
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Moiety effect on the luminescent property of star-shaped triphenylamine (TPA) derivatives as mechanochromic materials†
Ying Zhang *a,
Yao-Qin Fenga,
Jun-Hao Wangb,
Gaoyi Hana,
Miao-Yu Li*a,
Yaoming Xiaoa and
Zhen-Dong Fenga
*a,
Yao-Qin Fenga,
Jun-Hao Wangb,
Gaoyi Hana,
Miao-Yu Li*a,
Yaoming Xiaoa and
Zhen-Dong Fenga
aInstitute of Molecular Science, Innovation Center of Chemistry and Molecular Science, Shanxi University, Key Laboratory of Energy Conversation and Storage of Shanxi Province, Taiyuan 030006, PR China. E-mail: yzhang@sxu.edu.cn; limiaoyu@sxu.edu.cn
bInstitute of Crystalline Materials, Shanxi University, Taiyuan 030006, PR China
First published on 18th July 2017
Abstract
Two TPA-based star-shaped mechanochromic materials, namely ester-TPA and COOH-TPA, were designed and synthesized using TPA as the core and benzoic acid and methyl benzoate as the arms, and then importantly the moiety effect on the luminescent property of the materials was investigated. They displayed similar solvatochromic effects in different solvents for the D–π–A motif with charge transfer from TPA to the benzoic acid or methyl benzoate moiety, which was confirmed by the consistent UV-vis absorption and large differences displayed when dropped into different solvents as well as by orbital distribution analysis using density functional theory. However, they showed different mechanochromism performances, which were investigated by analysing the fluorescent spectrum, fluorescent quantum yield and fluorescence lifetime. The COOH-based emitter showed a better performance with a relatively obvious red-shift for CIE (0.17, 0.35) to (0.26, 0.49) together with transformation from crystalline to amorphous states, indicating the molecular packing pattern had been destroyed, which could be speculated from single-crystal structure analysis and powder X-ray diffraction analysis. In addition, COOH-TPA could also respond to alkalis, leading to a blue-shift and band broadening of the spectra, which might be ascribed to a reduced charge transfer from TPA to benzoic acid owing to deprotonation.
Introduction
Mechanochromic fluorescent materials (MCFs) possess an interesting property of the emission colour or/and intensity capable of being tuned with an external stimulus, such as through grinding, shearing or pressing, and recovering to their initial state by annealing and/or fuming with an organic solvent. They have attracted extensive investigation due to their potential applications in displays, security printing, data storage devices, etc.1 In the past several years, various MCFs have been reported based on polymers,2 metal complexes3 and, in particular, organic dyes for their flexible of options in molecule design and rich molecule interactions for tuning their emission properties with an external stimulus.4–12A twisted conformation is important for organic MCFs owing to their loose packing simultaneously allowing an easy change of the stacking mode in response to an external stimulus, which is better than MCFs with structure disruption caused by chemical bond breaking, and leads to better stability and lifetime. To date, several types of excellent packing mode variation dependent organic MCFs have been reported, including derivatives of tetraphenylethene,4 9,10-divinylanthracene4a,5 oligo(p-phenylene vinylene),6 β-diketone boron complexes,7 pyrene,8 cyanostyrene,9 pyran10 and onium salt.11 These MCF emitters are often investigated with regard to the tuning of their intermolecular interactions, such as π–π, C–H⋯π and H-bonding related to their transformation from crystalline to amorphous states, as well as changes with their polymorphs or intramolecular interactions caused by different molecular conformations.4–12 Thus, investigation of the fluorescence property of such emitters under an external stimulus can help us to better understand the relationship between the molecular structure and luminescent property, which is very important for mechanochromic analysis and therefore essential for designing novel luminescent materials with MCF activity.13
TPA is a typical electron-donor with high hole-mobility and a high fluorescent quantum yield; thus, it often serves as a hole-transport material, luminescent emitter or sensitizing dye in optical-electrical fields.14 Introducing some strong electron-withdrawing groups into the triphenylamine framework may form a class of typical donor–acceptor (D–A) or donor–π-conjugated bridge (D–π–A) structure, where a twisted structure is beneficial for twisted intramolecular charge transfer (TICT), which could promote efficient intramolecular charge transfer from the donor to acceptor under certain conditions, and to a certain extent increase the electronic delocalization of the whole system, thereby, for example, displaying solvatochromic effects with a fluorescence red-shift gradually with increasing the solvent polarity.
On the other hand, the three dimensional (3D) propeller-shaped unit of a TPA derivative with a non-coplanar structure is beneficial for a loose packing formation in the aggregate state, which is the basis for the active MCF property.
For the reasons mentioned above, various excellent TPA-containing luminescent materials have been designed and synthesized by introducing some electron-withdrawing groups, such as benzophenone,15 boron complex,7g dicyanvinyl,16 benzo-thiazole/-oxazole,17 tetraphenylethene4b or cyanostyrene18 etc. to form a D–π–A motif for MCF activity. However, the study of the effect of introducing a small moiety on the luminescent property of MCFs based on star-shaped TPA derivatives is still limited. Furthermore, the introduction of certain small moieties, such as formyl and/or –COOH, will form supra-molecular interactions (especially H-bonding) with different self-assemblies, sometimes giving excellent luminescent property.19 Herein, we synthesized and investigated two different star-shaped TPA derivatives denoted as ester-TPA and COOH-TPA (Scheme 1), respectively. Owing to their (i) potential high quantum yield and twisted molecular structure for the loose packing of TPA and (ii) benefit for intramolecular charge transfer for the D–π–A motif as well as H-bonding formation ability of –COOH and –COOCH3, using two such simple molecules by attaching –COOH and –COOCH3 on the end branch of TPA together with bridging by benzene ring was a good choice for investigation of the moiety effect on the MCF property.
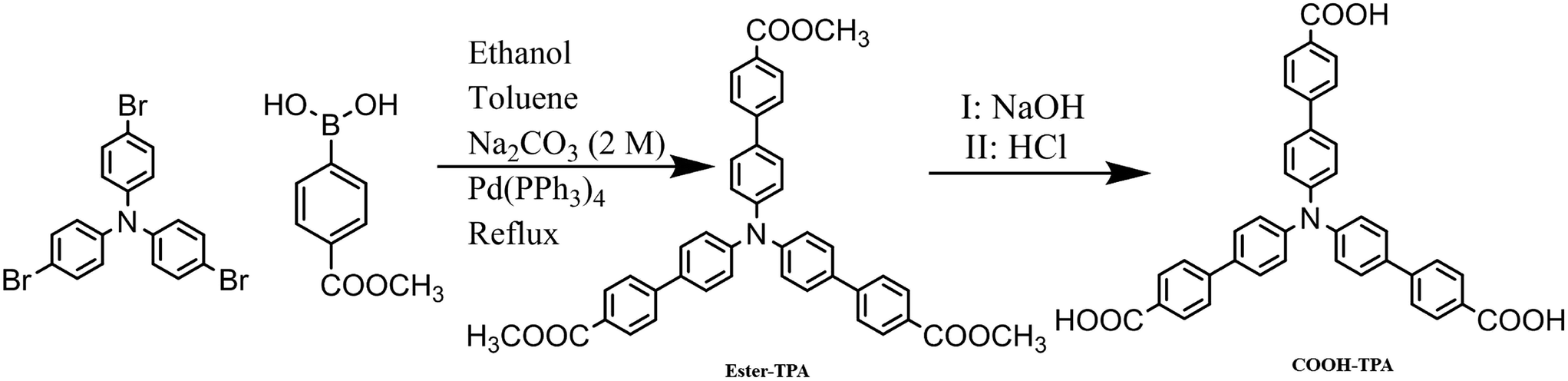 | ||
| Scheme 1 Synthesis route for ester-TPA and COOH-TPA using a Suzuki coupling reaction and hydrolysis. | ||
As expected, they displayed a typical D–π–A structure with an intramolecular charge transfer (ICT) character and showed similar solvatochromic behaviour in different solvents. By utilizing theory calculation, powder X-ray diffraction (PXRD), fluorescent/excitation spectrum, fluorescent lifetime and single-crystal structure analyses, we found that the moiety on the end branch does indeed play an important role in the different performances in terms of the MCF behaviour, whereby (1) for COOH-TPA, we found a relatively obvious red-shift with the CIE coordinate changing from (0.17, 0.35) to (0.26, 0.49) and a decrease in quantum yield from 14.53% to 7.53% after grinding. The decreasing quantum yield of COOH-TPA might be due to the damaged intermolecular hydrogen bond, which weakens the adjacent intermolecular bond so that it increases the non-radiation transition. Crystals of COOH-TPA with different colour emission (blue and green) could be obtained from different solvents (Fig. 2b). In addition, COOH-TPA could respond to ammonia vapour with an accompanying blue-shift of the emission with the spectrum profile broadening due to a deprotonation effect; (Fig. 4) (2) for ester-TPA, we found an increase in the quantum yield from 39.69% to 45.90% with a little red-shift of only about 8 nm after grinding, where the increase in the quantum yield may be ascribed to the increased co-planarity and expanded conjugated system after grinding, with the non-radiation transition then reduced.
Results and discussion
Photo-physical properties in different solvents
The UV-Vis absorption and fluorescence emission spectra of ester-TPA and COOH-TPA in different solvents are shown in Fig. 1. Two distinct absorption bands at ca. 255 nm and ca. 360 nm can be clearly seen for ester-TPA and COOH-TPA, which are attributed to π → π* transitions of the triphenylamine fragment and intramolecular charge transfer (ICT) from the donor of TPA to the acceptor of the moiety on the end branch, respectively (Fig. 1a and d). Fig. 1a and d show that the absorption spectra in different solvents have little difference, suggesting that solvent polarity cannot affect the ground state electronic structures of the two compounds.17b,d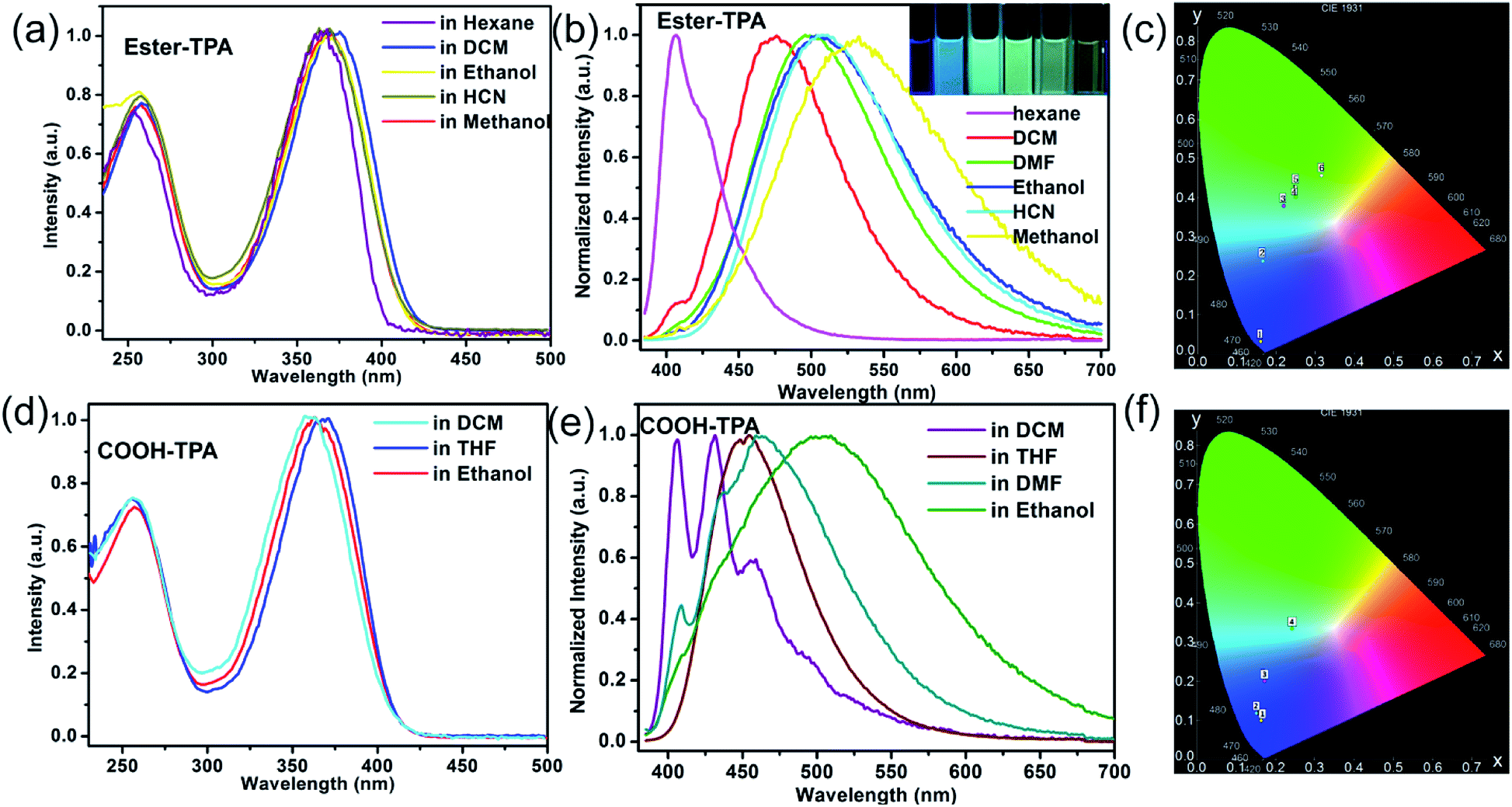 | ||
| Fig. 1 UV-Vis absorption spectra, fluorescence spectra and CIE chromaticity coordinates for ester-TPA (a, b, c) and COOH-TPA (d, e, f) in different solvents. Inset picture of (b) is a photo of ester-TPA excited under a 365 nm UV-hand lamp in the solvents hexane, DCM, DMF, ethanol, HCN, and methanol from left to right. | ||
Fig. 1b shows the fluorescence spectra of ester-TPA in different solvents, namely n-hexane, DCM, DMF, ethanol, HCN, and methanol. With the increasing solvent polarity from n-hexane to methanol, the maximum emission wavelength of ester-TPA displays an obvious red-shift from 405 nm to 532 nm, gradually changing from bluish violet to green-yellow, while the CIE chromaticity coordinates vary from (0.16, 0.03) to (0.32, 0.45) (Fig. 1b and c, and inset picture). In the low polarity solvent n-hexane, the emission spectrum profile is relatively narrow with a weak shoulder peak, while it becomes featureless and broad in highly polar solvents, such as HCN and methanol, because of the dipole–dipole interactions between the solute and solvents, with the full width at half-maximum (FWHM) increasing from 44 nm in n-hexane to 119 nm in ethanol and 138 nm in methanol, which resembles the behaviour observed in linear D–π–A compounds, thus indicating that the emission derives from an excited state with a polar nature.17b
COOH-TPA showed a solvatochromic phenomenon similar to that of ester-TPA. From the florescence spectra of Fig. 1e, we can see that there is a blue emission in DCM with well-resolved structured spectra, with the main peaks at 406 nm and 435 nm accompanied by a shoulder peak at 458 nm. However, in ethanol, COOH-TPA reveals a featureless and broad emission peak at 503 nm (FWHM: 156 nm), with the maximum emission wavelength red-shifted about 78 nm towards that in DCM, and the emission spectra band gradually broadens from dichloromethane to ethanol, with the CIE coordinates changing from (0.16, 0.09) to (0.24, 0.33) (Fig. 1f).
Above all, the absorption profiles and excitation spectra of both compounds varied little in different solvents (Fig. 1a and d, S2 in the ESI†), suggesting a solvent-polarity-independent ground state electronic structures, whereas the emissions exhibit an obvious bathochromic shift (Fig. 1b and e).17b,d The density functional theory (DFT) calculations performed (see more detail in the following theoretical calculation part) and the different distributions in the HOMO and LUMO suggest the formation of the ICT state. Thus, in the excited state, the polar solvents could promote the charge transfer from the donor of the TPA moiety to the acceptor of the small moiety on the end of the branch within the molecule, thereby affecting the HOMO and effectively giving a low-lying energy level, thus leading to a large red-shift of the emission spectra with increasing polarity. Such an ICT state will also be beneficial for color tuning in the solid state under external stimuli.19b
Mechanofluorochromic properties
TPA displays a special 3D propeller-shaped twisted structure due to the SP3 hybrid orbital of the N atom and phenyl rings, which is beneficial for the fluorescence response to external stimuli with tuning of the emission colour or intensity. Fig. 2 shows the MCF properties for both COOH-TPA and ester-TPA. As can be seen from Fig. 2b and the inset picture, grinding the initial powder of COOH-TPA with a mortar leads to a red-shift of the maximum emission peak from 485 nm to 503 nm, with the CIE chromaticity coordinates changing from (0.17, 0.35) to (0.26, 0.49), and subsequently the original emission spectra could be recovered by dropping DCM in to the sample. Furthermore, we could obtain crystals in a mixture of solvents with different ratios of DCM and ethanol, with different emission colours of blue and green peaking at 474 nm and 500 nm, respectively. Although we could not obtain crystals good enough for crystal analysis, from the excitation spectra (Fig. S3 in ESI†) for B-crystal and G-crystal, we could see a difference in the relative intensity in the long wavelength (425 nm for B-crystal and 434 nm for G-crystal) and short wavelength (328 nm for B-crystal and 287 nm for G-crystal), indicating that molecule packing in the solid state with different intermolecular interactions might be caused by a different conjugation of the twisted TPA, which would affect the luminescent property.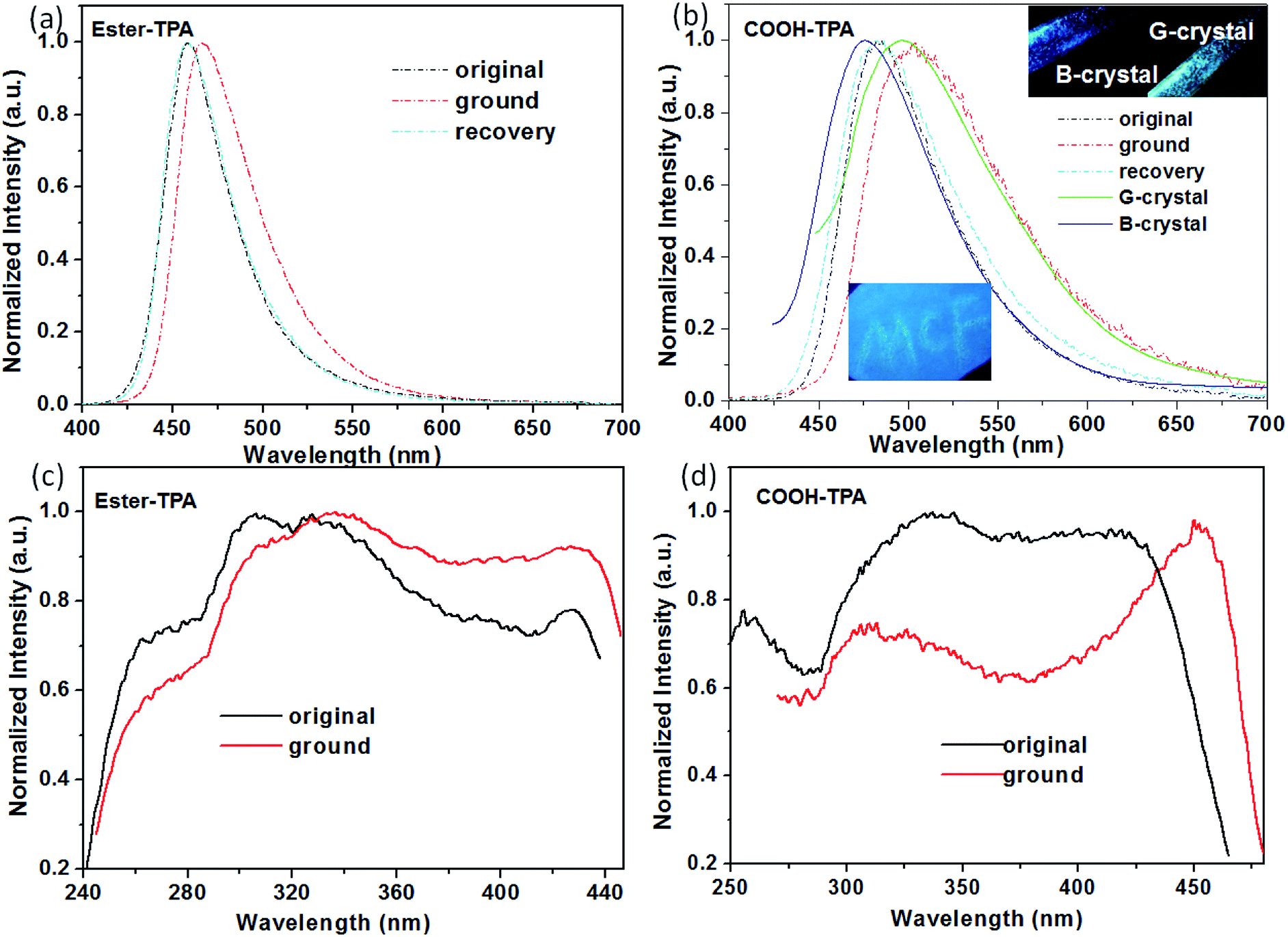 | ||
| Fig. 2 Normalized fluorescence emission (λex = 380 nm) and excitation spectra of the original, ground and recovered powders of ester-TPA (a and c) and COOH-TPA (b and d). | ||
However, by replacing the moiety of –COOH with –COOCH3, the obtained compound of ester-TPA showed only a relatively slight change with ∼8 nm red-shift from 458 to 466 nm, with the CIE chromaticity coordinates changing from (0.15, 0.12) to (0.14, 0.19) according to the original powder after grinding (Fig. 2a), indicating that the end moiety on the branch of the star-shaped TPA plays an important role in the MCF properties for the different fluorescent response ability of colour tuning towards external stimuli.
We also observed a difference in the MCF property for COOH-TPA and ester-TPA, which was also reflected in the fluorescent spectrum profile directly and showed a significantly decreased intensity accompanying an obvious red-shift for the ground sample of COOH-TPA, while an increased intensity and relatively smaller red-shift for the ground powder of ester-TPA (see Fig. S1 in the ESI,† Table 1). The pure fluorescent quantum yield for both the original powder and the ground powder of COOH-TPA was obtained by integrating sphere, showing a decrease from 14.53% to 7.56% upon grinding. In contrast, the emission of ester-TPA was enhanced, with the quantum yield increasing from 39.69% to 45.90%.
| τ1 (ns) | τ2 (ns) | τ1 Rel.% | τ2 Rel.% | χ2 | τ (ns) | λmax | ΦF (%) | Kr (s−1) | Knr (s−1) | |
|---|---|---|---|---|---|---|---|---|---|---|
| a ΦF = fluorescence quantum yield determined using a calibrated integrating sphere, τ = lifetime, Kr = radiative transition rate constant (Kr = ΦF/τ), Knr = non-radiative transition rate constant (Knr = (1 − ΦF)/τ). | ||||||||||
| Ester-TPA | 1.61 | 3.05 | 78.90 | 21.10 | 1.032 | 1.91 | 458 | 39.69 | 2.08 × 108 | 3.16 × 108 |
| Ester-TPA grinding | 2.11 | 6.68 | 98.98 | 1.02 | 0.925 | 2.16 | 466 | 45.90 | 2.13 × 108 | 2.51 × 108 |
| COOH-TPA | 1.12 | 2.30 | 69.31 | 30.69 | 0.848 | 1.48 | 485 | 14.53 | 9.82 × 107 | 5.78 × 108 |
| COOH-TPA grinding | 1.07 | 2.84 | 75.07 | 24.93 | 0.931 | 1.51 | 503 | 7.56 | 5.01 × 107 | 6.12 × 108 |
For better understanding the moiety effect on the MCF property, we investigated the PL performance together with the excitation spectrum, fluorescent lifetime and PXRD of the original and ground samples of the two compounds (Fig. 2c and d, 3, S4 and 5 in the ESI†). Knr and Kr were calculated from the fluorescent quantum yield and lifetime, and the summary data are shown in Table 1. After grinding COOH-TPA, the non-radiation transition rate increased from 5.78 × 108 s−1 to 6.12 × 108 s−1, while the radiation transition rate decreased from 9.82 × 107 s−1 to 5.01 × 107 s−1, which might be due to (1) the damaged intermolecular hydrogen bond caused by the mechanical force, which will be beneficial for free rotation of the benzene ring, with more excitation energy wasted in vibration. This can also be proven by the PXRD results, where it was found to turn into an amorphous phase, with H-bonding disrupting after grinding, but could be recovered into the crystalline state again with the H-bonding framework reforming by dropping DCM (Fig. 3b); (2) a more planar conformation versus twisted core will be beneficial for enhanced π–π interaction, leading to emission quenching, which is consistent with the obvious red-shift of the excitation spectrum from 422 nm to 452 nm (Fig. 2d).
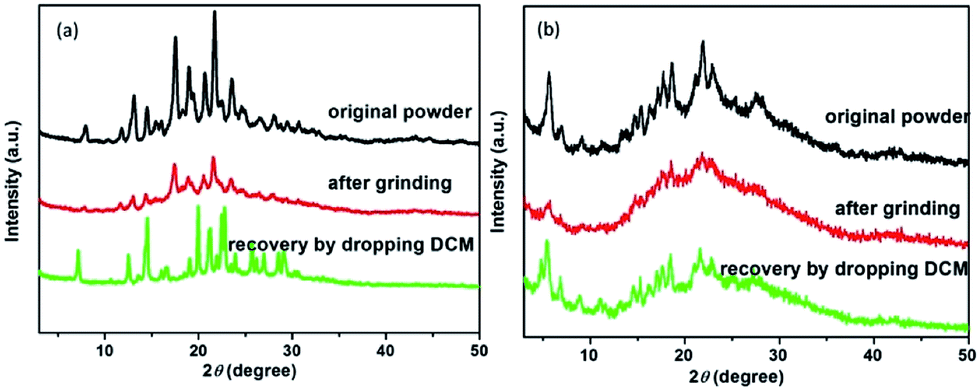 | ||
| Fig. 3 PXRD patterns of the original powder, ground and recovered by dropping into organic solvents the compounds ester-TPA (a) and COOH-TPA (b). | ||
For ester-TPA, the increased quantum yield of the ground powder may originate from the extended conjugation of the entire system being more efficient for emission, with the non-radiation transition rate decreasing from 3.16 × 108 s−1 to 2.51 × 108 s−1 (Table 1). In addition, there was nearly no shift of the excitation spectrum for the ground powder of ester-TPA, which prevents the strong π–π interaction induced emission quenching compared to that of COOH-TPA, and this is also consistent with the increased quantum yield and increased fluorescence lifetime from 1.91 to 2.16 ns after grinding.
The above results show a large difference between ester-TPA and COOH-TPA in terms of the MCF property despite there being only a small difference in moiety replacement. The mechanism of mechanochromism could be beautifully explained by powder X-ray diffraction (PXRD) in Fig. 3, which shows some difference in behaviour between the original powder for the ground sample of ester-TPA and that for COOH-TPA. After grinding, little changes in the patterns for ester-TPA can be observed with a well-resolved peak, while in contrast an obvious transformation into the amorphous state for COOH-TPA can be observed. This is consistent with the MCF property whereby ester-TPA showed just a little change in the maximum wavelength, while COOH-TPA showed an obvious colour tuning from blue to green. COOH-TPA undergoes an orderly accumulation in the crystal state, linked with the hydrogen bonds,14 which suppresses intramolecular vibration and rotation of the phenyl ring, reducing the non-radiative relaxation of the excited states, thus leading to a relatively strong fluorescence intensity. However, when the ordered crystal structure of COOH-TPA is destroyed by grinding, with the crystal phase transformed into the amorphous phase, and the close intermolecular packing is damaged accordingly, it puts the molecule in a poorly organized state, leading to a low quantum yield and an obvious red-shift of the emission spectrum.19b The original powder of ester-TPA exhibits much more intense and sharp diffraction peaks, yet the ground powders show relatively little change in the XRD pattern with relatively well-resolved diffraction, reflecting that molecular packing from the regular crystalline nature changes to a relatively disordered state, which could also be proven by the crystal analysis described in the following section. And it is reversible by dropping the dichloromethane into ground powder, the weak or disappeared peaks are gradually restored or even enhanced, implying the recovery to the initial ordered crystalline state. The much stronger mechanochromism of COOH-TPA compared with that of ester-TPA implies that it does make a difference to their MCF property by tuning the stacking mode with different intermolecular interactions caused by the replacement with the different moieties of –COOH and –COOCH3 on the star-shaped TPA.
Response to ammonia vapour
Analysis of the structure of COOH-TPA showed that there are three carboxylic acid units on the periphery of each molecule. The carboxylic acid unit always has protonated or deprotonated activity when placed in an acidic or alkaline environment. As is shown in Fig. 4, when it was fumed with ammonia, the fluorescence spectrum showed a slight blue-shift accompanying band widening of the emission spectrum. This change could be obviously observed on the CIE chromaticity coordinates from (0.17, 0.35) to (0.23, 0.32) (Fig. 4b). This may be attributed to the deprotonation of COOH-TPA, which exists as an alkaline structure when fumed with ammonia, which may weaken the adjacent intermolecular hydrogen bond, leading to relative conjugation reduction of the entire system, thus inhibiting effective charge transfer from triphenylamine to carboxylic acid, which can be reflected in a slight emission peak blue-shift.19c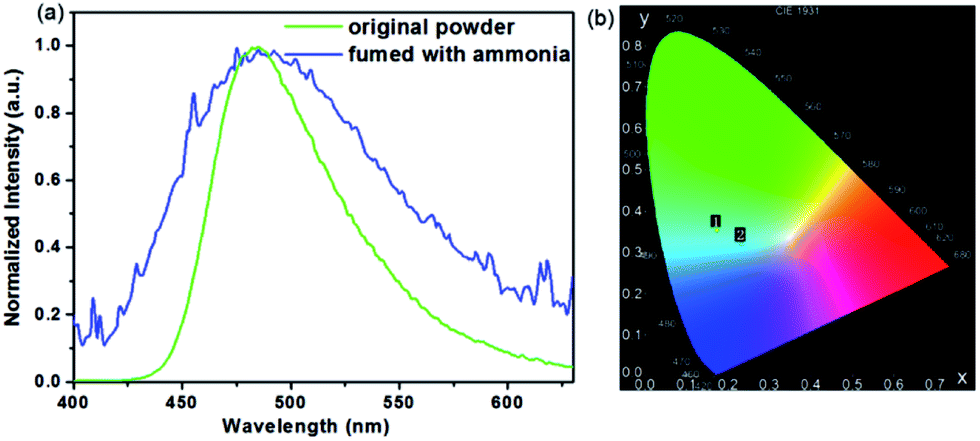 | ||
| Fig. 4 Normalized spectrum (a) and CIE coordinates (b) of COOH-TPA for the original powder and that fumed with ammonia. | ||
Crystal analysis
Yellow-green needle-like crystals of ester-TPA were obtained by slow evaporation of the dichloromethane solution at room temperature for one week. Single-crystal X-ray diffraction revealed that ester-TPA molecules were crystallized in the monoclinic system P21/c space group. As shown in Fig. 5b–f, there are three types of interactions observed, including (1) two types of C–H⋯π interactions (i) with a H atom from the methyl of the ester moiety linked in one row (3.14 Å) or between rows (2.88 Å) (Fig. 5f) or (ii) with a H atom from a benzene ring within a distance of 2.87–3.66 Å (Fig. 1c); (2) C–H⋯O interactions with a distance of 2.54 Å linked by H from methyl or benzene ring to the carbonyl of the ester group with distances of 2.54 Å and 2.49 Å, respectively; (3) as expected, π–π interactions are effectively suppressed owing to the twisted structure of TPA, such that there is only one type of π–π interaction between the phenyl ring of two adjacent molecule with a distance of 3.84 Å. Usually, the “A” group of one molecule is often located on the “D” group of another molecule in a dipole D–π–A molecule, which is beneficial for stability of the crystalline state. Those molecules with an “A” group placed on a “D” group are relatively rare as the D–π–A dipole–dipole interactions are mutually repulsive, which is sufficient for intermolecular excitonic coupling with quenched fluorescence.15b Such is the case for ester-TPA in this study (Fig. 5f); on the other hand, it displays a twisted structure with phenyls in the molecule adopting a different orientation (Fig. 5a). Thus, the net formed by the H-bonding/π–π interaction motif might be beneficial for a loose packing mode and interestingly, it showed an “off–on” mode, with the fluorescent quantum yield increased from 39.69% to 45.90% after grinding. This might be due to the more planar conjugation of the molecule after grinding, blocking the non-radiative deactivation pathways, which can also be proven by the lifetime and quantum yield measurement, with Knr decreasing from 3.16 × 10−8 to 2.51 × 10−8. Crystals with different emission colours of COOH-TPA (blue and green) were obtained in different ratio mixtures of DCM and ethanol (Fig. 2b and the inset picture); unfortunately, they were too small for measurement; however, it can be proven that the different moiety on the branch may affect the packing mode, thus giving a different mechanochromism behaviour.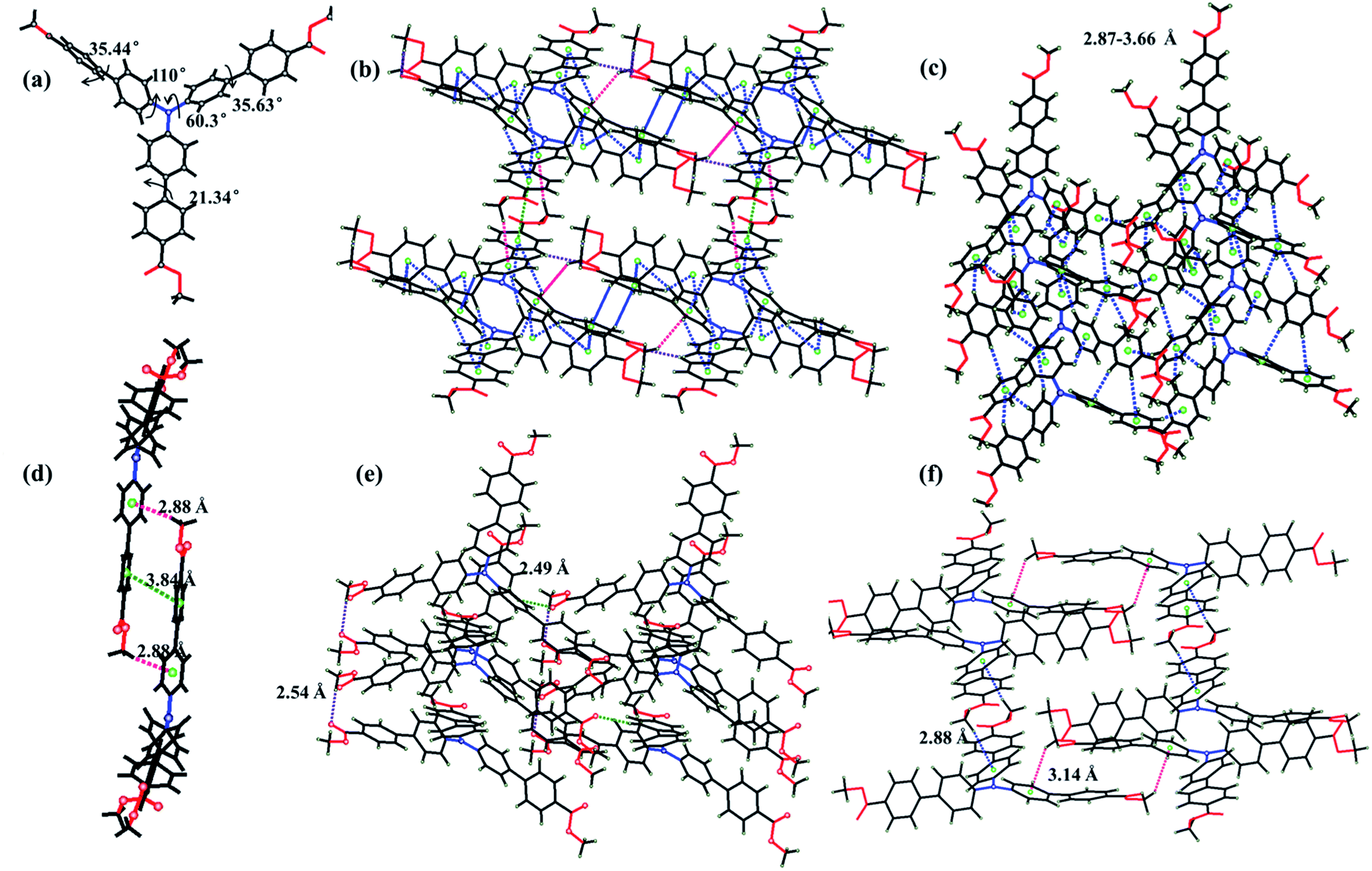 | ||
| Fig. 5 (a) Twisted structure of ester-TPA, (b) full analysis of the molecule interaction in the crystal structure, including π–π, C–H⋯π and C–H⋯O interactions, (c) clarified analysis of C–H⋯π interactions in which H is from the benzene ring, (d) π–π interactions in the crystal structure, (e) clarified analysis of C–H⋯O interactions, (f) clarified analysis of C–H⋯π interactions in which H is from the methyl. | ||
Theoretical calculations
To obtain further insights into the fluorescent property of ester-TPA and COOH-TPA, density functional theory (DFT) calculations were performed via Gaussian 09w at the B3LYP/6-31G level, using the structure of a single-crystal of ester-TPA obtained from the X-ray diffraction crystallographic data and the optimized molecule geometry of COOH-TPA obtained from Chemdraw 3D.Fig. 6 shows the spatial electron distributions of ester-TPA and COOH-TPA. In the highest occupied molecular orbital (HOMO), the electrons are mainly located on the electron-donating triphenylamine moiety and there is relatively little distribution on the π bridge of the benzene ring; However, in the lowest unoccupied molecular orbital (LUMO), the electrons are mainly located on the two branches of the star-shaped molecule, including the π bridge of the benzene ring and the moiety of –COOH and –COOCH3. For both of the compounds, the appropriate separation between HOMO and LUMO indicated that a substantial charge transfer from the donor of TPA to the acceptor of the small moiety on the end of the branch occurs within the molecule when the molecule is excited, which agrees with the absorption spectra and emission wavelength red-shift with increasing the polarity of the solvent, as shown in Fig. 1. In addition, grinding the compounds with a twisted structure may increase the ICT process as indicated from Fig. 6, thus leading to the red-shift of the maximum wavelength for both compounds.18b
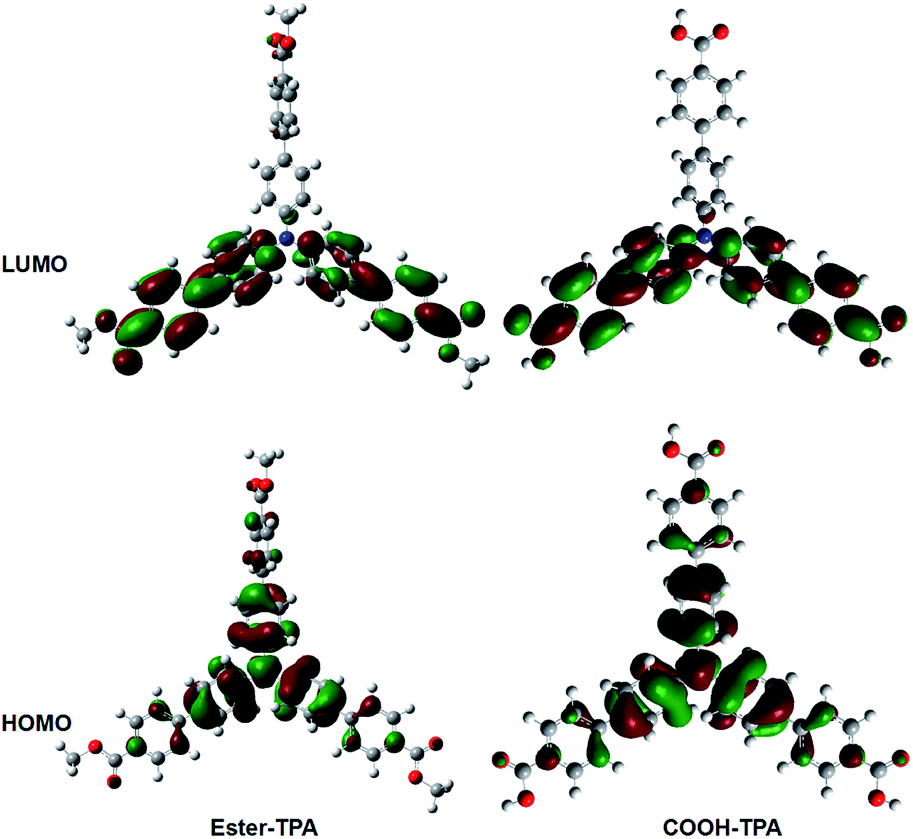 | ||
| Fig. 6 Spatial electron distributions of the HOMO and LUMO of ester-TPA and COOH-TPA. | ||
Experimental
Materials and instruments
All the solvents and chemicals were purchased from Alfa Aesar or TCI and used without further purification. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III-600 MHz spectrometer, using DMSO-d6 as the solvent and tetramethyl-silane (TMS) as the internal standard (1H NMR and 13C NMR spectra can be seen in Fig. S5 and 6 in the ESI†). Absorption spectra were recorded using a Cary 50 Bio UV-visible spectrometer with the samples in solution and a quartz cuvette (path length 1 cm). The steady state photoluminescence (PL) curves were measured with an Edinburgh Instruments FLS980 device, and the time-resolved photoluminescence (TRPL) decay curves were measured under the excitation of a hydrogen flash lamp with the wavelength at 377 nm (nF920, Edinburgh Instruments). Absolute fluorescence quantum yields in solid powder state (ΦF) were determined using an integrating sphere on Edinburgh Instruments FLS980. The powder X-ray diffraction (PXRD) patterns were recorded on Rigaku Ultima IV with the 2θ range from 2° to 50°. The single-crystal structure was measured with a Bruker D8 Venture X-ray diffraction system equipped with a CCD detector using graphite-monochromated Mo Kα radiation.Synthesis
The two compounds in this study have been previously reported as ligands for a metal–organic framework20 and here they were synthesized using the same methods reported previously in literature. Tris-(4′-carbomethoxybiphenyl)amine (ester-TPA) was synthesized by a Pd-catalyzed Suzuki coupling between tris-(4-bromophenyl)amine and 4-(methoxycarbonyl)phenylboronic acid, and then tris-(4-carboxybiphenyl)amine (COOH-TPA) was synthesized followed by a base-catalyzed hydrolysis reaction (shown in Scheme 1).Synthesis of tris-(4′-carbomethoxybiphenyl)amine (ester-TPA)
A mixture of tris-(4-bromophenyl)amine (2.41 g, 5 mmol), (4-(methoxycarbonyl)phenyl)boronic acid (3.6 g, 20 mmol), Pd(PPh3)4 (0.29 g, 0.25 mmol), aqueous Na2CO3 (2 M, 18 mL) in toluene (36 mL) and ethanol (12 mL) was heated to reflux under argon atmosphere for 24 h. The mixture was cooled to room temperature and extracted with dichloromethane. The extracts were dried with anhydrous Na2SO4 and concentrated by rotary evaporation. The residue was purified by column chromatography (petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 = 1:1) to obtain the pure product as a pale yellow powder. Yield: 3 g, 90%. 1H NMR (600 MHz, DMSO) δ 8.04 (d, J = 8.2 Hz, 6H), 7.81 (dd, J = 42.0, 8.4 Hz, 12H), 7.23 (d, J = 8.5 Hz, 6H), 3.88 (s, 9H). 13C NMR (151 MHz, DMSO) δ 166.33, 130.33(s), 128.75 (s), 126.90 (s), 124.84 (s), 55.40 (s).
:CH2Cl2 = 1:1) to obtain the pure product as a pale yellow powder. Yield: 3 g, 90%. 1H NMR (600 MHz, DMSO) δ 8.04 (d, J = 8.2 Hz, 6H), 7.81 (dd, J = 42.0, 8.4 Hz, 12H), 7.23 (d, J = 8.5 Hz, 6H), 3.88 (s, 9H). 13C NMR (151 MHz, DMSO) δ 166.33, 130.33(s), 128.75 (s), 126.90 (s), 124.84 (s), 55.40 (s).
Synthesis of tris-(4′-carboxybiphenyl)amine (COOH-TPA)
Tris-(4′-carbomethoxybiphenyl)amine (1.30 g) was added to a mixture of aqueous NaOH (1 M, 60 mL) and THF (60 mL), and the mixture was refluxed for 6 h. The solution was cooled to room temperature and the solvent was removed under vacuum evaporation; then, some H2O was added into the residue. The obtained yellow clear solution was stirred at room temperature for 2 h. Then, the pH value was adjusted to ∼2 using dilute HCl. The resulting yellow solid was collected by filtration, washed with water, and then dried under vacuum. Yield: 1.0 g, 82%. 1H NMR (600 MHz, DMSO) δ 12.93 (s, 3H), 8.01 (d, J = 7.8 Hz, 6H), 7.78 (dd, J = 28.7, 7.4 Hz, 12H), 7.21 (d, J = 7.3 Hz, 6H). 13C NMR (151 MHz, DMSO) δ 167.66 (s), 147.26 (s), 143.99 (s), 134.16 (s), 130.47 (s), 129.79 (s), 128.69 (s), 126.73 (s), 124.81 (s).Conclusion
In this study, we designed and synthesized TPA-based derivatives of ester-TPA and COOH-TPA and investigated the moiety effect on the luminescent property in both solution and the solid state. The experiment results revealed the existence of intramolecular charger transfer (ICT) from TPA to the carboxyl or ester moiety on the end branch, which could also be confirmed by the orbital separation of HOMO and LUMO. By attaching different moieties of COOH– or –COCH3 to TPA, COOH-TPA showed an obvious change of emission colour, with CIE coordinates from (0.17, 0.35) to (0.26, 0.49), together with the quantum yield decreasing from 14.53% to 7.53%, which is different from ester-TPA, exhibiting only an 8 nm red-shift after grinding, but with the quantum yield increasing from 36.69% to 45.90%. The emission could be recovered to the original state by dropping DCM into the ground sample. In different ratio mixtures of DCM and ethanol, we obtained crystal of COOH-TPA with different colour emission (B-crystal and G-crystal). Furthermore, COOH-TPA could respond to ammonia vapour with an emission blue-shift and broadening, with CIE coordinates changing from (0.17, 0.35) to (0.23, 0.32) owing to the deprotonation effect. By solid-state fluorescence, lifetime, XRD, and single-crystal structure analyses, we found that the mechanism of the MCF for COOH-TPA was the transformation from the crystal state to a disordered amorphous state. The increased quantum yield of ester-TPA after grinding may be attributed to the relative planarity conjugate configuration, which is beneficial for emission, with a reduction of the non-radiation transition rate. However, the decrease in the quantum yield of COOH-TPA may be ascribed to the destroyed intermolecular H-bonding, leading to strong π–π interactions, along with the reduction of the radiation transition rate and increase of the non-radiation transition. In summary, the experiment results proved that the moiety on the end branch does play an important role in the emission property, and this insight will help for designing MCFs with better emission performance.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (Project No. 21602127), and Natural Science Foundation of Shanxi Province (206626901001), Technological Innovation Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (206548901022), Scientific Research Startup Funds of Shanxi University (020354010, 231545003). We thank Scientific Instrument Center of Shanxi University for NMR and MS measurements.References
- (a) Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372 RSC; (b) K. Ariga, T. Mori and J. P. Hill, Adv. Mater., 2012, 24, 158 CrossRef CAS PubMed; (c) Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811 CrossRef CAS PubMed; (d) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376 RSC; (e) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS PubMed.
- (a) W. E. Lee, C. L. Lee, T. Sakaguchi, M. Fujiki and G. Kwak, Chem. Commun., 2011, 47, 3526 RSC; (b) B. M. McKenzie, R. J. Wojtecki, K. A. Burke, C. Y. Zhang, A. Jakli, P. T. Mather and S. J. Rowan, Chem. Mater., 2011, 23, 3525 CrossRef CAS.
- (a) A. L. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641 CrossRef CAS PubMed; (b) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044 CrossRef CAS PubMed; (c) S. Perruchas, X. F. Le Goff, S. Maron, I. Maurin, F. Guillen, A. Garcia, T. Gacoin and J. P. Boilot, J. Am. Chem. Soc., 2010, 132, 10967 CrossRef CAS PubMed; (d) G. G. Shan, H. B. Li, H. T. Cao, D. X. Zhu, P. Li, Z. M. Su and Y. Liao, Chem. Commun., 2012, 48, 2000 RSC.
- (a) W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Lam, X. Y. Shen, C. Feng, H. H. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837 CrossRef CAS PubMed; (b) C. Y. K. Chan, J. W. Y. Lam, Z. Zhao, S. Chen, P. Lu, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4320 RSC; (c) B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Chem. Sci., 2015, 6, 3236 RSC; (d) L. Zhao, Y. Lin, T. Liu, H. Li, Y. Xiong, W. Z. Yuan, H. H. Y. Sung, I. D. Williams, Y. Zhang and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 4903 RSC; (e) Y. Wang, I. Zhang, B. Yu, X. Fang, X. Su, Y. M. Zhang, T. Zhang, B. Yang, M. Li and S. X. A. Zhang, J. Mater. Chem. C, 2015, 3, 12328 RSC; (f) C. Ma, X. Zhang, Y. Yang, Z. Ma, L. Yang, Y. Wu, H. Liu, X. Jia and Y. Wei, J. Mater. Chem. C, 2016, 4, 4786 RSC; (g) Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005 CrossRef CAS.
- (a) Y. Sagara and T. Kato, Angew. Chem., 2011, 123, 9294 CrossRef; (b) Y. Dong, J. Zhang, X. Tan, L. Wang, J. Chen, B. Li, L. Ye, B. Xu, B. Zou and W. Tian, J. Mater. Chem. C, 2013, 1, 7554 RSC; (c) C. Niu, Y. You, L. Zhao, D. He, N. Na and J. Ouyang, Chem.–Eur. J., 2015, 21, 13983 CrossRef CAS PubMed; (d) K. C. Naeem, A. Subhakumari, S. Varughesec and V. C. Nair, J. Mater. Chem. C, 2015, 3, 10225 RSC; (e) Y. Matsunaga and J. S. Yang, Angew. Chem., Int. Ed., 2015, 54, 7985 CrossRef CAS PubMed.
- J. Sun, X. Lv, P. Wang, Y. Zhang, Y. Dai, Q. Wu, M. Ouyang and C. Zhang, J. Mater. Chem. C, 2014, 2, 5365 RSC.
- (a) G. Q. Zhang, J. W. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS PubMed; (b) X. X. Sun, X. P. Zhang, X. Y. Li, S. Y. Liu and G. Q. Zhang, J. Mater. Chem., 2012, 22, 17332 RSC; (c) G. R. Krishna, M. S. R. N. Kiran, C. L. Fraser, U. Ramamurty and C. M. Reddy, Adv. Funct. Mater., 2013, 23, 1422 CrossRef CAS; (d) Z. Zhang, P. Xue, P. Gong, G. Zhang, J. Peng and R. Lu, J. Mater. Chem. C, 2014, 2, 9543 RSC; (e) Z. Zhang, Z. Wu, J. Sun, B. Yao, G. Zhang, P. Xue and R. Lu, J. Mater. Chem. C, 2015, 3, 4921 RSC; (f) R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2015, 21, 7231 CrossRef CAS PubMed; (g) Z. Zhang, Z. Wu, J. Sun, B. Yao, P. Xue and R. Lu, J. Mater. Chem. C, 2016, 4, 2854 RSC; (h) F. Qi, J. Lin, X. Wang, P. Cui, H. Yan, S. Gong, C. Ma, Z. Liu and W. Huang, Dalton Trans., 2016, 7278 RSC; (i) Z. Zhang, Z. Wu, J. Sun, P. Xue and R. Lu, RSC Adv., 2016, 6, 43755 RSC; (j) D. E. Wu, X. L. Lu and M. Xia, New J. Chem., 2015, 39, 6465 RSC.
- (a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 1605 CrossRef PubMed; (b) E. Nagata, S. Takeuchi, T. Nakanishi, Y. Hasegawa, Y. Mawatari and H. Nakano, ChemPhysChem, 2015, 16, 3038 CrossRef CAS PubMed; (c) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520 CrossRef CAS PubMed; (d) Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 39, 12268 CrossRef PubMed.
- (a) S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed; (b) Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009 RSC; (c) W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H. Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837 CrossRef CAS PubMed; (d) X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334 CrossRef CAS; (e) Y. Xu, K. Wang, Y. Zhang, Z. Xie, B. Zou and Y. Ma, J. Mater. Chem. C, 2016, 4, 1257 RSC; (f) C. Feng, K. Wang, Y. Xu, L. Liu, B. Zou and P. Lu, Chem. Commun., 2016, 52, 3836 RSC.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. V. Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS PubMed.
- G. Li, F. Song, D. Wu, J. Lan, X. Liu, J. Wu, S. Yang, D. Xiao and J. You, Adv. Funct. Mater., 2014, 24, 747 CrossRef CAS.
- (a) M. Tanioka, S. Kamino, A. Muranaka, Y. Ooyama, H. Ota, Y. Shirasaki, J. Horigome, M. Ueda, M. Uchiyama, D. Sawada and S. Enomoto, J. Am. Chem. Soc., 2015, 137, 6436 CrossRef CAS PubMed; (b) L. Zhu, X. Li, Q. Zhang, X. Ma, M. Li, H. Zhang, Z. Luo, H. Ågren and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 5175 CrossRef CAS PubMed; (c) J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956 CrossRef CAS PubMed; (d) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS PubMed; (e) Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., 2015, 127, 6368 CrossRef; (f) R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Li and T. Yi, Chem. Sci., 2014, 5, 3922 RSC.
- G. Fan and D. Yan, Sci. Rep., 2014, 4, 4933 CrossRef CAS PubMed.
- (a) E. Shi, J. He, H. Zhuang, H. Liu, Y. Zheng, H. Li, Q. Xu, J. Zheng and J. Lu, J. Mater. Chem. C, 2016, 4, 2579 RSC; (b) Y. Zhang, S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, Z. C. Wen, G. Q. Zhang, S. T. Lee, H. L. Kwong and C. S. Lee, J. Mater. Chem., 2011, 21, 8206 RSC; (c) H. Choi, H. M. Ko and J. Ko, Dyes Pigm., 2016, 126, 179 CrossRef CAS; (d) Y. Zhang, S. L. Lai, Q. X. Tong, M. Y. Chan, Z. C. Wen, J. He, K. S. Jeff, X. L. Tang, W. M. Liu, C. C. Ko, P. F. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61 CrossRef.
- (a) H. Sun, Y. Zhang, W. Yan, W. Chen, Q. Lan, S. Liu, L. Jiang, Z. Chi, X. Chen and J. Xu, J. Mater. Chem. C, 2014, 2, 5812 RSC; (b) Y. Zhang, J. Sun, G. Zhuang, M. Ouyang, Z. Yu, F. Cao, G. Pan, P. Tang, C. Zhang and Y. Ma, J. Mater. Chem. C, 2014, 2, 195 RSC.
- (a) J. Wei, B. Liang, X. Cheng, Z. Zhang, H. Zhang and Y. Wang, RSC Adv., 2015, 5, 71903 RSC; (b) P. S. Hariharan, D. Moon and S. P. Anthony, J. Mater. Chem. C, 2015, 3, 8381 RSC.
- (a) V. S. Padalkar, D. Sakamaki, K. Kuwada, N. Tohnai, T. Akutagawa, K. Sakaid and S. Seki, RSC Adv., 2016, 6, 26941 RSC; (b) P. Xue, P. Chen, J. Jia, Q. Xu, J. Sun, B. Yao, Z. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569 RSC; (c) K. Wang, S. Huang, Y. Zhang, S. Zhao, H. Zhang and Y. Wang, Chem. Sci., 2013, 4, 3288 RSC; (d) Y. Gong, Y. Zhang, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Phys. Chem. C, 2014, 118, 10998 CrossRef CAS.
- (a) Y. Zhang, H. Li, G. Zhang, X. Xu, L. Kong, X. Tao, Y. Tian and J. Yang, J. Mater. Chem. C, 2016, 4, 2971 RSC; (b) Y. Zhang, K. Wang, G. Zhuang, Z. Xie, C. Zhang, F. Cao, G. Pan, H. Chen, B. Zou and Y. Ma, Chem.–Eur. J., 2015, 21, 2474 CrossRef CAS PubMed; (c) C. Feng, K. Wang, Y. Xu, L. Liu, B. Zou and P. Lu, Chem. Commun., 2016, 52, 3836 RSC.
- (a) Q. Chen, L. Kong, Y. Tian, X. Xu, L. M. Yang, G. Zhang, W. Jia and J. Yang, RSC Adv., 2014, 4, 18981 RSC; (b) P. S. Hariharan, N. S. Venkataramanan, D. Moon and S. P. Anthony, J. Phys. Chem. C, 2015, 119, 9460 CrossRef CAS; (c) L. Kong, Y. Liu, H. Wang, X. Tian, Q. Chen, Y. Tian, S. Li, Z. Xue and J. Yang, J. Mater. Chem. C, 2016, 4, 2990 RSC; (d) P. S. Hariharan, N. S. Venkataramanan, D. Moon and S. P. Anthony, J. Mater. Chem. C, 2016, 4, 2971 RSC.
- D. Shi, Y. Ren, H. Jiang, B. Cai and J. Lu, Inorg. Chem., 2012, 51, 6498 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1507841. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra03123h |
| This journal is © The Royal Society of Chemistry 2017 |