
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A one-step process employing various amphiphiles for an electrically insulating silica coating on graphite†
Yeongseon Kim,
Yingjie Qian,
Minjae Kim,
Jaechul Ju,
Sung-Hyeon Baeck and
Sang Eun Shim *
*
Department of Chemistry & Chemical Engineering, Inha University, Republic of Korea. E-mail: seshim@inha.ac.kr; Fax: +82 32 876 0327; Tel: +82 10 6272 6290
First published on 3rd May 2017
Abstract
Ceramic coatings endow carbon materials with electrically insulating properties. For graphite, it is unclear whether ceramic coatings applied via a one-step process under mild conditions can lead to superior coverage. This paper reports that a modified Stöber method with an appropriate choice of amphiphiles could yield an electrically insulating coating layer on graphite (d50 3–300 μm, d90 6–550 μm) within 24 h with high reproducibility. A silica coating mechanism involving functional groups on the edge of the graphite, Oswald ripening agents, and a bridgemer was investigated. The mechanism was based on the silica coating morphology, which depends on the amphiphile, and the correlation between coverage and surface resistivity was assessed. Amphiphile-assisted silica@graphite with a surface resistivity of 1012 ohm sq−1 was produced. The thermal conductivity of the silica-coated graphite (amphiphile-assisted silica@graphite)/TPEE composite reached values over 75% higher than that of the raw graphite/TPEE composite with electrically insulating properties.
1. Introduction
Graphite has excellent thermal performance (Λ∥: 800–2000 W mK−1, Λ⊥: 10–200 W mK−1), product reliability resulting from its convenience in extrusion feeding/operating, and functionality (i.e., a low coefficient of thermal expansion, light weight, density, lubricative properties, corrosion resistance, high dielectric constant, and EMI shielding/absorption properties).1–3 These properties have given it a dominant position in the materials fields for thermal management, such as in light-emitting diode (LED) housings, thermal spreaders for displays, heat sinks for computer microchips, thermal paste/grease, thermal tape, battery system packaging, and parts for high-technology devices.4,5The development of graphite-incorporated materials depends not only on the superior thermal performance of graphite, but also on its other properties. For example, the lubricative properties and thermal conductivity of graphite make it suitable for thermal grease and thermal lubricants. On the other hand, inappropriate combinations of these diverse properties can obstruct valuable attempts to explore more advanced and broader applications. Carbon-based materials, such as graphite and graphite-incorporated polymer composites, have a critical drawback—the surface resistivity of raw graphite is under 102 ohm sq−1. This limits their applicability as an additive to the coating solution for printed circuit boards (PCBs), an adhesive for the assembly of electronic chips, pigments for thermal paints, composites fillers for cable coverings, and packaging materials for electronics.6
Surface modification methods for carbon materials have helped them achieve superior potential and reliable properties (i.e., mechanical, electrical, and thermal properties) in carbon fillers. Methods for improving the physical and chemical affinity between fillers and polymer matrices affect the distribution/dispersion state of the filler networks. These improvement methods include surface functionalization (i.e., grafting functional groups on/from the surface of the carbon), dispersant treatment, coupling agent treatment, and the decoration method. The percolation threshold for the filler loading in a polymer matrix is shifted to a lower level.7–15 On the other hand, severe surface modification methods such as acid treatments and physical impacts degrade the original properties of the carbon, because these methods damage the conjugated sp2-structures of the planar moieties.
Coating a amphiphile-covered carbon substrate with a high-coverage ceramic layer is a suitable method for overcoming the abovementioned drawbacks of the carbon material, while avoiding the formation of defects via harsh modification methods. Such a method offers transforms to the electrical properties of carbon fillers, and they can possess thermally conductive and electrically insulating properties via this strategy.16–23 In previous studies, a dense silica layer was successfully coated onto graphite via a polyvinylpyrrolidone (PVP)-assisted sol–gel reaction under basic conditions. PVP played a crucial role in overcoming poor chemical affinity between the hydrophilic silica and hydrophobic planar moieties of the graphite surface. The PVP-assisted silica@graphite was categorized as an electrical insulator with a high surface resistivity of up to 1012 ohm sq−1; nevertheless, the PVP-assisted silica@graphite has a problem in terms of productivity. The process consists of two steps: the physical adsorption of PVP on graphite in H2O, and the formation of a silica layer on PVP@graphite in ethanol.18,19 A work-up process that included filtration and drying was involved in both steps. As a result, the two-step process required over 40 h for processing.
There exist a few results on the use of alumina and silica coated graphite for thermally conductive composites. However, the synthetic conditions, synthetic mechanism or genuine properties of the as-prepared materials, which strongly influence the thermal and electrical transporting characteristics, have not been scrutinized well to date.22,23
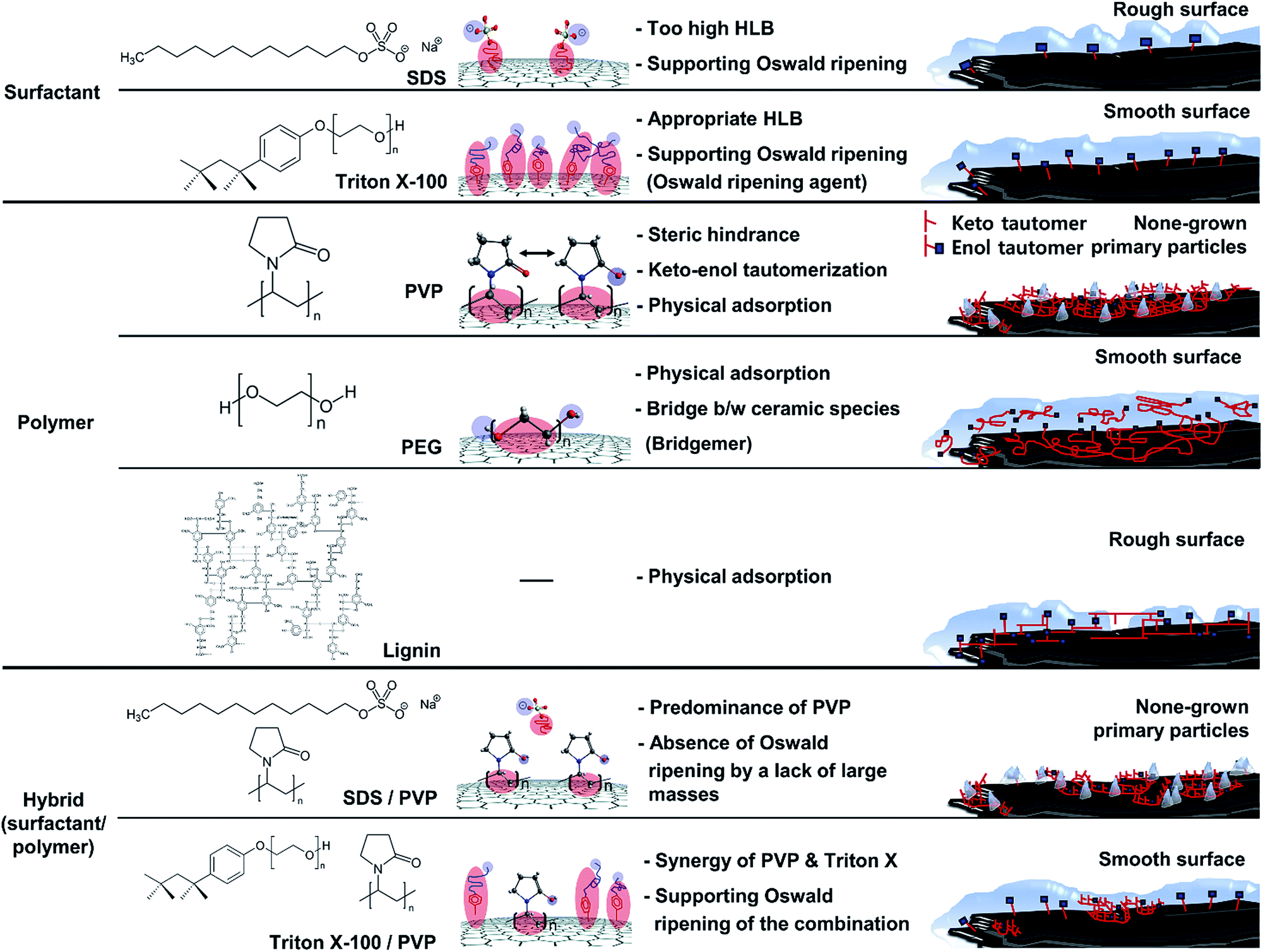
Herein, one-step processes that fully covers graphite (d50 3–300 μm, d90 6–550 μm) with a silica layer within 12–25 h, are suggested. Fig. 1 presents the basic concept of the amphiphile-assisted silica@graphite (Grasil), with a correlation between each silica coating procedure for silica coating and the resulting properties (i.e., thermal conductivity and surface resistivity). Although covering raw graphite with an amphiphile causes a minuscule decrease in its thermal conductivity, the amphiphile provides a hydrophobic basal plane with functional groups. The hydroxyl groups of the enol tautomer of PVP19 (Triton X-100, SDS,25 lignin,26 and PEG27) are appropriate for inducing a dehydration reaction in the silanol group of a silica precursor. The growth of polysilicate species under basic conditions results in branched structures, which have a broader distribution. In contrast, the growth of polysilicate species under acidic conditions is predisposed to form a linear structure.28 The abundance of growing points (i.e., secondary, tertiary) of nuclei has the advantage of the covering work; it can facilitate the growth of silica species in the immediate vicinity of existing nuclei. A thin silica layer coated on the outside is considered a thermal and electrical barrier (the thermal conductivity of silica is 1.5 W mK−1),29 which endows raw graphite with electrically insulating properties. Fig. 2 illustrates how to approach the modified one-step process, and reduce the processing time to less than 50% of existing two-step process. The media were unified as ethanol under basic conditions. There are several considerations for encouraging appropriate amphiphile absorption on graphite for silica growth. High-temperature treatment at 500 °C reduces drying time to less than 50% of its original value, and reinforces the mechanical properties of the silica layer (thermal sintering). PEG is applied to the modified one-step process, and helps reduce the coating-process time under one-fourth (ver. 2).
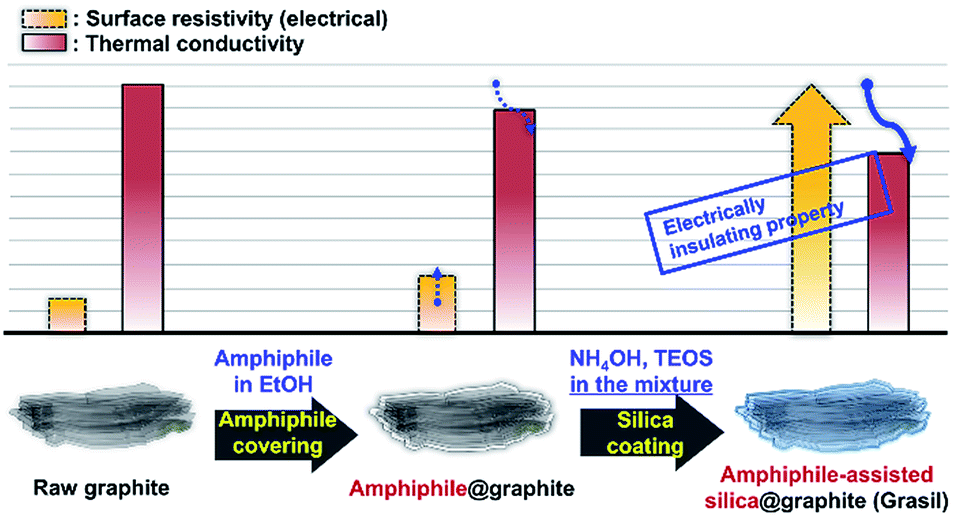 | ||
| Fig. 1 Basic concept for the amphiphile-assisted silica@graphite (Grasil) via the modified one-step process. | ||
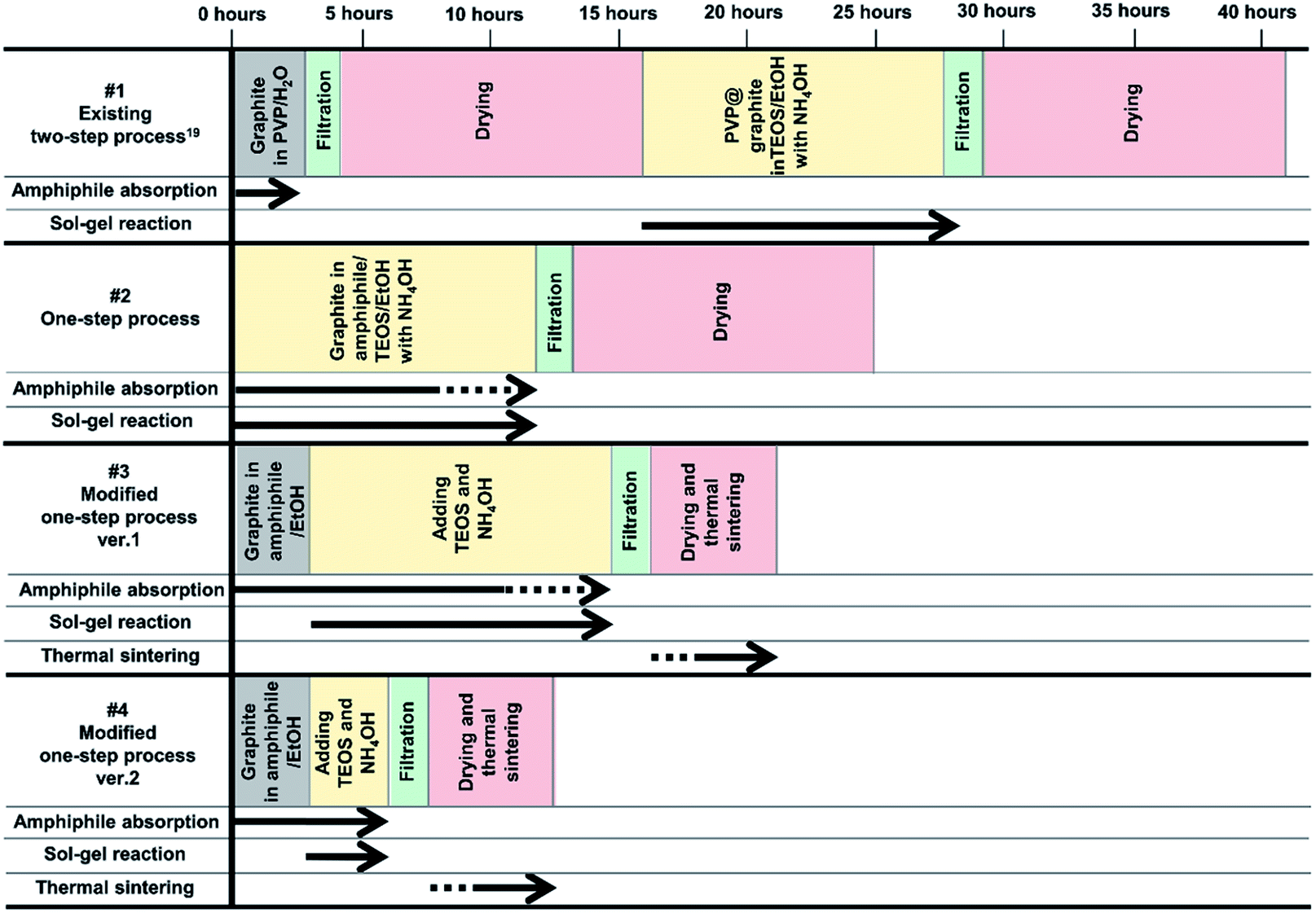 | ||
| Fig. 2 Four processes (existing two-step,19 one-step, modified one-step ver. 1 and ver. 2) for synthesizing amphiphile-assisted silica@graphite. | ||
This paper aimed to provide thermally conductive and electrically insulating filler via optimized process in terms of graphite size, suitable amphiphile, and processing time/steps. Generally, larger graphite/polymer composite possess higher thermal conductivity than that of smaller graphite, however, the larger graphite is less suitable to silica coated layer. The amphiphiles determine coated morphology, coating rate, growth model, and ultimately the success of the coating. The amphiphile-assisted silica@graphite that was synthesized under optimal conditions was compounded with a thermoplastic polyester elastomer (TPEE) to determine its electrically insulating properties after being subjected to shear force during high-content mixing of 50 wt% (100 phr). The main paragraph text follows directly on here.
2. Experimental
2.1. Materials
Five different sizes of synthetic graphite were supplied by Timcal (Switzerland): KS6 (d50: 3.4 μm, d90: 6.5 μm); KS44 (d50: 18.6 μm, d90: 45.4 μm); KS75 (d50: 23.1 μm, d90: 55.8 μm); KS150 (d50: approximately 100 μm,30 d90: approximately 140 μm); and KS500 (approximately 300 μm,30 d90: about 550 μm). Octylphenyl 9.5 ethoxylate (Triton X-100) was provided by Samchun (S. Korea), and polyvinylpyrrolidone (PVP, Mw: 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was supplied by Junsei. Lignin (alkali), polyethyleneglycol (PEG, Mn: 400, 2050, 4600, 10000 g mol−1), polyethyleneimine (PEI, Mw: 800 g mol−1), and sodium dodecyl sulfate (SDS) were supplied by Sigma-Aldrich (USA). Tetraethyl orthosilicate (TEOS, ≥95%) from Junsei Chemical Co., Ltd. (Japan), ammonium hydroxide (25 wt% NH3 in water) from OCI Co., Ltd. (S. Korea) and ethanol from Duksan Chemicals Co., Ltd. (S. Korea) were selected.
000 g mol−1) was supplied by Junsei. Lignin (alkali), polyethyleneglycol (PEG, Mn: 400, 2050, 4600, 10000 g mol−1), polyethyleneimine (PEI, Mw: 800 g mol−1), and sodium dodecyl sulfate (SDS) were supplied by Sigma-Aldrich (USA). Tetraethyl orthosilicate (TEOS, ≥95%) from Junsei Chemical Co., Ltd. (Japan), ammonium hydroxide (25 wt% NH3 in water) from OCI Co., Ltd. (S. Korea) and ethanol from Duksan Chemicals Co., Ltd. (S. Korea) were selected.
2.2. Sample preparation
:3 ratio. The TEOS sol–gel reaction was stirred at room temperature for 12 h. After coating, the mixture was filtered and washed with ethanol. Finally, the wet filler-cake was dried in an oven at 60 °C. In the thermal sintering work, the wet filler cake was sintered in a furnace at 500 °C for 5 h. Tables 1 and 2 show the additional experimental conditions (for the experimental conditions of the one-step process and modified one-step process, ver. 2, see ESI†).
| No. | Graphite | Amphiphile | NH4OH | TEOS | Media | Reaction time | |||
|---|---|---|---|---|---|---|---|---|---|
| d50/d90 (μm) | Mass (g) | Mass (g) | Mass% of amphiphile input compared with graphite | Mass ratio of NH4OH to TEOS | Mass (g) of 1st/2nd input | Mass% of amphiphile input compared with graphite | Volume (ml) of EtOH | Time (h) | |
| a Two different size of raw graphite, KS6 (d50 3.4 μm, d90 6.5 μm) and KS44 (d50 18.6 μm, d90 45.4 μm), were prepared with a mass ratio of 1:1.b The amphiphiles used in the preparation of samples #1–6 were: (1) Triton X-100, (2) PVP, (3) lignin, (4) SDS, (5) a hybrid of Triton X-100 and PVP with a mass ratio of 1:1, (6) a hybrid of SDS and PVP with a mass ratio of 1:1.c The amphiphile used in the preparation of samples #7–24 was Triton-X 100. |
|||||||||
| 1–6 | 12/25a | 5 | 0.50b | 10 | 1:3 |
6.0/0 | 120/0 | 75 | 12 |
| 7–18 | 12/25a | 5 | 0.25c | 5 | 1:3 |
1.0/0 | 20/0 | 75 | 12 |
| 0.25c | 5 | 2.0/0 | 40/0 | ||||||
| 0.25c | 5 | 4.0/0 | 80/0 | ||||||
| 0.25c | 5 | 6.0/0 | 120/0 | ||||||
| 0.50c | 10 | 1.0/0 | 20/0 | ||||||
| 0.50c | 10 | 2.0/0 | 40/0 | ||||||
| 0.50c | 10 | 4.0/0 | 80/0 | ||||||
| 0.50c | 10 | 6.0/0 | 120/0 | ||||||
| 2.00c | 40 | 1.0/0 | 20/0 | ||||||
| 2.00c | 40 | 2.0/0 | 40/0 | ||||||
| 2.00c | 40 | 4.0/0 | 80/0 | ||||||
| 2.00c | 40 | 6.0/0 | 120/0 | ||||||
| 19–24 | 12/25a | 5 | 0.50c | 10 | 1:3 |
4.0/2.0 | 80/40 | 75 | 3 |
| 6 | |||||||||
| 9 | |||||||||
| 10 | |||||||||
| 11 | |||||||||
| 12 | |||||||||
| No. | Graphite | Amphiphile | NH4OH | TEOS | Media | Reaction time | |||
|---|---|---|---|---|---|---|---|---|---|
| d50/d90 (μm) | Mass (g) | Mass (g) | Mass% of amphiphile input compared with graphite | Mass ratio of NH4OH to TEOS | Mass (g) of 1st/2nd input | Mass% of amphiphile input compared with graphite | Volume (ml) of EtOH | Time (h) | |
| a Two different size of raw graphite, KS6 (d50 3.4 μm, d90 6.5 μm) and KS44 (d50 18.6 μm, d90 45.4 μm), were used with a mass ratio of 1:1.b The amphiphiles used for samples #25–30 were: (25) Triton X-100, (26) PVP, (27) lignin, (28) SDS, (29) PEI, and (30) PEG (Mn 4600 g mol−1).c The amphiphile used for samples #31–39 was a PEG of Mn 4600 g mol−1.d Molecular weights (Mn) of PEG for samples #40–55 were (40–43) 400, (44–47) 2060, (48–51) 4600, and (52–55) 10000 g mol−1. |
|||||||||
| 25–30 | 100/140 | 5 | 0.50b | 10 | 1:3 |
2.7/1.3 | 53/27 | 75 | 12 |
| 31–39 | 100/140 | 5 | 0.25c | 5 | 1:3 |
0.7/0.3 | 7/13 | 75 | 12 |
| 0.25c | 5 | 1.3/0.7 | 27/13 | ||||||
| 0.25c | 5 | 2.7/1.3 | 53/27 | ||||||
| 0.50c | 10 | 0.7/0.3 | 7/13 | ||||||
| 0.50c | 10 | 1.3/0.7 | 27/13 | ||||||
| 0.50c | 10 | 2.7/1.3 | 53/27 | ||||||
| 1.00c | 20 | 0.7/0.3 | 7/13 | ||||||
| 1.00c | 20 | 1.3/0.7 | 27/13 | ||||||
| 1.00c | 20 | 2.7/1.3 | 53/27 | ||||||
| 40–59 | 12/25a | 5 | 0.5d | 10 | 1:3 |
2.7/1.3 | 53/27 | 75 | 12 |
| 23/55 | 0.5d | 10 | |||||||
| 100/140 | 0.5d | 10 | |||||||
| 300/550 | 0.5d | 10 | |||||||
| 12/25a | None | 0 | |||||||
| 23/55 | None | 0 | |||||||
| 100/140 | None | 0 | |||||||
| 300/550 | None | 0 | |||||||
2.3. Characterization
Field emission transmission electron microscopy (FE-TEM, CM 2000, Philips) and scanning electron microscopy (SEM, Hitachi S-4300) were used to observe the surface morphology of the coated silica on the graphite surface. Quantitative changes in the composition of the silica@graphite were calculated with thermogravimetric analysis (TGA, Diamond TG/DTA Lab System, Perkin Elmer Inc.) over a temperature range of 50–900 °C at a heating rate of 5 °C min−1 in air. Prior to TGA, the samples were dried overnight in a vacuum oven at 60 °C. A quick thermal conductivity meter (QTM-500, Kyoto Electronics) was used to measure the thermal conductivity of the composites. The surface resistivity of the fillers and their thermoplastic polyester elastomer (TPEE) composites was measured using a high-resistivity meter (Hiresta-UP, Mitsubishi Chemical Co.) and a low-resistivity meter (Loresta-GP, Mitsubishi Chemical Co.). A high-resistivity meter uses constant-voltage processing with a concentric ring probe and a measurement range of 104 to 1013 Ω, and a low-resistivity meter uses constant-current processing with a linear four-point probe and a measurement range of 10−3 to 107 Ω.3. Results and discussion
3.1. Morphology of the amphiphile-assisted silica@graphite
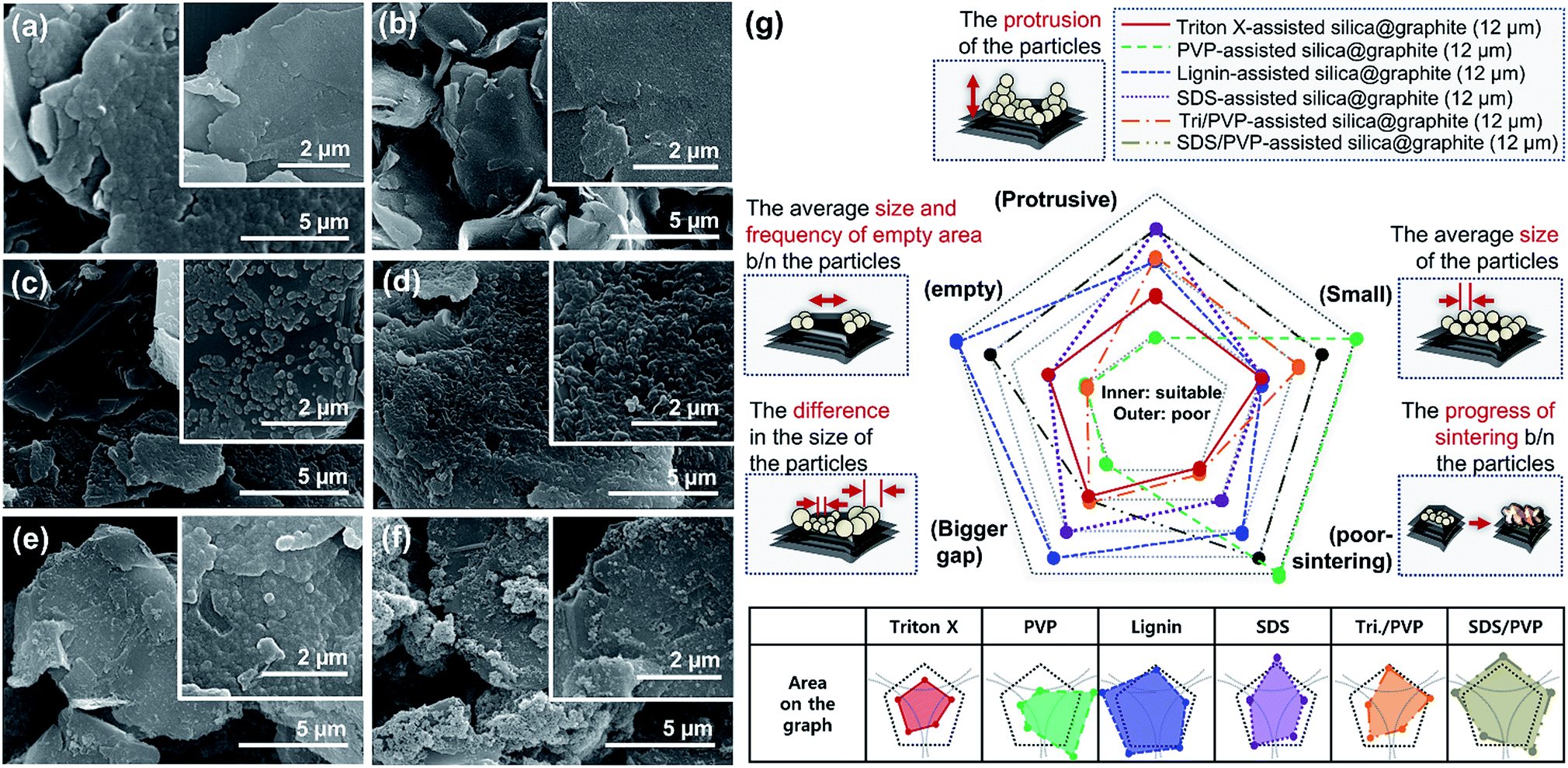 | ||
| Fig. 3 FE-SEM images of silica@graphite surfaces (d50 12 μm, d90 25 μm) prepared using (a) Triton-X, (b) PVP, (c) lignin, (d) SDS, (e) combination of Triton-X/PVP, (f) combination of SDS/PVP, and (g) morphology analysis. | ||
Fig. 4 shows SEM images of Triton X-assisted silica@graphite synthesized with two different variables: the amounts of Triton X-100 and TEOS. The optimal conditions for uniform silica coating on graphite were found to be 10 wt% of Triton X-100 and 120 wt% of TEOS with respect to the mass of graphite. Three facts regarding the silica-coating mechanism can be deduced from the coating tendencies. The formation and growth of nuclei occurs preferentially on the edge rather than the basal plane of graphite. The growth of nuclei tends to proceed via the connection of grown particles and absorption of adjacent embryos. In other words, few islands are separated from the grown silica species that are based on the edge. Triton X-100 helps the silica species grow toward the basal plane and fills the many channels with silica species.
 | ||
| Fig. 4 FE-SEM images of the surfaces of the Triton X-assisted silica@graphite (d50 12 μm, d90 25 μm) synthesized with different amounts of Triton X-100 and TEOS. | ||
Fig. 5 shows SEM images of silica@graphite synthesized with six different amphiphiles: Triton X-100, PVP, lignin, SDS, PEI, and PEG. The coating morphology was analyzed with five metrics, as shown in Fig. 5g. An item that abides by the 3rd line is shown only in Fig. 5e (PEG), which shows a smooth and compact coating-surface with full coverage, containing few cracks on the thick silica layer. In contrast to what is shown for the 12 μm Trion X-assisted silica@graphite, the growth of nuclei is suppressed, and there are no large particles to absorb embryos (see Fig. 5a, Triton X-100). Triton X-100 is a subsidiary amphiphile, which leads silica species to grow from the edge to the basal plane. Triton X-100 is unable to facilitate growth toward the Z-axis without large adjacent silica species. Therefore, the growth of the silica species on the middle portions of the wide basal plane in this system is not expected. Hong et al. suggested that a liquid crystal surfactant/silicate film is aligned with a hemimicelle-graphite structure. Micelles of cetyltrimethylammonium chloride (CTACl) were assembled in an aqueous medium under acidic conditions; the silica species grew from the outer hydrophilic heads of the micelles, which were stacked in tiers. The behavior facilitated growth toward the Z-axis.24 On the other hand, the existence of micelles is rare in an alcoholic medium containing approximately 5–10% water (25% ammonia solution and 95% ethanol were employed) under basic conditions. In the cases shown in Fig. 5b (PVP), Fig. 5c (lignin), Fig. 5d (SDS), and Fig. 5f (PEI), there were insufficient seed nuclei, meaning that there was a lack of amphiphile adsorption on the graphite. Four characteristics regarding the silica-coating mechanism can be deduced from the coating tendencies. (1) The hydrophobic portion of Triton X-100 binds to graphite more easily than that of SDS. (2) Triton X-100 cannot form islands with the silica species on the wide basal plane (graphite over 100 μm). (3) The PEG backbone has the advantage of physical adsorption onto graphite compared to PVP and PEI backbones. (4) In this system, lignin is an indefinite and vague amphiphile, with weak physical adsorption and uneven hydroxyl groups. As a result, PEG was selected as a suitable amphiphile for a silica coating of the 100 μm graphite.
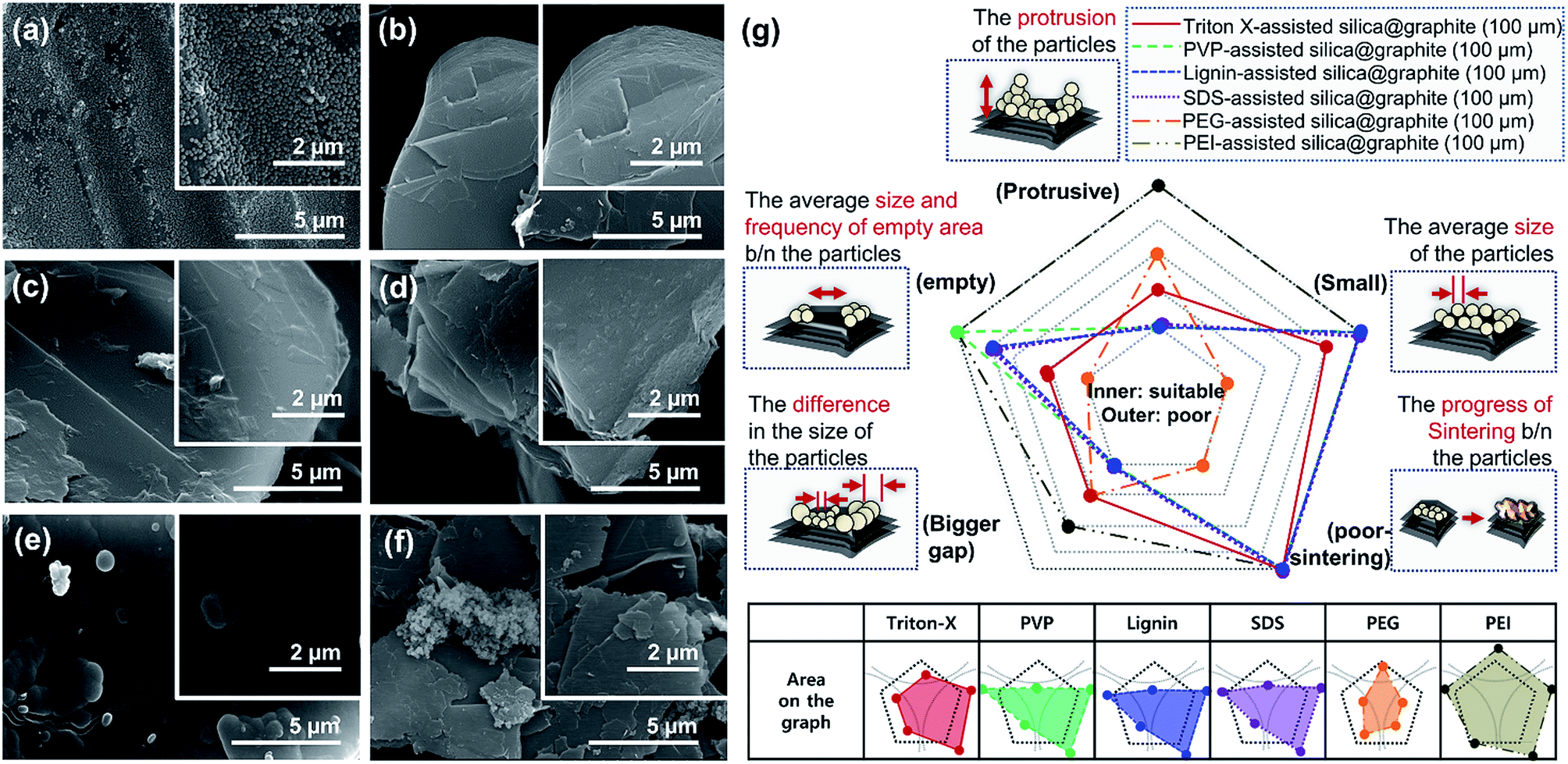 | ||
| Fig. 5 FE-SEM images of surfaces of silica@graphites (d50 100 μm, d90 140 μm) prepared using (a) Triton X, (b) PVP, (c) lignin, (d) SDS, (e) PEG, (f) PEI, and (g) morphology analysis. | ||
 | ||
| Fig. 6 FE-SEM images of surfaces of (a) the Triton X-assisted silica@graphite (12 μm) and (b) PEG-assisted silica@graphite (100 μm) depending on the sol–gel reaction time. | ||
 | ||
| Fig. 7 Mechanism of silica coating on graphite depending on amphiphile. | ||
The mechanism for surfactant (amphiphile)-assisted silica coating involves (1) the molecular structures of the surfactants, and (2) hydrophile(polar)–lipophile(non-polar) balance (HLB), given the weak VDW interaction resulting from very low molecular weight. The HLB values of Triton-X and SDS were calculated using the following equation.32
| HLB = ∑(hydrophilic group number) − n(group number per CH2 group) + 7 | (1) |
The SDS molecule has an HLB value of 40; it incorporates a linear carbon chain on its hydrophobic portion and a sulfate on its hydrophilic portion. Triton X-100 combines a linear carbon chain and a benzene ring on the hydrophobic portion with a long chain of polyoxyethylene on the hydrophilic portion. Although the molecule has a moderate HLB value of 13.5, its hydrophobic portion allows it to closely approach graphite. Just as “like dissolves like” in the world of solvents, the concept of “like attracts like” is considered in this system in terms of how the hydrophobic portion of the surfactant interacts with graphite. Compared to the HLB and molecular structure of SDS, Triton X-100 is more suitable for binding to graphite because of the more hydrophobic HLB and benzene ring. Because of the more bounded Triton X-100, Triton X-100@graphite can provide a larger number of hydrophilic-functional groups on graphite than SDS@graphite in the system for the same reaction time. This forms more seed-nuclei on the graphite than in the case of SDS with the same reaction time. In addition, the more seed-nuclei also facilitates the overlap of silica species-hemispheres (nuclei and island) on graphite. Consequently, Triton X-assisted silica@graphite has a smoother and more compact coating-surface than SDS-assisted silica@graphite. The amphiphile of the surfactant type (i.e., Triton X-100 and SDS) provides nuclei that are surrounded by functional groups forming embryos; the amphiphile facilitates the adsorption of embryos by the nuclei. Proceeding further, such behavior assists the formation of islands and nuclei layers via Oswald ripening. Therefore, amphiphiles are referred to as “Oswald ripening agents”.
The mechanism of the polymer (amphiphile)-assisted silica coating is under a dominant influence on the VDW interaction that resulting from the high molecular weight and the structures of the polymers. As mentioned in Section 3.1.1, the PVP covering disturbs the edge growth of silica species, which means that the PVP chains are firmly bound to the edge. In the case of surfactants, such as SDS and Triton X-100, the hydrophobic portions of the surfactants can also bind to edges, but no channels are observed on the edge of the surfactant-assisted silica@graphite. Three characteristics of this mechanism can be deduced from the results. The surfactants can escape the edge and adsorb to the basal plane. The physical adsorption of polymers by VDW interactions enables them to firmly bind to all areas (basal plane and edge) of the graphite. Therefore, polymer-assisted silica coating cannot support the edge growth of silica species in this system. In terms of the molecular structures of the polymers, hydroxyl groups are located on: the end of the pyrrolidone rings of the PVP enol-tautomer backbone,19 the irregular points of the lignin network that is intertwined with diverse functional groups, and the PEG terminals. Accordingly, in comparison to PVP and lignin backbone chains, PEG backbone chains can (advantageously) physically adsorb onto graphite. The steric hindrance that results from the pyrrolidone rings of the PVP enol tautomer prevents the hydroxyl groups from encountering the silanol group of the silica precursor molecule. Moreover, the keto–enol tautomerism of PVP is a reversible reaction under basic conditions; the PVP layer on graphite cannot always provide stable hydroxyl groups. Although a PEG chain has just two hydroxyl groups on both terminals, and the total number of hydroxyl groups per volume is lower than that of other amphiphiles, PEG chains can form ‘silica sol–PEG’ networks as cross-linking of polymers.33 Therefore, the ‘silica sol–PEG’ network reacts on (route 1) the hydroxyl groups of PEG chains adsorbed to the graphite, (route 2) the hydroxyl groups of PEG bound on silica species (i.e., nuclei, islands) on the graphite, and (route 3) the silanol groups of silica species (i.e., nuclei, islands) condensed on the graphite. Based on these routes, PEG facilitates the individual growth of silica species toward the Z-axis, without adjacent nuclei and islands on the basal plane and edges. Therefore, an amphiphile that (1) couples the sol masses and (2) binds the networks to the other material substrate can be called a “bridgemer”. As a result, PEG-assisted silica@graphite has a smoother and more compact coating-surface, while the nuclei of the PVP-assisted silica@graphite cannot grow into islands. Small (12 μm) lignin-assisted silica@graphite has large channels resulting from irregular islands and nuclei. Large (100 μm) lignin-assisted silica@graphite has no islands; the nuclei are few and minuscule.
With respect to the mechanism for a surfactant/polymer hybrid, the important qualities are its synergy effect and adverse effect, and a more-dominant amphiphile to bind it to the graphite. Although PVP cannot facilitate the growth of silica species on graphite, it provides 12 μm graphite in the system with functional groups for seed-nuclei. Few channels on the coating surface of the Triton X-assisted silica@graphite are filled with silica species through the assistance of seed-nuclei resulting from PVP, whereas SDS-assisted silica@graphite loses its rough surface, and therefore loses graphite edge space because of the physical adsorption of PVP chains. The difference in adsorption on graphite is due to their molecular structure and HLB. Triton X-100, which has an HLB of 13.5, can better bind to graphite than SDS, which has an HLB of 40. There are three explanations for this: (1) the Triton-X/PVP hybrid generates a synergy effect. Although Triton-X can escape from graphite, Triton X-100 disturbs the physical adsorption of the PVP chains, particularly on the edge of the graphite. (2) The SDS/PVP hybrid generates an adverse effect. The PVP chains bind dominantly to the graphite relative to SDS, and the PVP that is bound on the edges restrains the growth of silica species there. (3) The contribution of surfactants in the surfactant/polymer hybrid system depends on their molecular structure and HLB. Considering the interaction between surfactants and polymers from another point of view, some studies have described ceramic hollow microspheres using a pear-necklace SDS/PVP model, a core–shell model of SDS/polyacrylic acid (PAA), and a core–shell model of sodium dodecyl benzenesulfonate (SDBS)/PAA.27,34 On the other hand, the models depend on the micelle structures in an aqueous system. Consequently, the Triton-X/PVP-assisted silica@graphite has a more complementary full-coverage coating-surface than that of the SDS/PVP-assisted silica@graphite.
In summary, Sections 3.1.1–3.1.4, and Fig. 8 present the mechanism of silica species growth on graphite via (a) a non-amphiphile, (b) an Oswald ripening agent such as Triton-X, (c) a bridgemer, such as PEG, and (d) three strategies depending on the graphite size.
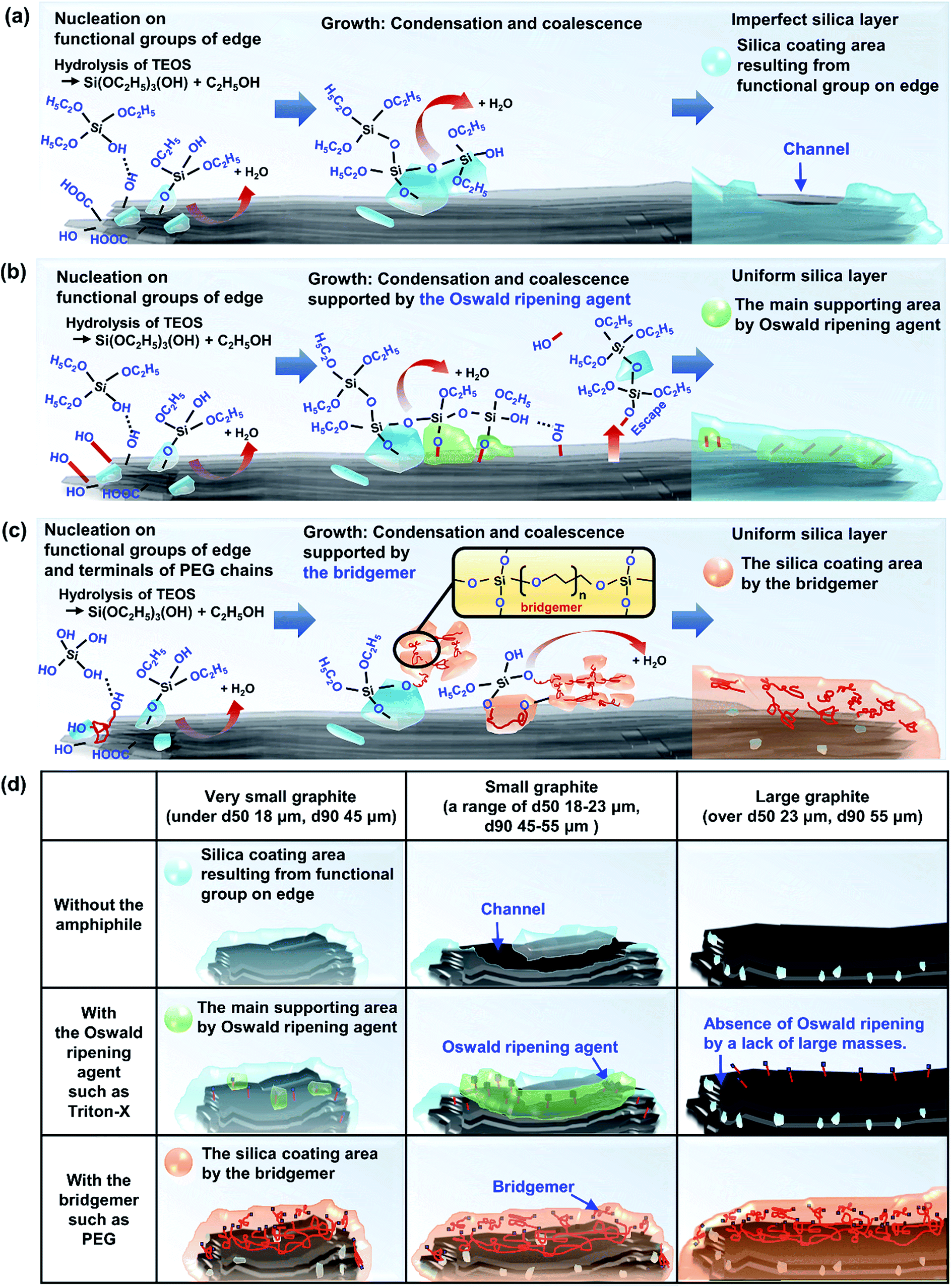 | ||
| Fig. 8 Mechanism of silica coating on graphite (a) in the absence of an amphiphile, and with the assistance of (b) an Oswald ripening agent, (c) a bridgemer, and (d) amphiphile strategies for silica coating depending on the size of the graphite. | ||
3.2. Surface resistivity of the amphiphile-assisted silica@graphite
From a broad point of view, Fig. 9 provides information on the most suitable amphiphile for an insulating graphite treatment (d50 sizes of 12, 23, 100, and 300 μm), and the inextricable correlation between surface resistivity and morphology. Fig. 9a shows the surface resistivity of an amphiphile-assisted silica@graphite (with sufficient TEOS applied for full coverage) synthesized with two different variables: the d50 size of the graphite and amphiphile. In contrast to the 2.13 × 1012 ohm sq−1 of PVP-assisted silica@graphite (d50 size = 12 μm) that was prepared via a two-step process18,19 the PVP-assisted silica@graphite resulting from the modified one-step process maintains low surface resistivity, in the range of 102 ohm sq−1 over all sizes of employed graphite. The additional physical adsorption work, which entails a drying time of 12 h, enables the PVP chains to adequately bind to the surface of the graphite. Additionally, the enol tautomer of the PVP, which is bound on the graphite via the two-step process, provides sufficient hydroxyl groups. The two-step process improves the physical absorption of PVP relative to the one-step process. Nevertheless, the PVP-assisted two-step process is not capable of coating a graphite surface larger than d50 100 μm (for the surface resistivity of the PVP-assisted silica@graphite, d50 size 100, 300 μm, via the two-step process, see ESI†). Considering that silica species cannot grow individually on a large basal-plane with Triton X-100, the surface resistivity of the Triton X-assisted silica@graphite depends on the size of the graphite. SDS-assisted silica@graphite and lignin-assisted silica@graphite, which have rough morphology and some channels, have surface resistances of 1011 and 108 ohm sq−1, respectively. The combination of SDS/PVP-assisted graphite, in which PVP has a more dominant position than SDS, has a surface resistivity of 102 ohm sq−1. This value is similar to that of the PVP-assisted silica@graphite. The Triton-X/PVP-assisted silica@graphite attains a higher surface resistivity (in the range of 1012 to 1013 ohm sq−1) through synergy than through Triton X-100. The PEG-assisted silica@graphite shows electrically insulating properties (1011 to 1012 ohm sq−1) in a broad range of graphite sizes: d50 3 to 300 μm, d90 6 to 550 μm.
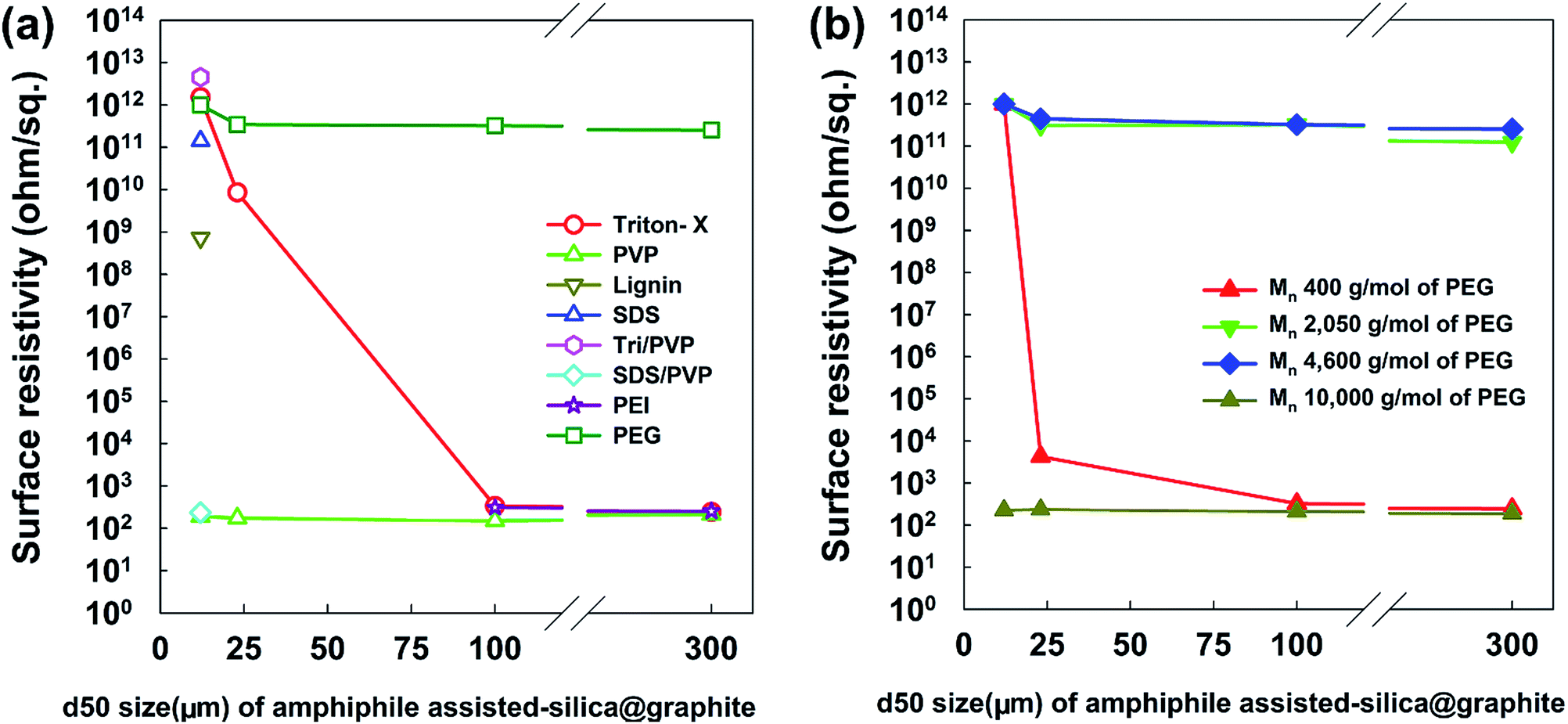 | ||
| Fig. 9 Surface resistivity of amphiphile-assisted silica@graphite depending on the amphiphile and size (d50) of graphite. | ||
Fig. 9b shows the most appropriate range of PEG molecular weights is Mn 2050–4600 g mol−1 (for FE-SEM images of the surfaces of PEG-assisted silica@graphite depending on the size of the graphite and Mn of PEG, see ESI†). PEG with a molecular weight of Mn 400 g mol−1 had very weak VDW interactions, while PEG with a molecular weight of Mn 10000 g mol−1 (lacking hydroxyl groups) cannot show satisfactory performance (it had a surface resistivity of 102 to 103 ohm sq−1 except in the case of Mn 400 g mol−1 and a d50 size of 12 μm). As a result, Triton X-100 was only selected as a suitable silica-coating amphiphile for the 12 μm graphite, while PEG (Mn 2050–4600 g mol−1) can be used as a coating over the broadest spectrum of graphite.
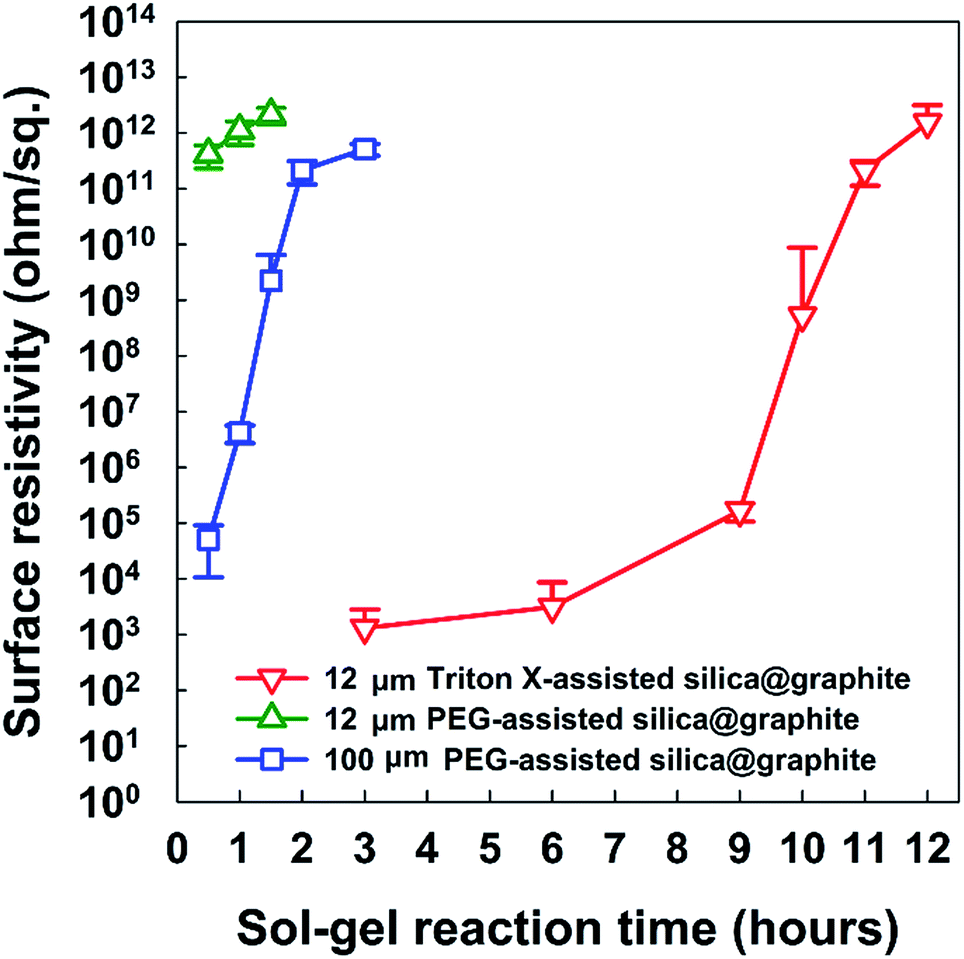 | ||
| Fig. 10 Surface resistivity of Triton X-assisted silica@graphite (12 μm), PEG-assisted silica@graphite (12 μm), and PEG-assisted graphite (100 μm) vs. sol–gel reaction time. | ||
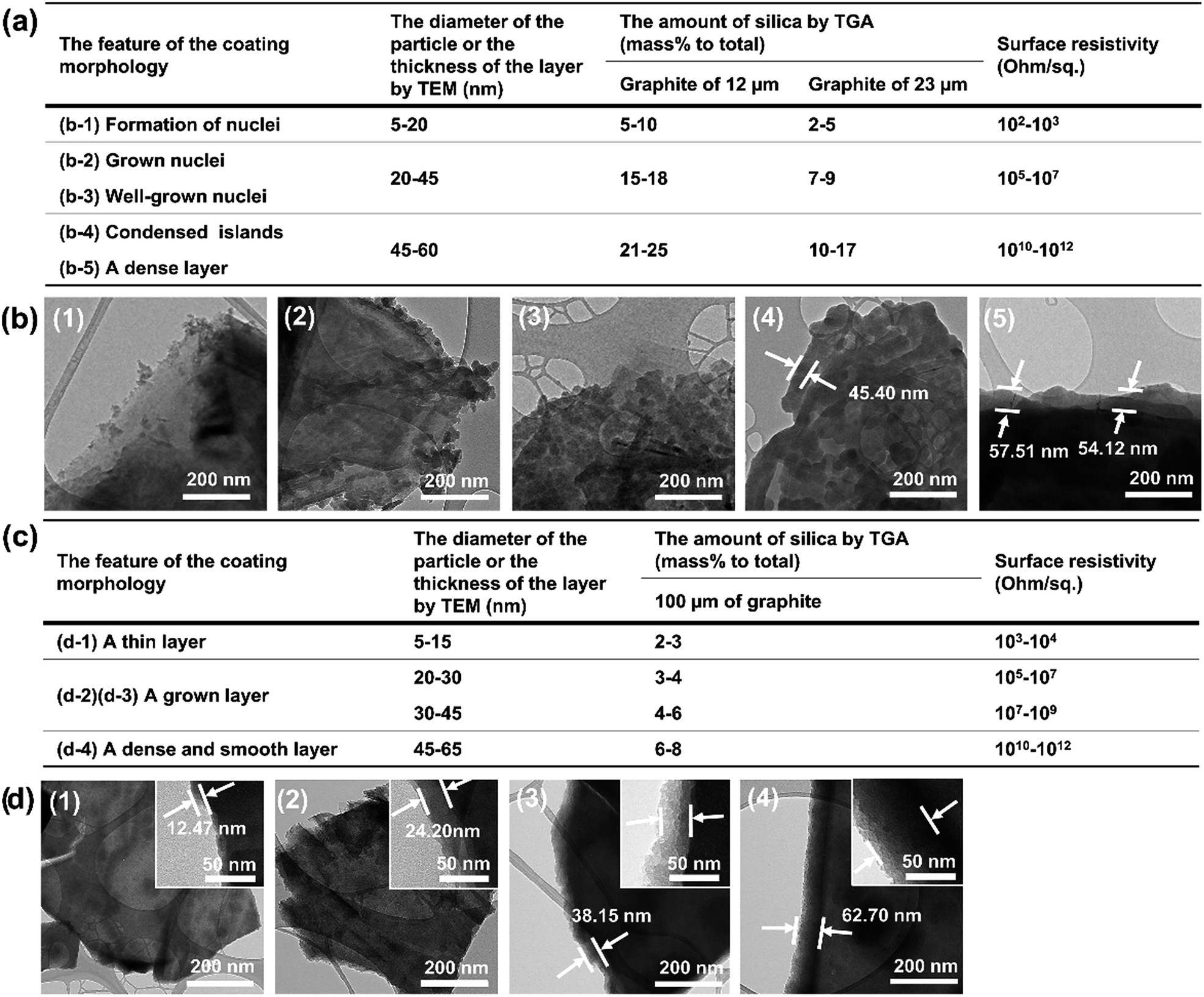 | ||
| Fig. 11 (a) Surface resistivity, coating morphology features, particle diameter or thickness, amount of silica for the Triton-X-100-assisted silica@graphite (12 and 23 μm) and (b) TEM images. (c) Surface resistivity, coating morphology features, particle diameter or thickness, amount of silica for the PEG-assisted silica@graphite (100 μm) and (d) TEM images (for original TGA graphs, see ESI†). | ||
Overall, to endow graphite with electrically insulating properties via a silica coating, the coated layer must have a surface devoid of channels and a thickness greater than 40–45 μm.
3.3. Thermal conductivity and surface resistivity of silica@graphite/TPEE composites
Fig. 12 shows the thermal conductivity and surface resistivity of both raw graphite/TPEE composites and amphiphile-assisted silica@graphite/TPEE composites with 50 wt% filler content. The silica layer on the graphite acts as a thermal and electrical barrier (the thermal conductivity of silica is 1.5 W mK−1 (ref. 29)).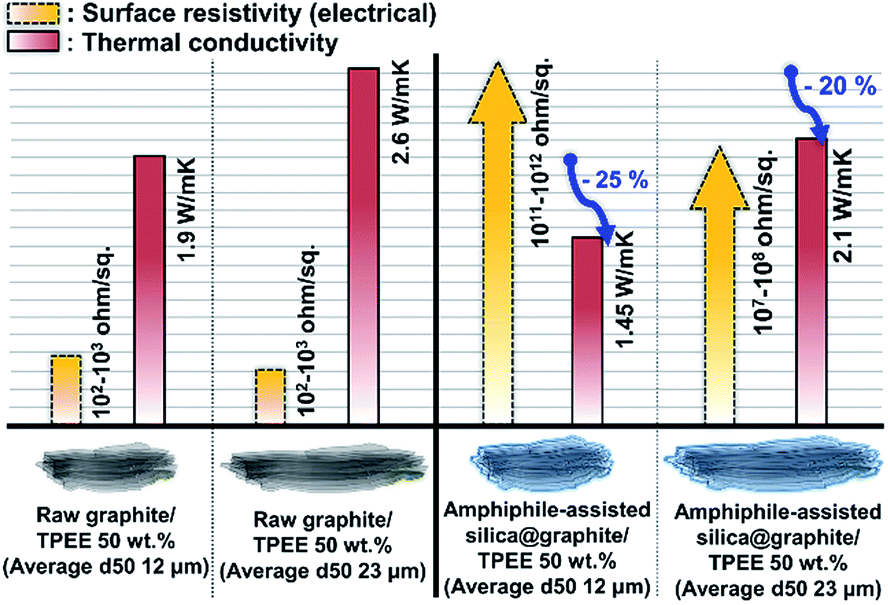 | ||
| Fig. 12 Thermal conductivity and surface resistivity of raw graphite (d50 size: 12 and 23 μm) and amphiphile-assisted silica@graphite (d50 size: 12 and 23 μm)/TPEE composites with 50 wt% filler content. | ||
The silica layer reduces the thermal conductivity of the composites to 70–75% of their original values (12 μm graphite: 70%, 23 μm graphite: 75%). The difference in reduction results from the overall number of silica species and the number of total barriers per volume of the composite. In the case of the 12 μm amphiphile-assisted silica@graphite/TPEE 50 wt% composite, electrically insulating properties were successfully attained (1011 to 1012 ohm sq−1); however, the 23 μm amphiphile-assisted silica@graphite/TPEE composite lost its insulating properties (107 to 108 ohm sq−1). This means that the silica layer on the graphite is ground against the molten polymer resin, which has a high melt viscosity during the compounding process. The 12 μm amphiphile-assisted silica@graphite has similar thickness to the 23 μm amphiphile-assisted silica@graphite, and the same silica species; however, there is an approximately twofold difference in the number of silica species. In the case of the 12 μm amphiphile-assisted silica@graphite/TPEE composite, the abundance of crushed splinters interrupts the electrical flow pathways between the damaged amphiphile-assisted silica@graphite in the polymer matrix.
4. Conclusion
Amphiphile-assisted silica@graphite with uniform full coverage was synthesized using an Oswald ripening agent, such as Triton X-100, and a bridgemer, such as PEG, via a modified one-step process. The mechanism of amphiphile-assisted silica formation on graphite was elucidated, and three suitable strategies were suggested, depending on the graphite size. The Oswald ripening agent provides nuclei surrounded by embryo-forming functional groups; it facilitates the absorption of embryos by the nuclei. This behavior assists the formation of islands and layers from the nuclei through Oswald ripening. For example, with Triton-X-100, the silica species grow from the edge toward the basal plane, and the assistance helps fill the many channels with silica species. The bridgemer couples the sol masses and binds the networks onto the other material substrates. For example, PEG can facilitate the growth of silica species toward the Z-axis, without the existence of adjacent nuclei and islands on the basal plane and edge. Consequently, the amphiphile-assisted silica@graphite can become electrically insulating (1011 to 1012 ohm sq−1). The TPEE composite containing 50 wt% filler shows electrically insulating properties (1011 to 1012 ohm sq−1) and 70–75% of the thermal conductivity of a composite made of equal parts raw graphite/TPEE. Finally, the amphiphile-assisted silica@graphite can be applied in various fields. Moreover, the coating strategies have potential to can coat not only graphite, but also other cabonaceous materials, such as graphene and carbon nanotubes.Acknowledgements
This work was supported by a grant [1415140523/10052976, 2015] from the Korea Ministry of Trade, Industry and Energy (MOTIE), Republic of Korea.Notes and references
- B. Lee, J. Liu, B. Sun, C. Shen and G. Dai, eXPRESS Polym. Lett., 2008, 2, 57–63 CrossRef.
- S. Ganguli, A. K. Roy and D. P. Anderson, Carbon, 2008, 46, 806–817 CrossRef CAS.
- K. Kalaitzidou, H. Fukushima and L. T. Drzal, Compos. Sci. Technol., 2007, 67, 2045–2051 CrossRef CAS.
- P. K. Schelling, L. Shi and K. E. Goodson, Mater. Today, 2005, 8, 30–35 CrossRef CAS.
- D. G. Cahill, W. K. Ford, K. E. Goodson, G. D. Mahan, A. Majumdar and H. J. Maris, et al., J. Appl. Phys., 2003, 93, 793–818 CrossRef CAS.
- W. Cui, F. Du, J. Zhao, W. Zhang, Y. Yang and X. Xie, et al., Carbon, 2011, 49, 495–500 CrossRef CAS.
- A. Guimont, E. Beyou, P. Alcouffe, P. Cassagnau, A. Serghei and G. Martin, et al., Polymer, 2014, 55, 22–28 CrossRef CAS.
- S. H. Song, K. H. Park, B. H. Kim, Y. W. Choi, G. H. Jun and D. J. Lee, et al., Adv. Mater., 2013, 25, 732–737 CrossRef CAS PubMed.
- L. Cao, W. Zhang, X. Zhang, L. Yuan, G. Liang and A. Gu, Ind. Eng. Chem. Res., 2014, 53, 2661–2672 CrossRef CAS.
- F. He, K. Lam, J. Fan and L. H. Chan, Carbon, 2014, 80, 496–503 CrossRef CAS.
- C. Min, D. Yu, J. Cao, G. Wang and L. Feng, Carbon, 2013, 55, 116–125 CrossRef CAS.
- Y. Kim, M. Kim, J. K. Choi and S. E. Shim, Polymer, 2015, 39, 136–143 CAS.
- T. Matsuda, D. Minami, F. Khoerunnisa, M. Sunaga, M. Nakamura and S. Utsumi, et al., Langmuir, 2015, 31, 3194–3202 CrossRef CAS PubMed.
- M. Kim, Y. Kim, S. H. Baeck and S. E. Shim, Carbon letters, 2015, 16, 34 CrossRef.
- J. Hong, J. Lee, C. K. Hong and S. E. Shim, J. Therm. Anal. Calorim., 2010, 101, 297–302 CrossRef CAS.
- K. Kim, Y. Kim, J. Nam, S.-H. Baeck, D. W. Park and S. E. Shim, Polymer, 2016, 40, 117–123 CAS.
- J. Nam, Y. Kim, K. Kim, S.-H. Baeck, D. W. Park and S. E. Shim, Polymer, 2016, 40, 109–116 CAS.
- S. Choi, Y. Kim, J. H. Yun, I. Kim and S. E. Shim, Mater. Lett., 2013, 90, 87–89 CrossRef CAS.
- S. Choi, K. Kim, J. Nam and S. E. Shim, Carbon, 2013, 60, 254–265 CrossRef CAS.
- S. Choi, J. Yang, Y. Kim, J. Nam, K. Kim and S. E. Shim, Compos. Sci. Technol., 2014, 103, 8–15 CrossRef CAS.
- M.-C. Hsiao, C.-C. M. Ma, J.-C. Chiang, K.-K. Ho, T.-Y. Chou and X. Xie, et al., Nanoscale, 2013, 5, 5863–5871 RSC.
- D. Tang, J. Su, Q. Yang, M. Kong, Z. Zhao and Y. Huang, et al., RSC Adv., 2015, 5, 55170–55178 RSC.
- Y. Noma, Y. Saga and N. Une, Carbon, 2014, 78, 204–211 CrossRef CAS.
- H. Yang, N. Coombs, I. Sokolov and G. A. Ozin, J. Mater. Chem., 1997, 7, 1285–1290 RSC.
- C. I. Zoldesi, P. Steegstra and A. Imhof, J. Colloid Interface Sci., 2007, 308, 121–129 CrossRef CAS PubMed.
- Y. Qu, Y. Tian, B. Zou, J. Zhang, Y. Zheng and L. Wang, et al., Bioresour. Technol., 2010, 101, 8402–8405 CrossRef CAS PubMed.
- W. Guo, Y. W. Sun, G. S. Luo and Y. J. Wang, Colloids Surf., A, 2005, 252, 71–77 CrossRef CAS.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- G.-W. Lee, M. Park, J. Kim, J. I. Lee and H. G. Yoon, Composites, Part A, 2006, 37, 727–734 CrossRef.
- Y.-S. Yoon, M.-H. Oh, A.-Y. Kim and N. Kim, J. Chem. Chem. Eng., 2012, 6, 515–519 CAS.
- M. A. Herman, W. Richter and H. Sitter, Epitaxy: Physical Principles and Technical Implementation, Springer-Verlag, Berlin, 2004, pp. 6–12 Search PubMed.
- J. T. Davies, A quantitative kinetic theory of emulsion type, I. Physical chemistry of the emulsifying agent, Proceedings of 2nd the International Congress of Surface Activity, Academic Press, New York, 1957, pp. 426–38 Search PubMed.
- B. Delmon, P. A. Jacobs, R. Maggi, J. A. Martens, P. Grange and G. Poncelet. Preparation of Catalysts VII., Louvain-la-Neuve, Belgium, 1998, pp. 618–623 Search PubMed.
- Y. Pan, Y. Guo, X. Zhao and Z. Wang, Chem. Res. Chin. Univ., 2012, 28, 737–742 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03049e |
| This journal is © The Royal Society of Chemistry 2017 |