
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication and characterization of hydrogels formed from designer coiled-coil fibril-forming peptides†
A. F. Dexter‡
*a,
N. L. Fletcher§
a,
R. G. Creasey b,
F. Filardo¶
a,
M. W. Boehmb and
K. S. Jack*c
b,
F. Filardo¶
a,
M. W. Boehmb and
K. S. Jack*c
aThe University of Queensland, Australian Institute for Bioengineering and Biotechnology, Qld 4072, Australia. E-mail: a.dexter@uq.edu.au
bThe University of Queensland, School of Chemical Engineering, Qld 4072, Australia
cThe University of Queensland, Centre for Microscopy and Microanalysis, Qld 4072, Australia. E-mail: k.jack@uq.edu.au
First published on 23rd May 2017
Abstract
Hydrogels are soft solids that represent attractive matrices for tissue engineering, wound healing and drug delivery. We previously reported an α-helical peptide, AFD19, that forms fibrils and hydrogels at pH 6, but precipitates under physiological conditions. We now show that a single targeted change in AFD19 yields peptide AFD36, which gels at physiological pH and in the presence of salt. Furthermore, we present a simple method for homogeneous sol–gel conversion through pH titration with sodium bicarbonate followed by loss of carbon dioxide. Chemical and thermal denaturation studies show AFD36 self-assembles to give stable α-helical structures, forming fibrils of 3.8–3.9 nm diameter at pH 4.0–7.0 as shown by small-angle X-ray scattering and atomic force microscopy. An AFD36 gel at 0.35% (w/v) showed an elastic modulus of 350 Pa. Mouse fibroblasts exhibited low cellular toxicity and spread morphologies when grown on the gel as a preliminary proof of principle towards cell culture studies. These peptide gels offer a molecularly simple, biodegradable alternative to polymer-based systems for biomedical applications.
Introduction
Hydrogels are an area of burgeoning research interest, with an increasing number of papers reporting the preparation and characterization of these highly-hydrated soft solids.1,2 High-value applications for hydrogels exist particularly in the biomedical field,3 where a range of chemistries have been tested for regenerative medicine,4–6 drug delivery7,8 and wound healing.9,10Two classes of hydrogels—chemical and physical—can be distinguished in terms of the cross-links between the structural components. In chemical (chemically cross-linked) hydrogels, gel formation involves covalent cross-linking of polymer chains into a three-dimensional network. Such gels typically possess a solid content in the range 5–15% (w/v), have high elasticity, and undergo an equilibrium swelling process.11–13 In contrast, physical (physically cross-linked) hydrogels rely on weak non-covalent interactions, such as hydrogen bonding, hydrophobic forces or charge–charge interactions, between structural components for network formation.14 The preparation of physical hydrogels involves conversion of a sol to a gel state, typically by mixing components or a change in temperature; such gels cannot be prepared by equilibrium swelling of a solid.14,15 Physical gels are commonly prepared at concentrations in the range 0.1–5% (w/v) and have lower strength than chemical gels.1,5,16–18 Physical gels often display shear thinning with self-healing, making them more suitable for soft tissue engineering or as injectable media.2,19,20
Peptides are favorable starting materials for the preparation of physical gels, given their biodegradability, stimuli-responsiveness21,22 and the possibility of rationally designing their self-assembly.16,23 The largest body of work to date on self-assembling peptide hydrogels has employed β-sheet motifs.17,18,24 β-sheet peptides form fibrils in which component peptides align perpendicular to the fibril axis, held together by hydrophobic and hydrogen bonding interactions. Control of fibril self-assembly and gel formation can be achieved by control of peptide molecular charge,18,19 salt composition25 and solvent quality,17 yielding robust gels that have been successfully applied in a number of tissue engineering model systems.26,27
β-sheet assemblies have been investigated and used extensively for clinical applications.28,29 However, the core structure of β-sheet fibrils resembles amyloid or prion structures that occur in Alzheimer's and Parkinson's diseases, as well as a number of other less well-known disease states in which normal proteins refold into pathogenic structures in the presence of seed elements.30,31 Some reports have demonstrated the cross-seeding of amyloidogenic sequences by dissimilar amyloid structures.32,33 While there remains controversy as to the generality of these results, in at least one case, fibrils of designed peptides with amyloid-like properties were reported to induce amyloidosis, illustrating the potential for initiation of disease states by β-sheet gels.34
Another pathway to peptide hydrogels employs α-helices that form extended coiled-coil fibrils in which the components align parallel to the fibril axis. Rational design work from the Woolfson group35 has yielded α-helical peptides with forced offsets that self-assemble into insoluble thickened fibrils. The same group has reported peptide sequences that form thinner fibrils and hydrogels that support cell growth.16 In addition, several other researchers, including ourselves, have reported α-helical peptides without forced offsets that form α-helical fibrils and in some cases hydrogels.36–38 We have attributed such fibril formation to permissive, rather than forced, overlap of self-similar peptide sequences,36 and believe fibril formation of this kind may be a more widespread property of coiled-coil peptides than previously recognized.
In this study, a coiled-coil peptide designed to gel at physiological pH is characterized. The 21-residue peptide AFD36 was generated by a single amino acid change from the previously reported peptide AFD19.36 It is shown that this targeted change shifts gelation from pH 6.0 to pH 7.4, allowing gel formation to proceed under conditions favorable to mammalian cell growth. Slow and uniform gelation was achieved by titration with bicarbonate, permitting a handling window for the gelling peptide solution. Small-angle X-ray scattering (SAXS) data collected under varying conditions of pH are consistent with the formation of higher-order coiled-coil fibrils as the basis of the gel, with fibril formation confirmed using atomic force microscopy (AFM). Chemical and thermal denaturation studies show that the fibrils are extremely stable, a result which is attributed to the strong hydrophobic driving force for self-assembly. Oscillatory shear rheology results show the hydrogels to possess soft-solid properties. As a proof of principle, the peptide hydrogels were used to support the growth of mouse fibroblasts, making these materials promising candidates for tissue engineering applications.
Experimental
Materials
Reagents were of the highest grade available. Water was purified using an Elga Purelab Classic and had a resistivity of >18.2 MΩ cm. Glassware was acid-cleaned as previously described.39 Peptides AFD19 (Ac-LKELAKV LHELAKL VSEALHA-CONH2, FW 2354) and AFD36 (Ac-LKELAKV LHELAKL VKEALHA-CONH2, FW 2395) were synthesized and purified by GenScript (Piscataway, New Jersey). The final purity was >95% in each case. The peptide content of the solid was determined by quantitative amino acid analysis (Australian Proteome Analysis Facility, Sydney). The charge on each peptide as a function of pH was estimated as described previously.36Plate reader assays
Spectrophotometric determination of pH used an Infinite M200 plate reader (Tecan, Männedorf, Switzerland) with 48-well plates and sample volumes of 200 µL. The acidity constant of phenol red in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) was determined by titration, with absorbance monitored at 433 nm. Reference A433 readings for fully protonated and fully deprotonated phenol red were obtained by addition of 10 mN HCl and 10 mN NaOH, respectively. A pKa of 7.70 was obtained as the y-intercept of a plot of pH vs. log((Aacid − A433)/(A433 − Abase)). The same pKa value was employed for peptide solutions in phosphate-buffered saline (140 mM NaCl, 5 mM KCl, 1.5 mM Na + phosphate, 60 µM phenol red).For gelling tests, 100 µL of an AFD36 stock solution (8.4 mM, pH ∼3) was mixed in a 48-well plate with 100 µL of an NaHCO3 solution in either 2× DMEM containing 80 µM phenol red or 2× phosphate-buffered saline (240 mM NaCl, 10 mM KCl, 3.0 mM Na + phosphate) containing 120 µM phenol red. UV-visible spectra (700–350 nm) were recorded at 3 min intervals over 60 min and single wavelength readings were extracted for analysis. Spectra showing an isosbestic point at 477 nm were used for pH calculations, while samples showing light scattering were excluded. For greater sensitivity, the pH of gelling samples was determined at 558 nm, with pH calculated as 7.70 + log(A558 − Aacid)/(Abase − A558).
Small-angle X-ray scattering
SAXS data were collected using an Anton Paar SAXSess instrument which uses a monochromated Cu sealed anode operating at 40 kV and 50 mA, a line focus (Kratky) geometry and a CCD detector at a fixed sample-to-detector distance. Peptide samples were prepared at 5 mg mL−1 in 10 mM Na + acetate (pH 4.0 or 5.0) or 2-(N-morpholino)ethanesulfonate (MES; pH 6.0 or 7.0) with addition of NaCl to maintain an ionic strength of 0.01. Samples were housed in a silica capillary maintained at 20 °C during the measurements. SAXS data were measured over a range of q = 0.007 to 0.2 Å−1 (where q is the momentum transfer vector) and the data were reduced by standard procedures to remove the dark current and background measured from the buffer, after accounting for the relative transmission of the sample and background. The data were placed on an absolute intensity scale using the known cross section of water as a primary standard.40 The data presented represent the average of 1800 exposures collected for 10 s each to ensure that there were no changes occurring during exposure to the X-ray beam.Analysis of the X-ray data was carried out using the NCNR analysis macros in Igor Pro. For the data obtained here, a flexible cylinder model was used to determine both the mean radius and persistence length of the fibrils as a function of pH. In these models the electron density for the buffer was calculated to be 9.45 × 10−6 Å−2, while that for AFD36 was estimated to be 1.22 × 10−5 Å−2 based on chemical composition.
Atomic force microscopy
AFM micrographs were collected using an Asylum MFP3D SPM (Oxford Instruments) using NanoWorld FM probes (nom k = 2.8 N m−1, nom f = 75 kHz) in tapping mode under ambient conditions. Peptide samples were freshly prepared at 20 mg mL−1 in water, then diluted to a final concentration of 5 mg mL−1 in MES buffer with final concentration of 10 mM (pH 6). 5 µL of sample was quickly spotted onto mica and immediately removed, and allowed to dry in air overnight before imaging. Image analysis was undertaken using freeware Gwyddion (http://gwyddion.net) v2.41 and ImageJ v1.5 (http://imagej.nih.gov/ij/).Electronic circular dichroism (ECD)
ECD spectra were recorded on a Jasco J-815 spectropolarimeter in 0.1–1 mm quartz cuvettes. AFD36 spectra were recorded at 20 °C using 1.75 mM peptide self-buffered at pH 7.0 (gel) or 3.1 (sol). Heating-cooling studies were carried out at 20–90 °C at a heating rate of 1.5 °C min−1, using 0.84 mM AFD36 in 10 mM MES pH 7.0. Peptide solutions for chemical denaturation studies were prepared at 250 µM peptide in 10 mM Na + acetate pH 5.0 (AFD19) or 10 mM MES pH 6.0 (AFD36) containing 0–8 M guanidinium chloride (G.HCl), and were equilibrated at 20 or 50 °C before recording ellipticity at 222 nm. Raw ellipticity was converted to mean residue ellipticity by normalizing for path length and the concentration of peptide bonds.Gel melting
A ball drop test was conducted using a stainless steel ball (6 mm diameter; 1.0 g) placed on the surface of a AFD36 gel heated in a water bath. The gel was prepared by mixing 4.2 mM AFD36 in 10 mM MES with 22.5 mM NaHCO3 (final concentrations), and was allowed to stand overnight before the heating test to allow loss of CO2 and complete setting. The gel was heated from 30 to 80 °C in 10 °C steps, and was held for 15 min at each temperature before being photographed. A separate test was conducted on an AFD36 gel prepared in phosphate-buffered saline at the same concentration. Gel melting (Tm) was recorded at the temperature where the ball dropped completely through the gel.Guanidinium chloride denaturation
Thermodynamic analysis of peptide self-assembly in the presence of denaturant was based on an approach by Fairman,41 adapted for a higher-order coiled-coil assembly. Various schemes were tested using pentamer, hexamer or heptamer states for the minimum coiled-coil assembly of AFD36 at 20 °C, as well as a dodecamer based on end-to-end stacking of two hexamers. Given that similar thermodynamic results were obtained on a per-monomer basis for each of these oligomer models, only a hexamer model was used for comparison of AFD19 at 20 °C and AFD36 at 50 °C.Assembly of monomers to higher-order oligomers in the absence of denaturant was treated as a two-state system in which Kn,1 is the equilibrium constant for disassembly of the coiled coil (n = 5, 6, 7 or 12):
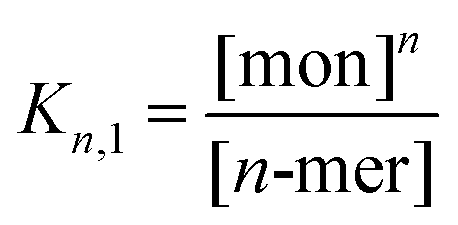 | (1) |
ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKn,1 lnKn,1
| (2) |
The presence of guanidinium chloride (G.HCl) was taken to modify the free energy of peptide self-assembly in a linear fashion:
| ΔG = ΔG(H2O) + m[G.HCl] | (3) |
For electronic circular dichroism, the contribution of monomers and n-mers to the ellipticity at a single wavelength (in this case, 222 nm) is given by:
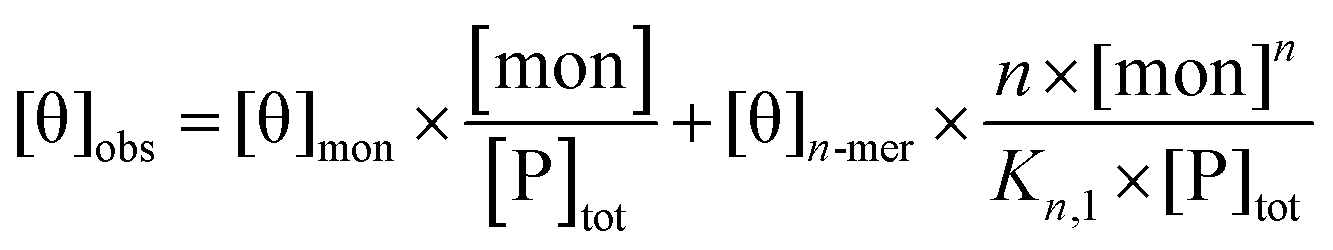 | (4) |
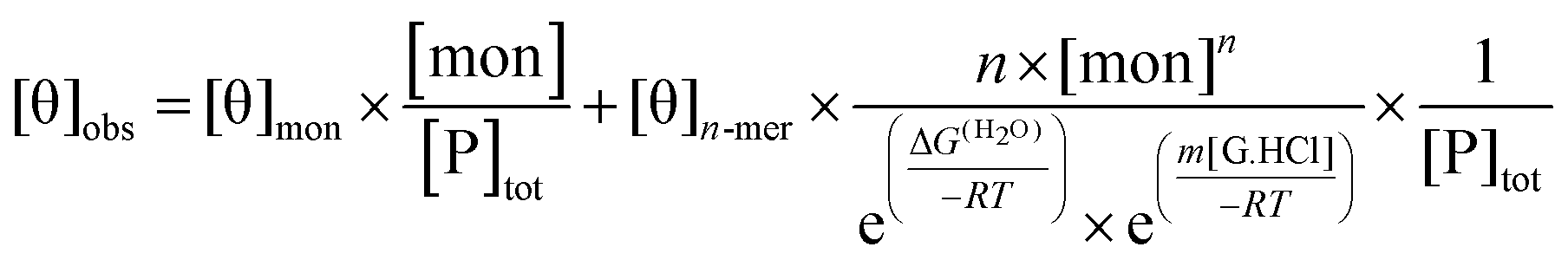 | (5) |
For test values of Kn,1 and m for each value of n, values for the free monomer concentration [mon] are obtained by numerical fitting to the mass balance equation for the total peptide in solution, [P]tot, allowing assessment of the least-squares fit to the data:
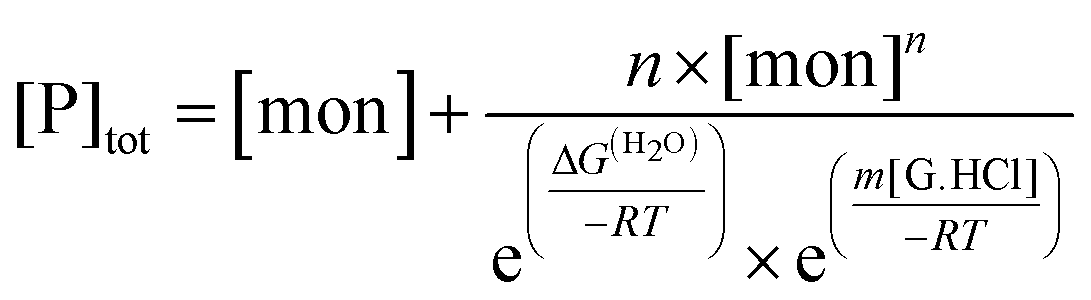 | (6) |
Best-fit values of ΔG(H2O), m, [θ]mon and [θ]n-mer obtained for each test value of n are given in Table S1 of the ESI.†
Gel rheology
Rheological measurements were carried out at 23 °C on a Thermo Scientific Haake MARS III stress-controlled rheometer using parallel plates (35 mm titanium) at a gap of 500 µm; the top plate had emery paper attached to limit slip. To prepare a gel, 500 µL 2.8 mM AFD36 pH 3 was mixed with 500 µL 25 mM NaHCO3, 2× DMEM and loaded onto the lower plate using a micropipette, after which the upper plate was lowered. To limit evaporation, drops of water were placed in the solvent trap around the outer edge of the lower plate and a hood was lowered to cover the plates; the water drops were not in contact with the sample. A transient oscillatory test was used to capture development of structure as the pH rose with loss of CO2 from the sample, permitting gelation. The angular frequency was set to 6.28 rad s−1 for quick measurements and the oscillatory shear stress was set to 0.1 Pa, which gave strain less than 0.005. Relative to the final gel, an oscillatory shear stress of 0.1 Pa was well within the linear viscoelastic region. The oscillation time test was run for 3600 seconds and measurements commenced within 5 minutes of peptide mixing.An oscillatory stress sweep at an angular frequency of 6.28 rad s−1 was then run on the same sample, to determine the linear viscoelastic region. Using an oscillatory shear stress of 0.1 Pa, an oscillatory frequency sweep was also run from 0.29 to 19.9 rad s−1.
Mammalian cell culture
NIH/3T3 cells (CRL-1658, ATCC) were maintained in DMEM (Gibco, Life Technologies) supplemented with 10% (v/v) newborn calf serum (Ausgenex, Australia). Cultures were kept at 37 °C in a humidified 5% CO2 incubator.To remove residual by-products from solid-phase peptide synthesis, solutions of AFD36 were stirred with 8 M urea and 3 mg mL−1 activated charcoal for 24 hours. Charcoal was removed by centrifugation at 20000 g for 45 minutes and filtering (0.2 µm). The solution was then dialyzed against 0.1 M NaCl (24 h) and water (2 × 24 h). Recovered peptide (2.8 mM) was adjusted to pH 3 and sterile-filtered (0.20 µm) then stored at −80 °C until use. To prepare gels for cell culture, 50 µL acidic peptide solution was mixed thoroughly with 50 µL 25 mM NaHCO3, 2× DMEM (initially bicarbonate free) in individual wells of a 48-well tissue culture plate. Samples were allowed to gel for 1 h at RT then transferred to an incubator and soaked for 2 × 2 days in serum-free DMEM (500 µL) before seeding with NIH/3T3 cells at 104 cells per well. Control wells contained NIH/3T3 cells seeded onto tissue culture polystyrene (TCPS) alone. Cell growth was quantified using AlamarBlue (Life Technologies) at days 1–4 (n = 3). Fluorescence readings used a Tecan Infinite M200 plate reader with λex 560 nm and λem 590 nm. Fluorescence values for cell-free wells were subtracted from the values of cell-containing wells. Statistical analyses used Student's t-test, with differences considered significant for p values of <0.05. Cell images were taken at 10× magnification at day 3.
Results and discussion
Peptide design
We have previously reported the design of a 21 residue pH-responsive α-helical peptide, AFD19 (Ac-LKELAKV LHELAKL VSEALHA-CONH2), that forms self-healing gels at pH 6.0 and weight fractions as low as 0.1%. Here, it is shown that a single designed change in AFD19 yields a new peptide, AFD36 (Ac-LKELAKV LHELAKL VKEALHA-CONH2), that gels under physiological conditions of pH and salt.Fig. 1 illustrates the peptide AFD36 as a higher order coiled-coil n-mer. Canonical coiled coils are based on an abcdefg heptad repeat45,46 in which positions a and d comprise the hydrophobic core of the coil and are typically occupied by aliphatic amino acid residues such as leucine (L), valine (V) or isoleucine (I). AFD19 and AFD36 are three-heptad peptides conforming to this general pattern, where the flanking positions e and g are populated with additional aliphatic amino acids, including helix promoting alanine (A). This yields an expanded hydrophobic core that provides a strong driving force for self-assembly, leading to a highly thermodynamically stable coiled coil.47–49 At the same time, an expanded hydrophobic core tends to lead to formation of higher-order (tetramer and larger) coiled coils,45 as suggested for AFD19.36
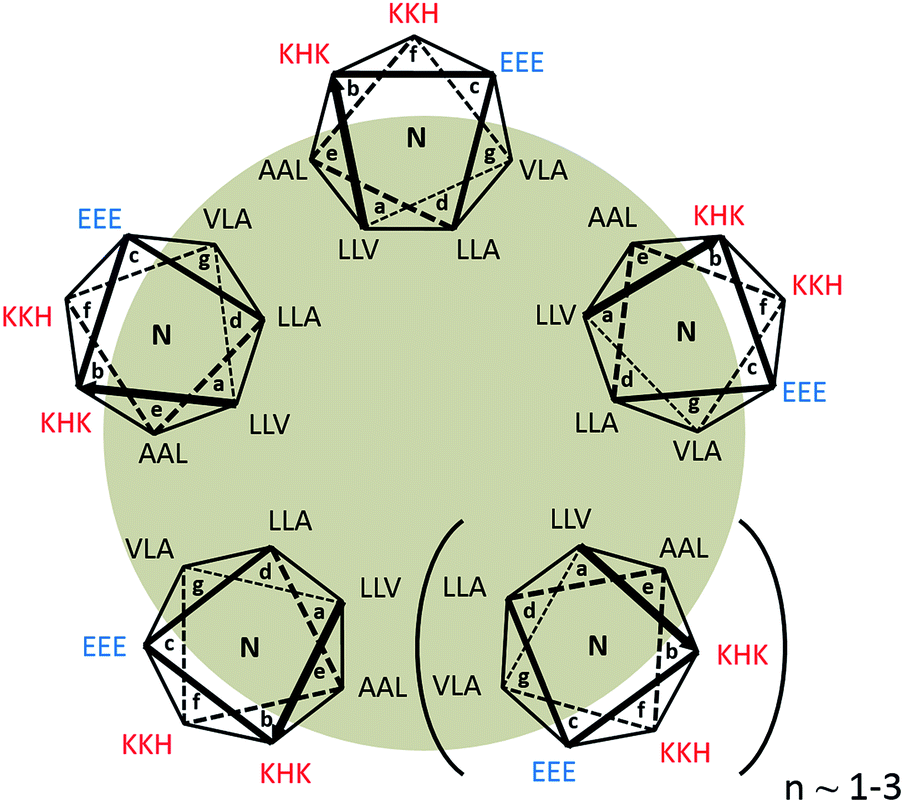 | ||
| Fig. 1 Structure of AFD36 modeled as a parallel coiled-coil n-mer. L, V and A residues comprise an expanded hydrophobic core at positions a, d, e and g of the abcdefg heptad repeat. Charged residues occupy positions b, c, and f, which face the aqueous medium. For clarity, cationic residues are in red, anionic in blue, and the hydrophobic core is shaded. | ||
For geometric reasons, higher-order coiled coils contain an internal tubular cavity, as observed in heptamer,47,50 hexamer,49 pentamer51 and even tetramer51,52 structures.
The outward-facing surface of the AFD36 coiled coil at positions b, c, and f is occupied by hydrophilic residues, primarily helix-favoring cationic lysine (K) and anionic glutamate (E) residues, but also histidine residues (H; pKa ∼6) intended to confer pH-responsiveness in self-assembly. The sole difference between AFD19 and AFD36 is the presence of a lysine rather than an uncharged serine residue (S) at position 16, meaning that at physiological pH, AFD36 carries a single positive charge, sufficient to prevent large aggregate formation while permitting gelation based on the physical association of fibrils.
α-Helices in which e and g sites are populated by oppositely-charged residues are generally expected to form parallel coiled coils due to charge–charge repulsion in the antiparallel arrangement.53 While the e and g sites in AFD19 and AFD36 are occupied by non-polar residues, a similar argument may be applied to the b and c flanking positions,54 making it likely that these peptides assume a parallel structure.
In the absence of more structural information AFD36 is illustrated as a parallel coiled coil containing 5–7 base units in cross-section. This is consistent with the propensity for α-helices with an expanded hydrophobic face to form higher-order coiled coils and the positioning of charged residues at b and c positions favoring a parallel arrangement. An oligomerization state larger than a heptamer is thought to be unlikely as higher oligomerization states have not been observed in designed peptide systems.45 This structure is also consistent with the observed fibril diameter and cooperativity of assembly of ∼6 determined for the parent peptide AFD19.36
The designs reported herein complement the work reported by the Woolfson group, which has shown that designer coiled-coil peptides form elongated fibrils if elements are included that force end-to-end overlapping of α-helices. This approach has been used to prepare insoluble α-helical fibrils with controllable morphologies55–57 and more recently, thin fibrils that form self-supporting gels.16 A similar approach, based on placement of polar residues within the coiled-coil core, has been reported by Zimenkov et al.58,59 Another system showing offsetting of helices was reported by Liu et al.,47 who observed the peptide GCN4-pAA to form a heptameric coiled coil with a single-residue shift in registry between adjacent helices, resulting in an offset of a full heptad between helices 1 and 7. Helix offsetting is likely driven by preferential packing of hydrophobic residues at positions a, d, e and g, as well as a ring of hydrogen bonding residues within the coiled-coil core. Xu et al.50 more recently reported redesign of this peptide to give 7HSAP1. This peptide forms staggered coiled coils which are able to stack via end-to-end overlap of helices to give coiled-coil fibrils.
We have previously proposed another approach to coiled-coil fibril formation that is driven by permissive rather than specified helix offsetting.36 Included in this class are peptides AFD19 and AFD36 as well as the fibril-forming sequences of Kojima et al.,60 Dong et al.,61 Potekhin et al.48 and Melnik et al.37 which possess some broad similarities. These designed peptides lack forced offsets and have a high degree of self-similarity in the constituent heptad repeats. Importantly, the net intermolecular interactions of an α-helix with self-similar heptad repeats are likely to be energetically similar for both an in-register arrangement and a staggered fibril with heptad offsets. Designs based on self-similar heptad repeats avoid the thermodynamically destabilizing52 placement of polar residues in the coiled-coil core, thereby facilitating development of gels composed of short single peptides, as opposed to paired peptides or longer sequences that may be expensive to synthesize. As exact repeats are not required for fibril assembly, this design approach still allows the incorporation of specific residues to control molecular charge, as was done here to achieve gelation at physiological pH. It also allows for diversity of residue composition, which may be useful in the context of peptide bioproduction.
Self-assembly of coiled-coils and gels
AFD36 self-assembles into a coiled coil as judged by electronic circular dichroism (ECD) spectroscopy. Fig. 2 shows ECD spectra for an AFD36 solution at pH 3.1 and a gelled sample at pH 7.0. The double minimum at 208 and 222 nm and maximum at 192 nm are characteristic of an α-helix. Unlike AFD19, which gels at pH 6.0 but forms aggregates at pH 7.0, the redesigned peptide forms transparent gels at pH 7.0. In the gelled state, the mean residue ellipticity at 222 nm is −30100 deg cm2 dmol−1, implying an α-helical content close to 91% for AFD36 under these conditions.43 In contrast to AFD19, which has a similar helical content at pH 3.0 and pH 6.0 at low millimolar concentrations (comparable to those used here),36 AFD36 is less helical at pH 3.1 than at pH 7.0. The difference may be due to the higher molecular charge in AFD36 (+6 for AFD36 vs. +5 for AFD19 at pH 3), leading to destabilization of the coiled coil by a greater degree of electrostatic repulsion.
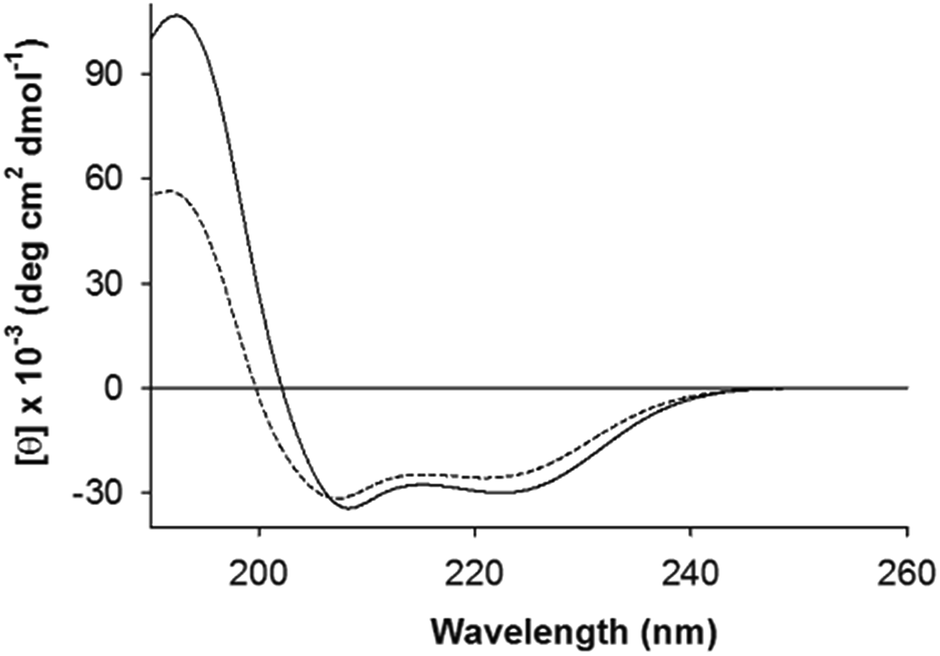 | ||
| Fig. 2 Electronic circular dichroism (ECD) spectra of self-buffered AFD36 in gelled and non-gelled states. Solid line, pH 7.0 (gel); dashed line, pH 3.1 (sol). The peptide concentration was 1.6 mM. | ||
Formation of a gel at low weight concentrations strongly suggests the formation of fibrils (as opposed to globular structures) as the basis of the gel. The shape of the ECD double minimum for AFD36 at pH 7.0 is consistent with the formation of fibrils that are not thickened by lateral association. While early reports62,63 that coiled coils could be distinguished from monomeric α-helices by a higher [θ]222/[θ]208 ratio appear to have been over-generalized,43,64 high [θ]222/[θ]208 ratio are indeed seen in cases of fibril thickening, likely due to light scattering.16,55 No such spectral changes are seen for AFD36; the formation of optically clear gels at physiological pH also favors the interpretation that the fibrils are not aggregated.
Fibril structure
To further investigate fibril self-assembly by AFD36, SAXS data was obtained for AFD36 samples prepared at pH 4.0–6.0 (non-gelled) and 7.0 (gelled). The data, with fits to a flexible cylinder model, are shown in Fig. 3. Fitted diameters and persistence lengths are given in Table 1 and are consistent with the formation of fibrils at each pH, even though gelation is not seen below pH 7.0. The fibrils have similar properties across the entire pH range, with a diameter of 3.8–3.9 nm. From the current SAXS data, it is evident that the fibril diameter for AFD36 (<4 nm) is similar to that of observed using negative staining transmission electron microscopy (TEM) of AFD19 fibrils at acidic pH,36 and that bundling or thickening of AFD36 fibrils in solution does not occur in the pH range from 4.0 to 7.0. For comparison, Banwell et al.16 have reported designer peptides with zero net charge that form bundled fibrils (as judged by electron microscopy and ECD) as well as opaque gels. It appears that the molecular charge on AFD36 at the pH values tested here prevents fibril bundling.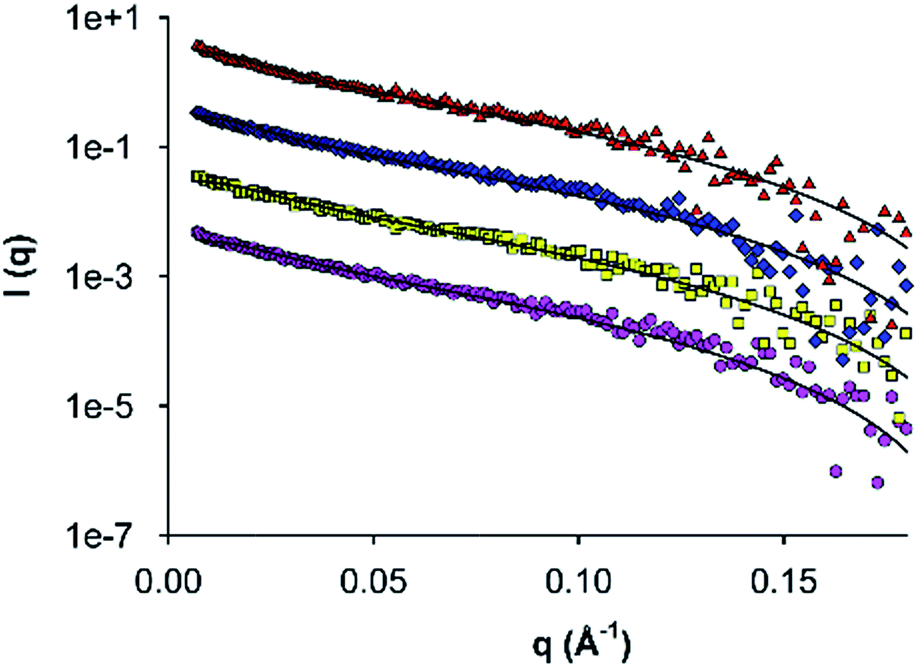 | ||
| Fig. 3 Small-angle X-ray scattering by self-assembled AFD36 fibrils. Data were collected for 0.5% (w/v) peptide at pH 4.0 (pink circles), 5.0 (yellow squares), 6.0 (blue diamonds) and 7.0 (red triangles). Solid lines indicate fits to a flexible cylinder model. Data sets above pH 4.0 are displaced upwards progressively by a factor of ten for clarity. | ||
| pH | Fibril diameter (nm) | Persistence length (nm) |
|---|---|---|
| 4.0 | 3.91 ± 0.05 | 10.6 ± 1.4 |
| 5.0 | 3.82 ± 0.05 | 10.7 ± 2.1 |
| 6.0 | 3.79 ± 0.05 | 14.0 ± 0.3 |
| 7.0 | 3.78 ± 0.05 | 12.4 ± 3.5 |
To better characterize fibril structure and dimensions, AFM was performed on samples prepared at pH 6. As shown in Fig. 4, fibrils were observed at pH 6 on the mica surface. AFD36 fibrils had a height of 3.5 ± 0.9 nm (N = 51) as measured by AFM. It is documented that the AFM imaging process may reduce the apparent height of biological molecules and in addition, the fibrils are in a dehydrated state.65 Hence, the AFM data is consistent with SAXS data.
 | ||
| Fig. 4 AFM 3D micrographs of AFD36, prepared from 5 mg mL−1 concentration under fibril-forming conditions (10 mM MES; pH 6) at (A) micron (5 µm x-scale, 20 nm z-scale) and (B) nanometer scale (125 nm x-scale, 3 nm z-scale). | ||
Comparison of AFD36 fibril diameters with reported higher-order coiled coils shows observed sizes to be consistent with the proposed coiled-coil structure. Data from X-ray crystallography or nuclear magnetic resonance spectroscopy give approximate diameters of 3.1 nm for the designed peptide GCN4-pAA (seven strands; 2HY6),47 3.0 nm for the M2 segment of the acetylcholine receptor (five strands; 1EQ8),66 3.3 nm for the designed peptide CC-Hex (six strands; 3R48)49 and 2.9 nm for cartilage oligomeric matrix protein (COMP, five strands; 1VDF).67 Redesign of GCN4-pAA has yielded peptide 7HSAP1, which forms fibrils with a proposed heptamer structure and a diameter of 3.0 nm as measured by X-ray diffraction.50 The AFD36 fibrils, at 3.8–3.9 nm as measured by SAXS and 3.5 nm as measured by AFM, are slightly larger than these reference systems. The diameter of an α-helix is not constant but instead dependent on residue composition. Therefore, the larger observed fibril diameter may in part be due to the higher content of larger aliphatic residues in AFD36 than are present in the comparison systems. Nonetheless, the diameter is still consistent with a fibril structure comprising unbundled higher-order coiled coils of at least a pentamer. In an effort to more precisely determine the minimum oligomer state of the peptide size exclusion chromatography was attempted under acidic conditions. At lower pH, high molecular charge was expected to prevent fibril assembly; however, conditions were not able to be identified that gave discrete coiled-coil assembly in the absence of fibril formation. Chemical denaturation studies (below) also fail to distinguish between different oligomer states, but confirm that the fibrils are highly thermodynamically stable, apparently as a result of a strong hydrophobic driving force for self-assembly.
The persistence lengths of AFD36 fibrils as determined by SAXS may also be compared to those of other biomolecular systems. In the pH range from 4.0 (sol) to 7.0 (gel), AFD36 fibrils show a persistence length of 10–14 nm, indicating a flexible structure that does not alter over the sol–gel transition. For comparison, the persistence length of double-stranded DNA, with a hydrodynamic diameter of 3 nm, lies in the range 50–100 nm,68 indicating that the DNA double helix has less flexibility than AFD36 fibrils. Fibrils of 1–6 nm diameter formed from the milk protein β-lactoglobulin have a persistence length of 90 nm to 4 µm depending on β-sheet content,69 while amyloid-forming designer β-sheet peptides have persistence lengths of up to 70 µm at a diameter of 8 nm.70 AFD36 fibrils are thus relatively flexible, which may relate to the non-specific character of hydrophobic interactions between the subunits in the fibril. Fibril lengths were also measured by AFM, with fibrils identified from <100 nm to >4 um in length (average length 914.4 ± 838 nm, N = 81). Many fibrils were intertwined or were not fully visible within an image area (Fig. 4A); nonetheless, AFD36 fibrils show a very high aspect ratio.
Charge-based control of gelation
Formation of physical hydrogels involves a delicate balance between solubility of the main components and association between them. We have previously reported a “phase diagram” for formation of sol, gel and aggregate states by AFD19 as a function of pH,36 and have accounted for transitions between these states in terms of changes in the molecular charge. Fig. 5 shows a similar schematic for AFD36, with the charge curve for AFD19 included for comparison. At acidic pH, both peptides form low-viscosity solutions, which convert to transparent hydrogels on titration with base. For AFD19, gel formation occurs at pH 6.0, where the molecular charge lies close to +1. For AFD36, a similar charge is only reached at around pH 7.0, allowing gelation to proceed at physiological pH. Both peptides form precipitates at pH values where the predicted molecular charge lies close to zero, but this occurs at pH 7.0 for AFD19, while the equivalent state is not reached until pH 10.3 for AFD36. Both peptides form a second gel phase at high pH (data not shown), where the molecular charge lies close to −1; Aggeli et al.18 have similarly reported that a molecular charge of +1 or −1 is compatible with the gelation of β-sheet peptides.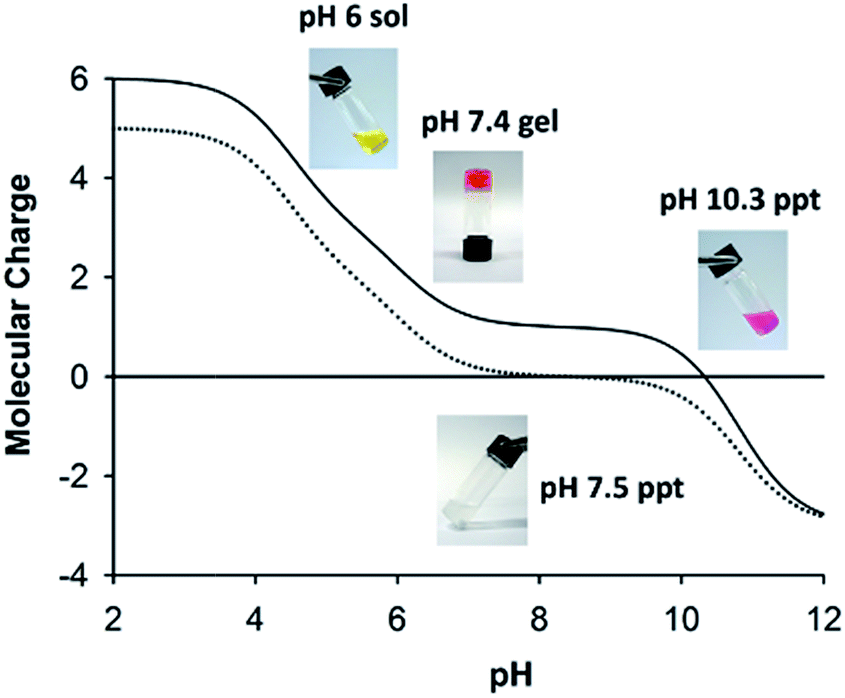 | ||
| Fig. 5 Gelation as a function of pH and molecular charge for AFD36 and parent sequence AFD19. Calculated charge curves are given for AFD36 (solid line) and AFD19 (dotted line). Inset: photographs of sol, gel and aggregate states for AFD36 (top) and aggregate state for AFD19 (bottom). Phenol red has been included in the self-buffered AFD36 samples as a visual indicator of pH. The peptide concentration was 2.0 mM for AFD36 and 1.2 mM for AFD19; above critical gelling concentration. | ||
Although we have not relied on intermolecular charge–charge interactions in the design of AFD19 and AFD36, we consider net molecular charge to be an important factor in control of fibril–fibril interactions, allowing inter-fibril association to drive gel formation while preventing fibril bundling that could lead to loss of solubility. Results with both AFD19 and AFD36 show that gel transitions occur at pH values close to a molecular charge of +1 or −1, while loss of solubility occurs at a pH where the molecular charge approaches zero (Fig. 5). The morphology of the AFD19 or AFD36 aggregates formed at zero charge have not been examined, but it is reasonable to assume that they comprise bundles of the fibrils seen at nearby pH values, possibly resembling the fibril bundles formed by offset peptide designs with a zero net charge.55–57 Development of a method that provides a handling window for pH-responsive gelation is an important element in potential biomedical application of gels in this class, as it permits the formation of evenly-mixed gels, which can be otherwise difficult to prepare in fast-gelling systems.
Interestingly, both AFD19 and AFD36 gels appeared to self-heal after mechanical disruption on a time scale similar to the original gel formation. Gelation of AFD19 or AFD36 is also reversible with pH indicating that they may be suited for use in injectable gels.
Uniform gelation via transient buffering
One disadvantage of pH-responsive gelling systems is a tendency to mix poorly due to local gelation on addition of acid or base. This was observed with AFD36 gels prepared in the presence of pH indicator dyes (Fig. 6, inset). On addition of NaOH to an acidic peptide solution containing phenol red, a basic gel forms locally (crimson) that is surrounded by acidic medium (yellow). pH equilibration occurs slowly, leading to a non-uniform structure and lower gel strength; in extreme cases, gel formation may fail completely. To overcome this problem, a method of slow pH adjustment was developed based on transient buffering by carbon dioxide. The method gives a handling window for gelation of concentrated peptide solutions, while allowing uniform mixing to maximize gel strength. To achieve this, an acidic peptide solution is titrated with the weak base sodium bicarbonate leading to the formation of carbonic acid, which itself dissociates to give a slightly acidic solution pH. Carbonic acid is in equilibrium with atmospheric carbon dioxide, meaning that it is gradually lost from the gelling solution, leading to a slow pH rise that can be observed using an indicator dye.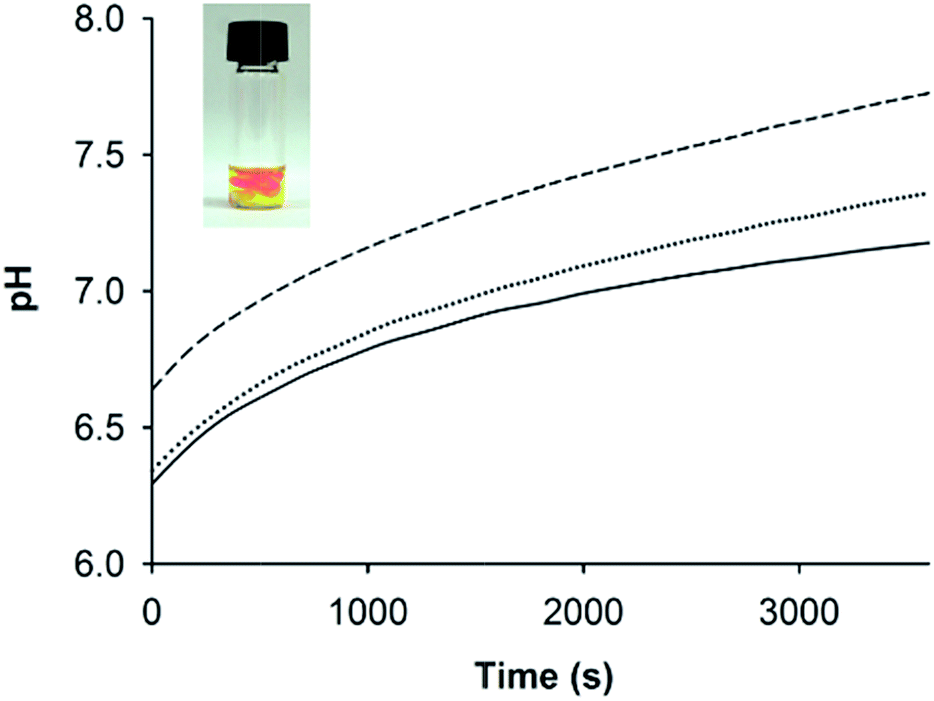 | ||
| Fig. 6 pH changes over time for AFD36 gels prepared in model serum salts upon titration with different concentrations of bicarbonate. Solid line, 10.0 mM NaHCO3; dotted line, 11.2 mM NaHCO3; dashed line, 12.5 mM NaHCO3. Inset: local gelling with pH inhomogeneity on titration of AFD36 with NaOH. The peptide concentration in each case was 2.1 mM. | ||
UV-visible spectrophotometry and phenol red were used to track pH changes in AFD36 samples prepared in phosphate-buffered saline and titrated with different amounts of sodium bicarbonate; results are shown in Fig. 6. As would occur on titration with a strong base, the final pH depends on the amount of base added, but following addition of bicarbonate, pH change occurs as an initial step change followed by a slow drift upwards, with equilibration in a 200 µL volume taking more than an hour. Clear gels are obtained at pH values up to 8.2 (not shown), but light scattering is observed at higher pH, consistent with fibril bundling as the molecular charge on the peptide begins to approach zero. Gel formation occurs before the pH change is complete, but the slow rate of pH change allows a handling window of several minutes for the peptide solutions prior to gelation, a property that may be useful in clinical settings. Overnight exposure of the gels to a 5% CO2 atmosphere in a mammalian cell incubator lowered the pH by approximately 0.4–0.5 pH units (as observed by color change of phenol red), but an appropriate concentration of bicarbonate could be chosen to allow the preparation of gels with a pH suited to cell culture studies. Similar kinetic results were obtained for AFD36 gels prepared by the addition of bicarbonate in Dulbecco's modified Eagle medium (DMEM), allowing sample preparation for tissue culture experiments. Similarly, gel preparation using 10 mM MES-bicarbonate buffer allowed sample characterization by ECD (below) while minimizing interference by salt.
Thermal stability of AFD36 helices and gels
Thermal denaturation studies were carried out to determine the heat stability of self-assembled AFD36 structures. This was done both in terms of primary (coiled coil) and secondary (gel) assembly, and showed gel melting to occur independently to α-helix denaturation. Fig. 7 shows the effects of temperature on AFD36 helix content, as determined by ECD. At 20 °C, the mean residue ellipticity at 222 nm is −30100 deg cm2 dmol−1 for a gelled sample at pH 7.0. On heating to 90 °C, the mean residue ellipticity decreases to −25500 deg cm2 dmol−1, indicating that the temperature of half-denaturation (“melting temperature”) is well in excess of 100 °C. Cooling to 20 °C results in recovery of most, but not all, of the original helical content, giving a final value of [θ]222 of −28800 deg cm2 dmol−1. Compared to the helical content, the macroscopic gel structure of AFD36 is less stable. A ball drop test showed a gel–sol transition temperature (Tm) of close to 80 °C for an AFD36 gel prepared in 10 mM MES buffer, although this increased to 90 °C for a gel prepared in phosphate-buffered saline at higher ionic strength. The disparity between the ECD and gel transition temperatures suggests that the gel–sol transition may occur by loss of the physical cross-links between the fibrils, as opposed to loss of the helical structure. Alternately, the slight decrease in helix content may lead to shortening of the fibril segments with loss of an overall network structure. In either case, the thermal transition for the AFD36 gel lies well above physiological temperatures, which is promising for potential biomedical applications.
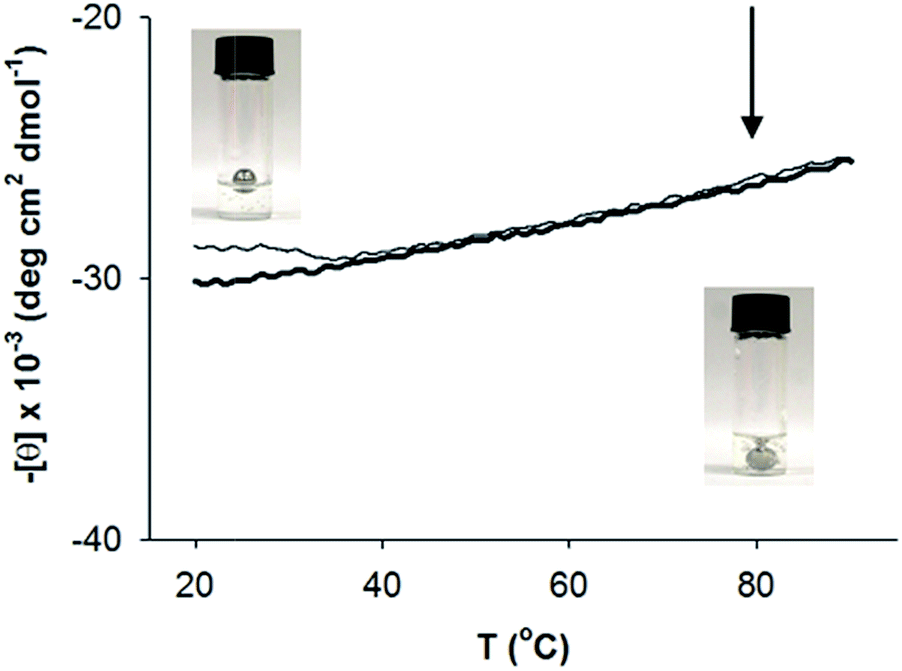 | ||
| Fig. 7 Thermal stability of secondary structure for 0.84 mM AFD36 in 10 mM MES pH 7.0. Thick line, heating curve; thin line, cooling curve. Inset: determination of the gel melting temperature via a ball drop test, with photos taken at 30 °C (upper left) and 80 °C (lower right). Arrow indicates Tm. The peptide concentration was 4.2 mM. | ||
Thermodynamics of coiled-coil assembly
Guanidinium chloride (G.HCl) denaturation studies on AFD36 and AFD19 were used to quantify the stability of these two peptide systems under weak gel-forming conditions (i.e., low concentration at pH 5.0 for AFD19 and pH 6.0 for AFD36). Details of these experiments are in the ESI.† Thermodynamic parameters for peptide self-assembly were determined by fitting helical content (measured by ECD; Fig. S1†) to monomer-oligomer models. These data showed both AFD36 and AFD19 to have extremely high thermodynamic stabilities of 9.3 and 9.5 k cal mol−1 per monomer at 20 °C for a hexamer model, respectively. Furthermore, variations in the assumed oligomerization state from n = 5–12, used in the thermodynamic modelling, resulted in only small changes in calculated stability, from 9.1 to 9.5 k cal mol−1 per monomer for AFD36 (Table S1†). This indicated calculated stability values to be relatively insensitive to assumed oligomerization state. While this means such analysis is unable to explicitly determine coiled–coiled oligomerization state, even in the absence of such knowledge calculated stability values may be compared to previous published findings. These values are larger than reported for comparable systems,41 likely due to high average helix propensity. In addition, the expansion of the hydrophobic core to include positions e and g is a key factor in the designs of AFD19 and AFD36 and likely contributes to the high thermodynamic stability by increasing the hydrophobic driving force for assembly. This is further supported by the increased stability of 10.6 k cal mol−1 per monomer of AFD36 when G.HCl denaturation was conducted at 50 °C (Table S1†), where such hydrophobic forces may be enhanced.Gel rheology
Rheology was used to confirm the formation of robust gels using sodium bicarbonate titration. Furthermore, it has been shown that cell growth and function are impacted by the mechanical properties of the underlying substrate.71–73 The storage (G′) and loss (G″) moduli of a gelling peptide solution were recorded using oscillatory shear rheology as the pH rose with loss of CO2 from the sample following addition of bicarbonate, allowing observation of the rate of network formation (Fig. 8A). A viscoelastic solid (G′ > G″) was formed in the time taken to mix and load the sample and structure development then followed approximately first order kinetics. After 60 minutes the value of G′ was 350 Pa, indicating mechanical properties similar to soft tissues such as brain and lymph node.74 Few hydrogels based on coiled coils have been reported and only a small number have been investigated using rheology.16 Comparison of AFD36 gels to published β-sheet peptide systems show comparable properties at similar or lower weight concentrations.75,76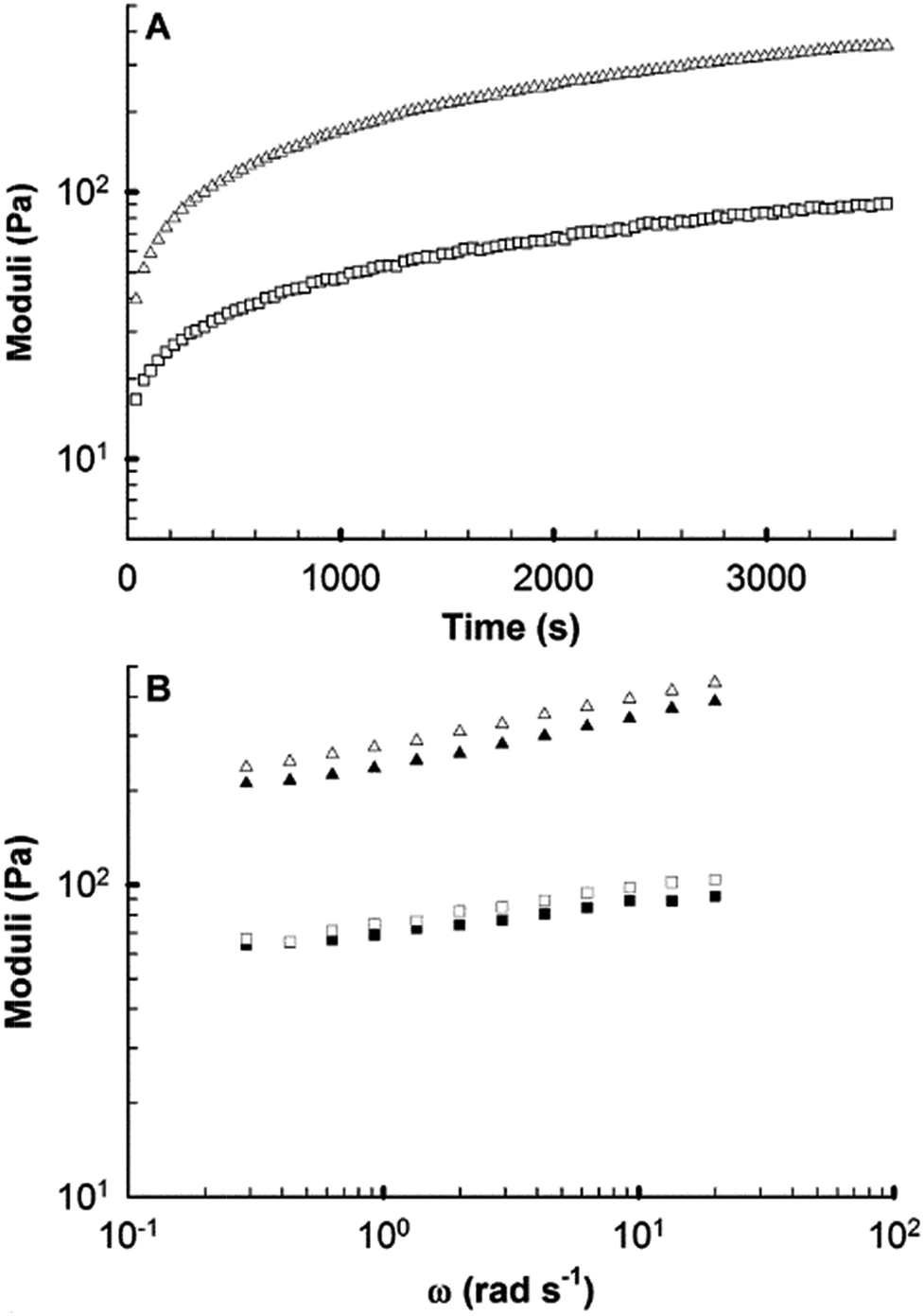 | ||
| Fig. 8 Rheological characterization of a 0.35% (w/v) AFD36 hydrogel (A) over time during gelation (x axis is time from measurement initialization) and (B) as a function of frequency (two samples). In both plots G′; triangles, G″; squares. | ||
After one hour, the formed gel showed moderate frequency dependence of G′ and G″ (Fig. 8B). However, no cross-over point was observed, with G′ being greater than G″ by at least at order of magnitude at all frequencies tested. At the completion of testing, visual observations of pH-dependent color change showed the peptide sample to be lower than physiological pH, likely due to limitations to the loss of CO2 at the plate edge. The results obtained are thus likely to represent a low-end estimate of the rate of gelation and the final gel strength.
Mammalian cell culture on peptide gels
As a proof of concept to assess the ability of AFD36 hydrogels to support mammalian cell growth, NIH/3T3 cells were cultured for 4 days on either tissue culture polystyrene (TCPS) or AFD36 gels pre-equilibrated with serum-free growth medium. Cell growth was quantified by AlamarBlue assay at each day, with cell images recorded at day 3 (Fig. 9). The fibroblasts proliferated well on AFD36 hydrogels, with cell growth at 68% of TCPS levels at day 4, a level of growth similar to that seen with β-hairpin peptide gels.77,78 The fibroblasts adopted spread morphologies on AFD36 gels similar to the TCPS surface. Adhesion and spreading may be due to non-specific protein adsorption from serum onto the surface of the hydrogels, providing cellular attachment points. The lack of toxicity and observation of cellular proliferation suggest that the peptide gels have potential as biomaterials for tissue engineering applications. | ||
| Fig. 9 Growth of 3T3 fibroblasts on AFD36 gels and TCPS controls. Representative images of cells cultured on (A) TCPS control surface or (B) AFD36 hydrogel at 3 days (scale bar 100 µm). (C) Cell growth measured via AlamarBlue fluorescence assay on TCPS (black bars) and AFD36 hydrogels (grey bars) at 1–4 days ± SD (n = 3). *p values ≤ 0.05, relative to the control, as determined by Student's t-test. | ||
Conclusions
In the present work, we show that simple design algorithms yield a short α-helical peptide that forms responsive fibrils and hydrogels at physiological pH and salt. The generality of the fibril design approach reported here has not yet been fully tested, but it is clearly broad. Suitable thermodynamic analysis may be able to distinguish between sequences that will or will not support fibril formation, and we are currently developing a predictive model to this end.Low-weight gels were formed uniformly by utilizing a simple methodology of transient buffering by carbonic acid to obtain physiological pH. The coiled-coil state was confirmed by ECD, with fibril formation investigated via SAXS and AFM. Nanofibrils were observed possessing diameters consistent with a cross-section of 5–7 monomers per coiled-coil, with a low persistence length and high aspect ratio. The resulting gels are highly thermostable, with melting temperatures exceeding 80 °C as shown by ECD and ball drop tests. Characterization of the peptide hydrogel's mechanical properties via rheology show a weak viscoelastic solid comparable with soft tissues. Fibroblast cell culture showed spread morphologies with some cellular proliferation, providing a proof of principle for the capability of these peptide hydrogels for use as a mammalian cell growth platform. The peptide hydrogels offer a potential pathway for drug and/or cell delivery in a biocompatible matrix, in addition to providing insights into protein aggregation, which in turn may provide for greater understanding of related disease states.
Acknowledgements
This research was facilitated by access to the Australian Proteome Analysis Facility and the Queensland node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and micro-fabrication facilities for Australia's researchers. This work was carried out in part in the Centre for Microscopy and Microanalysis at The University of Queensland, the Queensland node of the Australian Microscopy and Microanalysis Research Facility (AMMRF). Financial support was provided by the Australian Research Council (Grant IDs FT0991283, DP0987407 and DP150100268).References
- A. M. Jonker, D. W. P. M. Lowik and J. C. M. van Hest, Chem. Mater., 2012, 24, 759–773 CrossRef CAS
.
- Y. L. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef
- T. Delair, Carbohydr. Polym., 2012, 87, 1013–1019 CrossRef CAS
- D. Macaya and M. Spector, Biomed. Mater., 2012, 7, 22 Search PubMed
- E. R. Aurand, K. J. Lampe and K. B. Bjugstad, Neurosci. Res., 2012, 72, 199–213 CrossRef CAS PubMed
- E. Lallana, F. Fernandez-Trillo, A. Sousa-Herves, R. Riguera and E. Fernandez-Megia, Pharm. Res., 2012, 29, 902–921 CrossRef CAS PubMed
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS PubMed
- B. Balakrishnan, M. Mohanty, P. R. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS PubMed
- W. K. Loke, S. K. Lau, L. L. Yong, E. Khor and C. K. Sum, J. Biomed. Mater. Res., 2000, 53, 8–17 CrossRef CAS PubMed
- F. Yang, C. G. Williams, D. A. Wang, H. Lee, P. N. Manson and J. Elisseeff, Biomaterials, 2005, 26, 5991–5998 CrossRef CAS PubMed
- N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward and J. Zhang, Annu. Rev. Biomed. Eng., 2000, 2, 9–29 CrossRef CAS PubMed
- A. N. Buxton, J. Zhu, R. Marchant, J. L. West, J. U. Yoo and B. Johnstone, Tissue Eng., 2007, 13, 2549–2560 CrossRef CAS PubMed
- Y. Q. Tang, C. L. Heaysman, S. Willis and A. L. Lewis, Expert Opin. Drug Delivery, 2011, 8, 1141–1159 CrossRef CAS PubMed
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS PubMed
- A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS PubMed
- A. Aggeli, M. Bell, L. M. Carrick, C. W. G. Fishwick, R. Harding, P. J. Mawer, S. E. Radford, A. E. Strong and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619–9628 CrossRef CAS PubMed
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC
- K. Chockalingam, M. Blenner and S. Banta, Protein Eng., Des. Sel., 2007, 20, 155–161 CrossRef CAS PubMed
- C. Y. Xu, V. Breedveld and J. Kopecek, Biomacromolecules, 2005, 6, 1739–1749 CrossRef CAS PubMed
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed
- S. G. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS
- M. R. Caplan, P. N. Moore, S. G. Zhang, R. D. Kamm and D. A. Lauffenburger, Biomacromolecules, 2000, 1, 627–631 CrossRef CAS PubMed
- R. G. Ellis-Behnke, Y. X. Liang, S. W. You, D. K. C. Tay, S. G. Zhang, K. F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054–5059 CrossRef CAS PubMed
- D. A. Narmoneva, O. Oni, A. L. Sieminski, S. G. Zhang, J. P. Gertler, R. D. Kamm and R. T. Lee, Biomaterials, 2005, 26, 4837–4846 CrossRef CAS PubMed
- J. J. Panda and V. S. Chauhan, Polym. Chem., 2014, 5, 4431 RSC
- C. Vepari and D. L. Kaplan, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef CAS PubMed
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed
- F. E. Cohen and S. B. Prusiner, Annu. Rev. Biochem., 1998, 67, 793–819 CrossRef CAS PubMed
- M. E. Oskarsson, J. F. Paulsson, S. W. Schultz, M. Ingelsson, P. Westermark and G. T. Westermark, Am. J. Pathol., 2015, 185, 834–846 CrossRef CAS PubMed
- K. Hartman, J. R. Brender, K. Monde, A. Ono, M. L. Evans, N. Popovych, M. R. Chapman and A. Ramamoorthy, PeerJ, 2013, 1, e5 Search PubMed
- P. Westermark, K. Lundmark and G. T. Westermark, PLoS One, 2009, 4, e6041 Search PubMed
- D. N. Woolfson, Biopolymers, 2010, 94, 118–127 CrossRef CAS PubMed
- N. L. Fletcher, C. V. Lockett and A. F. Dexter, Soft Matter, 2011, 7, 10210–10218 RSC
- T. N. Melnik, V. Villard, V. Vasiliev, G. Corradin, A. V. Kajava and S. A. Potekhin, Protein Eng., 2003, 16, 1125–1130 CrossRef CAS PubMed
- T. Takei, K. Hasegawa, K. Imada, K. Namba, K. Tsumoto, Y. Kuriki, M. Yoshino, K. Yazaki, S. Kojima, T. Takei, T. Ueda and K.-I. Miura, Biochemistry, 2013, 52, 2810–2820 CrossRef CAS PubMed
- A. F. Dexter, Langmuir, 2010, 26, 17997–18007 CrossRef CAS PubMed
- C. A. Dreiss, K. S. Jack and A. P. Parker, J. Appl. Crystallogr., 2006, 39, 32–38 CrossRef CAS
- R. Fairman, H. G. Chao, L. Mueller, T. B. Lavoie, L. Y. Shen, J. Novotny and G. R. Matsueda, Protein Sci., 1995, 4, 1457–1469 CrossRef CAS PubMed
- J. K. Myers, C. N. Pace and J. M. Scholtz, Protein Sci., 1995, 4, 2138–2148 CrossRef CAS PubMed
- J. M. Scholtz, H. Qian, E. J. York, J. M. Stewart and R. L. Baldwin, Biopolymers, 1991, 31, 1463–1470 CrossRef CAS PubMed
- P. J. Gans, P. C. Lyu, M. C. Manning, R. W. Woody and N. R. Kallenbach, Biopolymers, 1991, 31, 1605–1614 CrossRef CAS PubMed
- D. N. Woolfson, G. J. Bartlett, M. Bruning and A. R. Thomson, Curr. Opin. Struct. Biol., 2012, 22, 432–441 CrossRef CAS PubMed
- D. A. D. Parry, R. D. B. Fraser and J. M. Squire, J. Struct. Biol., 2008, 163, 258–269 CrossRef CAS PubMed
- J. Liu, Q. Zheng, Y. Q. Deng, C. S. Cheng, N. R. Kallenbach and M. Lu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15457–15462 CrossRef CAS PubMed
- S. A. Potekhin, T. N. Melnik, V. Popov, N. F. Lanina, A. A. Vazina, P. Rigler, A. S. Verdini, G. Corradin and A. V. Kajava, Chem. Biol., 2001, 8, 1025–1032 CrossRef CAS PubMed
- N. R. Zaccai, B. Chi, A. R. Thomson, A. L. Boyle, G. J. Bartlett, M. Bruning, N. Linden, R. B. Sessions, P. J. Booth, R. L. Brady and D. N. Woolfson, Nat. Chem. Biol., 2011, 7, 935–941 CrossRef CAS PubMed
- C. F. Xu, R. Liu, A. K. Mehta, R. C. Guerrero-Ferreira, E. R. Wright, S. Dunin-Horkawicz, K. Morris, L. C. Serpell, X. B. Zuo, J. S. Wall and V. P. Conticello, J. Am. Chem. Soc., 2013, 135, 15565–15578 CrossRef CAS PubMed
- J. Liu, Q. Zheng, Y. Q. Deng, N. R. Kallenbach and M. Lu, J. Mol. Biol., 2006, 361, 168–179 CrossRef CAS PubMed
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CAS
- B. Apostolovic, M. Danial and H. A. Klok, Chem. Soc. Rev., 2010, 39, 3541–3575 RSC
- C. Vu, J. Robblee, K. M. Werner and R. Fairman, Protein Sci., 2001, 10, 631–637 CrossRef CAS PubMed
- M. J. Pandya, G. M. Spooner, M. Sunde, J. R. Thorpe, A. Rodger and D. N. Woolfson, Biochemistry, 2000, 39, 8728–8734 CrossRef CAS PubMed
- D. Papapostolou, A. M. Smith, E. D. T. Atkins, S. J. Oliver, M. G. Ryadnov, L. C. Serpell and D. N. Woolfson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10853–10858 CrossRef CAS PubMed
- M. G. Ryadnov and D. N. Woolfson, Nat. Mater., 2003, 2, 329–332 CrossRef CAS PubMed
- Y. Zimenkov, V. P. Conticello, L. Guo and P. Thiyagarajan, Tetrahedron, 2004, 60, 7237–7246 CrossRef CAS
- Y. Zimenkov, S. N. Dublin, R. Ni, R. S. Tu, V. Breedveld, R. P. Apkarian and V. P. Conticello, J. Am. Chem. Soc., 2006, 128, 6770–6771 CrossRef CAS PubMed
- S. Kojima, Y. Kuriki, T. Yoshida, K. Yazaki and K. Miura, Proc. Jpn. Acad., Ser. B, 1997, 73, 7–11 CrossRef
- H. Dong, S. E. Paramonov and J. D. Hartgerink, J. Am. Chem. Soc., 2008, 130, 13691–13695 CrossRef CAS PubMed
- S. Y. M. Lau, A. K. Taneja and R. S. Hodges, J. Biol. Chem., 1984, 259, 3253–3261 Search PubMed
- N. E. Zhou, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1992, 267, 2664–2670 CAS
- B. Heitmann, G. E. Job, R. J. Kennedy, S. M. Walker and D. S. Kemp, J. Am. Chem. Soc., 2005, 127, 1690–1704 CrossRef CAS PubMed
- A. R. Kirby, V. J. Morris and A. Patrick Gunning, in Atomic Force Microscopy for Biologists, Imperial College Press, 2nd edn, 2012 Search PubMed
- S. J. Opella, F. M. Marassi, J. J. Gesell, A. P. Valente, Y. Kim, M. Oblatt-Montal and M. Montal, Nat. Struct. Biol., 1999, 6, 374–379 CrossRef CAS PubMed
- V. N. Malashkevich, R. A. Kammerer, V. P. Efimov, T. Schulthess and J. Engel, Science, 1996, 274, 761–765 CrossRef CAS PubMed
- Y. Lu, B. Weers and N. C. Stellwagen, Biopolymers, 2002, 61, 261–275 CrossRef CAS PubMed
- C. C. vandenAkker, M. F. M. Engel, K. P. Velikov, M. Bonn and G. H. Koenderink, J. Am. Chem. Soc., 2011, 133, 18030–18033 CrossRef CAS PubMed
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS PubMed
- S. R. Peyton, C. M. Ghajar, C. B. Khatiwala and A. J. Putnam, Cell Biochem. Biophys., 2007, 47, 300–320 CrossRef CAS PubMed
- M. D. Stevenson, H. Piristine, N. J. Hogrebe, T. M. Nocera, M. W. Boehm, R. K. Reen, K. W. Koelling, G. Agarwal, A. L. Sarang-Sieminski and K. J. Gooch, Acta Biomater., 2013, 9, 7651–7661 CrossRef CAS PubMed
- A. S. Rowlands, P. A. George and J. J. Cooper-White, Am. J. Physiol.: Cell Physiol., 2008, 295, C1037–C1044 CrossRef CAS PubMed
- J. H. Collier, J. S. Rudra, J. Z. Gasiorowski and J. P. Jung, Chem. Soc. Rev., 2010, 39, 3413–3424 RSC
- B. Ozbas, K. Rajagopal, J. P. Schneider and D. J. Pochan, Phys. Rev. Lett., 2004, 93, 268106 CrossRef PubMed
- C. Cunha, S. Panseri, O. Villa, D. Silva and F. Gelain, Int. J. Nanomed., 2011, 6, 943–955 CrossRef CAS PubMed
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS PubMed
- J. K. Kretsinger, L. A. Haines, B. Ozbas, D. J. Pochan and J. P. Schneider, Biomaterials, 2005, 26, 5177–5186 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Thermodynamics of coiled-coil assembly. See DOI: 10.1039/c7ra02811c |
| ‡ Present address: The University of Queensland, School of Agriculture and Food Science, Brisbane QLD 4072, Australia. |
| § Present address: The University of Queensland, Centre for Advanced Imaging, Australian Institute for Bioengineering and Nanotechnology and the ARC Centre of Excellence in Convergent BioNano Science and Technology, Brisbane QLD 4072, Australia. |
| ¶ Present address: Department of Agriculture and Fisheries, Brisbane QLD 4102, Australia. |
| This journal is © The Royal Society of Chemistry 2017 |