
All the structures have been fully optimized using the Becke-3 for exchange and Lee–Yang–Parr for correlation (B3LYP)34–37 functional with the standard 6-311G (d,p) basis set in solvent phases. To model the solvent, we used polarizable continuum model (PCM)38 considering methanol (ε = 32.613). All the stationary states were confirmed by frequency calculations. All the above-mentioned calculations were performed using the Gaussian 09 suite of programs.391.6. Cytotoxicity assay
1.7. Cellular assays
The cytotoxicity of compounds 4a–g was assessed by following the reduction of MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) to formazan blue.30 Briefly, cells were seeded into 96-well plates at a density of 10000 cells per well for 24 h and then incubated for 48 h with or without the compounds. Cells were then washed twice with warm PBS and incubated with MTT (0.5 mg mL−1) for 2 h at 37 °C. Incubation medium was thereafter discarded and the blue formazan crystals were solubilized by adding 100 μL DMSO per well. The color solution was subsequently read at 550 nm. Results are expressed as % of MTT reduction compared to untreated control conditions. The calculation of IC50 values were performed by using GraphPad Prism software (San Diego, CA, USA).
Conclusions
In summary, in the present work we have described results on the synthesis of novel 2-acyl-3-(3,4,5-trimethoxyanilino)-1,4-naphthoquinones of interest as potential anticancer agents. The synthetic approaches were based on direct amination of 2-acyl-1,4-naphthoquinones with TMA and on transamination reactions of TMA with 2-acyl-3-anilino- and 2-acyl-3-(4-nitroanilino)-1,4-naphthoquinones. The results reveal that the reactions of TMA with 2-acyl-1,4-naphthoquinones proceed through N–C and C–C bonding formation to give benzophenanthridinequinone and benzocarbazole derivatives, together with 2-acyl-3-(3,4,5-trimetoxyanilino)-1,4-naphthoquinones as the minor products. A density functional theory analysis performed on the stabilities of intermediates and products in these reactions are in good agreement with the experimental results on these competitive processes. Preliminary results on the reactions of TMA with 2-acyl-3-anilino-1,4-naphthoquinones 6a, 6b, 6c that yield 2-acyl-3-(3,4,5-trimetoxyphenyl)amino-1,4-naphthoquinones 4a and 4d, demonstrates the potential use of this strategy for the synthesis of 2-acyl-1,4-naphthoquinones containing highly substituted anilino groups at 3-position. Studies on the scope of the transamination reactions of 2-anilino-3-acyl-1,4-naphthoquinones with substituted anilines are currently underway.Acknowledgements
We thank Fondo Nacional de Ciencia y Tecnología, Chile (Grant No. 1114 0544 and 114 1307) for financial support to this study.Notes and references
- R. H. Thompson, Naturally Occurring Quinones IV: Recent Advances, Blackie Academic & Professional, London, UK, 4th edn, 1997 Search PubMed.
- K. Maruyama and Y. Naruta, Chem. Lett., 1977, 8, 847–850 CrossRef.
- H. J. Uno, J. Org. Chem., 1986, 51, 350–358 CrossRef CAS.
- M. A. Brimble and S. M. Lynds, J. Chem. Soc., Perkin Trans. 1, 1994, 5, 493–496 RSC.
- G. A. Kraus and H. Maeda, Tetrahedron Lett., 1994, 35, 9189–9190 CrossRef CAS.
- P. A. Waske, J. Mattay and M. Oelgemöller, Tetrahedron Lett., 2006, 47, 1329–1332 CrossRef CAS.
- J. A. Valderrama, D. Pessoa-Mahana, R. A. Tapia, A. Rojas de Arias, H. Nakayama, S. Torres, J. Miret and M. E. Ferreira, Tetrahedron, 2001, 57, 8653–8658 CrossRef CAS.
- J. A. Valderrama, J. Benites, M. Cortés, D. Pessoa-Mahana, E. Prina and A. Fournet, Tetrahedron, 2002, 58, 881–886 CrossRef CAS.
- J. A. Valderrama, C. Zamorano, M. F. González, E. Prina and A. Fournet, Bioorg. Med. Chem., 2005, 13, 4153–4159 CrossRef CAS PubMed.
- J. A. Valderrama, M. F. González, D. Pessoa-Mahana, R. A. Tapia, H. Fillion, F. Pautet, J. A. Rodríguez, C. Theoduloz and G. Schmeda-Hishmann, Bioorg. Med. Chem., 2006, 14, 5003–5011 CrossRef CAS PubMed.
- J. A. Valderrama and D. Vásquez, Tetrahedron Lett., 2008, 49, 703–706 CrossRef CAS.
- D. Vásquez, J. A. Rodríguez, C. Theoduloz, P. Buc and J. A. Valderrama, Eur. J. Med. Chem., 2010, 45, 5234–5242 CrossRef PubMed.
- V. Delgado, A. Ibacache, C. Theoduloz and J. A. Valderrama, Molecules, 2012, 17, 7042–7056 CrossRef CAS PubMed.
- W. Schäfer, A. Aguado and U. Sezer, Angew. Chem., Int. Ed., 1971, 10, 406–407 CrossRef.
- M. Pardo, K. Joos and W. Schäfer, Liebigs Ann. Chem., 1979, 4, 503–521 CrossRef.
- D. Ríos, J. Benites, J. A. Valderrama, M. Farias, R. C. Pedrosa, J. Verrax and P. Buc Calderon, Curr. Top. Med. Chem., 2012, 12, 2094–2102 CrossRef.
- H. Buff and U. Kuckländer, Tetrahedron, 2000, 56, 5137–5145 CrossRef CAS.
- G. R. Allen Jr and M. J. Weiss, J. Org. Chem., 1968, 33, 198–200 CrossRef.
- J. A. Valderrama, M. F. González, P. Colonelli and D. Vásquez, Synlett, 2006, 17, 2777–2780 CrossRef.
- J. A. Valderrama, D. Ríos, G. G. Muccioli, P. Buc Calderon, I. Brito and J. Benites, Tetrahedron Lett., 2015, 56, 5103–5105 CrossRef CAS.
- D. Ríos, J. Benites, F. Torrejón, C. Theoduloz and J. A. Valderrama, Med. Chem. Res., 2014, 23, 4149–4155 CrossRef.
- M. S. Farias, C. T. Pich, N. C. Falcão, K. B. Felipe, M. R. Kviecinski, F. Ourique da Silva, T. M. Fisher, D. Ríos, J. Benites, J. A. Valderrama, P. Buc Calderon and R. Curi Pedrosa, Mol. Med. Rep., 2014, 10(1), 405–410 CAS.
- M. K. Hadden, S. A. Hill, J. Davenport, R. L. Matts and B. S. Blagg, Bioorg. Med. Chem., 2009, 17, 634–640 CrossRef CAS PubMed.
- C. D. S. Lisboa, V. G. Santos, B. G. Vaz, N. C. de Lucas, M. N. Eberlin and S. J. Garden, J. Org. Chem., 2011, 76, 5264–5273 CrossRef CAS PubMed and references cited therein.
- Sh. A. Patil, R. Patil and D. D. Miller, Curr. Org. Chem., 2008, 12, 691–717 CrossRef CAS.
- J.-L. Bernier, J.-P. Henichart, C. Vaccher and R. Houssain, J. Org. Chem., 1980, 45, 1493–1496 CrossRef CAS.
- B. Dotzauer, R. Grunert, P. J. Bednarski, H. Lanig, J. Landwehr and R. Troschutz, Bioorg. Med. Chem., 2006, 14, 7282–7292 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev. [Sect.] B, 1964, 136, 864–879 CrossRef.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, USA, 1989 Search PubMed.
- W. Schäfer, H. W. Moore and A. Aguado, Synthesis, 1974, 30–32 CrossRef.
- T. J. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- J. Benites, D. Ríos, P. Díaz and J. A. Valderrama, Tetrahedron Lett., 2011, 52, 609–611 CrossRef CAS.
- K. Kobayashi, M. Suzuki, H. Takeuchi, A. Konishi, H. Sakurai and H. Suginome, J. Chem. Soc., Perkin Trans. 1, 1994, 8, 1099–1104 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- S. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and J. D. J. Cioslowski Fox, Gaussian 09 Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |