DOI:
10.1039/C7RA02889J
(Paper)
RSC Adv., 2017,
7, 22360-22368
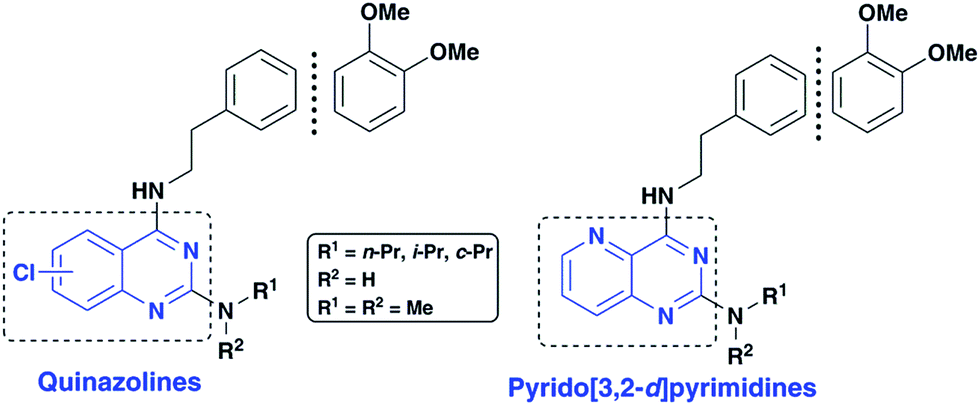
Application of quinazoline and pyrido[3,2-d]pyrimidine templates to design multi-targeting agents in Alzheimer's disease†
Received
9th March 2017
, Accepted 17th April 2017
First published on 24th April 2017
Abstract
A quinazoline and pyrido[3,2-d]pyrimidine based compound library was designed, synthesized and evaluated as multi-targeting agents aimed at Alzheimer's disease (AD). The SAR studies identified compound 8h (8-chloro-N2-isopropyl-N4-phenethylquinazoline-2,4-diamine) as a potent inhibitor of Aβ40 aggregation (IC50 = 900 nM) which was 3.6-fold more potent compared to the reference agent curcumin (Aβ40 IC50 = 3.3 μM). It also exhibited dual ChE inhibition (AChE IC50 = 8.6 μM; BuChE IC50 = 2.6 μM). Compound 9h (8-chloro-N4-(3,4-dimethoxyphenethyl)-N2-isopropylquinazoline-2,4-diamine) was identified as the most potent Aβ42 aggregation inhibitor (IC50 ∼ 1.5 μM). Transmission electron microscopy (TEM) imaging demonstrates their anti-Aβ40/Aβ42 aggregation properties. Compound 8e was identified as a potent BuChE inhibitor (BuChE IC50 = 100 nM) which was 36-fold more potent compared to donepezil (BuChE IC50 = 3.6 μM). The pyrido[3,2-d]pyrimidine bioisostere 10b (N2-isopropyl-N4-phenethylpyrido[3,2-d]pyrimidine-2,4-diamine) exhibited good anti-Aβ activity (Aβ40 IC50 = 1.1 μM), dual ChE inhibition and iron-chelating properties (23.6% chelation at 50 μM). These investigations demonstrate the usefulness of either a quinazoline or a pyrido[3,2-d]pyrimidine based ring scaffold in the design of multi-targeting agents to treat AD.
1 Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder with no cure in sight. It is anticipated that with an aging population and increasing lifespan, the social and economic burden is going to multiply in the years to come. Current AD research indicates that many factors are involved in its pathophysiology. Due to the ever-growing complexity of AD, it is no surprise that the shift toward multi-targeting small molecule candidates which can counter the various facets of the disease, as the next generation AD treatments is increasing.1–3 While current pharmacotherapy options rely on cholinesterase inhibitors (donepezil, rivastigmine and galantamine) that reduce the degradation of the neurotransmitter acetylcholine (ACh) and an N-methyl-D-aspartate antagonist (memantine), these were not designed as multi-targeting agents and provide only symptomatic relief.4,5 The role of beta-amyloid (Aβ) which tends to form neurotoxic aggregates, cholinesterases (ChE) that degrade ACh which facilitates neurotransmission and oxidative stress including metal-induced neurotoxicity in AD disease pathophysiology, highlights the need to design and develop novel therapies to target multiple pathways.6–10
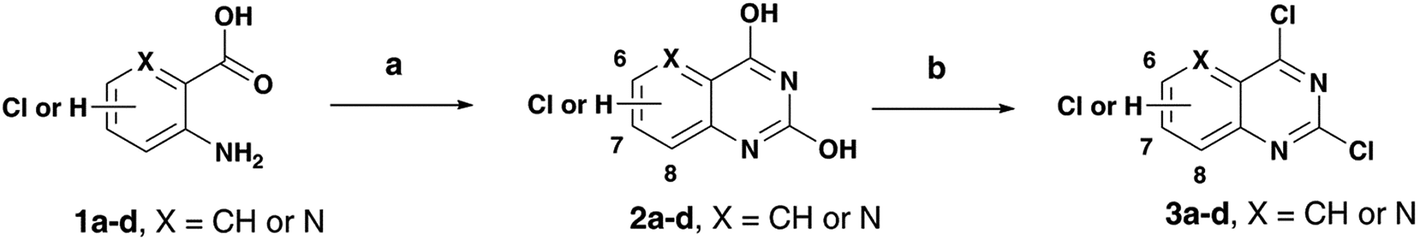
As part of our research program aimed at designing multi-targeting small molecules, we have investigated the development of novel agents including quinazolines that exhibit Aβ aggregation inhibition, dual acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibition and possess radical scavenging activity.11,12 In this regard, dual inhibition of both AChE and BuChE is known to be beneficial as BuChE can process ACh in advance stages of AD.13 Building on our recent efforts on multi-targeting small molecules, herein we present the structure-activity relationship (SAR) studies of substituted quinazolines and its bioisostere, pyrido[3,2-d]pyrimidine ring template (Fig. 1). Several compounds from this series exhibited desirable multi-targeting ability including dual Aβ/ChE inhibition and iron chelation properties. The road toward this library of 32 derivatives (8–15) started by first reacting the substituted-chloro-2-aminobenzoic or picolinic acids (1a–d, Scheme 1) with urea to afford the quinazoline or pyrido[3,2-d]pyrimidine ring template (2a–d), followed by 2,4-dichlorination with POCl3 to obtain 2,4,6-, 2,4,7- or 2,4,8-trichloroquinazolines (3a–c) or 2,4-dichloropyrido[3,2-d]pyrimidine (3d) respectively (65–80% yield, Scheme 1).12
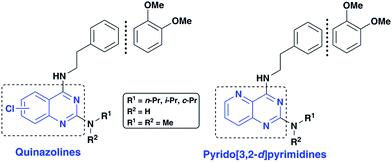 |
| | Fig. 1 SAR studies of quinazoline and pyrido[3,2-d]pyrimidines as multi-targeting anti-AD agents. | |
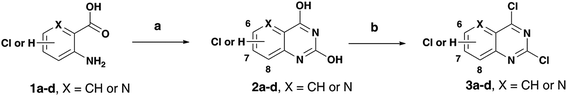 |
| | Scheme 1 Reagents and conditions: (a) urea, pressure vial, 150–155 °C, 2 h; (b) toluene, N,N-diethylaniline, POCl3, 0–105 °C, reflux, 14–16 h. | |
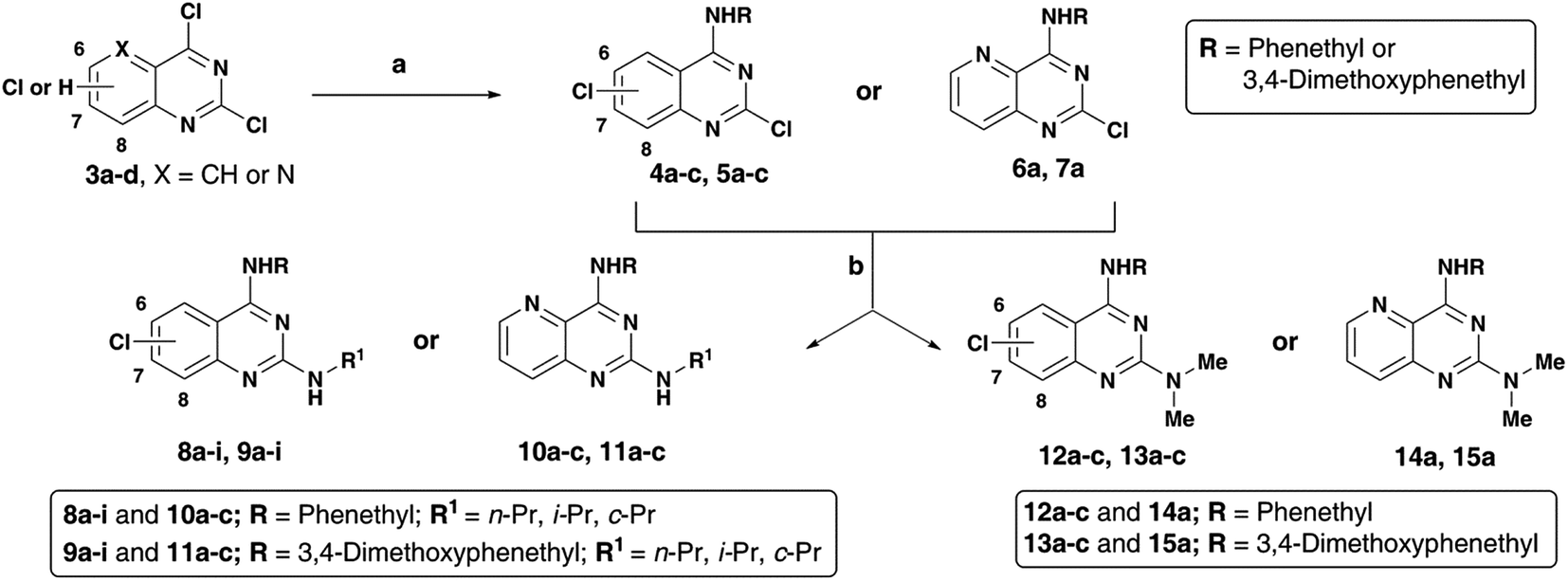
The N4-substituted dichloroquinazolines (4a–c and 5a–c) or N4-substituted-2-chloropyrido[3,2-d]pyrimidine (6a and 7a) intermediates were obtained in moderate to good yields (∼75–90%) by coupling 3a–d with phenethylamine or 3,4-dimethoxyphenethylamine in the presence of N,N-diisopropylethylamine (DIPEA) under reflux for 4 hours (Scheme 2). Displacement of the C2 chlorine in 4a–c, 5a–c, 6a and 7a was readily achieved by heating with respective amines (n-propylamine, isopropylamine, cyclopropylamine or N,N-dimethylamine) with 1,4-dioxane in the presence of DIPEA, in a pressure vial for 2 h to afford the quinazoline (Qnz) and pyrido[3,2-d]pyrimidine (Ppd) based compound library 8a–i, 9a–i, 10a–c, 11a–c, 12a–c, 13a–c, 14a and 15a with yields ranging from 68–78% (Scheme 1). The synthesized quinazoline derivatives were evaluated for their ability to modulate the aggregation kinetics of Aβ40 and Aβ42 using the thioflavin T (ThT) fluorescence assay.12,14 Their cholinesterase (hAChE/hBuChE) inhibition activity was determined using the Ellman's protocol.12,15 We identified few chloroquinazoline and pyrido[3,2-d]pyrimidines that exhibited excellent Aβ aggregation inhibition, dual ChE inhibition and iron-chelation properties thereby demonstrating multi-targeting ability.
 |
| | Scheme 2 Reagents and conditions: (a) phenethylamine or 3,4-dimethoxyphenethylamine, DIPEA, ethanol, reflux, 80–85 °C, 4 h; (b) alkylamine, DIPEA, 1,4-dioxane, 150–155 °C, pressure vial, 2 h. | |
2 Results and discussion
2.1 Inhibition of Aβ aggregation
The results of the SAR investigation on the anti-Aβ aggregation properties of chloroquinazolines and pyrido[3,2-d]pyrimidines is shown in Table 1. Examining the SAR of C4 phenethylamine-based chloroquinazolines (8a–i), shows that chlorine placement on the quinazoline ring had an effect on anti-Aβ activity with the 8-Cl placement exhibiting superior Aβ aggregation inhibition. The activity order was 8-Cl > 7-Cl > 6-Cl with respect to both Aβ40/Aβ42 inhibition. While 8a–c was ineffective toward Aβ42, others in the group showed inhibition ranging from ∼3.9–12 μM, with 8h being the most potent Aβ42 inhibitor. Significant Aβ40 aggregation inhibition was observed with compounds 8g–i (∼900 nM to 3.7 μM), with 8h (8-Cl, C2 i-Pr) being the most potent inhibitor (Aβ40 IC50 = 900 nM) and was 3.6-fold more potent compared to the reference agent curcumin (Aβ40 IC50 = 3.3 μM, Table 1). The addition of a C4 3,4-dimethoxylphenethylamine substituent in compounds (9a–i) generally provided dual inhibition of both Aβ40 and Aβ42 aggregation except for the 7-chloro derivative 9e (Table 1). Compound 9h (8-chloro, C2 i-Pr) was identified as a good inhibitor of both Aβ40 and Aβ42 (Aβ40 IC50 ∼ 4.0 μM; Aβ42 IC50 ∼ 1.5 μM). Significantly, it was ∼6.6-fold and 10-fold more potent toward Aβ42 compared to reference agents resveratrol (Aβ42 IC50 = 15.3) and curcumin (Aβ42 IC50 = 9.9 μM). Replacing the quinazoline ring template with a pyrido[3,2-d]pyrimidine bioisostere was generally ineffective. In this group of compounds, the C4 phenethylamine derivatives 10a–c were better than the corresponding C4 3,4-dimethoxyphenethylamine derivatives 11a–c with compound 10b identified as the most potent Aβ40 aggregation inhibitor (IC50 ∼ 1.1 μM). In the next step, the impact of a tertiary dimethylamine moiety at the C2-position was investigated. The presence of a C4 phenethylamine, increased potency toward Aβ40 12a–c; 6-Cl < 7-Cl < 8-Cl while only 12c was active toward Aβ42. On the other hand, with a 3,4-dimethoxyphenethylamine at the C4 position, potency toward Aβ40 ranged from ∼2–4 μM with chlorine placement at C6 being equipotent to C8 placement. The Aβ42 aggregation inhibitory potency increased from 7-Cl (13b, IC50 ∼ 9.4 μM) to 6-Cl (13a, IC50 ∼ 5.6 μM) to 8-Cl (13c, IC50 ∼ 1.8 μM). Combining the C2 dimethylamine group with a C4 phenethylamine or 3,4-dimethoxyphenethylamine on a pyrido[3,2-d]pyrimidine scaffold resulted in either weak or no inhibition of Aβ aggregation (compounds 14a and 15a, Table 1). With respect to the Aβ aggregation inhibition SAR, ∼22% of the derivatives screened surpassed the activity level of curcumin toward Aβ40, while ∼44% surpassed the activity level of curcumin toward Aβ42. Derivative 8h (8-chloro-N2-isopropyl-N4-phenethylquinazoline-2,4-diamine) was the most potent Aβ40 aggregation inhibitor (IC50 ∼ 900 nM), while 9h (8-chloro-N4-(3,4-dimethoxyphenethyl)-N2-isopropylquinazoline-2,4-diamine) was the most potent Aβ42 aggregation inhibitor (IC50 ∼ 1.5 μM).
Table 1 Amyloid-β (Aβ40/Aβ42) inhibition activity of quinazolines (8, 9, 12 and 13) and pyrido[3,2-d]pyrimidines (10, 11, 14 and 15)
| Compd |
R1 |
Chloride substitution |
IC50a,b (μM) |
| Aβ40 |
Aβ42 |
| IC50 values were calculated using the ThT-based fluorescence spectroscopy assay (excitation = 440 nm, emission = 490 nm). Values are mean of triplicate readings for three independent experiments. NA = not active. |
| 8a |
n-Pr |
C6 |
>25 |
>25 |
| 8b |
i-Pr |
C6 |
8.6 ± 0.7 |
>25 |
| 8c |
c-Pr |
C6 |
8.7 ± 0.9 |
>25 |
| 8d |
n-Pr |
C7 |
6.9 ± 1.4 |
4.6 ± 0.9 |
| 8e |
i-Pr |
C7 |
6.1 ± 1.3 |
7.2 ± 1.6 |
| 8f |
c-Pr |
C7 |
4.8 ± 1.0 |
11.7 ± 2.0 |
| 8g |
n-Pr |
C8 |
3.7 ± 0.5 |
6.5 ± 0.4 |
| 8h |
i-Pr |
C8 |
0.9 ± 0.1 |
3.9 ± 0.5 |
| 8i |
c-Pr |
C8 |
1.3 ± 0.2 |
6.8 ± 1.3 |
| 9a |
n-Pr |
C6 |
4.9 ± 0.5 |
10.7 ± 1.1 |
| 9b |
i-Pr |
C6 |
5.8 ± 0.7 |
7.7 ± 0.8 |
| 9c |
c-Pr |
C6 |
8.9 ± 0.9 |
9.8 ± 0.8 |
| 9d |
n-Pr |
C7 |
3.1 ± 0.3 |
12.4 ± 1.1 |
| 9e |
i-Pr |
C7 |
10.6 ± 0.9 |
>25 |
| 9f |
c-Pr |
C7 |
5.2 ± 0.6 |
22.5 ± 1.9 |
| 9g |
n-Pr |
C8 |
4.9 ± 0.5 |
2.3 ± 0.3 |
| 9h |
i-Pr |
C8 |
4.0 ± 0.4 |
1.5 ± 0.2 |
| 9i |
c-Pr |
C8 |
4.6 ± 0.5 |
2.2 ± 0.2 |
| 10a |
n-Pr |
— |
NA |
13.7 ± 2.0 |
| 10b |
i-Pr |
— |
1.1 ± 0.1 |
>25 |
| 10c |
c-Pr |
— |
6.8 ± 1.0 |
11.5 ± 1.5 |
| 11a |
n-Pr |
— |
NA |
>25 |
| 11b |
i-Pr |
— |
NA |
>25 |
| 11c |
c-Pr |
— |
>25 |
>25 |
| 12a |
— |
C6 |
6.5 ± 0.9 |
>25 |
| 12b |
— |
C7 |
5.1 ± 1.7 |
>25 |
| 12c |
— |
C8 |
1.9 ± 0.3 |
>5.8 ± 0.7 |
| 13a |
— |
C6 |
2.5 ± 0.3 |
5.6 ± 0.7 |
| 13b |
— |
C7 |
4.3 ± 0.4 |
9.4 ± 0.9 |
| 13c |
— |
C8 |
2.3 ± 0.2 |
1.8 ± 0.3 |
| 14a |
— |
— |
NA |
12.7 ± 2.0 |
| 15a |
— |
— |
>25 |
>25 |
| Resveratrol |
— |
— |
1.1 ± 0.1 |
15.3 ± 1.9 |
| Curcumin |
— |
— |
3.3 ± 0.4 |
9.9 ± 0.4 |
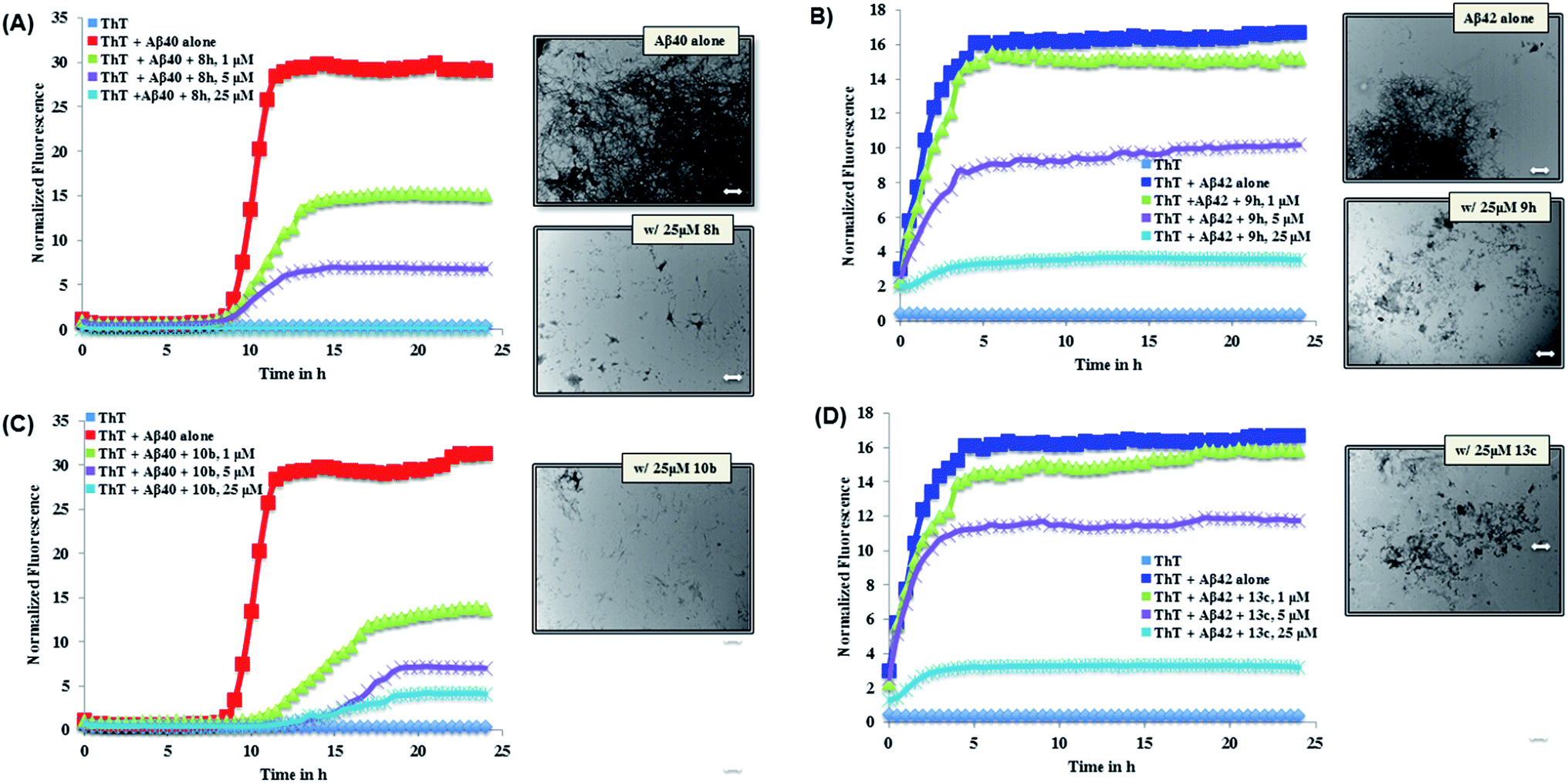
The ThT-based Aβ40 aggregation kinetics data in the presence of the most active inhibitors (8h and 10b for Aβ40) is shown in Fig. 2. This reveals that, their modes of aggregation inhibition do differ from one another. As seen in Fig. 2 (panel A), 8h was exhibiting a concentration-dependent reduction in the aggregation with complete inhibition of Aβ40 aggregation seen at 25 μM. On the other hand, 10b (Fig. 2, panel C) was also exhibiting a concentration-dependent reduction in aggregation. In addition, unlike compound 8h, it was able to delay the onset of aggregation by delaying the lag phase further. Investigating the Aβ morphology by TEM imaging further confirms the ability of both compounds 8h and 10b to prevent Aβ aggregation (Fig. 2). Analysis of the Aβ42 aggregation kinetics in the presence of the most active inhibitors (9h and 13c for Aβ42, Fig. 2) revealed similar observations. Both derivatives, which were nearly equipotent against Aβ42 aggregation, shared similar modes of action. As seen in Fig. 2 (panel B), 9h was able to reduce the overall load of Aβ42 fibril formation in a concentration dependent manner with almost complete inhibition seen at 25 μM. A similar trend was seen with 13c (Fig. 2, panel D). Furthermore, TEM morphology studies corroborate aggregation kinetics data observed (Fig. 2).
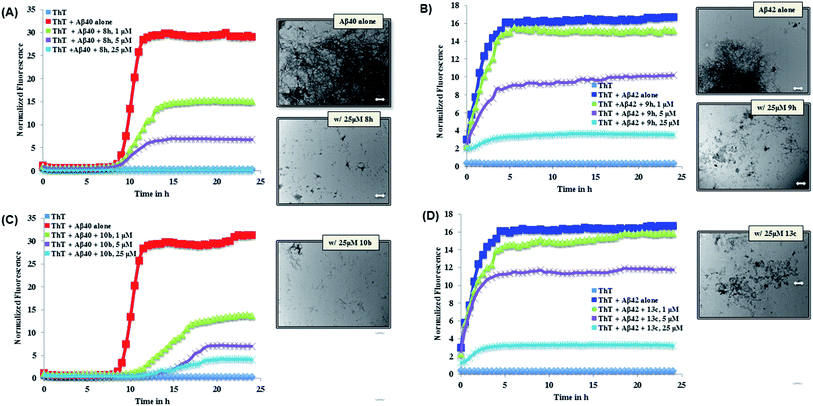 |
| | Fig. 2 Aβ40/Aβ42 (5 μM) aggregation kinetics study with or without 1, 5 or 25 μM of 8h (panel A), 9h (panel B), 10b (panel C) or 13c (panel D) along with corresponding TEM morphology in the presence of 25 μM of 8h, 9h, 10b or 13c. Aggregation kinetics were monitored by ThT-fluorescence spectroscopy (excitation = 440 nm, emission = 490 nm) for 24 h at 37 °C in phosphate buffer at pH 7.4. TEM image scale: white bars represent 500 nm. | |
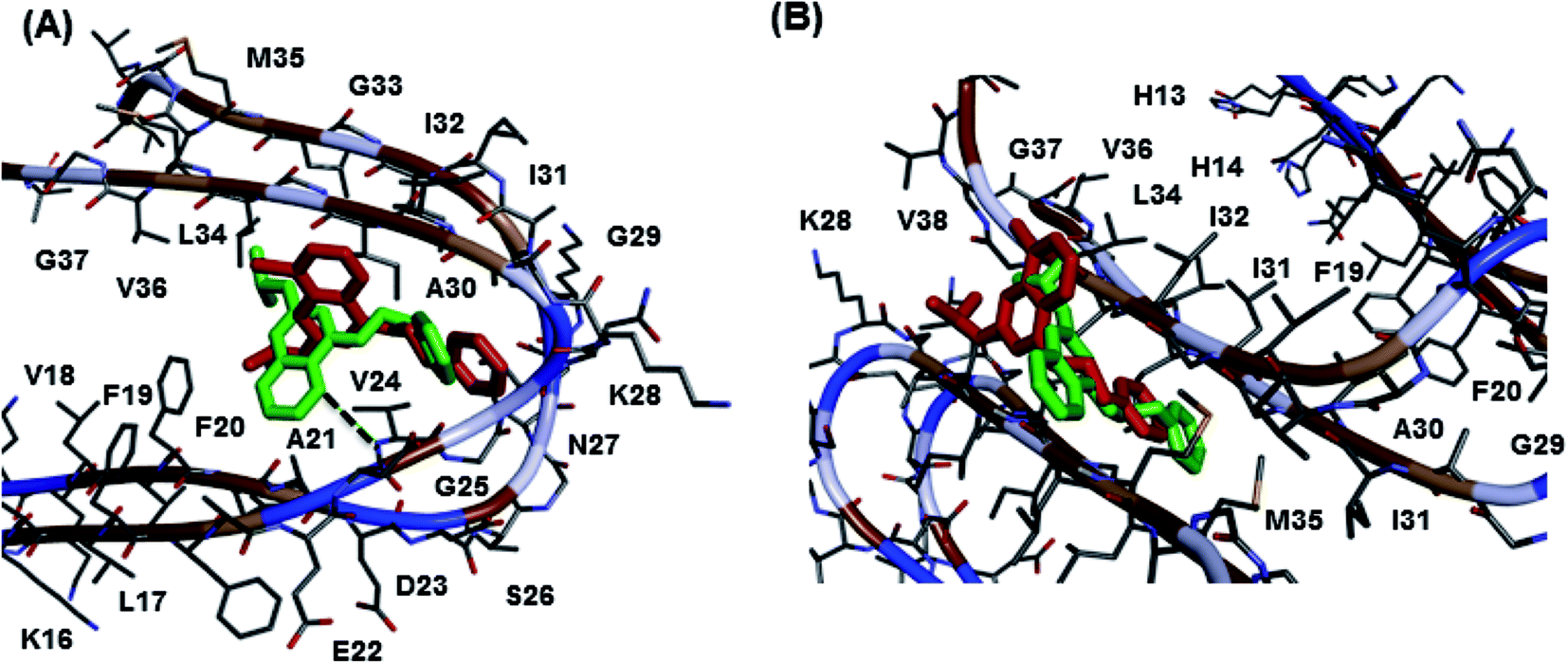
The molecular docking study of best two Aβ40 aggregation inhibitors, 8h and 10b, in the Aβ dimer model (Fig. 3, panel A) derived from the solution structure of Aβ fibrils16 (pdb id:2LMN) indicated that both the 8-chloroquinazoline and the pyrido[3,2]pyrimidine templates were oriented between C- and N-terminal region whereas the C4-phenethylamine substituent was closer to the turn region Asp23-Gly29 (distance ∼ 6–8 Å). Hydrophobic interactions were the dominating force involved. In the pyrido[3,2-d]pyrimidine compound 10b, the presence of an additional ring nitrogen (N5) led to a hydrogen bonding contact with the amide backbone of Val24 (distance ∼ 3 Å). In the fibril model, Fig. 3, panel B, both 8h and 10b had similar binding modes where the C4-phenethylamine group was oriented toward the center region of the fibril core (Ile32-Met35, distance ∼ 5–8 Å). Both core templates were undergoing hydrophobic interactions with Ile31-Val38 (distance ∼ 5–8 Å) at the C-terminal end. These studies support the ability of 8h and 10b to reduce fibril aggregation.
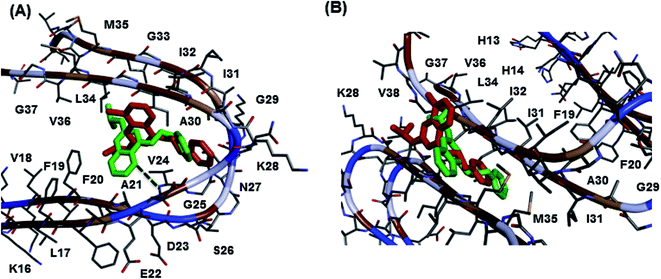 |
| | Fig. 3 Aβ molecular docking studies. Binding modes of the quinazoline compound 8h (red) and pyrido[3,2-d]pyrimidine compound 10b (green) in Aβ-dimer model (panel A) and Aβ-fibril model (panel B). Hydrogen atoms were removed for clarity. | |
2.2 Inhibition of cholinesterases
With respect to cholinesterase inhibition, the SAR data is shown in Table 2. While examining the first cluster of C4 phenethylamine-based chloroquinazolines (8a–i), it became evident that chlorine placement at either C6, C7 or C8 was able to modulate the ChE inhibitory activity with 7-Cl exhibiting better ChE inhibition. The ChE inhibition can be ranked as 7-Cl > 6-Cl > 8-Cl with respect to both ChEs. Most noteworthy was the outcome from 8e (AChE IC50 ∼ 6.6 μM, BuChE IC50 ∼ 100 nM), which was the most potent AChE inhibitor in this group, and was the most potent BuChE inhibitor overall (about 36-fold more potent compared to donepezil, IC50 = 3.60 μM). The addition of the 3,4-dimethoxyphenethyl substituent in the second cluster of derivatives (9a–i) exhibited similar AChE inhibition range as per compounds 8a–i. However, it was detrimental for BuChE inhibition, although one key derivative stood out from the pack (9e, BuChE IC50 ∼ 4 μM) and was equivalent to donepezil. It is interesting to point out that both compounds 8e and 9e that exhibited significant BuChE inhibition possessed a C2 isopropylamine substituent which promoted BuChE inhibition. Replacing the quinazoline template with a pyrido[3,2-d]pyrimidine template (10a–c and 11a–c), led to weak or no inhibitory activity toward BuChE. With respect to AChE inhibition, they were on par with the chloroquinazolines (8a–i and 9a–i) with activity range AChE IC50 ∼ 7–8 μM, Table 2. The incorporation of a N,N-dimethylamine at the C2 position was explored to investigate the role of a tertiary amine instead of a secondary amine toward ChE inhibition (12a–c and 13a–c). The chlorine placement played a role as 12b (7-Cl, BuChE IC50 ∼ 1.5 μM) was ∼3.6-fold more potent compared to its 6-chloro isomer (12a). In addition, 12b was more potent compared to compound 13b (BuChE IC50 > 50 μM). This trend also extended to the AChE profiles, where 12b (AChE IC50 ∼ 5.8 μM) was ∼2.3-fold more potent compared to 13b, it was also ∼1.3-fold more potent compared to its 6- and 8-chloro isomers (12a and 12c). These studies show that combining the N,N-dimethylamine group at C2 with phenethylamine or 3,4-dimethoxyphenethylamine on a pyrido[3,2-d]pyrimidine scaffold resulted in equipotent and selective AChE inhibition (14a and 15a, AChE IC50 ∼ 6.7 μM, BuChE IC50 > 50 μM, Table 2). The quinazoline derivative 8e (7-chloro-N2-isopropyl-N4-phenethylquinazoline-2,4-diamine) was the most potent BuChE inhibitor with an IC50 ∼ 100 nM, while 12b (7-chloro-N2,N2-dimethyl-N4-phenethylquinazoline-2,4-diamine) was the most potent AChE inhibitor with an IC50 ∼ 5.8 μM.
Table 2 Cholinesterase inhibition (AChE and BuChE) of quinazolines (8, 9, 12 and 13) and pyrido[3,2-d]pyrimidines (10, 11, 14 and 15)
| Compd |
R1 |
Chloride substitution |
IC50a (μM) |
| hAChE |
hBuChE |
| IC50 values were an average ± SD of triplicate readings for two to three independent experiments. |
| 8a |
n-Pr |
C6 |
8.9 ± 0.6 |
4.8 ± 0.3 |
| 8b |
i-Pr |
C6 |
8.7 ± 0.5 |
1.5 ± 0.1 |
| 8c |
c-Pr |
C6 |
8.7 ± 0.9 |
9.1 ± 0.7 |
| 8d |
n-Pr |
C7 |
7.2 ± 0.7 |
4.4 ± 0.4 |
| 8e |
i-Pr |
C7 |
6.6 ± 0.6 |
0.1 ± 0.02 |
| 8f |
c-Pr |
C7 |
7.3 ± 0.6 |
3.5 ± 0.4 |
| 8g |
n-Pr |
C8 |
7.7 ± 0.7 |
6.0 ± 0.6 |
| 8h |
i-Pr |
C8 |
8.6 ± 0.9 |
2.6 ± 0.2 |
| 8i |
c-Pr |
C8 |
7.6 ± 0.7 |
10.9 ± 0.9 |
| 9a |
n-Pr |
C6 |
6.2 ± 0.4 |
>50 |
| 9b |
i-Pr |
C6 |
6.8 ± 0.7 |
20.3 ± 1.5 |
| 9c |
c-Pr |
C6 |
8.0 ± 0.7 |
>50 |
| 9d |
n-Pr |
C7 |
7.2 ± 0.7 |
>50 |
| 9e |
i-Pr |
C7 |
7.9 ± 0.8 |
3.9 ± 0.3 |
| 9f |
c-Pr |
C7 |
8.7 ± 0.8 |
>50 |
| 9g |
n-Pr |
C8 |
6.5 ± 0.5 |
>50 |
| 9h |
i-Pr |
C8 |
6.8 ± 0.7 |
19.3 ± 1.1 |
| 9i |
c-Pr |
C8 |
6.6 ± 0.7 |
>50 |
| 10a |
n-Pr |
— |
8.1 ± 0.8 |
30.3 ± 2.8 |
| 10b |
i-Pr |
— |
7.8 ± 0.8 |
29.3 ± 2.0 |
| 10c |
c-Pr |
— |
7.6 ± 0.6 |
>50 |
| 11a |
n-Pr |
— |
7.2 ± 0.8 |
>50 |
| 11b |
i-Pr |
— |
7.4 ± 0.7 |
>50 |
| 11c |
c-Pr |
— |
7.2 ± 0.5 |
>50 |
| 12a |
— |
C6 |
7.7 ± 0.8 |
5.4 ± 0.6 |
| 12b |
— |
C7 |
5.8 ± 0.7 |
1.5 ± 0.1 |
| 12c |
— |
C8 |
7.5 ± 0.8 |
>50 |
| 13a |
— |
C6 |
7.2 ± 0.7 |
31.2 ± 3.4 |
| 13b |
— |
C7 |
13.4 ± 1.1 |
>50 |
| 13c |
— |
C8 |
8.1 ± 0.8 |
>50 |
| 14a |
— |
— |
6.8 ± 0.7 |
>50 |
| 15a |
— |
— |
6.7 ± 0.5 |
>50 |
| Donepezil |
— |
— |
0.03 ± 0.002 |
3.6 ± 0.4 |
| Galantamine |
— |
— |
2.6 ± 0.6 |
>50 |
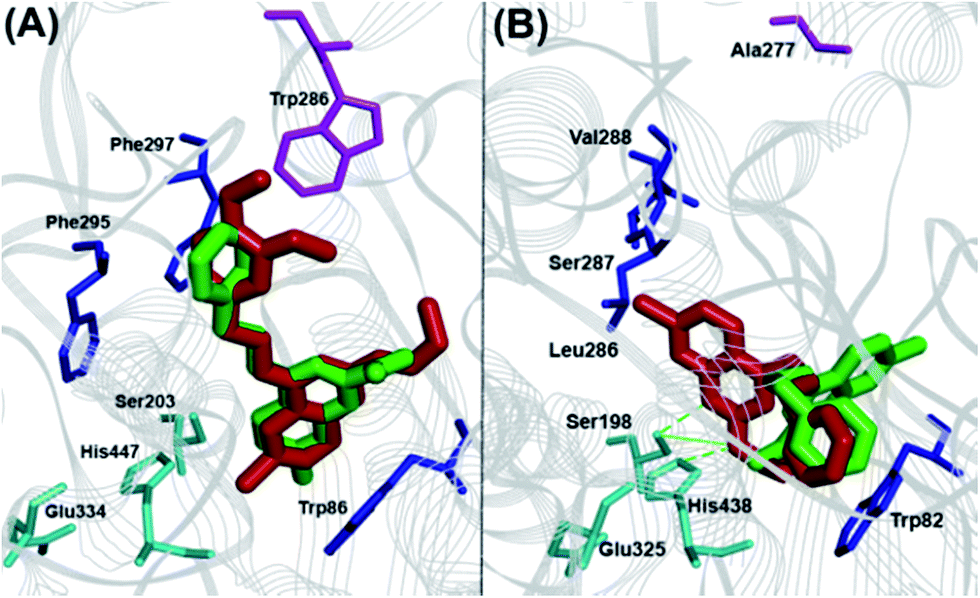
The binding interaction of quinazoline derivatives was investigated by using the X-ray crystal structures of human AChE (pdb id:1B41) and BuChE (pdb id:1P0I) enzymes.17,18 The binding modes of two best quinazoline based AChE inhibitors 9a and 12b in hAChE (Fig. 4, panel A) revealed quite similar binding interactions, explaining their near-equipotent inhibition of the AChE (IC50 ∼ 6 μM). In both derivatives, the 7-chloroquinazoline ring scaffold was stacked parallel against Trp86 (distance ∼ 4–6 Å) and the C4-benzylamino groups were extending from the catalytic site toward the peripheral anionic site (PAS), where the 3,4-dimethoxy moiety of compound 9a was undergoing favorable interactions with Trp286 (distance ∼ 3–4 Å). The C6 and C7 chlorine atoms were oriented toward the catalytic triad (distance ∼ 6–8 Å), while the C2-alkylamine groups underwent hydrophobic interactions with Trp86 (distance ∼ 3–5 Å). The binding orientation of compounds 8e and 12b that exhibited superior inhibitory potency toward BuChE was quite different (Fig. 4, panel B). The most potent BuChE inhibitor 8e underwent three hydrogen-bonding interactions centered around the C2 isopropylamine-NH, the 7-chloroquinazoline-N1 with His447 and Ser203 duo (distance ∼ 2.5–3.5 Å). The core ring scaffold was also oriented in the catalytic triad, with the C7 chlorine oriented toward the acyl pocket (Leu286-Val288, distance ∼ 4–5 Å). The C4-phenethylamine substituent was oriented perpendicular to Trp82 (Fig. 4, panel B). On the other hand, derivative 12b had its core scaffold and C4-phenethylamine substituent directly interacting with Trp82 (distance ∼ 3–4 Å), while the N,N-dimethylamine was oriented toward the catalytic triad (distance ∼ 4–5 Å) which explains their superior inhibition profile.
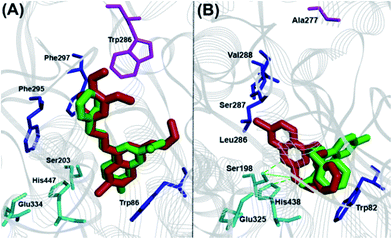 |
| | Fig. 4 Cholinesterase molecular docking studies. Panel (A): Binding modes of 9a (red) and 12b (green) in hAChE. Panel (B): Binding modes of 8e (red) and 12b (green) in hBuChE. Hydrogen atoms removed for clarity. | |
2.3 Iron chelation properties
The presence of an additional ring nitrogen in the pyrido[3,2-d]pyrimidine class of compounds (10a–c, 11a–c, 14a and 15a) provides a chelation center.19,20 Due to the role of iron in promoting oxidative stress in AD21–23 and its contribution in the formation of neurotoxic Aβ plaques and neurofibrillary tangles (NFTs), we assessed the Fe2+-chelation potential of the pyrido[3,2-d]pyrimidines (Table 3) in ferrozine based assay. The known iron chelators clioquinol and deferroxamine were used as reference agents. These studies show that, the C4 phenethylamine substituted compounds 10a–c exhibited better chelation capacity compared to their C4 3,4-dimethoxyphenethyl dimethoxyphenethylamine counterparts 11a–c, 14a and 15a (inactive at 50 μM).
Table 3 Iron (Fe2+) chelation studies of pyrido[3,2-d]pyrimidines
| Compd |
R |
R1 |
% Fe2+ Chelationa |
| % Fe2+ chelation values were an average ± SD of duplicate readings for two independent experiments. The ferrozine absorbance was measured at 562 nm. |
| 10a |
Phenethyl |
n-Pr |
29.3 ± 4.3 |
| 10b |
Phenethyl |
i-Pr |
23.6 ± 4.0 |
| 10c |
Phenethyl |
c-Pr |
37.0 ± 5.6 |
| 11a |
3,4-DiOMe-phenethyl |
n-Pr |
24.1 ± 3.6 |
| 11b |
3,4-DiOMe-phenethyl |
i-Pr |
21.8 ± 3.3 |
| 11c |
3,4-DiOMe-phenethyl |
c-Pr |
26.7 ± 4.0 |
| 14a |
Phenethyl |
— |
22.9 ± 3.4 |
| 15a |
3,4-DiOMe-phenethyl |
— |
23.0 ± 3.0 |
| Clioquinol |
— |
— |
39.0 ± 5.8 |
| Deferoxamine |
— |
— |
86.7 ± 11.0 |
Overall, these pyrido[3,2-d]pyrimidine compounds exhibited weak to moderate (22–37% chelation) iron-chelation capacity. Interestingly, the presence of a C2 cyclopropyl substituent provided the best chelation capacity compared to other C2 substituents explored with compound 10c exhibiting equipotent iron-chelation capability (37% chelation at 50 μM) compared to the reference agent clioquinol (39% inhibition at 50 μM, Table 3). More significantly, the corresponding quinazoline class of compounds did not exhibit any iron-chelation capacity highlighting the requirement of a pyrido[3,2-d]pyrimidine ring scaffold to display iron-chelation properties.
3 Conclusions
In summary, we synthesized and optimized the structure of quinazoline based ring scaffold as multi-targeting agents aimed at the amyloid, cholinergic, and oxidative stress pathways of AD pathophysiology. A number of agents synthesized exhibited dual Aβ, ChE inhibition and iron-chelation capacity. The SAR acquired shows that compound 8h was the most potent Aβ40 aggregation inhibitor (IC50 = 900 nM) and compound 8e provided dual Aβ (IC50 Aβ40 = 6.1 μM; Aβ42 IC50 = 7.2 μM) and ChE inhibition (IC50 AChE = 6.6 μM; BuChE IC50 = 100 nM). The presence of a pyrido[3,2-d]pyrimidine bioisostere instead of a quinazoline was essential to exhibit iron-chelation properties (22–37% chelation) with compounds 10b and 10c exhibiting multi-targeting activity toward Aβ, ChE and iron-chelation properties. The data presented herein highlights the dual anti-Aβ40/β42 aggregation, ChE inhibition, and metal-chelation capabilities of quinazoline and pyrido[3,2-d]pyrimidine class of compounds as multi-targeting agents to treat AD.
4 Materials and methods
4.1 General information
All chemicals and reagents used were purchased from either Sigma-Aldrich, USA or Alfa Aesar, USA and was used without further purification. Melting points were determined using a Fisher-Johns melting point apparatus and are uncorrected. 1H NMR (300 MHz) and 13C NMR spectra (100 MHz) were recorded on a Bruker Avance NMR spectrometer in DMSO-d6. Coupling constants (J values) were recorded in hertz (Hz) and the following abbreviations were used to represent multiplets of NMR signals: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad. Carbon multiplicities (C, CH, CH2 and CH3) were assigned by DEPT 90/135 experiments. Low-resolution mass spectra (LRMS) was obtained using an Agilent 6100 series single quad LCMS whereas high-resolution mass spectra (HRMS) were recorded on a Thermo Scientific Q Exactive™ mass spectrometer with an ESI source, Department of Chemistry, University of Waterloo. Compound purity was assessed (∼95% purity) using an Agilent 6100 series single quad LCMS equipped with an Agilent 1.8 μm Zorbax Eclipse Plus C18 (2.1 × 50 mm) running 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water/ACN with 0.1% FA at a flow rate of 0.5 mL min−1 with detection at 254 nm by UV. Compounds 3a–d and 4a were previously reported.24–26
:50 water/ACN with 0.1% FA at a flow rate of 0.5 mL min−1 with detection at 254 nm by UV. Compounds 3a–d and 4a were previously reported.24–26
4.1.1 General procedure for the synthesis of compounds 3a–d. These compounds were synthesized as per previously reported method.24,26 In a 350 mL round pressure vial 16–18 g urea was heated at 150–155 °C till it melted. To the liquid urea solution 0.1 eq. of 1a, or 1b, or 1c or 1d was added. The pressure vial was sealed and heated at 150–155 °C for 2 h, cooled to room temperature, followed by the addition of water (100 mL) after which the reaction mixture was heated at 100–105 °C for 1 h. After cooling to room temperature, the reaction mixture was diluted with ∼80 mL EtOAc and washed with brine solution (50 mL × 4). The combined aqueous layers were washed with ∼35 mL EtOAc, dried over MgSO4 and the organic solvent was removed in vacuo. The solid obtained (compounds 2a–d) was straightaway carried to the next step (yield 70–85%) without further purification. In a 250 mL RBF, 29.24 mmol of either 2a or 2b or 2c or 2d was suspended in 25 mL of anhydrous toluene and allowed to stir on an ice bath. To this, 5 eq. of POCl3 was added in small aliquots followed by the slow addition of 5 eq. of DEA. The solution was kept on the ice bath for 10 min before moving to room temperature and allowed to stir for 1 h prior to refluxing at 105–110 °C for 14–16 h. Upon cooling to room temperature, the reaction mixture was added in small aliquots to a double-ice-water bath while stirring. The quenching solution was left stirring at room temperature for 5 h before vacuum filtering the yellowish-grey precipitate. The precipitate was stirred for 1 h in saturated NaHCO3 solution and was filtered. This neutralization process was carried out 2–3 times until the bicarbonate solution maintains a neutral to slight basic pH. The final precipitate was dissolved in DCM and purified by a silica gel column chromatography using 100% DCM as the eluent to afford white to light grey solids 3a–d (65–80%).
4.1.2 General procedure for the synthesis of compounds 4a–c, 5a–c, 6a and 7a. These compounds were synthesized as per previously reported method.27,28 To a 30 mL solution of ethanol in a 100 mL round-bottom flask on ice, ∼21.46–25.13 mmol of either 3a or 3b or 3c or 3d was added followed by slow addition of 1.3 eq. (27.90–32.66 mmol) of the corresponding primary amine. Contents were stirred on an ice bath while 2.0 eq. of diisopropyl-ethylamine (DIPEA, 42.92–50.25 mmol) was added in drop wise fashion. The solution was then heated at 80–85 °C under reflux for 3–4 h. The reaction contents were cooled to room temperature and precipitated residues were vacuum-filtered with EtOAc. The organic supernatant was concentrated in vacuo followed by two rounds of extraction using EtOAc and saturated brine solution (40–50 mL each respectively). The combined organic layers were dried over MgSO4, evaporated in vacuo and purified (1–2 times) using silica gel column chromatography with 5:1 EtOAc:MeOH as the elution solvent to afford 4a–c, 5a–c, 6a and 7a as white to beige solids with yields ranging from 75–90%.
4.1.3 General procedure for the synthesis of compounds 8a–i, 9a–i, 10a–c, 11a–c, 12a–c, 13a–c, 14a and 15a. The target compounds were synthesized as per previously reported method.12,28 Briefly in a 50 mL pressure vial (PV), 0.25 g of 2,6-dichloro, 2,7-dichloro or 2,8-dichloro-N-substituted quinazolin-4-amine or 2-chloro-N-substituted pyrido[3,2-d]pyrimidine-2,4-diamine (∼0.66–0.83 mmol) was combined with 2 eq. (∼1.32–1.66 mmol) of primary amine (methyl-, ethyl-, n-propyl-, isopropyl- or cyclopropylamine) or dimethylamine which was dissolved in 5 mL of 1,4-dioxane followed by the addition of 3 eq. of DIPEA (∼1.98–2.40 mmol). Pressure vial was sealed and stirred in an oil bath at 150–155 °C for 2 h. Upon completion and cooling to room temperature, the reaction mixture was diluted with ∼40 mL of EtOAc and washed with brine solution (25 mL × 2). The combined aqueous layer was washed with ∼25 mL of EtOAc. The combined EtOAc layers were dried over MgSO4 before removing EtOAc in vacuo to yield a solid product that was purified by silica gel column chromatography using 5:1 EtOAc:MeOH as the eluent to afford pale yellow to brown solids (yield ∼ 68–78%).
4.2 Experimental procedure for cholinesterase (hAChE/hBuChE) inhibition
Quinazoline and pyrido[3,2-d]pyrimidine derivatives (8a–i, 9a–i, 10a–c, 11a–c, 12a–c, 13a–c, 14a and 15a) were evaluated for ChE inhibitory activity using Ellman's method.12,15,29 The test compounds compete with substrates acetylthiocholine iodide (ATChI) and S-butyrylthiocholine iodide (BuTChI) for either human AChE (Sigma-Aldrich, St. Louis, MO) or human BuChE (Sigma-Aldrich, St. Louis, MO), respectively. Known ChE inhibitors donepezil and rivastigmine were used as reference agents for comparison. Quinazoline and pyrido[3,2-d]pyrimidine derivatives were prepared in DMSO (maximum concentration used 1% v/v) and 10 μL each (0.001–25 μM final concentration range), was incubated for 5 minutes at room temperature with 160 μL of 1.5 mM DTNB, 50 μL 0.22 U mL−1 AChE (in 50 mm Tris·HCl, pH 8.0, 0.1% w/v bovine serum albumin, BSA) or 50 μL 0.12 U mL−1 BuChE (in 50 mm Tris·HCl, pH 8.0, 0.1% w/v bovine serum albumin, BSA). After the incubation period, 30 μL of ATChI (15 mM, prepared in ultra pure water) or BuTChI (15 mM, prepared in ultra pure water) were added to 96-well plates. The ChE inhibition was measured at 412 nm wavelength using a microplate reader (BioTek Synergy H1 microplate reader) at various time intervals (t = 0, 1, 2, 3, 4 and 5 min). Appropriate controls, without the test compounds and ChE were included. The inhibitory concentration (IC50 values) was calculated from the concentration-inhibition dose response curve on a logarithmic scale based on two independent experiments run in triplicates.
4.3 Experimental procedure for thioflavin T based Aβ aggregation kinetics
The anti-Aβ aggregation activity of test compounds (8a–i, 9a–i, 10a–c, 11a–c, 12a–c, 13a–c, 14a and 15a), was evaluated using the ThT-based fluorescence assay.11,14 The Aβ40 and Aβ42 hexafluoro-2-propanol (HFIP) (rPeptide, Geogia, USA) stock solution was prepared by dissolving in 1% NH4OH solution, to a 1 mg mL−1 stock solution, followed by dilution to 50 μM in phosphate buffer (215 mM, pH 7.4). Stock solutions of test compounds were prepared in DMSO, diluted in phosphate buffer (pH 7.4), and were sonicated for 30 min. The final DMSO concentration per each well was 1% v/v or lower. The ThT fluorescent dye stock solution (15 μM) was prepared in 50 mM glycine buffer (pH 7.4). The aggregation kinetics assay was carried out using a Corning® 384-well flat, clear bottom black plates. Each well contains 44 μL of ThT, 20–35 μL of phosphate buffer (pH 7.4), 8 μL of test compounds in different concentrations (1, 5, 10 and 25 μM, final concentration) and 8 μL of either Aβ40 or Aβ42 (5 μM final concentration). The plate was incubated at 37 °C with a plate cover under shaking and fluorescence was measured every 5 min using a BioTek Synergy H1 microplate reader multimode plate reader (excitation = 440 nm and emission = 490 nm) over a period of 24 h. Appropriate control experiments that contain either Aβ40/42 and test compound alone were evaluated. The known Aβ aggregation inhibitors curcumin and resveratrol were used as reference agents. The IC50 value (μM) was calculated using the equation 100% control – [(IFi–IFo)] where 100% control indicates no inhibitor, IFi and IFo are the fluorescence intensities in the presence and absence of ThT. The results were expressed as IC50 values ± SD based on two separate experiments in triplicate measurements.
4.4 Experimental procedure for transmission electron microscopy (TEM)
In Costar 96-well, round-bottom plates were added 80 μL of 215 mM phosphate buffer, 20 μL of 10× test compound dilutions (250 μM – prepared in the same way as for the ThT assay) and 100 μL of Aβ40 or Aβ42 respectively (50 μM each). For the control wells, 2 μL of DMSO and 18 μL of phosphate buffer was added. Final Aβ: test compound ratio was 1:1 (25 μM:25 μM). Plates were incubated on a Fisher plate incubator set to 37 °C and the contents were shaken at 730 cpm for 24 h. To prepare the TEM grids, ∼20 μL droplet was added using a disposable Pasteur pipette over the formvar-coated copper grids (400 mesh). Grids were air-dried for about 3 h before adding two droplets (∼40 μL, using a disposable Pasteur pipette) of ultra-pure water and using small pieces of filter paper to wash out precipitated buffer salts. After air-drying for ∼15–20 min, the grids were negatively stained by adding a droplet (∼20 μL, using a disposable Pasteur pipette) of 2% phosphotungstic acid (PTA) and immediately after the grids were dried using small pieces of filter paper. Grids were further air-dried overnight. The scanning was carried out using a Philips CM 10 transmission electron microscope at 60 kV (Department of Biology, University of Waterloo) and micrographs were obtained using a 14-megapixel AMT camera.11,30
4.5 Experimental procedure for molecular docking studies
The modeling experiments were carried out using Discovery Studio (DS), Structure-Based-Design version 4.0, software program from BIOVIA Inc, San Diego, USA. Quinazoline and pyrido[3,2-d]pyrimidine derivatives 8e, 8h, 9a, 10b and 12b were built using the small molecules module in DS. For ChE docking studies, the X-ray coordinates of human AChE (pdb id:1B41) and BuChE (pdb id:1P0I) were obtained from protein data bank. The enzymes were prepared for docking using the macromolecules module in DS. The ligand binding site was defined by a 12 Å sphere for both AChE and BuChE.29,31 Docking simulation was carried out using the LibDock algorithm. During the docking simulation, CHARMm force field was used. The docked poses were evaluated using CDOCKER energy, CDOCKER interaction energy in kcal mol−1 and by considering the type and number of polar and nonpolar contacts.32 The molecular docking of test compounds with the Aβ assembly was carried out by using the NMR solution structure (pdb id:2LMN). The Aβ dimer and Aβ fibril assemblies were built using the macromolecules module in DS.32 Ligand binding site was defined by selecting a 15 Å radius sphere for both Aβ dimer and fibril assembly. Molecular docking was performed using the receptor–ligand interactions module in DS. The LibDock algorithm was used to find the most appropriate binding modes of quinazoline/pyrido[3,2-d]pyrimidine derivatives (8h and 10b) using CHARMm force field. The docked poses obtained were ranked based on the LibDock scores and the binding modes were analyzed by evaluating all the polar and nonpolar contacts between the ligands and Aβ-dimer and fibril regions.
4.6 Experimental procedure for iron [Fe(II)] chelation assay
Determined using the ferrozine (Sigma-Aldrich, USA) based competitive colorimetic assay.23 Test compounds were initially dissolved in anhydrous methanol to 10 mM and diluted down to 105 μM using 100 mM tris buffer (pH 7.4). Then 95 μL of test compound solutions (final concentration of 50 μM in each well) were added to clear 96-well plates, followed by a 10 μL aliquot of iron sulphate (FeSO4·7H2O) stock solution (from 800 μM stock solution prepared in methanol). After a 5 minute incubation period at room temperature, 95 μL ferrozine solution (from 210 μM stock solution prepared in tris buffer) was added. After incubating at room temperature for 30 minutes the absorbance was measured at 562 nm and subtracted from compound blanks (95 μL of compound solutions + 105 μL of tris buffer) and compared to the ferrozine-only positive control (95 μL of tris buffer + 10 μL of iron sulphate + 95 μL of ferrozine). The results obtained were compared with known iron chelators; clioquinol (50 μM) and desferoxamine (50 μM). The results were reported as average % iron-chelation ± SD in triplicate measurements based on two independent experiments.
Acknowledgements
The authors would like to thank the Faculty of Science, Office of Research, the School of Pharmacy at the University of Waterloo, Ontario Mental Health Foundation (graduate scholarship for TM), NSERC-USRA (for MM), NSERC-Discovery (RGPIN: 03830-2014), Canada Foundation for Innovation (CFI-JELF), Ontario Research Fund (ORF) and Early Researcher Award, Ministry of Research and Innovation, Government of Ontario, Canada (PR) for financial support of this research project.
References
- A. Cavalli, M. L. Bolognesi, A. Minarini, M. Rosini, V. Tumiatti, M. Recanatini and M. Carlo, J. Med. Chem., 2008, 51, 347–372 CrossRef CAS PubMed.
- J. J. Lu, W. Pan, Y. J. Hu and Y. T. Wang, PLoS One, 2012, 7, e40262 CAS.
- R. E. Hughes, K. Nikolic and R. R. Ramsay, Front. Neurosci., 2016, 10, 177 CrossRef PubMed.
- J. Birks, Cochrane Database Syst. Rev., 2006, CD005593 CAS.
- D. Lo and G. T. Grossberg, Expert Rev. Neurother., 2011, 11, 1359–1370 CrossRef CAS PubMed.
- P. T. Francos, A. M. Pamer, M. Snape and G. K. Wilcock, J. Neurol., Neurosurg. Psychiatry, 1999, 66, 137–147 CrossRef.
- M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS PubMed.
- M. Rosini, E. Simoni, A. Mielli, A. Minarini and C. J. Melchiorre, J. Med. Chem., 2014, 57, 2821–2831 CrossRef CAS PubMed.
- A. I. Bush, J. Alzheimer's Dis., 2013, 33, S277–S281 Search PubMed.
- T. Mohamed and P. P. N. Rao, Curr. Med. Chem., 2011, 18, 4299–4320 CrossRef CAS PubMed.
- T. Mohamed, A. Shakeri, G. Tin and P. P. N. Rao, ACS Med. Chem. Lett., 2016, 7, 502–507 CrossRef CAS PubMed.
- T. Mohamed and P. P. N. Rao, Eur. J. Med. Chem., 2017, 126, 823–843 CrossRef CAS PubMed.
- S. Darvesh, D. A. Hopkins and C. Geula, Nat. Rev. Neurosci., 2003, 4, 131–138 CrossRef CAS PubMed.
- H. Levine, Protein Sci., 1993, 2, 404–410 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres Jr and R. M. Feather-Stone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- A. T. Petkova, W. M. Yau and R. Tycko, Biochemistry, 2006, 45, 498–512 CrossRef CAS PubMed.
- G. Kryger, M. Harel, K. Giles, L. Toker, B. Velan, A. Lazar, C. Kronman, D. Barak, N. Ariel, A. Shafferman, I. Silman and J. L. Sussman, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 1385–1394 CrossRef CAS.
- Y. Nicolet, O. Lockridge, P. Masson, J. C. Fontecilla-Camps and F. Nachon, J. Biol. Chem., 2003, 278, 41141–41147 CrossRef CAS PubMed.
- J. S. Choi, J. J. Braymer, R. P. Nanga, A. Ramamoorthy and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21990–21995 CrossRef CAS PubMed.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS PubMed.
- M. P. Horowitz and J. T. Greenamyre, J. Alzheimer's Dis., 2010, 20, S551–S568 CrossRef PubMed.
- L. X. Yang, K. X. Huang, H. B. Li, J. X. Gong, F. Wang, Y. B. Feng, Q. Tao, Y. H. Wu, X. K. Li, X. M. Wu, S. Zeng, S. Spencer, Y. Zhao and J. Qu, J. Med. Chem., 2009, 52, 7732–7752 CrossRef CAS PubMed.
- M. Karamac, Int. J. Mol. Sci., 2009, 10, 5485–5497 CrossRef CAS PubMed.
- R. A. Smits, I. J. de Esch, O. P. Zuiderveld, J. Broeker, K. Sansuk, E. Guaita, M. Adami, E. Haaksma and R. Leurs, J. Med. Chem., 2008, 51, 7855–7865 CrossRef CAS PubMed.
- Z. Li, D. Wu and W. Zhong, Heterocycles, 2012, 85, 1417–1426 CrossRef CAS.
- A. Tikad, S. Routier, M. Akssira, J. M. Leger, C. Jarry and G. Guillaumet, Synlett, 2006, 12, 1938–1942 Search PubMed.
- T. Mohamed, J. C. Yeung, M. S. Vasefi, M. A. Beazely and P. P. N. Rao, Bioorg. Med. Chem. Lett., 2012, 22, 4707–4712 CrossRef CAS PubMed.
- T. Mohamed, A. Assoud and P. P. N. Rao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o554 CAS.
- T. Mohamed, X. Zhao, L. K. Habib, J. Yang and P. P. N. Rao, Bioorg. Med. Chem., 2011, 19, 2269–2281 CrossRef CAS PubMed.
- V. L. Anderson, T. F. Ramlal, C. C. Rospigliosi, W. W. Webb and D. Eliezer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18850–18855 CrossRef CAS PubMed.
- G. Tin, T. Mohamed, N. Gondora, M. B. Beazely and P. P. N. Rao, MedChemComm, 2015, 6, 1907–2044 RSC.
- P. P. N. Rao, T. Mohamed, K. Teckwani and G. Tin, Chem. Biol. Drug Des., 2015, 86, 813–820 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Includes 1H NMR spectra. See DOI: 10.1039/c7ra02889j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a