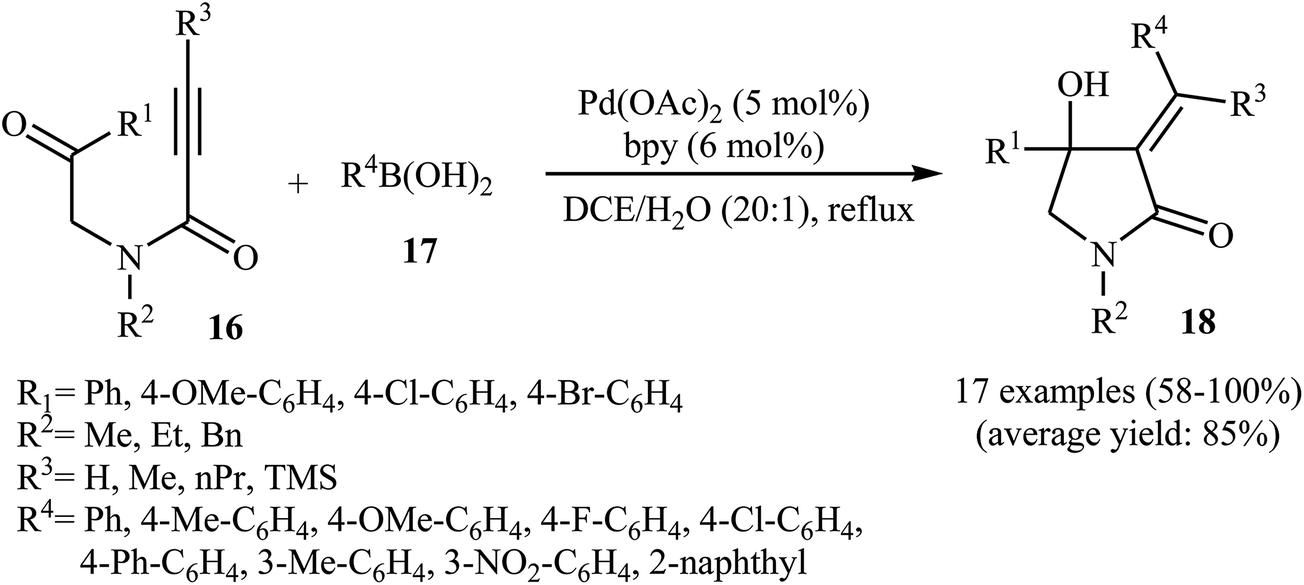DOI:
10.1039/C7RA03075D
(Review Article)
RSC Adv., 2017,
7, 28407-28418
Intramolecular cyclization of N-allyl propiolamides: a facile synthetic route to highly substituted γ-lactams (a review)
Received
15th March 2017
, Accepted 4th May 2017
First published on 30th May 2017
Abstract
The development of simple and efficient methods for construction of substituted γ-lactams is an important synthetic goal because such ring skeletons are present in numerous natural compounds that display diverse biological activities. Intramolecular cyclization of N-allyl propiolamides is an efficient, economic, and operationally simple strategy for the synthesis of the titled compounds. In the present review we will discuss recent advances in the synthesis of functionalized γ-lactam derivatives from these easily accessible and versatile building blocks with the emphasis on the mechanistic aspects of the reactions.
 Somayeh Soleimani-Amiri | Somayeh Soleimani-Amiri was born in Tehran, Iran, in 1975. She received her B.S. degree in Pure Chemistry from Shahid Beheshti University, Tehran, Iran, and her M.S. degree in organic chemistry from Alzahra University, Tehran, Iran, in 2002 under the supervision of Prof. S. H. Abdi Oskooie and Prof. M. M. Heravi. She completed his Ph.D. degree in 2009 under the supervision of Prof. M. Z. Kassaee. Now she is working at Karaj Branch, Islamic Azad University as Assistant Professor. Her research interests include Computational Organic Chemistry, Nano Chemistry, Synthesis of Organic Compounds. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor of Organic Chemistry. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
 Mirzaagha Babazadeh | Mirzaagha Babazadeh is Associate Professor of Organic Chemistry in Department of Chemistry at Tabriz Branch, Islamic Azad University, Tabriz, Iran. He was born in Tabriz, Iran, in 1973. He received his B.S. degree in Applied Chemistry (1996) and his M.S. degree in Organic Chemistry from University of Tabriz, Tabriz, Iran (1999) under the supervision of Prof. A. A. Entezami and Prof. H. Namazi. He completed his Ph.D. degree under the supervision of Prof. K. D. Safa in University of Tabriz, Tabriz, Iran (2005). His research interests focus on organic synthesis, catalyst, polymer, green chemistry, drug delivery systems, organosilicon compounds and nanocomposites. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed his Ph.D. degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Assistant Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
 Ladan Edjlali | Ladan Edjlali was born in Tabriz, Iran, in 1960. She received her B.S. degree in Applied Chemistry from University of Tabriz, Iran, and her M.S. degree in organic chemistry from University of Tabriz, Tabriz, Iran, in 1993 under the supervision of Prof. Y. Mirzaei. She completed his Ph.D. degree in 2000 under the supervision of Prof. Y. Mirzaei and Prof. S. M. Golabi. Now, she is working at Islamic Azad University, Tabriz Branch as Associate Professor. Her research interests include organic synthesis and new methodologies in organic synthesis. |
1. Introduction
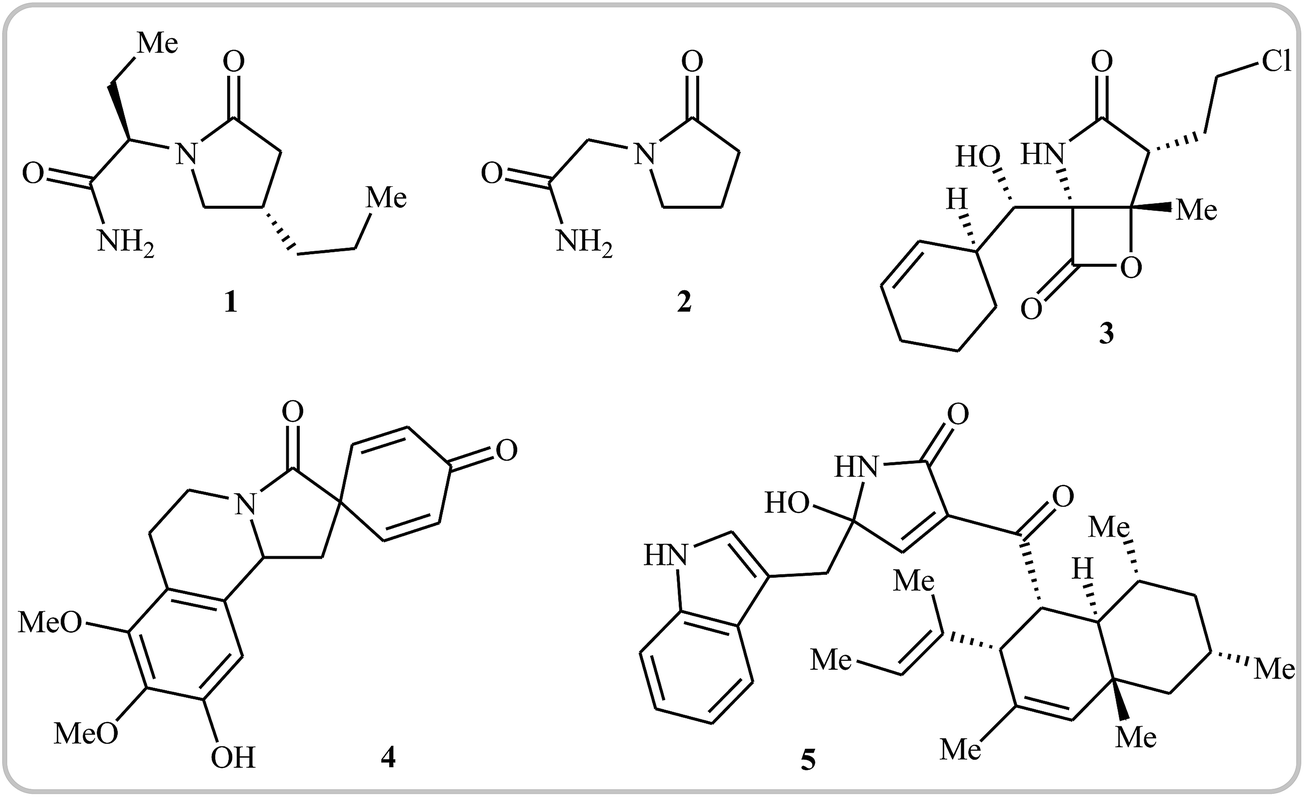
Needless to say, γ-lactams are at the heart of a number of medicinally and biologically important compounds, as well as a large array of natural products. For instance, brivaracetam 1, with the brand name Briviact, is an anticonvulsant drug marketed worldwide for the treatment of partial onset seizures.1 Piracetam 2 (also known as a smart drug) is a nootropic supplement that has the ability to enhance memory and learning ability. It also used as an adjunctive treatment for myoclonus of cortical origin.2 Marizomib 3 is a naturally-occurring salinosporamide, isolated from the marine actinomycete Salinospora tropica. This compound is a promising drug candidate for the treatment of multiple myeloma and mantle cell lymphoma.3,4 Annosqualine 4, which has good antibacterial activity, is a natural product isolated from the stems of Annona Squamosa.5,6 Codinaeopsin 5, which was isolated from an endophytic fungus collected in Costa Rica, has significant antimalarial activity.7 This biological activity has made the synthesis of γ-lactams quite attractive, and a large number of straightforward and robust methods for the construction of these cores has been established.8–12 More recently, Rivas and Ling published an interesting review paper that covers most of the recent advances in the synthesis of the titled compounds.12 However, synthesis of these cores through intramolecular cyclization of simple and easily accessible N-allyl propiolamide derivatives was omitted, while this synthetic strategy has recently attracted much attention because of its high efficiency, atom economy, and operational simplicity (Fig. 1).
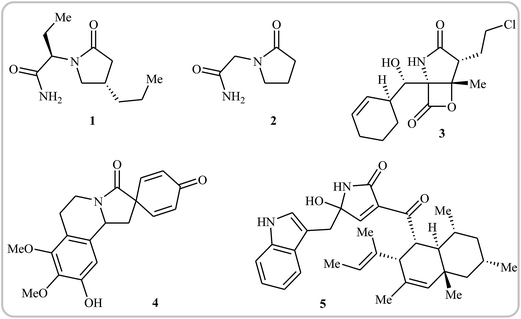 |
| | Fig. 1 Selected examples of bioactive γ-lactams. | |
In connection with our series of review papers on the synthesis of nitrogen based heterocycles from N-propargylamine/amide derivatives,13–22 we summarize here a variety of protocols for the synthesis of γ-lactam cores from readily accessible N-allyl propiolamides. The review is divided into two major sections. The first section focuses exclusively on transition metal-catalyzed cyclization of N-allyl propiolamides, while the second covers radical promoted cyclizations. It should be noted that we have not discussed synthesis of indolinones (benzene-fused γ-lactams), since it has recently been described in another publication.13
2. Transition metal-catalyzed cyclizations
2.1 Palladium
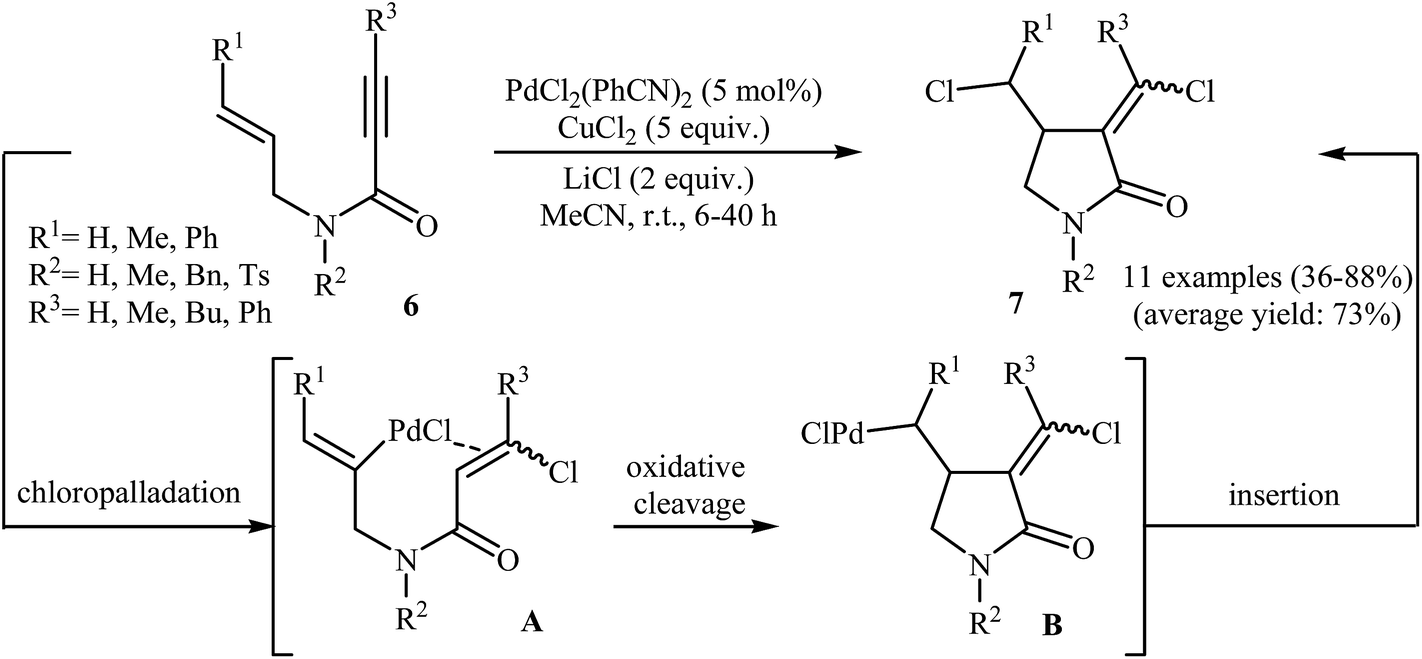
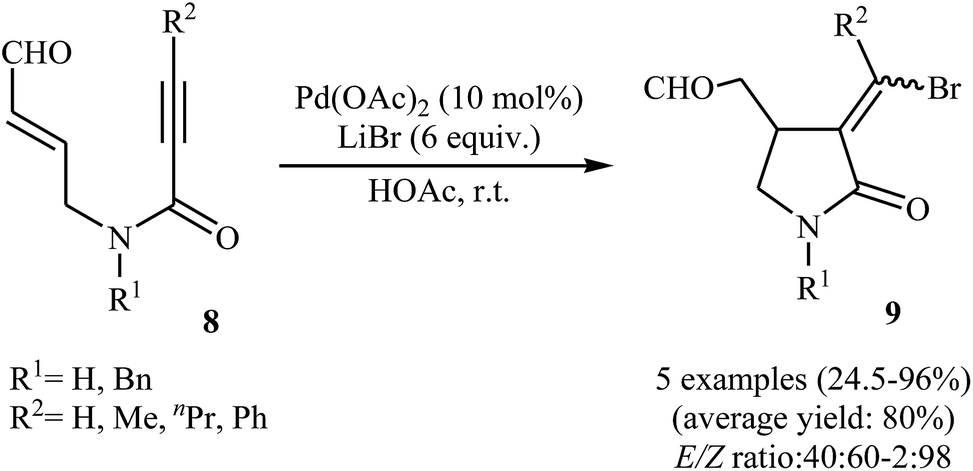
Synthesis of γ-lactams via Pd-catalyzed intramolecular cyclization of propiolamide derivatives has been the subject of a number of papers. One of the earliest report on this chemistry have been published by Lu and co-workers in 1996. They showed that treatment of N-allyl propiolamides 6 with the PdCl2(PhCN)2/CuCl2/LiCl system in acetonitrile directly gave corresponding α-chloromethylene-β-chloromethyl-γ-lactams 7 after 6–40 hours at room temperature (Scheme 1). According to the author proposed mechanism, this transformation proceeded via a chloropalladation/oxidative cleavage/insertion sequential process.23 In a closely related study, the same group found that N-(3′-formylallylic)propiolamides 8 were converted to the corresponding α-bromomethylene-β-formylmethyl-γ-lactams 9, via Pd(II)-catalyzed intramolecular cyclization using LiBr as bromide source in HOAc at room temperature (Scheme 2).24
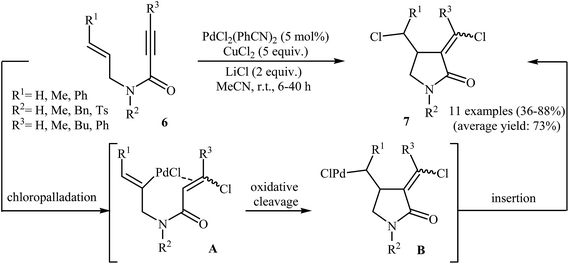 |
| | Scheme 1 Lu's synthesis of α-chloromethylene-β-chloromethyl-γ-lactams 7. | |
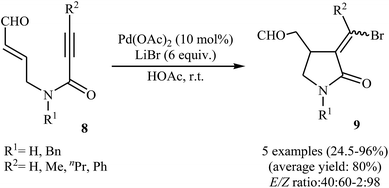 |
| | Scheme 2 Pd(II)-catalyzed cyclization of N-(3′-formylallylic)propiolamides 8 reported by Lu. | |
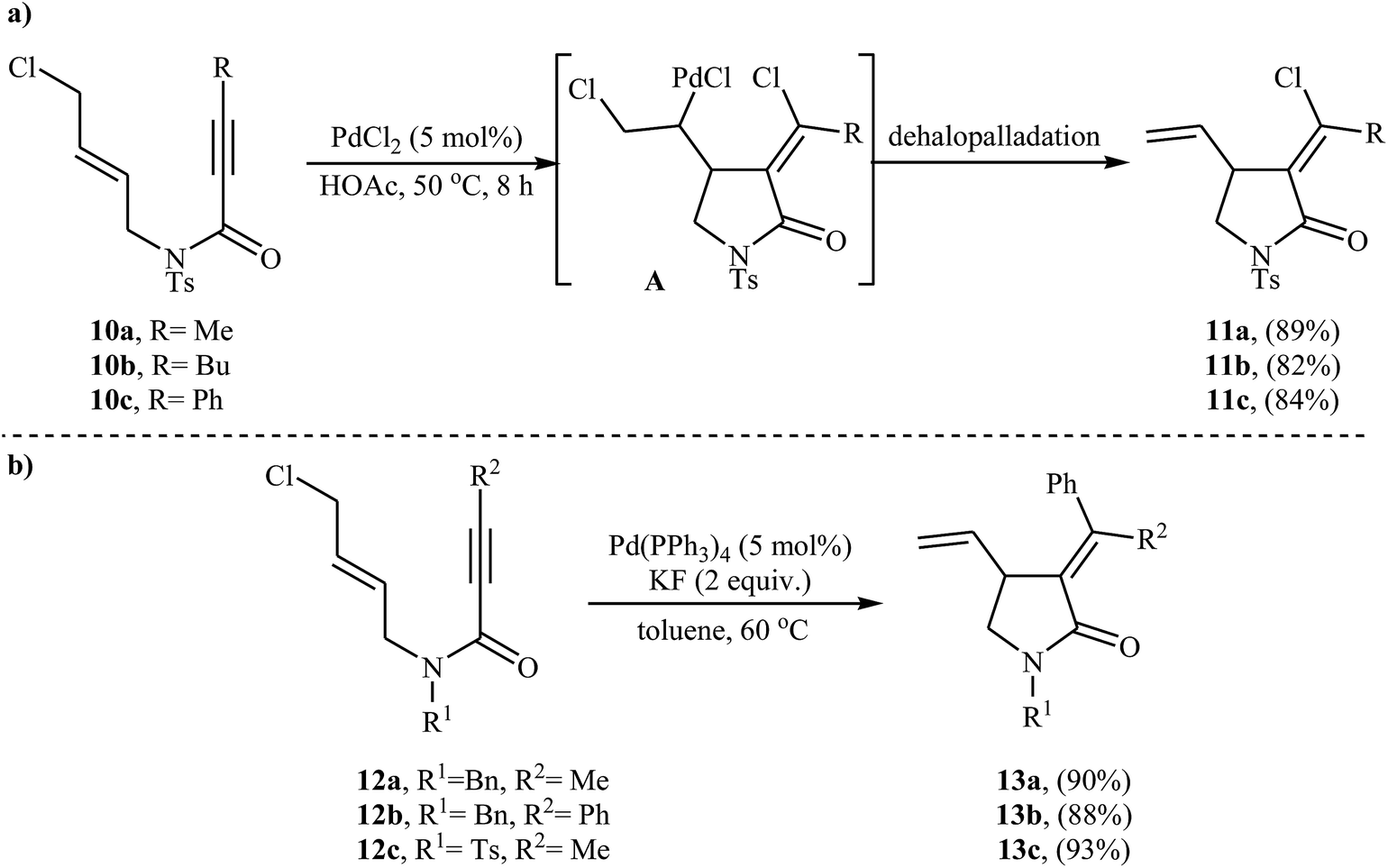
In 2005, Zhu and Zhang described the first PdCl2-catalyzed cis-chloropalladation-cyclization reaction of N-allyl propiolamides. Thus, the treatment of 1,6-enyne substrates 10 with 5 mol% of PdCl2 in HOAc at 50 °C afforded corresponding (E)-α-halomethylene-γ-lactams 11 in high isolated yields (Scheme 3a). The mechanism proposed for the formation of γ-lactams 11 involves the key intermediate A, which underwent dehalopalladation to produce the cyclized product.25 Subsequently, the same authors extended this chemistry to synthesis of α-phenylmethylene-γ-lactams 13 via a beautiful Pd(0)-catalyzed tandem cyclization/Suzuki coupling reaction of N-allyl propiolamides 12 with phenylboronic acid.26 Several catalysts, bases and solvents were tested, and the system Pd(PPh3)4/KF/toluene was found to be superior.27 Under optimized conditions, the reaction tolerates both aryl and alkyl substituted internal propiolamides and gave the corresponding lactams 13 in high yields (Scheme 3b).
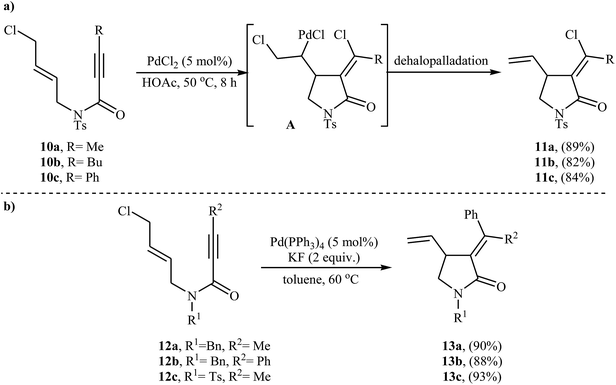 |
| | Scheme 3 (a) PdCl2-catalyzed cis-chloropalladation-cyclization of N-allyl propiolamides 10; (b) Pd(0)-catalyzed tandem cyclization/Suzuki coupling reaction of N-allyl propiolamides 12 with phenylboronic acid. | |
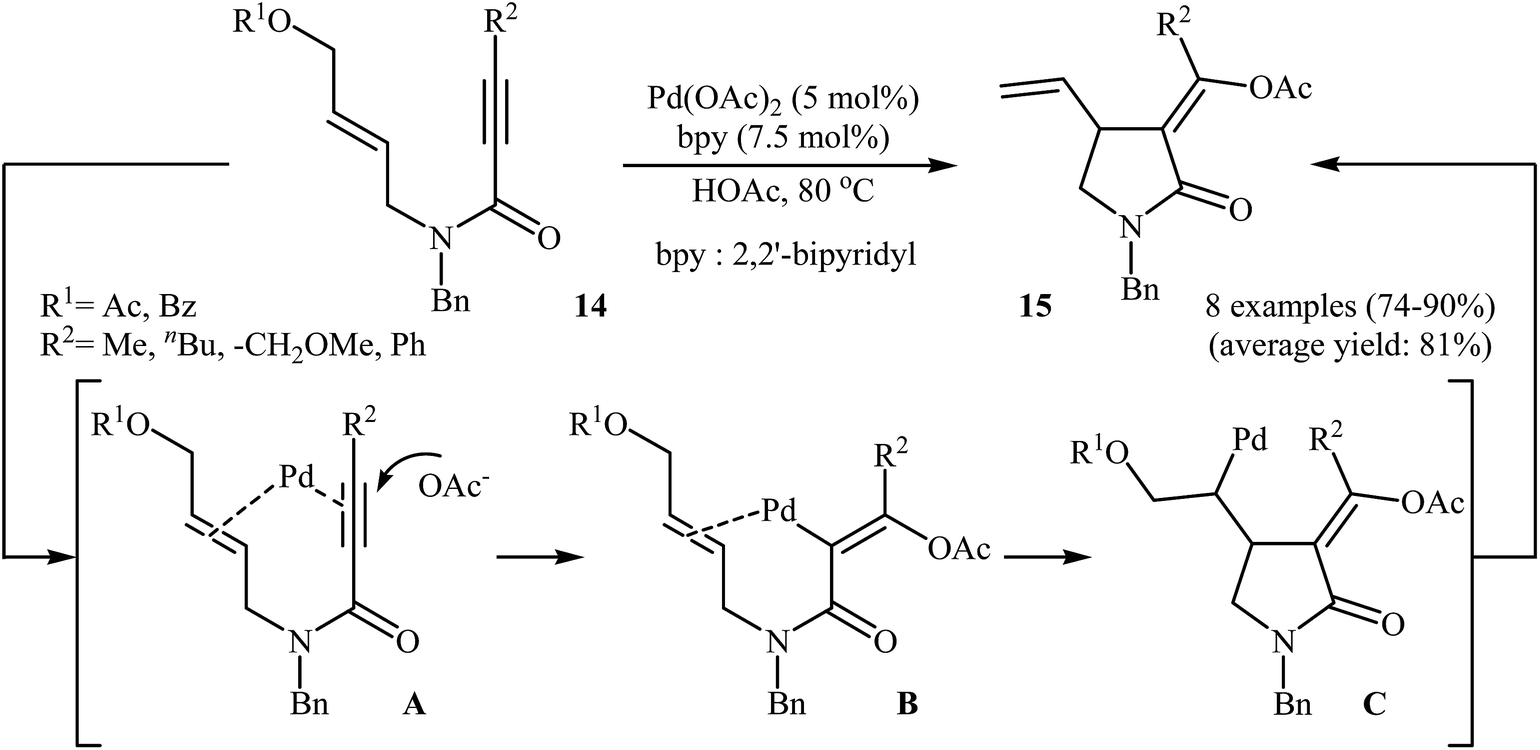
Shortly afterward, the group of Lu successfully synthesized a series of (Z)-α-alkylidene-γ-butyrolactams 15 from corresponding (Z)-N-allyl propiolamides 14 in good yields, as a single isomer, using 5 mol% of Pa(OAc)2 as catalyst, and 7.5 mol% of bpy as ligand in acetic acid (Scheme 4). Mechanistically, the reaction proceeded via a coordination/intramolecular insertion/trans-acetoxypalladation/β-heteroatom elimination sequential process. The author found that when the reaction was carried out in the presence of chiral nitrogen-containing ligands, an asymmetric version of this protocol with moderate enantioselectivity was established.28
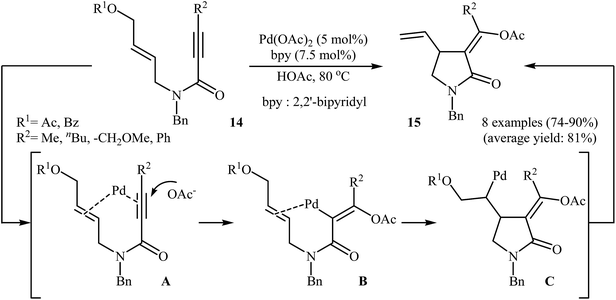 |
| | Scheme 4 Pd(II)-catalyzed asymmetric synthesis of (Z)-α-alkylidene-γ-butyrolactams 15 from corresponding (Z)-N-allyl propiolamides 14. | |
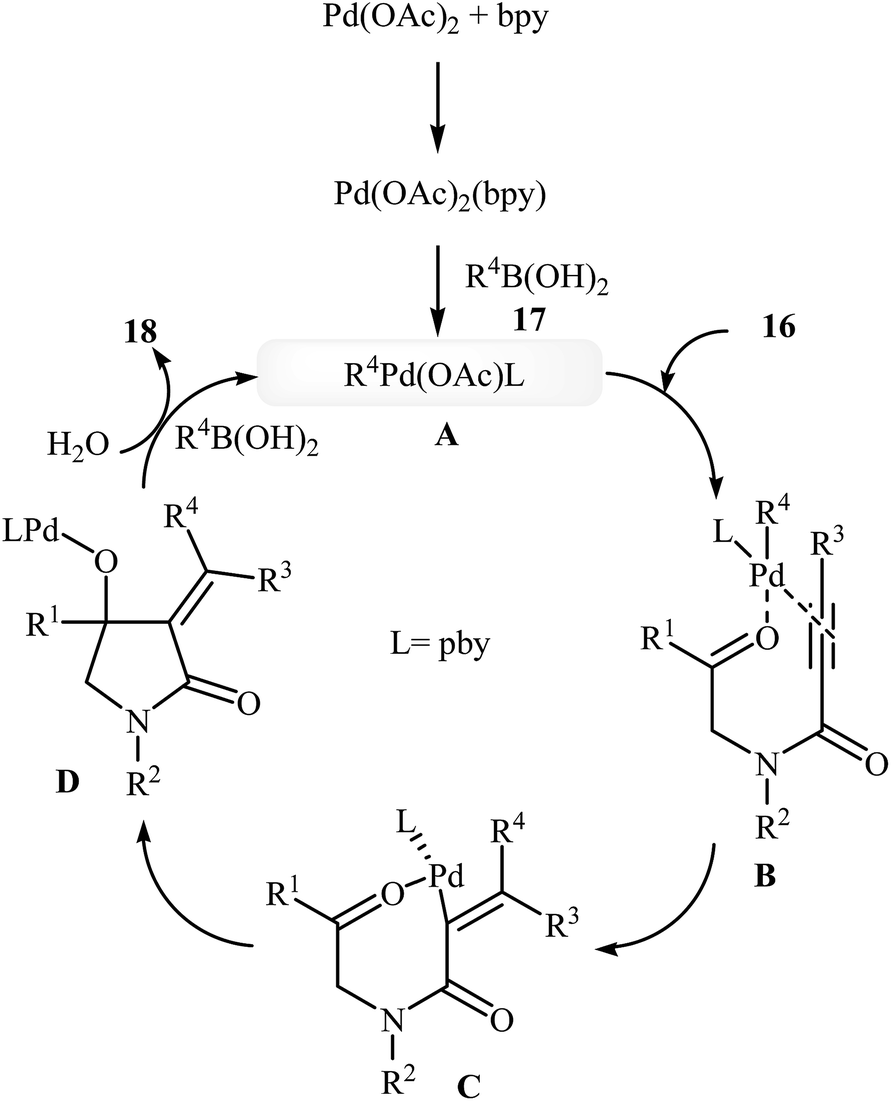
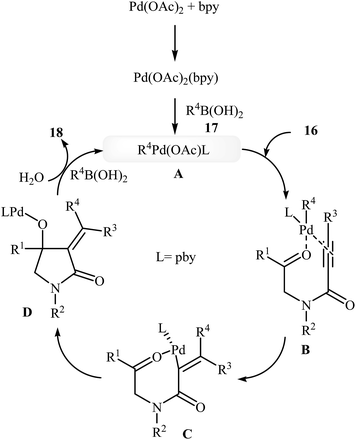
In 2010, the same group reported an elegant approach for the synthesis of α-alkylidene-β-hydroxy-γ-lactams 18 via Pd(II)-catalyzed cascade reaction of propiolamides 16 and arylboronic acids 17. The optimum conditions for this reaction utilize Pd(OAc)2 as the catalyst, bpy as ligand and DCE/H2O (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as solvent. Under optimized conditions, the reaction tolerates both terminal and alkyl substituted propiolamides and gave corresponding γ-lactams 18 in moderate to excellent yields, but extension of the reaction to aryl substituted alkynes and N-arylpropiolamides was failed (Scheme 5). The postulated reaction mechanism is displayed in Scheme 6. The reaction starts with in situ generation of bpyPd(OAc)2 by reaction between the Pd(OAc)2 and bpy. Its reaction with arylboronic acid 17 leads to an arylpalladium specie A that, after coordination with the triple bond and the carbonyl group of substrate 16, affords intermediate B. The insertion of the triple bond to arylpalladium species in B furnishes vinylpalladium intermediate C. Next, an intramolecular 1,2-addition of the vinylpalladium species to the carbonyl group in C takes place to form intermediate D which finally furnishes the final product 18 by a rapid protodemetalation and regeneration of the palladium(II) species.29
:1) as solvent. Under optimized conditions, the reaction tolerates both terminal and alkyl substituted propiolamides and gave corresponding γ-lactams 18 in moderate to excellent yields, but extension of the reaction to aryl substituted alkynes and N-arylpropiolamides was failed (Scheme 5). The postulated reaction mechanism is displayed in Scheme 6. The reaction starts with in situ generation of bpyPd(OAc)2 by reaction between the Pd(OAc)2 and bpy. Its reaction with arylboronic acid 17 leads to an arylpalladium specie A that, after coordination with the triple bond and the carbonyl group of substrate 16, affords intermediate B. The insertion of the triple bond to arylpalladium species in B furnishes vinylpalladium intermediate C. Next, an intramolecular 1,2-addition of the vinylpalladium species to the carbonyl group in C takes place to form intermediate D which finally furnishes the final product 18 by a rapid protodemetalation and regeneration of the palladium(II) species.29
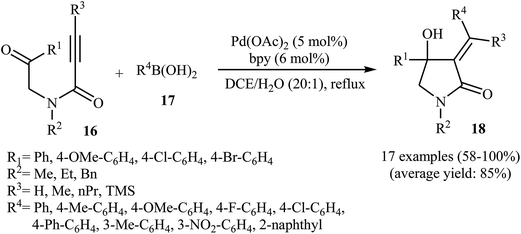 |
| | Scheme 5 Synthesis of α-alkylidene-β-hydroxy-γ-lactams 3 via Pd(II)-catalyzed annulation of propiolamides 16 with arylboronic acids 17. | |
 |
| | Scheme 6 Mechanism that accounts for the formation of 18. | |
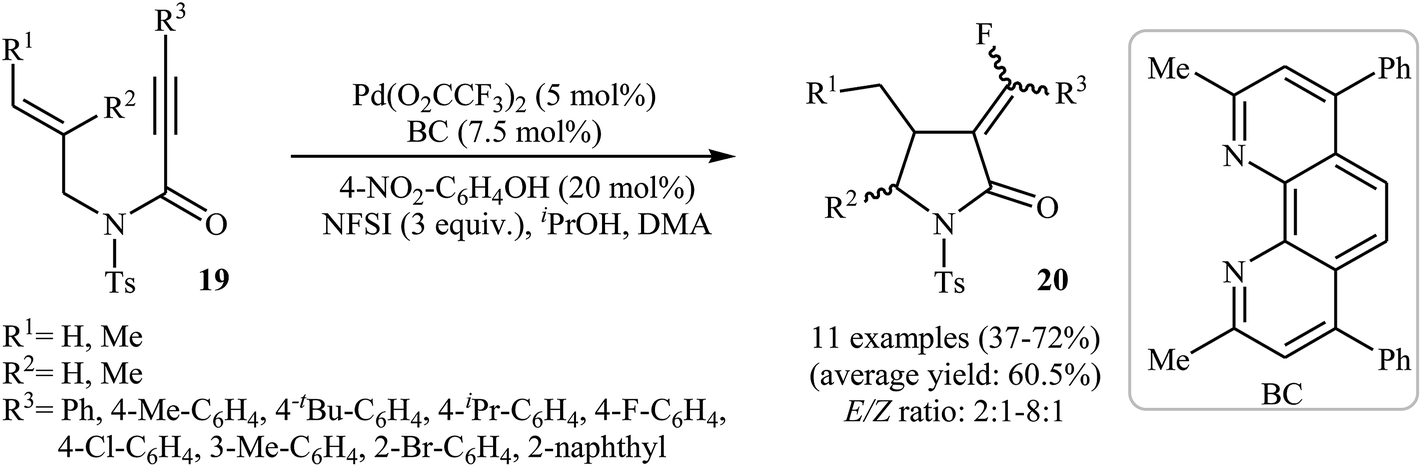
Inspired by these works, Peng and Liu reported a beautiful palladium(II)-catalyzed tandem fluorination and cyclization of N-allyl propiolamides 19 to prepare α-fluoromethylene γ-lactams 20. It was found that upon treatment with N-fluorobenzenesulfonimide (NSFI) as fluorinating reagent, Pd(TFA)2/BC as catalytic system and isopropyl alcohol and 4-nitrophenol as additives, N-allyl propiolamides 19 could be converted into the corresponding fluorinated γ-lactams 20 in moderate to good yields and high (E)-selectivity (Scheme 7). However, substrates bearing alkyl groups in the alkyne terminus failed to participate in the reaction. The results also demonstrated that the substrates with disubstituted alkene afforded only trace of γ-lactams.30
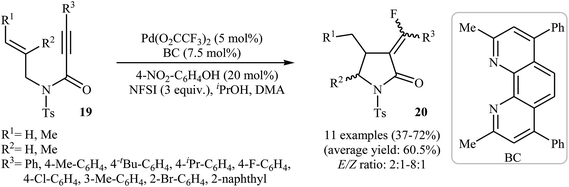 |
| | Scheme 7 Pd(II)-catalyzed tandem fluorination and cyclization of N-allyl propiolamides 19 developed by Liu. | |
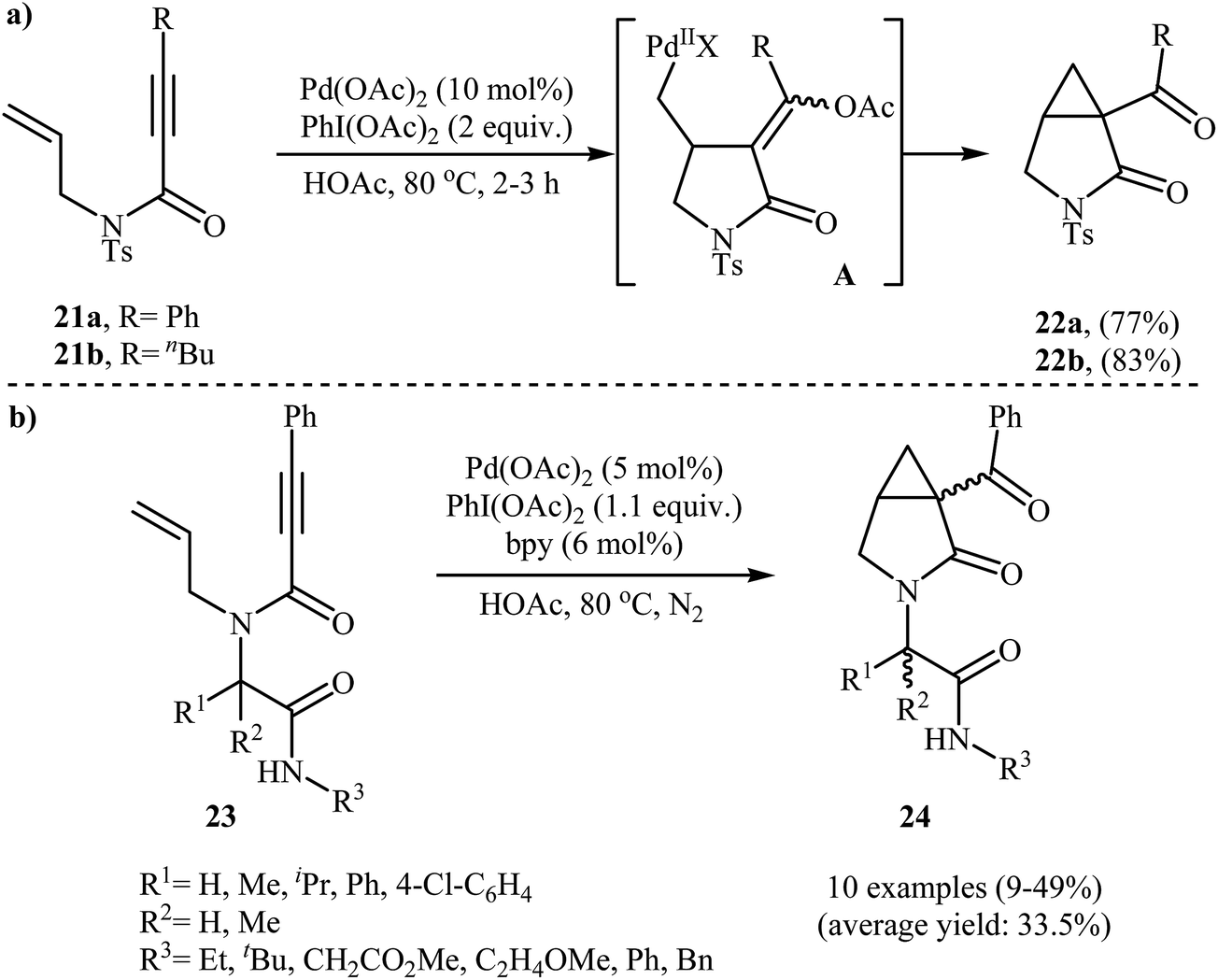
An interesting domino procedure for the synthesis of 3-aza-bicyclo[3.1.0]hexan-2-ones 22 from N-allyl propiolamides 21 through a 5-exo-trig cyclization and SN2 C–O formation process, in which three new C–C bonds are formed, was developed by Tse and co-workers. The reaction was performed in the presence of Pd(OAc)2/PhI(OAc)2 combination as catalytic system in acetic acid and resulted in the formation of desired products 22 in good yields (Scheme 8a).31 In a closely related investigation, the group of Welsch reported the cyclization of a series of Ugi-adducts 23 to construction of bicyclic lactams 24 employing Pd(OAc)2/PhI(OAc)2/bipy as catalytic system in HOAc. The 3-aza-bicyclo[3.1.0]hexan-2-ones 24 were formed with low to moderate yields (Scheme 8b). Unexpectedly, the authors did not notice any yield improvement when microwave irradiation was applied.32
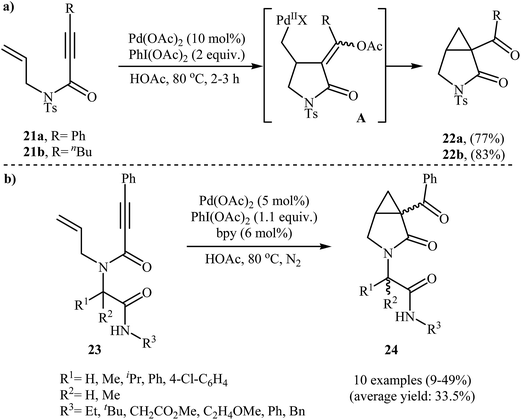 |
| | Scheme 8 (a) Tse's synthesis of 3-aza-bicyclo[3.1.0]hexan-2-ones 22; (b) synthesis of bicyclic lactams 24 from Ugi-adducts 23. | |
2.2 Rhodium
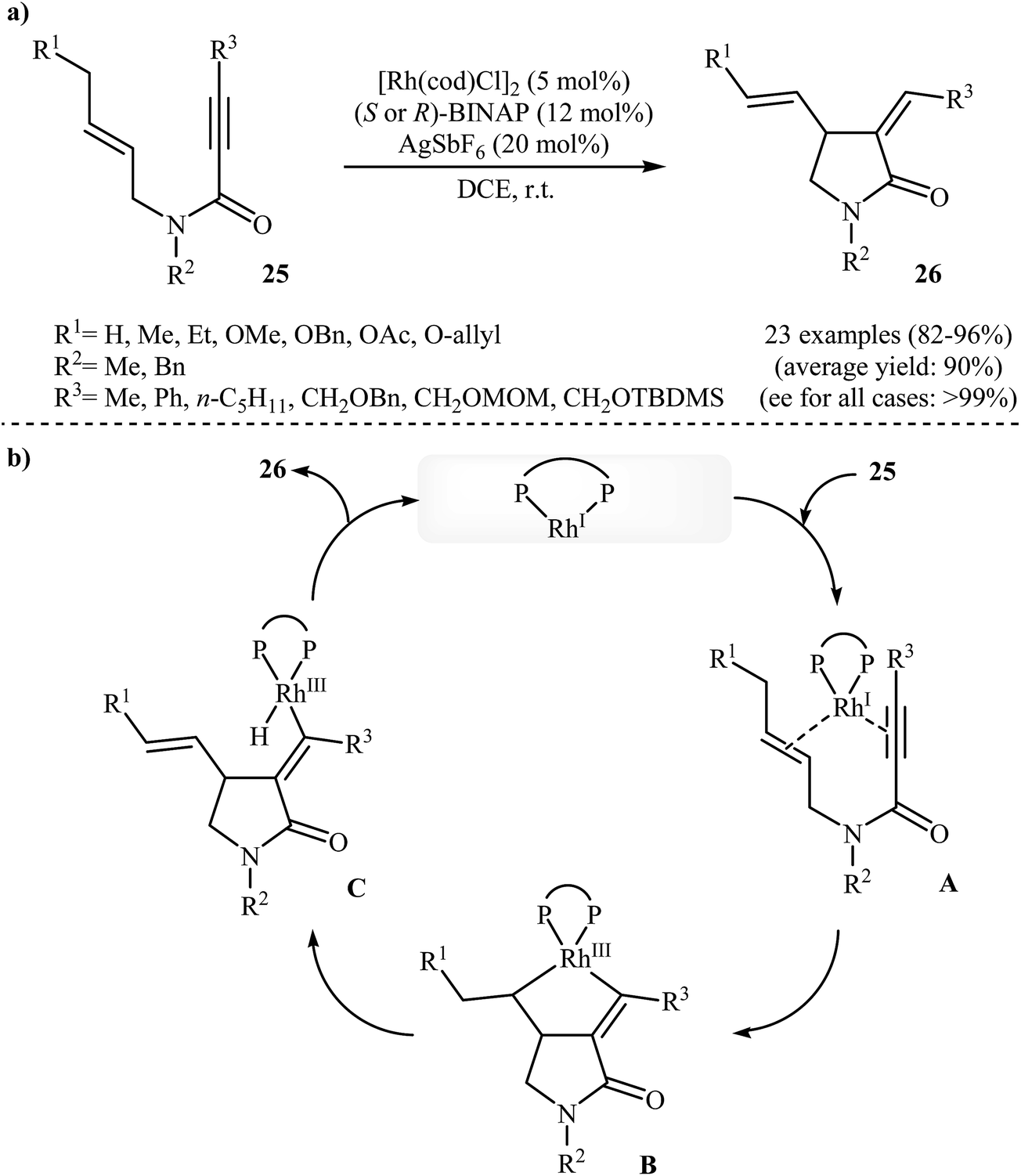
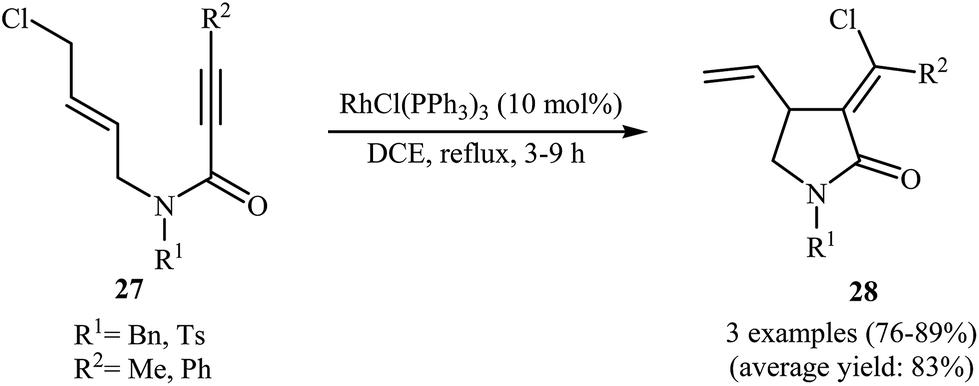
In 2002, the group of Zhang have shown that a commercially available Rh(I) catalyst [Rh(cod)Cl]2 could be used in combination with BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) and AgSbF6 to transform N-allyl propiolamides 25 into functionalized γ-lactams 26 (Scheme 9a). This enantioselective cycloisomerization proved to be highly efficient (82–96%) and allows general and practical access to a variety of functionalized γ-lactams under very mild reaction conditions. The excellent ee value (up to 99%) obtained in these reactions was especially remarkable. These results are consistent with a mechanism proceeding by the coordination of N-allyl propiolamides 25 with a RhI species to form intermediate A, followed by an intramolecular oxidative cyclization to form metallacyclopentane B. The Rh–H species C would be formed via β-H elimination of the metallacyclopentane B. The reductive elimination of this Rh–H species C would then give products 26 and generate RhI species (Scheme 9b).33 Interestingly, when N-allyl propiolamides 27 having a halogen on the allylic terminus were subjected to cyclizations, another type of Rh(I)-catalyzed ene reaction was observed. In these cases an unusual intramolecular halogen shift happened to give α-halomethylene-γ-butyrolactones 28 as product (Scheme 10).34–36
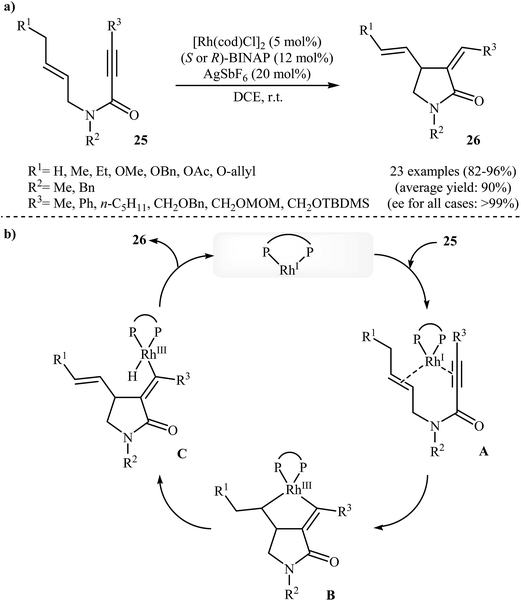 |
| | Scheme 9 (a) Enantioselective Rh-catalyzed cycloisomerization of propiolamides 25 into γ-actams 26; (b) plausible mechanism for the formation of 26. | |
 |
| | Scheme 10 Rh(II)-catalyzed cycloisomerization of N-allyl propiolamides 27 with an intramolecular halogen shift. | |
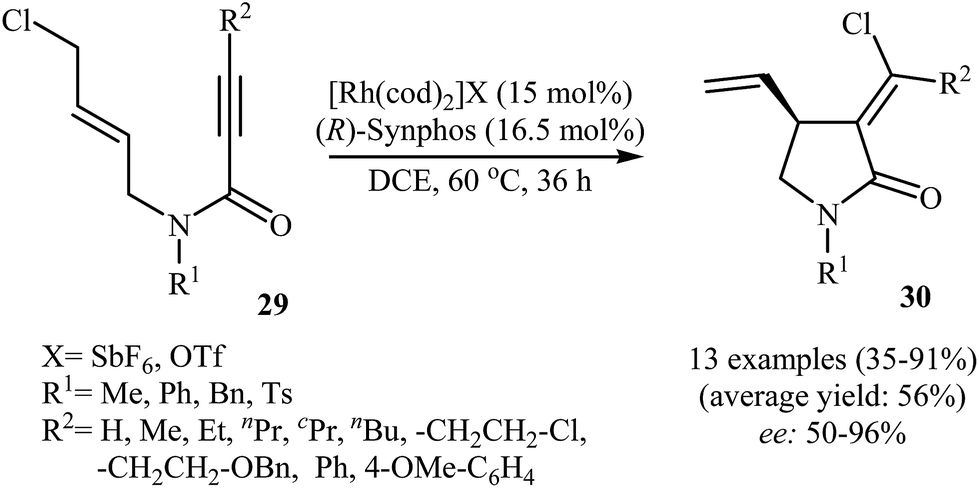
In 2012, Zhang and Ratovelomanana-Vidal along with their co-workers were able to demonstrate that a cationic Rh–Synphos catalyst can efficiency catalyze the cycloisomerization of N-allyl propiolamides 29 with an intramolecular halogen shift to the corresponding enantiomerically enriched α-chloromethylene-γ-butyrolactams 30 in moderate to high yields and good to very high enantiomeric excesses (Scheme 11). The reaction showed good functional group tolerance and could be applied for synthesis of various functionalized γ-lactams with potential biological activities.37
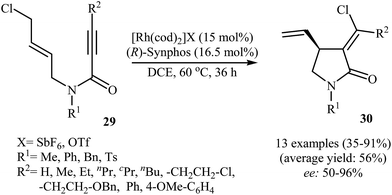 |
| | Scheme 11 Enantioselective Rh-catalyzed synthesis of α-chloromethylene-γ-butyrolactams 30 from N-allyl propiolamides 29. | |
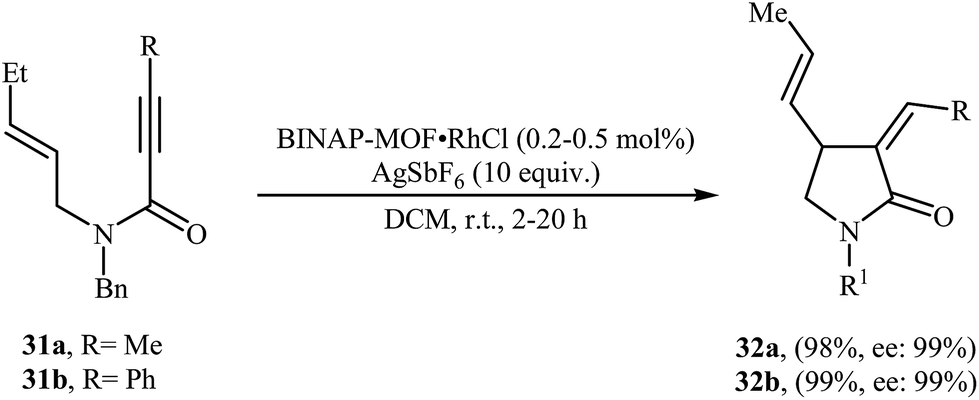
Recently, Lin and co-workers have described the synthesis of BINAP-based metal–organic framework BINAP–MOF (formula Zr6(OH)4O4(L1)6) via a solvothermal reaction between 4,4′-bis(4-carboxyphenylethynyl)BINAP (H2L1), ZrCl4, and trifluoroacetic acid in DMF, which can be metalated with rhodium complex [Rh(nbd)Cl]2 to provide highly active and enantioselective single-site catalyst BINAP–MOF RhCl for the asymmetric Alder-ene cycloisomerization reactions of N-allyl propiolamides 31 to provide the corresponding γ-lactams 32 in excellent yields and outstanding enantioselectivity (Scheme 12).38
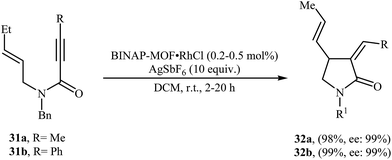 |
| | Scheme 12 Lin's synthesis of γ-lactams 32. | |
2.3 Gold
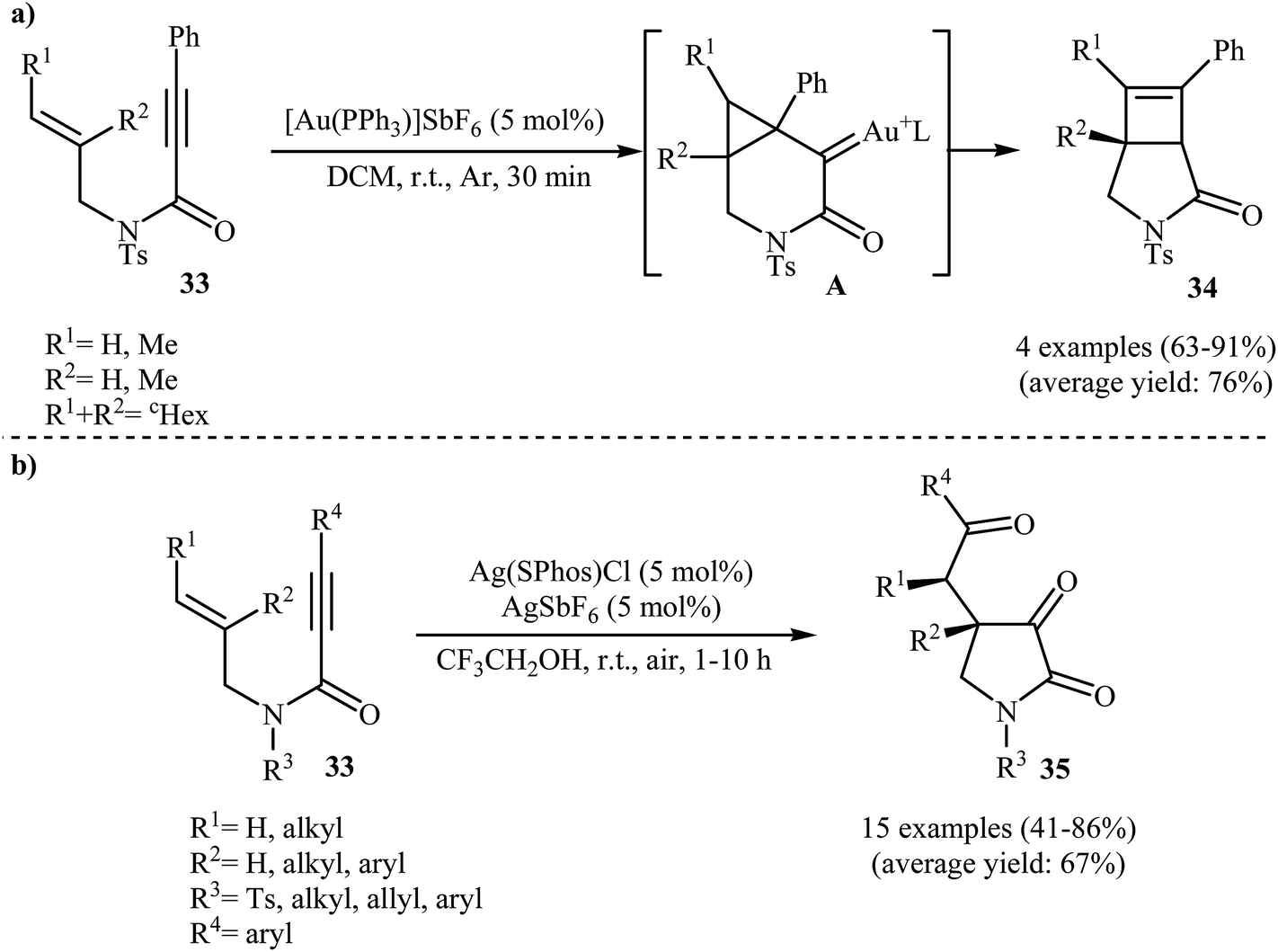
In 2009, the group of Kang and Chung demonstrated for the first time the usefulness of gold catalysts for the cycloisomerization reaction of N-allyl propiolamides. Thus, in the presence of 5 mol% of [Au(PPh3)]SbF6 in dichloromethane at room temperature, N-allyl propiolamides 33 undergo rapid intramolecular cycloisomerization to give the corresponding aza-bicyclo[3.2.0]hept-6-en-2-ones 34 in good to high yields (Scheme 13a). The mechanism of this cyclization was proposed based on density functional theory (DFT) calculations, determining that the reaction proceeds via tandem formal [2 + 2] cycloaddition/skeletal rearrangement.39 Curiously, when the reaction was carried out under air atmosphere in 2,2,2-trifluoroethanol, the tricarbonyl compounds 35 were obtained in yields ranging from 41 to 86% instead of the desired bicyclic γ-lactam products (Scheme 13b).40
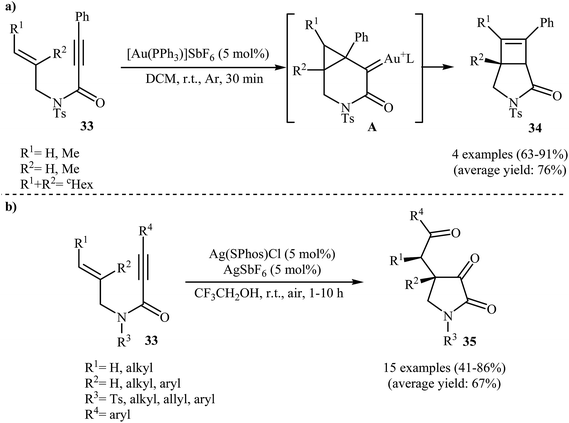 |
| | Scheme 13 (a) Au(I)-catalyzed cycloisomerization reaction of N-allyl propiolamides 33 to corresponding aza-bicyclo[3.2.0]hept-6-en-2-ones 34; (b) Synthesis of tricarbonyl compounds 35 through Au-catalyzed cyclization of N-allyl propiolamides 33 under air atmosphere. | |
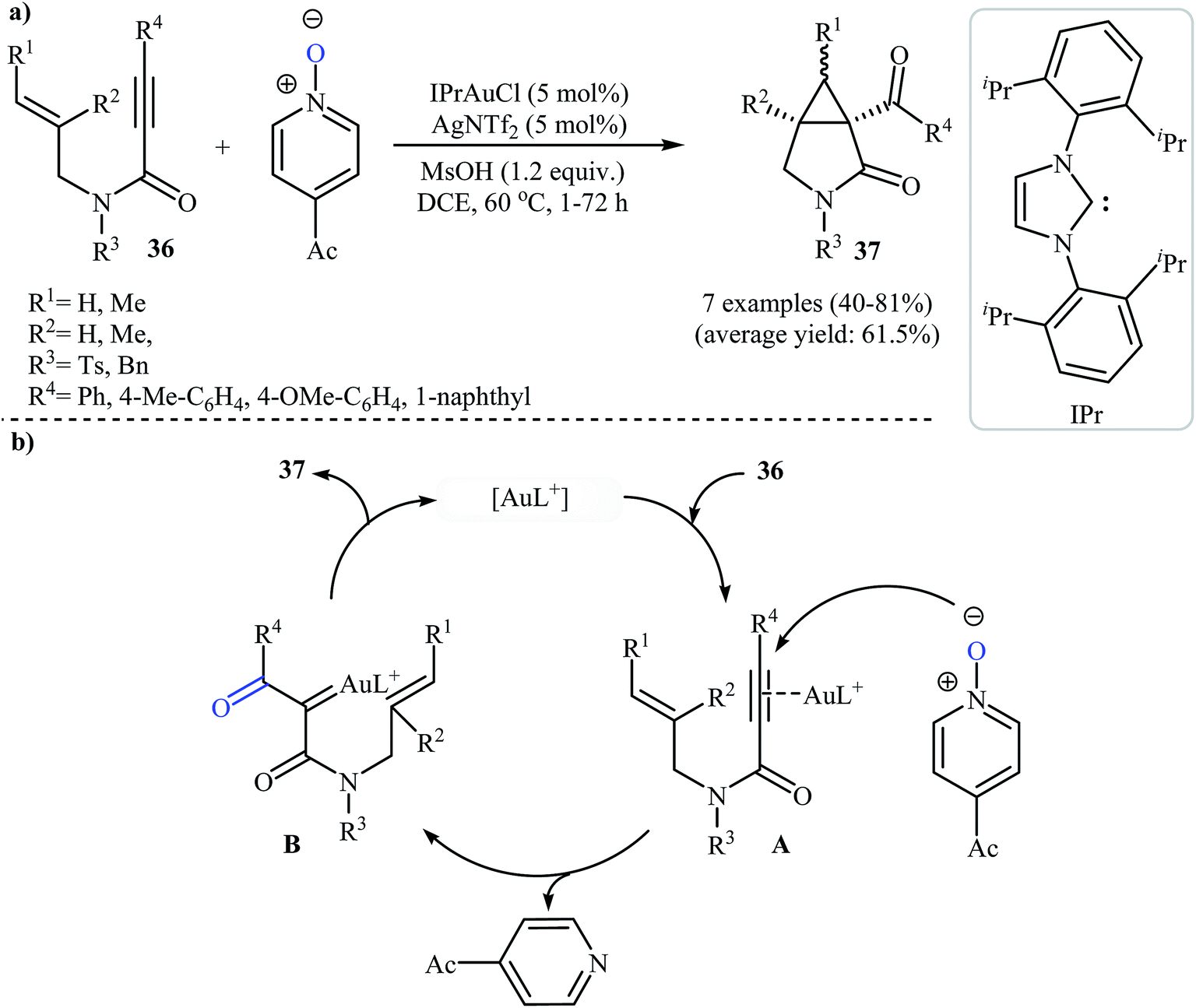
In 2011, Qian and Zhang published an efficient protocol for the synthesis of 3-aza bicyclo[3.1.0]hexan-2-ones 37 via gold(I)-catalyzed intramolecular oxidation–cyclopropanation sequence of N-allyl propiolamides 36 (Scheme 14a). Thus, the optimized reactions revealed that the optimum condition for this reaction was the combination of [IPrAuCl] (5 mol%) and AgNTf2 (5 mol%) as catalytic system, 4-acetyl-pyridine N-oxide (2.0 equiv.) as external oxidant, and MsOH (1.2 equiv.) as additive using DCM as the solvent, at 60 °C. Under optimized conditions, the reaction tolerated aryl substituted internal N-propargylamines 36 and gave final products 37 in moderate to high yields. However, the reaction fails for substrates bearing alkyl groups in the alkyne terminus. The author proposed mechanistic pathway for this reaction starts with the formation of π-complex A from the enyne 36 and gold species, followed by its oxidation by 4-acetyl-pyridine N-oxide to give α-oxo gold carbenoid B. Finally, an intramolecular cyclopropanation of intermediate B leads to the observed γ-lactams 37 (Scheme 14b).41
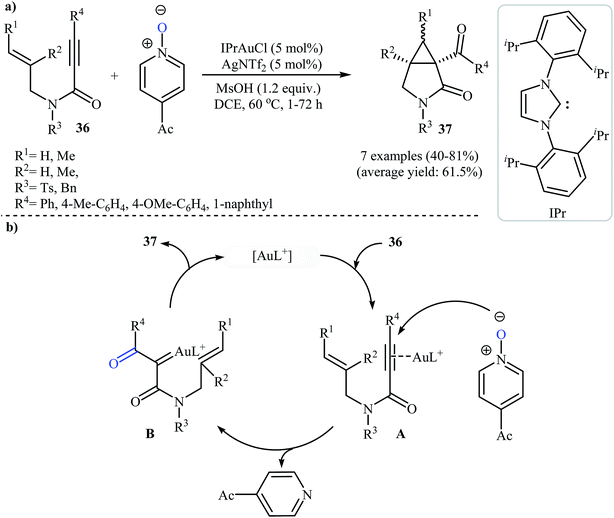 |
| | Scheme 14 (a) Synthesis of 3-aza-bicyclo[3.1.0]hexan-2-ones 37 via Au-catalyzed oxidative cyclopropanations of N-allyl propiolamides 36 with pyridine N-oxide; (b) mechanistic proposal for the reaction in (a). | |
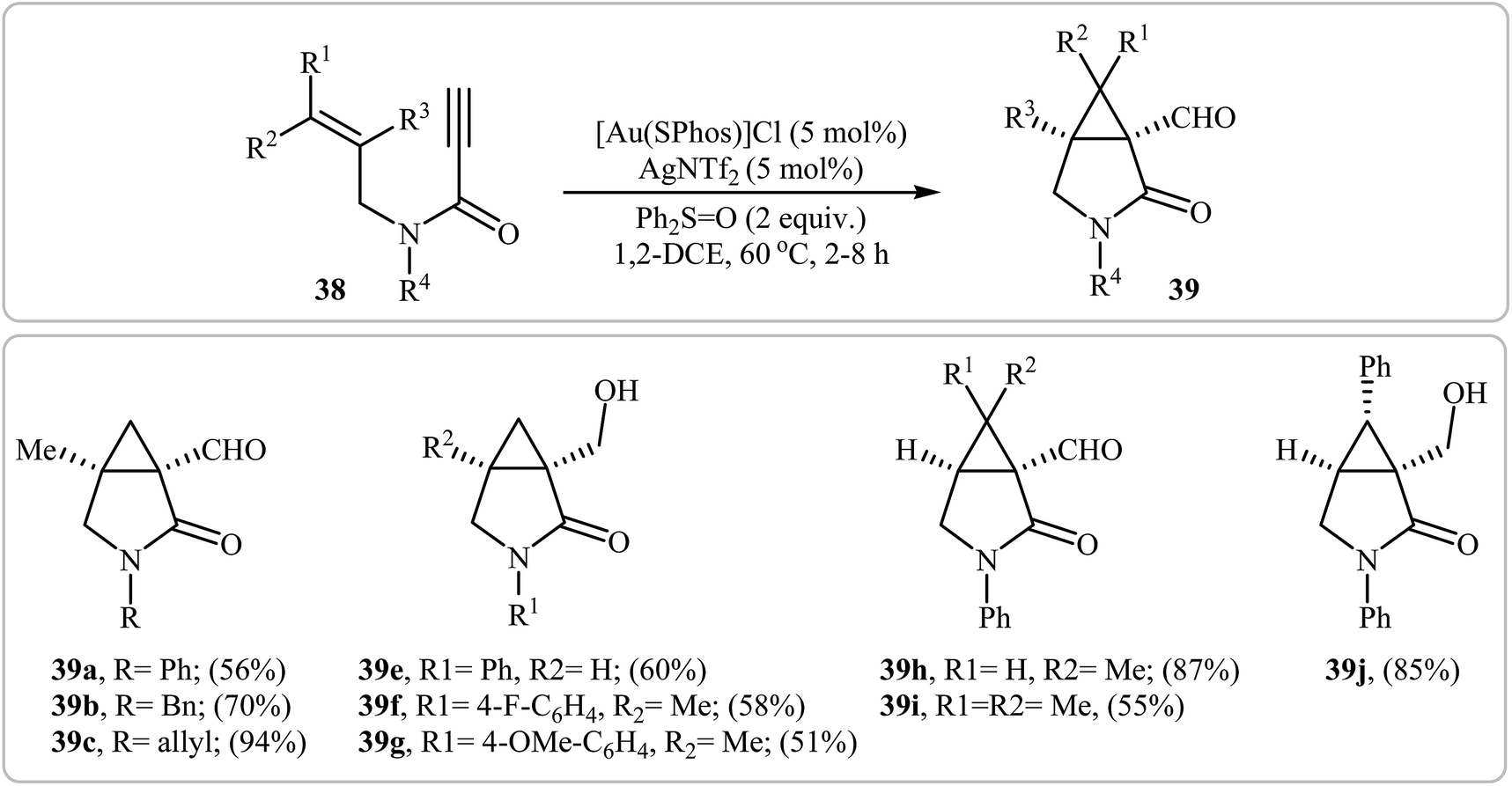
In a closely related study, Yeom and Shin also found that terminal N-allyl propiolamides 38 were converted to the corresponding cyclopropane-fused γ-lactams 39 via Au(I)-catalyzed oxidative cyclopropanation using [Au(SPhos)]Cl/AgNTf2 combination as catalytic system and diphenyl sulfoxide as an oxidant in 1,2-dichloroethane at 60 °C (Scheme 15).42
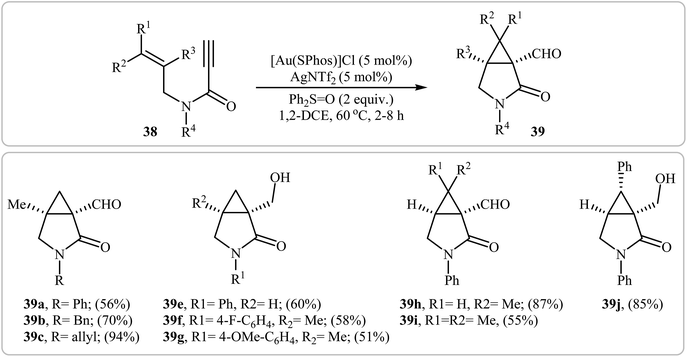 |
| | Scheme 15 Synthesis of cyclopropane-fused γ-lactams 39 via Au(I)-catalyzed oxidative cyclopropanations of N-allyl propiolamides 38 with diphenyl sulfoxide. | |
2.4 Silver
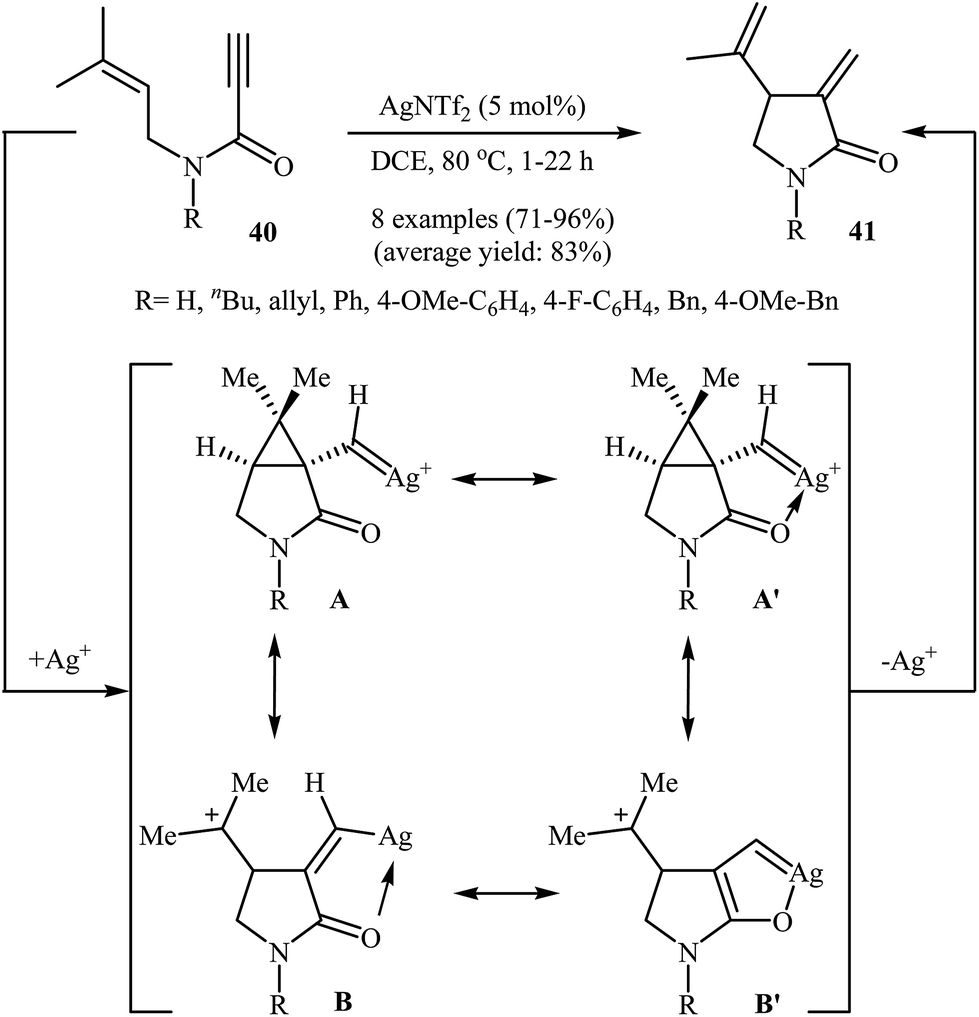
In 2013, Shin and co-workers have established a novel method for the preparation of Alder-ene-type 1,4-dienes. Thus in 1,2-dichloroethane, a AgNTf2-catalyzed cycloisomerization of N-allyl propiolamides 40 furnished 5-exo-dig 1,4-diene products 41 in good to excellent yields (Scheme 16). Other silver catalysts such as AgSbF6, AgOTf, and AgBF4 were also found to promote the reaction, however, in lower yields. The authors claimed that the presence of C(5) carbonyl group in combination with Ag salts is essential for the selective formation of 5-exo-dig products.43 Interestingly, when the reaction was carried out in the presence of an Au(I) catalyst, the 6-endo-dig mode is preferred over 5-exo-dig.44
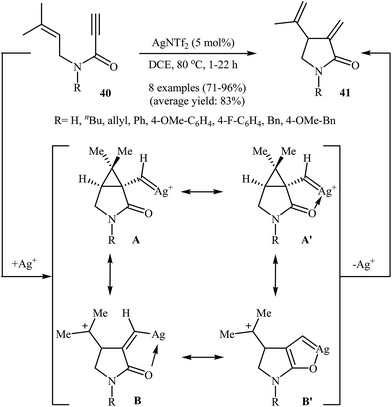 |
| | Scheme 16 Ag(I)-catalyzed cycloisomerization of N-allyl propiolamides 40. | |
2.5 Copper
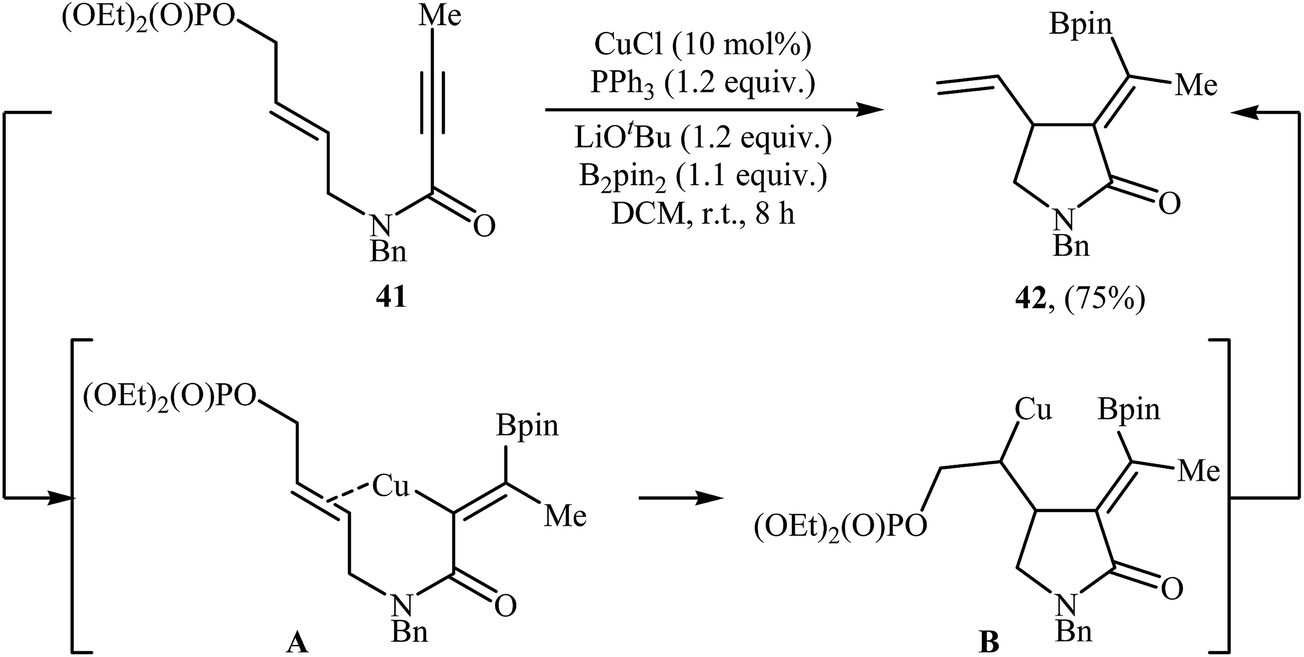
Copper-catalyzed cyclization of N-allyl propiolamides into γ-lactams has been scarcely studied; in fact, only one example of such a reaction was reported in the literature. In 2017, Bai and Zhu along with their co-workers have demonstrated the formation of α-alkylidene-γ-lactam 42 by room temperature CuCl/PPh3-catalyzed borylative cyclization of 1,6-enynyl phosphate 41 with B2Pin2 as the borylation reagent (Scheme 17). Among the various solvents like THF, DMF, 1,4-dioxane, toluene, DCM, DCE, MeCN; DCM was the most efficient for this borylative cyclization. As shown in Scheme 1, the reaction starts with the formation of an alkenylcopper intermediate A via addition of the in situ generated borylcopper species (CuBpin) to the C–C triple bond of 41, followed by addition of the alkenyl C–Cu bond to the intramolecular C–C double bond to generate alkylcopper species B. Finally, a β-elimination step leads to the final product.45
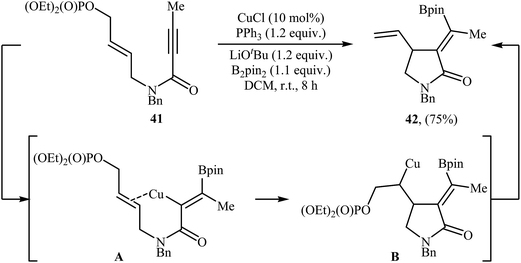 |
| | Scheme 17 Cu(I)-catalyzed cyclization of 1,6-enynyl phosphate 41 reported by Bai and Zhu. | |
3. Radical mediated cyclizations
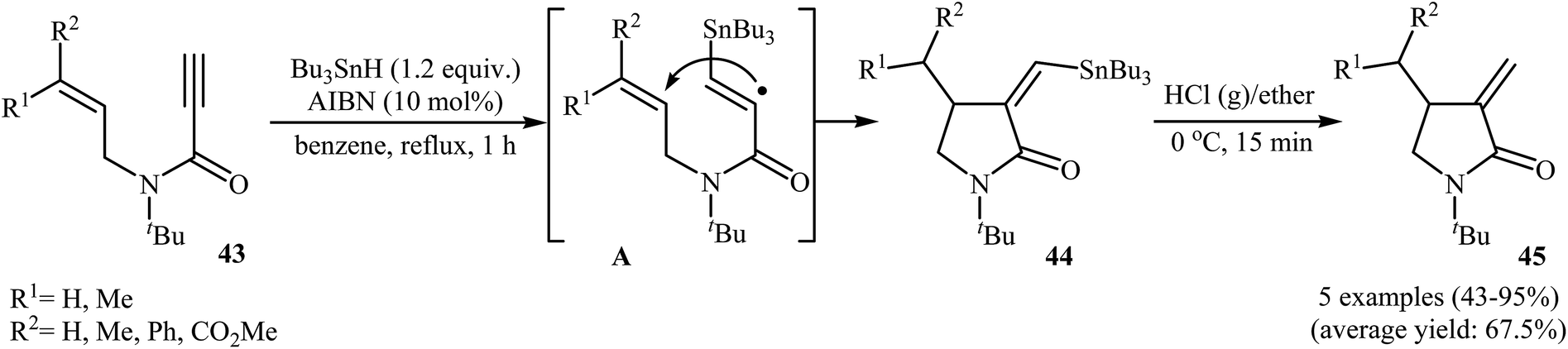
Synthesis of γ-lactams through radical mediated cyclization of propiolamide derivatives have been rarely studied. The earliest report on this chemistry have been published by Lee and Kang in 1993. They showed that treatment of N-allyl propiolamides 43 with 1.2 equiv. of Bu3SnH and 10 mol% of AIBN in refluxing benzene gave α-stannylmethylene γ-lactams 44 via 5-exo cyclization of (α-aminocarbonyl-β-atannyl)viny radicals A that formed by the addition of stannyl radicals to the propiolamides triple bonds. The following destannylation reactions using HCl/ether system afforded moderate to excellent yields of corresponding α-methylene γ-lactams 45 (Scheme 18).46 One main difference between this procedure and metal catalyzed reactions is that this protocol gives the products that having one C–C double bond in their structures while the latter affords the lactam derivatives that having two C–C double bond in their structures (compare Schemes 16 and 18).
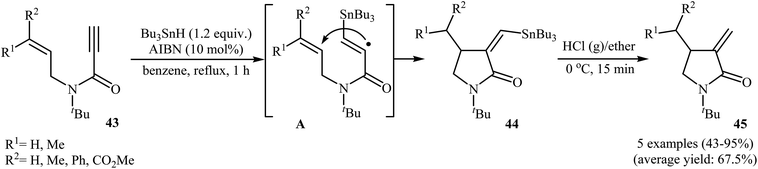 |
| | Scheme 18 Radical mediated synthesis of α-methylene γ-lactams 45 from N-allyl propiolamides 43. | |
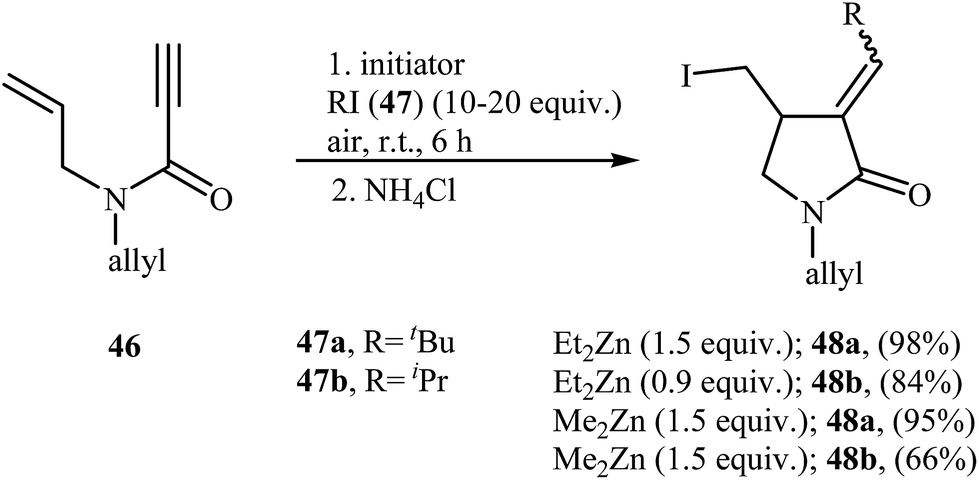
Fifteen years later, Feray and Bertrand reported the use of dialkylzinc mediated atom-transfer radical addition cyclization of N,N-diallylpropiolamides 46 into α-alkylidene-γ-lactams 48. Good to excellent isolated yields of γ-lactams 48 were obtained when propiolamides 46 was treated with either diethyl or dimethylzinc in the presence of alkyl iodides 47 and oxygen at room temperature (Scheme 19).47
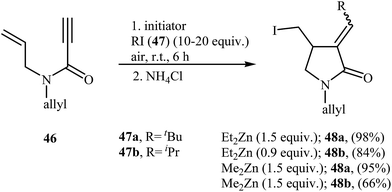 |
| | Scheme 19 Bertrand's synthesis of α-alkylidene-γ-lactams 48. | |
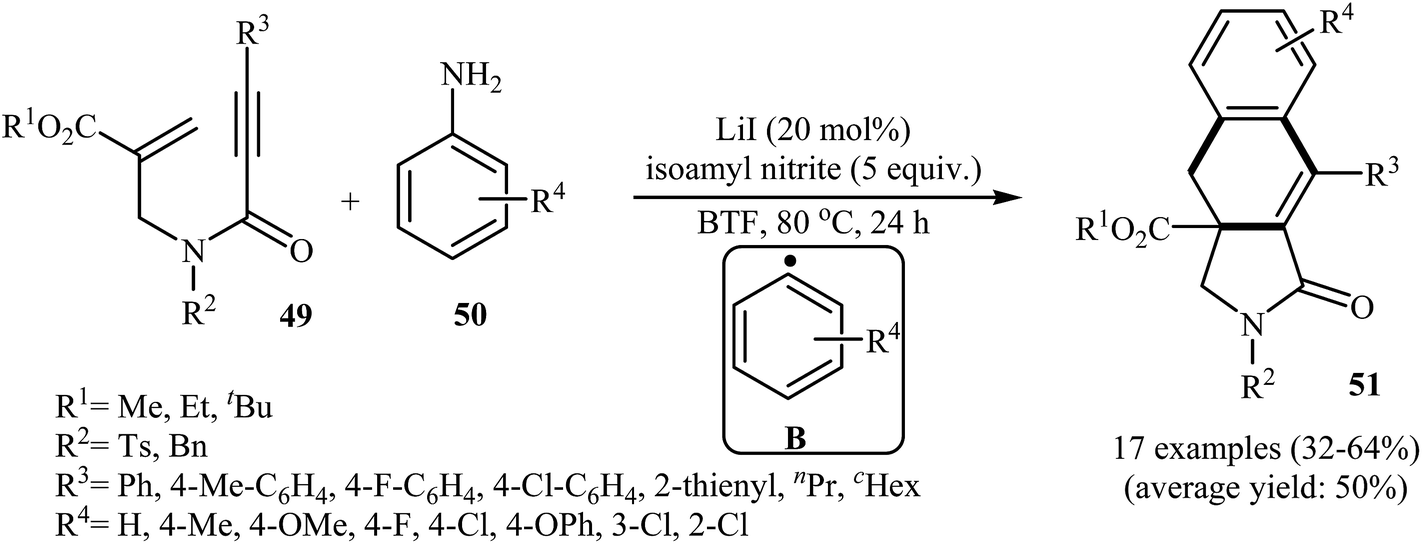
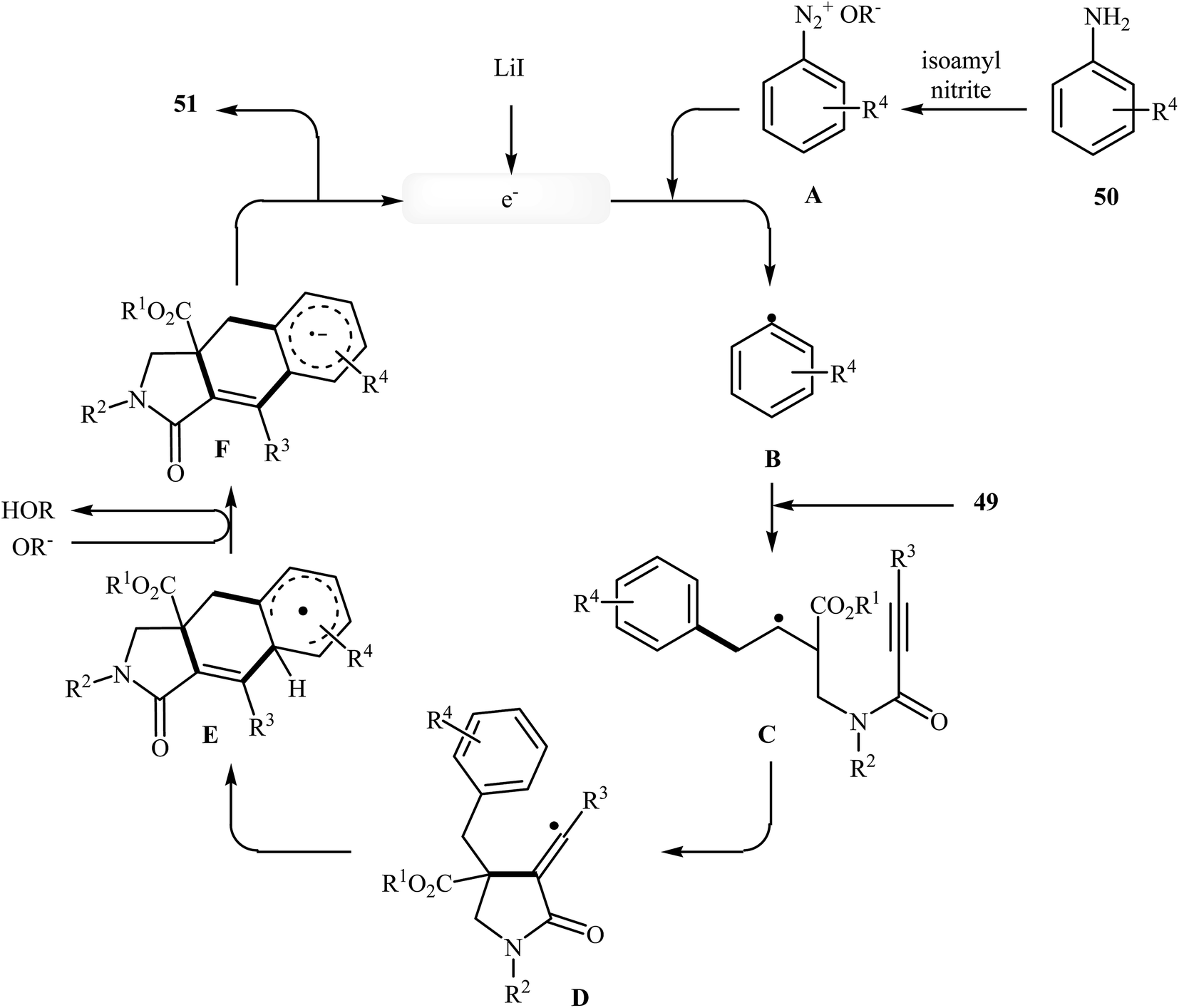
More recently, an interesting cascade radical cyclization of N-allyl propiolamides 49 toward the synthesis of polycyclic γ-lactams 51 was reported by Studer et al. Thus, the reaction of N-allyl propiolamides 49 with aryl radicals B, generated in situ from commercially available anilines 50, in benzotrifluoride afforded biologically important γ-lactams 51 in moderate yields (Scheme 20). The cyclization shows good functional group tolerance, including methoxy, chloro, flouro, and ester functionalities. It is noted that the strategy could be efficiently extended to the synthesis of polycyclic pyrrole and γ-butyrolactone derivatives. The mechanism shown in Fig. 2 is proposed for this transformation. It consists of the following key steps: (i) initial formation of diazonium salt A via reaction of aniline 50 with isoamyl nitrite; (ii) interaction of A with LiI to afford the aryl radical B; (iii) chemoselective radical addition of B to the activated alkene of N-allyl propiolamides 49 to give tertiary alkyl radical C; (iv) 5-exo cyclization of radical C leading to vinyl radical D; (v) intramolecular cyclization of radical D onto the arene produces the cyclohexadienyl radical E; (vi) deprotonation of E with alcoholate derived from isoamyl nitrite to afford arene radical anion F; and (vii) oxidation by liberating an electron gives polycyclic γ-lactam product 51.48
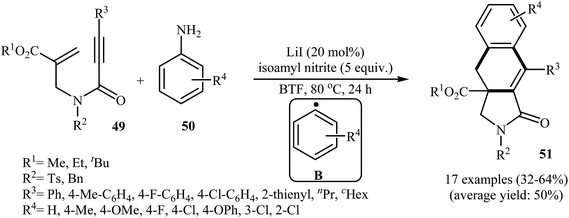 |
| | Scheme 20 Construction of polycyclic γ-lactams 51 via cascade radical cyclization of N-allyl propiolamides 49. | |
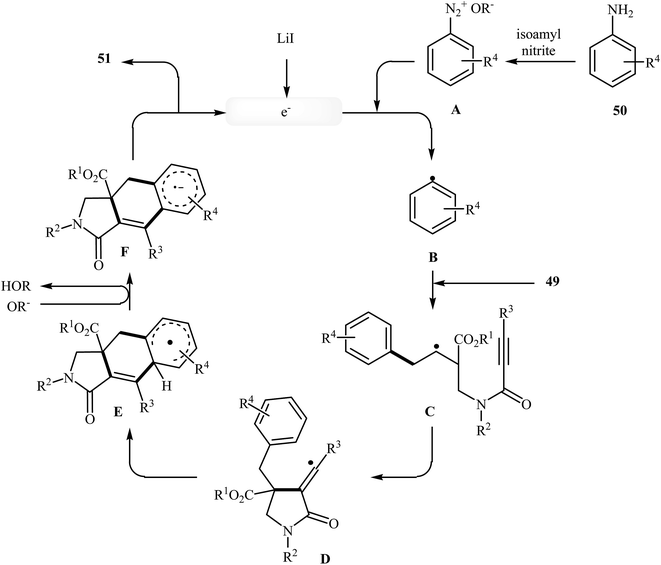 |
| | Fig. 2 Mechanistic proposal for the formation of 51. | |
4. Conclusion
γ-Lactams are at the heart of a number of natural products and synthetic pharmaceuticals that display an impressive variety of biological properties including antibacterial, antimicrobial, anticonvulsant, antidepressant, anti-inflammatory, antitumor, antimalarial, and antidiabetic properties. Several commercially available drugs, including brivaracetam, piracetam, levetiracetam, oxiracetam, seletracetam, aniracetam, ans phenylpiracetam are derived from γ-lactam-core entities. This biological activity has made the synthesis of γ-lactams quite attractive, and a large number of straightforward and robust methods for the construction of these cores established. In modern organic synthesis, intramolecular cyclization reactions are particularly important tools allowing the generation of at least one cycle in a single step with high atom economy, cost efficiency, and operational simplicity. As illustrated, the intramolecular cyclization of easily available N-allyl propiolamides into γ-lactam derivatives has gained a great deal of interest in recent years as useful alternative procedures. High atom and step economy, simplicity of operation, and good yields are the salient features of these reactions. Hopefully, this procedure will be employed in the synthesis of biologically important and complex natural γ-lactam-based compounds in future studies.
Acknowledgements
The authors gratefully thank to Dr Ahmadreza Bekhradnia for his useful comments and suggestions to improve our paper.
References
- P. von Rosenstiel, Neurotherapeutics, 2007, 4, 84–87 CrossRef CAS PubMed.
- B. Winblad, CNS Drug Rev., 2005, 11, 169–182 CrossRef CAS PubMed.
- M. Nett, T. A. Gulder, A. J. Kale, C. C. Hughes and B. S. Moore, J. Med. Chem., 2009, 52, 6163–6167 CrossRef CAS PubMed.
- D. Chauhan, L. Catley, G. Li, K. Podar, T. Hideshima, M. Velankar, C. Mitsiades, N. Mitsiades, H. Yasui and A. Letai, Cancer Cell, 2005, 8, 407–419 CrossRef CAS PubMed.
- Y. L. Yang, F. R. Chang and Y. C. Wu, Helv. Chim. Acta, 2004, 87, 1392–1399 CrossRef CAS.
- H. Shigehisa, J. Takayama and T. Honda, Tetrahedron Lett., 2006, 47, 7301–7306 CrossRef CAS.
- R. Kontnik and J. Clardy, Org. Lett., 2008, 10, 4149–4151 CrossRef CAS PubMed.
- A. Lebedev, Chem. Heterocycl. Compd., 2007, 43, 673–684 CrossRef CAS.
- G. Martelli, M. Orena and S. Rinaldi, Curr. Org. Chem., 2014, 18, 1373–1481 CrossRef CAS.
- A. Albrecht, Ł. Albrecht and T. Janecki, Eur. J. Org. Chem., 2011, 2747–2766 CrossRef CAS.
- J. Caruano, G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 CAS.
- F. Rivas and T. Ling, Org. Prep. Proced. Int., 2016, 48, 254–295 CrossRef CAS.
- E. Vessally, R. Hosseinzadeh-Khanmiri, E. Ghorbani-Kalhor, M. Es'haghi and A. Bekhradnia, RSC Adv., 2017, 7, 19061–19072 RSC.
- E. Vessally, A. Bekhradnia, A. Hosseinian, M. Sobati and S. Arshadi, J. CO2 Util., 2017, 19, 120–129 CrossRef.
- E. Vessally, A. Hosseinian, L. Edjlali, R. Hosseinzadeh-Khanmir and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638 CrossRef PubMed.
- S. Arshadi, E. Vessally, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmir, RSC Adv., 2017, 7, 13198–13211 RSC.
- E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, Curr. Org. Synth., 2017, 14, 557–567 CrossRef CAS.
- E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and A. Bekhradnia, RSC Adv., 2017, 7, 7079–7091 RSC.
- E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 99781–99793 RSC.
- E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 49730–49746 RSC.
- E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 71662–71675 RSC.
-
(a) E. Vessally, RSC Adv., 2016, 6, 18619–18631 RSC;
(b) E. Vessally and M. Abdoli, J. Iran. Chem. Soc., 2016, 13, 1235–1256 CrossRef CAS;
(c) E. Vessally, H. Saeidian, A. Hosseinian, L. Edjlali and A. Bekhradnia, Curr. Org. Chem., 2017, 21, 249–271 CrossRef CAS.
- H. Jiang, S. Ma, G. Zhu and X. Lu, Tetrahedron, 1996, 52, 10945–10954 CrossRef CAS.
- X. Xie and X. Lu, Synlett, 2000, 707–709 CAS.
- G. Zhu and Z. Zhang, J. Org. Chem., 2005, 70, 3339–3341 CrossRef CAS PubMed.
- G. Zhu, X. Tong, J. Cheng, Y. Sun, D. Li and Z. Zhang, J. Org. Chem., 2005, 70, 1712–1717 CrossRef CAS PubMed.
- G. Zhu and Z. Zhang, Org. Lett., 2003, 5, 3645–3648 CrossRef CAS PubMed.
- W. Xu, A. Kong and X. Lu, J. Org. Chem., 2006, 71, 3854–3858 CrossRef CAS PubMed.
- H. Wang, H. Xiuling and X. Lu, Tetrahedron, 2010, 66, 9129–9134 CrossRef CAS.
- H. Peng and G. Liu, Org. Lett., 2011, 13, 772–775 CrossRef CAS PubMed.
- X. Tong, M. Beller and M. K. Tse, J. Am. Chem. Soc., 2007, 129, 4906–4907 CrossRef CAS PubMed.
- S. J. Welsch, M. Umkehrer, G. Ross, J. Kolb, C. Burdack and L. A. Wessjohann, Tetrahedron Lett., 2011, 52, 6295–6297 CrossRef CAS.
- A. Lei, J. P. Waldkirch, M. He and X. Zhang, Angew. Chem., Int. Ed., 2002, 41, 4526–4529 CrossRef CAS PubMed.
- X. Tong, Z. Zhang and X. Zhang, J. Am. Chem. Soc., 2003, 125, 6370–6371 CrossRef CAS PubMed.
- X. Tong, D. Li, Z. Zhang and X. Zhang, J. Am. Chem. Soc., 2004, 126, 7601–7607 CrossRef CAS PubMed.
- J. Wang, X. Xie, F. Ma, Z. Peng, L. Zhang and Z. Zhang, Tetrahedron, 2010, 66, 4212–4217 CrossRef CAS.
- O. Jackowski, J. Wang, X. Xie, T. Ayad, Z. Zhang and V. Ratovelomanana-Vidal, Org. Lett., 2012, 14, 4006–4009 CrossRef CAS PubMed.
- T. Sawano, N. C. Thacker, Z. Lin, A. R. McIsaac and W. Lin, J. Am. Chem. Soc., 2015, 137, 12241–12248 CrossRef CAS PubMed.
- Y. T. Lee, Y. K. Kang and Y. K. Chung, J. Org. Chem., 2009, 74, 7922–7934 CrossRef CAS PubMed.
- D. V. Patil, H.-S. Park, J. Koo, J. W. Han and S. Shin, Chem. Commun., 2014, 50, 12722–12725 RSC.
- D. Qian and J. Zhang, Chem. Commun., 2011, 47, 11152–11154 RSC.
- H.-S. Yeom and S. Shin, Org. Biomol. Chem., 2013, 11, 1089–1092 CAS.
- J. Koo, H.-S. Park and S. Shin, Tetrahedron Lett., 2013, 54, 834–839 CrossRef CAS.
- S. I. Lee, S. M. Kim, S. Y. Kim and Y. K. Chung, Synlett, 2006, 2256–2260 CAS.
- F. Zhang, S. Wang, Z. Liu, Y. Bai and G. Zhu, Tetrahedron Lett., 2017, 58, 1448–1452 CrossRef CAS.
- S. K. Tae, Bull. Korean Chem. Soc., 1993, 14, 431–432 Search PubMed.
- L. Feray and M. P. Bertrand, Eur. J. Org. Chem., 2008, 3164–3170 CrossRef CAS.
- J. Xuan, C. G. Daniliuc and A. Studer, Org. Lett., 2016, 18, 6372 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Mirzaagha Babazadeh
c,
Akram Hosseinian
d and
Ladan Edjlali
c
b,
Mirzaagha Babazadeh
c,
Akram Hosseinian
d and
Ladan Edjlali
c