DOI:
10.1039/C7RA02720F
(Paper)
RSC Adv., 2017,
7, 29103-29111
Synthesis and antiproliferative activity of 2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazol derivatives as microtubule-destabilizing agents†
Received
6th March 2017
, Accepted 9th May 2017
First published on 5th June 2017
Abstract
A series of 2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazols were designed as analogs of substituted methoxybenzoyl-aryl-thiazole (SMART) under the consideration of geometric features. The target compounds were synthesized via concise and efficient processes including microwave-assisted cyclization, and were evaluated for their antiproliferative activity against three human cancer cell lines. Most compounds exhibited moderate antiproliferative activity with IC50 values in the micromolar to sub-micromolar range. Tubulin polymerization and immunofluorescence studies demonstrated that (Z)-9a was a potent microtubule-destabilizing agent and disrupted the polymerization dynamics. Moreover, (Z)-9a significantly induced accumulation of cells in the G2/M phase and caused microtubule destabilization. Molecular modeling studies showed that (Z)-9a probably binds to the colchicine site of tubulin.
Introduction
Microtubules, a vital part of the cytoskeleton, play a crucial role in mitosis.1 Disturbing microtubule arrangement and rearrangement, a dynamic equilibrium process, has been proved to be useful in the development of anticancer therapeutics.2 Microtubule targeted agents can be classified into two major types: (1) microtubule-stabilizing agents, including taxanes and epothilones that stimulate tubulin polymerization; (2) microtubule-destabilizing agents, including vinca alkaloids and colchicine (1, Fig. 1) that inhibit tubulin polymerization.3
 |
| | Fig. 1 Structures of reported CBSIs and designed new CBSIs. | |
Given the success of the taxanes and vinca alkaloids,4 which have established tubulin as a valid target in cancer therapy, research efforts have been focusing on the development of targeted agents to the colchicine site of microtubules, known as colchicine binding site inhibitors (CBSIs).5 During the past decades, a large number of CBSIs have been reported,6 such as combretastatin A-4 (CA-4 2, Fig. 1), 4-substituted methoxybenzoyl-aryl-thiazole (SMART 3, Fig. 1) and 2-aryl-4-benzoyl-imidazole (ABI 4, Fig. 1).7–9 SMART, a basic three ring (A, B and C) scaffold based on the ring geometry, demonstrates potent antiproliferative activity. Moreover, a range of further structural diversification molecules derived from the three rings has been developed to affect tubulin polymerization.10 In our previous study, a set of SMART analogs (5, Fig. 1) with the oxadiazole moiety as B-ring were reported to exhibit promising antiproliferative activities.11
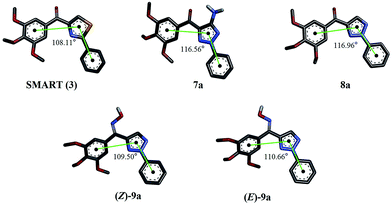 |
| | Fig. 2 Optimized structures of SMART and the target compounds 7a, 8a, (Z)-9a and (E)-9a. Black points represent the geometric centers of A, B and C rings, respectively. | |
1,2,3-Triazole has gained enormous interest due to their broad spectrum of pharmaceutical and therapeutic applications.12,13 Furthermore, this heterocycle has a high dipole moment and is capable for hydrogen bonding, which could be favorable in the binding to biomolecular targets.14 A 1,2,3-triazol-based CA-4 analog (6, Fig. 1) was found to display potent antiproliferative and antitubulin activities.15
In this study, we envisaged an optimization effort around the bioisosteric B ring of SMART by replacing the thiazole ring with triazole. We hypothesized that the degree of the angle between the geometric centers of A, B and C rings (∠ABC) is important for the activity of SMART and its derivatives (Fig. 2). Thus, to investigate the similarity between the designed compounds and SMART, density functional theory (DFT) calculation was applied in our case for the molecule design.16–18 Calculational results showed the similar values of ∠ABC for (Z)-9a and SMART, which were 109.50° and 108.11°, respectively. Therefore, a series of 2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazole derivatives (7–9) were designed and the effects of substitutions on the C5 position of the triazole ring and changing the carbonyl linker into oxime were evaluated.
Results and discussion
Chemistry
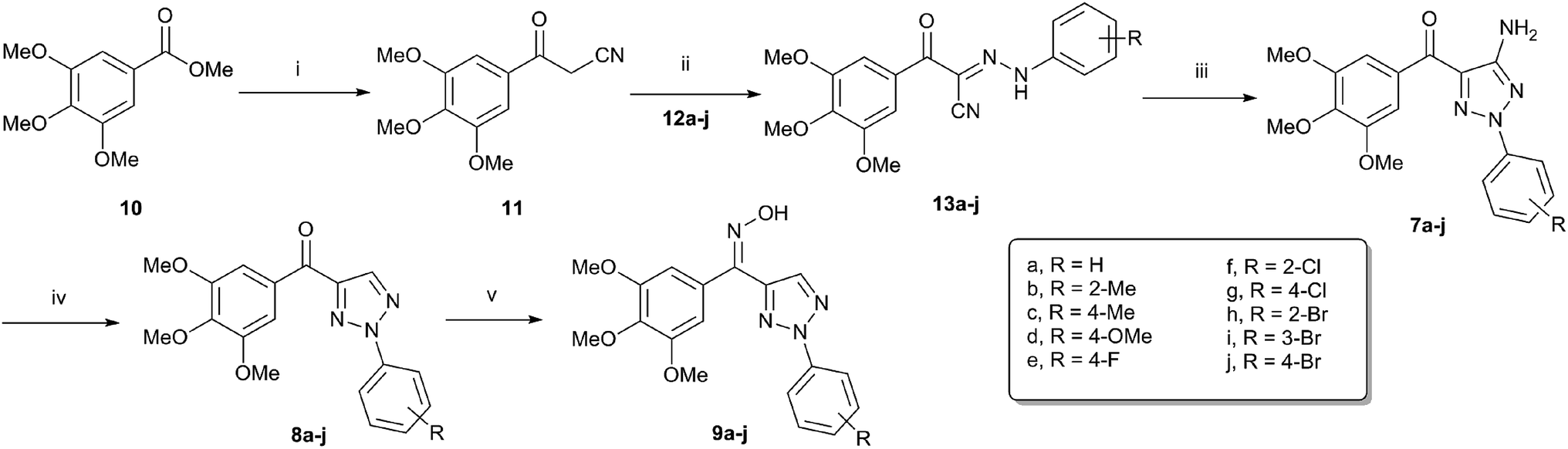
The synthetic routes for 2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazol derivatives (7–9) are outlined in Scheme 1. The starting material methyl 3,4,5-trimethoxybenzoate (10) was converted into the 3-oxo-3-(3,4,5-trimethoxyphenyl)propanenitrile (11) by using sodium hydride and acetonitrile in THF.19 Coupling the diazonium salt of different aromatic amines (12a–j) with compound 11 afforded the corresponding hydrazone derivatives 13a–j.20
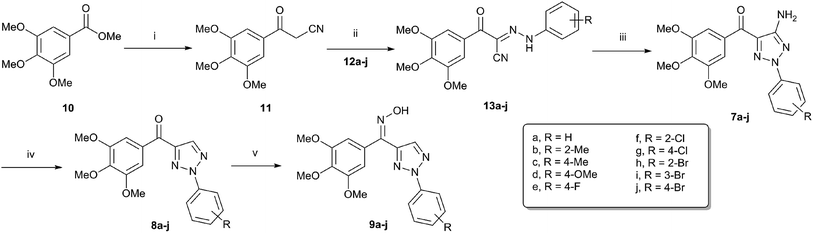 |
| | Scheme 1 Reagents and conditions: (i) CH3CN, NaH (70%), THF, reflux, 3 h; (ii) NaOAc, EtOH/H2O, 0 °C, 1 h; (iii) NH2OH·HCl, CH3COONa, DMF, 160 °C, MW, 7 min. (iv) Isopropyl nitrite, THF, reflux, 1 h; (v) NH2OH·HCl, CH3COONa, EtOH, reflux, 2 h. | |
Under conventional heating conduction, the reaction of 13a with hydroxylamine hydrochloride in the presence of sodium acetate to generate 7a suffered from long reaction time and low yields (entries 1–5).21 As a useful tool in organic synthesis, microwave irradiation could generally accelerate the reaction rate and improve the product's yield.22–24 When microwave irradiation was incorporated into synthesis of 7a, we found that the reaction rate was greatly improved. We systematically screened the influence of reaction temperature and time on the yield of 7a under microwave conditions. As shown in Table 1, the optimized condition was confirmed to be microwave irradiation at 160 °C for 7 min (entry 9). Accordingly, all of the cyclized products 7a–j were obtained smoothly with good to excellent yields 81–93%.
Table 1 The optimization of conditions for the preparation of compound 7a
| Entry |
Heating |
Temp (oC) |
Time (min) |
Yielda (%) |
| Isolated yield. MW: microwave. |
| 1 |
Oil bath |
110 |
30 min |
32 |
| 2 |
Oil bath |
110 |
60 min |
52 |
| 3 |
Oil bath |
110 |
120 min |
65 |
| 4 |
Oil bath |
110 |
240 min |
75 |
| 5 |
Oil bath |
110 |
360 min |
82 |
| 6 |
MWb |
110 |
5 min |
74 |
| 7 |
MW |
130 |
5 min |
81 |
| 8 |
MW |
160 |
5 min |
86 |
| 9 |
MW |
160 |
7 min |
93 |
| 10 |
MW |
160 |
9 min |
93 |
Subsequently, the amino group was removed using isoamyl nitrite in anhydrous THF under reflux conditions to yield the corresponding compounds 8a–j,25 which were then converted into oxime isomers (9a–j) under excess amount of hydroxylamine hydrochloride and sodium acetate by refluxing in anhydrous ethanol for 2 h.26
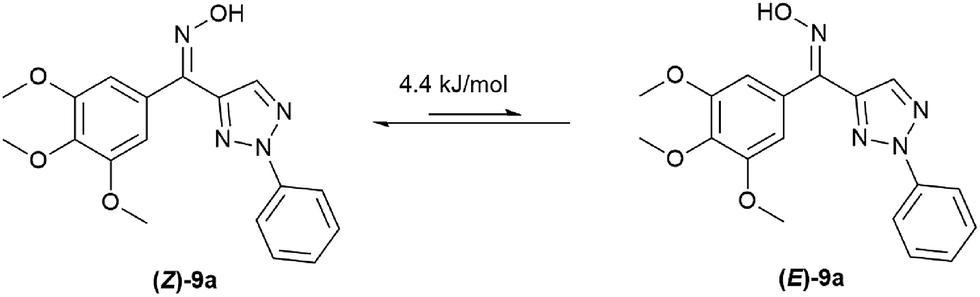
It is worth to note that there are two configurations for compounds 9a–9j (Z and E) and the isomer mixtures were evaluated without separation in our study due to the Z/E tautomerism even at room temperature. Generally, the ratio of two isomers were obtained by HPLC analysis. In addition, DFT calculation of 9a showed that Z-isomer should be the predominant one in the mixture (Fig. 3).
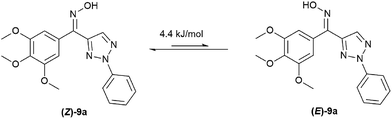 |
| | Fig. 3 Calculation of (Z/E)-9a tautomerismby DFT. | |
Biological evaluation
In vitro antiproliferative activity. The synthesized compounds (7–9) were investigated for their ability to inhibit cancer cells proliferation by the MTT method using three human carcinoma cell lines (gastric adenocarcinoma SGC-7901 cells, lung adenocarcinoma A549 cells and fibrosarcoma HT-1080 cells). The results were compared with SMART (3) and ABI (4) as the positive controls (Table 2). Of the enlisted compounds, almost all of the target compounds displayed moderate antiproliferative activity against different cancer cells with IC50 values in the micromolar to sub-micromolar range, which indicated the potential of the triazole ring. Among them, (Z)-9a showed the best antiproliferative activity against the SGC-7901 cell lines with IC50 value of 0.12 ± 0.02 μM, which was less cytotoxic than SMART (IC50 = 0.019 ± 0.008 μM) but higher cytotoxic than ABI (IC50 = 0.81 ± 0.08 μM). Compounds 7a–j possessing an amino group at the C5 position of B ring were found to be more potent for A549 and HT-1080 cell lines, compared with the corresponding C5 unsubstituted compounds 8a–j. Interestingly, the oxime linkage compounds 9a–j were slightly better than carbonyl linkage compounds 8a–j, which was in accorded with the calculational result that value of ∠ABC in 9a was closer to SMART than that of 7a and 8a. Furthermore, substitutions on C ring of the tested compounds (7h–j, 8h–j, 9h–j) exhibited an order of potency being para- > meta- > ortho-substituted in generally, and the types of substituted groups had no significant influence on antiproliferative activity.
Table 2 In vitro antiproliferative activities of the target compounds (7–9) on three human carcinoma cell lines
| Compound |
IC50 (μM) ± SDa |
| SGC-7901 |
A549 |
HT-1080 |
| IC50: concentration of the compound (μM) producing 50% cell growth inhibition after 72 h of drug exposure, as determined by the MTT assay. Each experiment was carried out in triplicate. Used as a positive control. Used as mixture without separation. |
| 7a |
0.35 ± 0.02 |
0.28 ± 0.04 |
0.70 ± 0.03 |
| 7b |
5.6 ± 0.4 |
8.1 ± 0.9 |
9.3 ± 1.1 |
| 7c |
3.3 ± 0.2 |
0.26 ± 0.02 |
0.41 ± 0.05 |
| 7d |
1.3 ± 0.1 |
3.4 ± 0.4 |
0.38 ± 0.04 |
| 7e |
1.8 ± 0.07 |
2.8 ± 0.2 |
0.28 ± 0.02 |
| 7f |
9.3 ± 0.8 |
5.3 ± 0.4 |
1.5 ± 0.1 |
| 7g |
0.70 ± 0.05 |
1.6 ± 0.1 |
0.24 ± 0.01 |
| 7h |
16.2 ± 1.5 |
10.1 ± 1.2 |
3.2 ± 0.2 |
| 7i |
0.48 ± 0.02 |
1.8 ± 0.2 |
2.4 ± 0.2 |
| 7j |
0.35 ± 0.04 |
0.58 ± 0.06 |
0.14 ± 0.02 |
| 8a |
0.87 ± 0.09 |
7.1 ± 0.6 |
5.2 ± 0.8 |
| 8b |
3.4 ± 0.4 |
8.6 ± 0.6 |
11.4 ± 1.2 |
| 8c |
1.5 ± 0.2 |
7.8 ± 0.7 |
4.3 ± 0.2 |
| 8d |
1.51 ± 0.09 |
6.3 ± 0.5 |
7.9 ± 0.9 |
| 8e |
2.9 ± 0.3 |
6.7 ± 0.8 |
2.4 ± 0.3 |
| 8f |
6.1 ± 0.6 |
19.2 ± 3.1 |
35.8 ± 8.3 |
| 8g |
1.8 ± 0.2 |
3.7 ± 0.4 |
0.68 ± 0.8 |
| 8h |
3.0 ± 0.4 |
11.0 ± 1.0 |
9.8 ± 1.6 |
| 8i |
2.4 ± 0.2 |
10.4 ± 1.1 |
8.2 ± 0.7 |
| 8j |
0.92 ± 0.07 |
2.6 ± 0.2 |
1.8 ± 0.1 |
| (Z)-9a |
0.12 ± 0.02 |
0.52 ± 0.3 |
0.34 ± 0.4 |
| (E)-9a |
0.61 ± 0.05 |
1.23 ± 0.07 |
0.98 ± 0.1 |
| 9ac |
0.19 ± 0.04 |
0.71 ± 0.05 |
0.52 ± 0.06 |
| 9bc |
4.9 ± 0.3 |
7.2 ± 0.7 |
16 ± 1.4 |
| 9cc |
0.32 ± 0.04 |
2.4 ± 0.3 |
0.54 ± 0.05 |
| 9dc |
0.47 ± 0.06 |
0.31 ± 0.03 |
6.1 ± 0.7 |
| 9ec |
0.42 ± 0.04 |
2.1 ± 0.2 |
2.1 ± 0.3 |
| 9fc |
3.1 ± 0.2 |
6.8 ± 0.4 |
37.9 ± 6.2 |
| 9gc |
0.38 ± 0.06 |
1.6 ± 0.09 |
0.49 ± 0.02 |
| 9hc |
1.7 ± 0.1 |
12.1 ± 2.0 |
41.3 ± 6.4 |
| 9ic |
1.3 ± 0.2 |
10.2 ± 0.9 |
8.5 ± 0.7 |
| 9jc |
0.69 ± 0.05 |
0.85 ± 0.06 |
0.83 ± 0.06 |
| SMART (3)b |
0.019 ± 0.008 |
0.029 ± 0.009 |
0.028 ± 0.016 |
| ABI (4)b |
0.81 ± 0.08 |
0.98 ± 0.11 |
0.15 ± 0.05 |
In order to elucidate selectivity of compounds to cancer cell lines and non-cancer cell lines, we further investigated the effect of three compounds with high antiproliferative activity (7a, 7j, (Z)-9a) toward normal fibroblasts L929 cells. Results are presented in Table 3. The selectivity indexes SIA (for SGC-7901 cell line), SIB (for A549 cell line) and SIC (for HT-1080 cell line) were calculated. The higher selectivity index means higher therapeutic safety margin. (Z)-9a showed apparent selectivity to three cancer cell lines and normal fibroblasts L929 cells. Surprisingly, both 7a and 7j displayed very slight effect on L929 cells, which mean these compounds have a higher selectivity than (Z)-9a.
Table 3 In vitro antiproliferative activities of 7a, 7j and (Z)-9a on normal fibroblasts L929 cells
| Compound |
L929 |
SIAa |
SIBb |
SICc |
| SIA = IC50(L929)/IC50(SGC-7901). SIB = IC50(L929)/IC50(A549). SIC = IC50(L929)/IC50(HT-1080). |
| 7a |
>100 |
>285.7 |
>357.1 |
>142.9 |
| 7j |
45.9 ± 3.2 |
131.1 |
79.1 |
327.9 |
| (Z)-9a |
3.8 ± 0.5 |
31.7 |
7.3 |
11.2 |
Tubulin polymerization. With the goal of investigating the functionary mechanism on (Z)-9a, the effects of (Z)-9a on the tubulin polymerization were assessed in vitro. CA-4 (2), one of well-known CBSIs, and Taxol were applied as the positive and negative controls, respectively. (Z)-9a caused a concentration-dependent inhibition of tubulin assembling (Fig. 4). The inhibitory activity of (Z)-9a (IC50 = 5.02 μM) towards tubulin polymerization was slightly weaker than the positive control CA-4 (2). The tubulin data suggested that (Z)-9a exerted similar influence on microtubule depolymerization as CA-4.
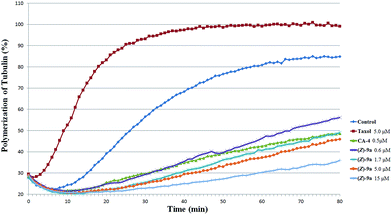 |
| | Fig. 4 Effects of (Z)-9a on tubulin polymerization. Tubulin had been pre-incubated for 1 min with (Z)-9a at 0.6 μM, 1.7 μM, 5 μM and 15 μM, CA-4 (2) at 0.5 μM, taxol at 5 μM or vehicle DMSO. | |
Immunofluorescence studies. To investigate the antiproliferative effect of (Z)-9a was derived from interactions with tubulins, SGC-7901 cancer cells were treated with (Z)-9a at 0.12 μM for 24 h and the cellular microtubule structures were visualized by indirect immunofluorescence using an anti-α-tubulin monoclonal antibody. Date in Fig. 5 indicated that compound (Z)-9a inhibited microtubule assembly and disrupted cytoskeleton similarly to CA-4 (2).
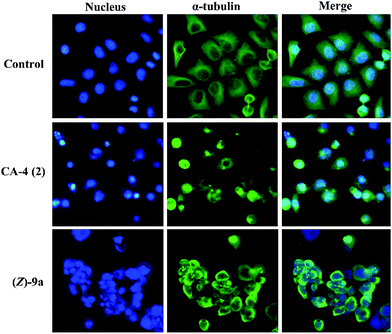 |
| | Fig. 5 (Z)-9a and CA-4 (2) induced depolymerization of the microtubule networks of SGC-7901 cancer cells. Untreated cells (control) and cells treated with CA-4 or (Z)-9a for 24 h, were fixed and stained with anti-α-tubulin-FITC specific antibodies followed by DAPI. Microtubules and unassembled tubulin are shown in green, and nuclei, which were stained with DAPI, are shown in blue. | |
Cell cycle analysis. The cell growth inhibitory potency of the tested compounds prompted us to evaluate their effects on the cell mitosis. For cell cycle progression analysis, the effect of the (Z)-9a at various concentrations on cell cycle progression was investigated in SGC-7901 cells via flow cytometry. As shown in Fig. 6, (Z)-9a arrested efficiently the cell cycle progression in G2/M phase, increasing the percentage of cells in G2/M phase in a concentration-dependent manner and time-dependent manner by 40–84% and 40–80%, respectively.
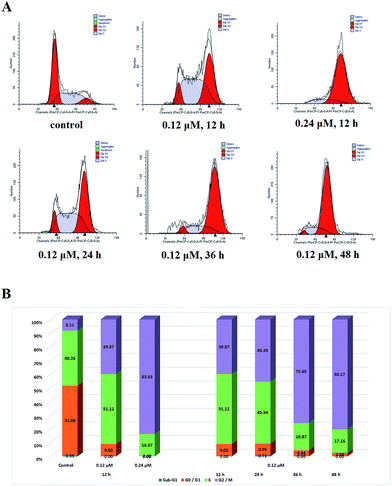 |
| | Fig. 6 Effects of (Z)-9a on cell mitosis. (A) Cell cycle distribution of SGC-7901 cancer cells treated with different concentrations of (Z)-9a. (B) The statistical graph of cell cycle distribution. | |
Molecular modeling study. To investigate the possible binding of target compounds to the colchicine site of tubulin, the Z-isomer of compound 9a, as a predominant one in the mixture, was investigated by using CDOCKER program in Discovery Studio 3.0 software.27 The docking obtained that the pose of (Z)-9a was tightly embedded into the interface of α/β tubulin subunits (Fig. 7). In the binding model, the binding orientations of (Z)-9a adopted a twisted geometry, which may be the key for its bioactivity. The two oxygen atoms of the 3,4,5-trimethoxy of (Z)-9a contributed to the hydrogen bonding interactions (O⋯HeN: 2.44 Å) with the sulfhydryl group of β-CYS 241, which were consistent with other colchicine site agents reported by Massarotti et al.28 The docking study was a beneficial complement and explanation to the above tubulin polymerization and immunofluorescence studies.
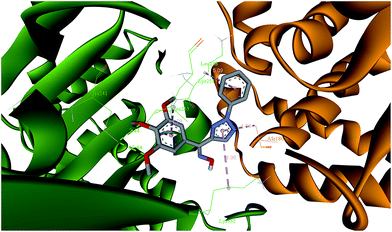 |
| | Fig. 7 The binging mode between the (Z)-9a and the α/β tubulin subunits. Location of (Z)-9a at the interface of α/β tubulin subunits (α-tubulin, yellow; β-tubulin, green). Binding mode between (Z)-9a and amino acid residues near colchicine site. The green lines represent hydrogen bonds. The pink lines represent hydrophobic interaction. | |
Conclusion
A series of 2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazols characterized by a carbonyl/oxime linker, combined with/without amino substituent on the C5 position of the triazole ring, were developed under the strategy of the bioisosteric replacement of B ring of SMART. The target compounds displayed moderate antiproliferative activity against three human carcinoma cell lines. Among them, (Z)-9a exhibited best antiproliferative activity against the SGC-7901 cell lines with IC50 value of 0.12 ± 0.02 μM. Mechanism of action studies confirmed that (Z)-9a was a microtubule-destabilizing agent that induced the accumulation of cells in the G2/M phase and caused cell apoptosis. Structure–activity relationship between 7, 8 and 9 indicated that ∠ABC might be a considerable factor for design of SMART analogs. (Z)-9a was discovered as the most potent compound and further structural optimization and activity test will be reported in the future.
Experimental section
Chemistry
All reagents were commercially available and were used without further purification. The silica gel plate (HSGF-254) and silica gel (H, 200–300 mesh) from Yantai Jiangyou silicone Development Co., Ltd. was used for preparative TLC and column chromatography, respectively. Visualization was made with UV light (254 nm and 365 nm). Melting points were tested with Micro-Melting point apparatus (X-4, Shanghai jing science instrument Co., Ltd.) and were uncorrected. Mass spectra (MS) were obtained from Agilent Co. Ltd. on an Agilent 1100-sl mass spectrometer with an electrospray ionization source. 1H-NMR and 13C-NMR spectra were measured in CDCl3 or d6-DMSO with TMS as the internal reference on a Bruker AVANCE spectrometer operating at 400 MHz or 600 MHz (1H at 400 or 600 MHz, 13C at 100 MHz). HPLC (HPLC-LC-20AT, Shimadzu) using SPD-20A UV as the detector, C18 (4.6 × 250 mm, 0.45 μm) as HPLC analysis column. Method A: gradient: 60% methanol in water as mobile elution, flow rate: 1.0 mL min−1; method B: gradient: 80% methanol in water as mobile elution, flow rate: 1.0 mL min−1. The microwave reactions were carried out in a single mode cavity microwave synthesizer (CEM Corporation, NC, USA).
General synthetic procedures for 3-oxo-3-(3,4,5-trimethoxyphenyl)propanenitrile (11)
Mixed methyl 3,4,5-trimethoxybenzoate (5.34 g, 23.6 mmol) with NaH (47.2 mmol, 1.62 g) in boiling tetrahydrofuran (20 mL) and then followed by dropwise addition of solution of acetonitrile (23.6 mmol, 0.98 g, 1.4 mL) in tetrahydrofuran (1 mL). After refluxing for 3 h the reaction was entirely finished (TLC control). Then the mixture was cooled to room temperature and diluted by water. The precipitated sodium salt was filtered and washed with diethylether. After the evaporation of the solvent, HCl (1 mol L−1) was added to acidize the compound to pH 2 and the solid was obtained, giving pale yellow solid, yield 84%. The crude 11 was utilized directly for the upcoming step without any extra purification.
General synthetic procedures for 3-oxo-3-(3,4,5-trimethoxyphenyl)-2-(phenylhydrazono) propanenitrile (13a–j)
To a solution of the arylamine was added hydrochloric acid (5.8 mmol in 4 mL, 6 M HCl) at 0 °C, a solution of sodium nitrite (6.4 mmol, 0.44 g dissolved in 2 mL water) was added dropwise over a period of 20 min, and stirring at 0 °C for 30 min to form the corresponding aryldiazonium salt (12a–j). Then the resulting solution of aryldiazonium salt was dropwise added to a solution of 3-oxo-3-(3,4,5-trimethoxyphenyl)propanenitrile (1.5 g, 6.4 mmol) in ethanol mixture (10 mL) involving sodium acetate trihydrate (0.95 g, 11.6 mmol) at 0 °C. Then the mixture was stirred at room temperature for 1 h and the resulting solid was collected by filtration, washed with water and ethanol. The crude 13a–j was utilized directly for the upcoming step without any extra purification.
General synthetic procedures for 5-amino-2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazol (7a–j)
Independent mixtures of 13a–j (1 mmol) containing hydroxylamine hydrochloride (0.35 g, 5 mmol) and anhydrous sodium acetate (0.41 g, 5 mmol) in DMF (5 mL) were stirred at irradiated in a microwave reactor for 7 min at 160 °C. The reaction mixtures were cooled to room temperature and poured into ice cold water. The formed solids were collected by filtration, washed with water. The residual crude product was purified by column chromatography on silica gel (200–300 mesh) with petroleum ether/AcOEt (v/v = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
5-Amino-2-phenyl-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7a). Compound 7a was obtained from 13a as a yellow solid (0.31 g, 93%); mp 166–170 °C; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 7.9 Hz, 2H), 7.83 (s, 2H), 7.49 (t, J = 7.6 Hz, 7.9 Hz, 2H), 7.37 (t, J = 7.6 Hz, 1H), 3.99 (s, 6H), 3.97 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.9, 152.9 (×2), 147.5, 143.1, 139.5, 138.8, 131.3, 129.5 (×2), 128.6, 119.2 (×2), 108.0 (×2), 61.0, 56.3 (×2) ppm; MS (ESI): [M + H]+ = 355.1, [M + Na]+ = 377.1.
5-Amino-2-(o-tolyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7b). Compound 7b was obtained from 13b as a yellow solid (0.32 g, 86%); mp 185–187 °C; 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 2H), 7.63 (d, J = 7.6 Hz, 1H), 7.36 (m, 3H), 3.94 (s, 3H), 3.92 (s, 6H), 2.48 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.6, 152.8 (×2), 149.5, 143.6, 142.6, 139.1, 132.2, 132.1, 131.8, 129.1, 126.7, 125.0, 107.9 (×2), 60.9, 56.2 (×2), 19.2 ppm; MS (ESI): [M + H]+ = 369.1, [M + Na]+ = 391.1.
5-Amino-2-(p-tolyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7c). Compound 7c was obtained from 13c as a yellow solid (0.33 g, 89%); mp 173–176 °C; 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.5 Hz, 2H), 7.83 (s, 2H), 7.28 (d, J = 8.5 Hz, 2H), 3.98 (s, 6H), 3.97 (s, 3H), 2.41 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.4, 156.5, 152.8 (×2), 142.4, 137.9, 137.2, 132.0, 131.3, 129.9 (×2), 118.5 (×2), 107.7 (×2), 61.0, 56.2 (×2), 21.0 ppm; MS (ESI): [M + H]+ = 369.1, [M + Na]+ = 391.1.
5-Amino-2-(4-methoxyphenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7d). Compound 7d was obtained from 13d as a yellow solid (0.31 g, 81%); mp 201–203 °C; 1H NMR (400 MHz, CDCl3) δ 7.94 (m, 2H), 7.82 (s, 2H), 6.99 (m, 2H), 3.98 (s, 6H), 3.96 (s, 3H), 3.86 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.4, 159.3, 156.5, 152.8 (×2), 142.3, 133.1, 132.0, 131.1, 120.1 (×2), 114.5 (×2), 107.7 (×2), 61.0, 56.2 (×2), 55.6 ppm; MS (ESI): [M + H]+ = 385.1, [M + Na]+ = 407.1.
5-Amino-2-(4-fluorophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7e). Compound 7e was obtained from 13e as a yellow solid (0.34 g, 91%); mp 193–195 °C; 1H NMR (400 MHz, CDCl3) δ 8.00 (m, 2H), 7.80 (s, 2H), 7.18 (m, 2H), 3.97 (s, 6H), 3.97 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.5, 161.9 (d, J = 246.7 Hz), 156.6, 152.8 (×2), 142.5, 135.7, 131.8 (×2), 131.6, 120.3 (d, J = 8.2 Hz, ×2), 116.3 (d, J = 23.0 Hz, ×2), 107.7 (×2), 61.0, 56.2 (×2) ppm; MS (ESI): [M + H]+ = 373.1, [M + Na]+ = 395.1.
5-Amino-2-(2-chlorophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7f). Compound 7f was obtained from 13f as a yellow solid (0.35 g, 90%); mp 181–184 °C; 1H NMR (600 MHz, CDCl3) δ 8.21 (s, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.82 (s, 2H), 7.48 (d, J = 7.9 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 3.98 (s, 6H), 3.97 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.3, 156.4, 152.8 (×2), 142.6, 140.1, 131.9, 131.6, 130.6, 130.5, 123.0, 121.8, 116.9, 107.7 (×2), 60.9, 56.1 (×2) ppm; MS (ESI): [M + H]+ = 389.1, [M + Na]+ = 411.1.
5-Amino-2-(4-chlorophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7g). Compound 7g was obtained from 13g as a yellow solid (0.34 g, 87%); mp 168–170 °C; 1H NMR (600 MHz, CDCl3) δ 7.90 (d, J = 8.6 Hz, 2H), 7.79 (s, 2H), 7.61 (d, J = 8.6 Hz, 2H), 3.97 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.5, 156.5, 152.8 (×2), 138.3, 133.7, 132.5 (×2), 131.9, 131.8, 121.3, 120.0 (×2), 107.7 (×2), 61.0, 56.2 (×2) ppm; MS (ESI): [M + H]+ = 389.1, [M + Na]+ = 411.1.
5-Amino-2-(2-bromophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7h). Compound 7h was obtained from 13h as a yellow solid (0.36 g, 83%); mp 153–159 °C; 1H NMR (600 MHz, CDCl3) δ 8.30 (s, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.69 (s, 2H), 7.53 (d, J = 7.5 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 3.98 (s, 3H), 3.96 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.7, 156.4, 152.9, 142.6, 139.2, 134.4 (×2), 132.1, 131.8, 130.4, 128.2, 127.6, 117.5, 107.8 (×2), 60.9, 56.4 (×2), 21.9 ppm; MS (ESI): [M + H]+ = 433.0.
5-Amino-2-(3-bromophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7i). Compound 7i was obtained from 13i as a yellow solid (0.37 g, 86%); mp 172–176 °C; 1H NMR (600 MHz, CDCl3) δ 7.84 (s, 2H), 7.69 (m, 1H), 7.58 (m, 1H), 7.41 (m, 2H), 3.94 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.6, 156.3, 152.8, 142.5, 137.4, 132.1, 131.7, 131.3 (×2), 129.9, 128.2, 127.6, 127.0, 107.7 (×2), 60.9, 56.2 (×2), 21.9 ppm; MS (ESI): [M + H]+ = 433.0, [M + Na]+ = 455.0.
5-Amino-2-(4-bromophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (7j). Compound 7j was obtained from 13j as a yellow solid (0.40 g, 92%); mp 182–186 °C; 1H NMR (600 MHz, CDCl3) δ 7.96 (m, 2H), 7.79 (s, 2H), 7.46 (m, 2H), 3.97 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.5, 156.6, 152.8 (×2), 142.6, 138.3, 132.5 (×2), 131.9, 131.8, 121.4, 120.1 (×2), 107.7 (×2), 61.0, 56.2 (×2), 21.9 ppm; MS (ESI): [M + H]+ = 433.0, [M + Na]+ = 455.1.
General synthetic procedures for 2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazol (8a–j)
To a solution of compound 7a–j (1 mmol) in anhydrous THF (5 mL) was added isopropyl nitrite (2 mmol). The original material was completely depleted after one hour refluxing. After the evaporation of the solvent, the residue was purified by column chromatography on silica gel (200–300 mesh) with petroleum ether/AcOEt (v/v = 3:1) or pure CH2Cl2.
2-Phenyl-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8a). Compound 8a was obtained from 7a as a light yellow solid (0.27 g, 81%); mp 138–140 °C; 1H NMR (400 MHz, CDCl3): δ 8.42 (s, 1H), 8.16 (d, J = 7.9 Hz, 2H), 7.77 (s, 2H), 7.54 (dd, J = 7.5 Hz, 7.9 Hz, 2H), 7.44 (t, J = 7.5 Hz, 1H), 3.98 (s, 3H), 3.97 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 185.7, 156.7, 153.0 (×2), 142.6, 139.5, 132.0, 131.8, 129.5 (×2), 128.0, 118.8 (×2), 107.9 (×2), 61.1, 56.4 (×2) ppm; MS (ESI): [M + H]+ = 340.1, [M + Na]+ = 362.1.
2-(o-Tolyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8b). Compound 8b was obtained from 7b as a light yellow solid (0.29 g, 83%); mp 146–148 °C; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 7.76 (s, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.38 (m, 3H), 3.96 (s, 3H), 3.94 (s, 6H), 2.48 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.9, 152.9 (×2), 147.4, 143.0, 139.1, 138.3, 132.4, 131.8, 131.3, 129.3, 126.8, 125.1, 107.9 (×2), 60.9, 56.2 (×2), 19.1 ppm; MS (ESI): [M + H]+ = 354.1, [M + Na]+ = 376.1.
2-(p-Tolyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8c). Compound 8c was obtained from 7c as a light yellow solid (0.28 g, 80%); mp 149–152 °C; 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1H), 8.02 (d, J = 8.2 Hz, 2H), 7.77 (s, 2H), 7.33 (d, J = 8.2 Hz, 2H), 3.98 (s, 3H), 3.97 (s, 6H), 2.43 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 184.0, 152.9 (×2), 147.3, 143.0, 138.7, 138.6, 137.3, 131.4, 130.1 (×2), 119.1 (×2), 108.0 (×2), 61.0, 56.3 (×2), 21.1 ppm; MS (ESI): [M + H]+ = 354.1, [M + Na]+ = 376.1.
2-(4-Methoxyphenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8d). Compound 8d was obtained from 7d as a light yellow solid (0.25 g, 69%); mp 168–172 °C; 1H-NMR (600 MHz, CDCl3) δ 8.69 (d, J = 2.7 Hz, 1H), 8.41 (s, 1H), 8.33 (dd, J = 2.7 Hz, 8.0 Hz, 1H), 7.75 (s, 2H), 7.27 (d, J = 8.0 Hz, 1H), 4.06 (s, 3H), 3.98 (s, 3H), 3.97 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.9, 159.8, 152.9 (×2), 147.1, 142.9, 138.5, 133.1, 131.4, 120.7 (×2), 114.5 (×2), 108.0 (×2), 61.0, 56.2 (×2), 55.6 ppm; MS (ESI): [M + H]+ = 370.1, [M + Na]+ = 392.1.
2-(4-Fluorophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8e). Compound 8e was obtained from 7e as a light yellow solid (0.27 g, 74%); mp 158–161 °C; 1H NMR (600 MHz, CDCl3) δ 8.37 (s, 1H), 8.10 (m, 2H), 7.71 (s, 2H), 7.20 (m, 2H), 3.96 (s, 3H), 3.94 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.8, 162.3 (d, J = 247.5 Hz), 152.9 (×2), 147.5, 143.1, 138.8 (×2), 135.7 (d, J = 2.9 Hz), 131.2, 121.0 (d, J = 8.3 Hz, ×2), 116.4 (d, J = 23.2 Hz, ×2), 107.9 (×2), 60.9, 56.2 (×2) ppm; MS (ESI): [M + H]+ = 358.1, [M + Na]+ = 380.1.
2-(2-Chlorophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8f). Compound 8f was obtained from 7f as a light yellow solid (0.28 g, 76%); mp 161–164 °C; 1H NMR (600 MHz, CDCl3) δ 8.42 (s, 1H), 8.35 (t, J = 1.9 Hz, 8.1 Hz, 1H), 8.10 (m, 1H), 7.75 (s, 2H), 7.56 (m, 1H), 7.41 (t, J = 1.9 Hz, 8.1 Hz, 1H), 3.99 (s, 3H), 3.98 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.7, 153.0 (×2), 147.8, 143.2, 140.3, 139.1, 131.5, 131.1, 130.9, 123.2, 122.5, 117.7, 108.0 (×2), 61.0, 56.3 (×2) ppm; MS (ESI): [M + H]+ = 374.1.
2-(4-Chlorophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8g). Compound 8g was obtained from 7g as a light yellow solid (0.30 g, 81%); mp 136–140 °C; 1H NMR (600 MHz, CDCl3) δ 8.40 (s, 1H), 8.11 (m, 2H), 7.72 (s, 2H), 7.50 (m, 2H), 3.98 (s, 3H), 3.96 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3): δ = 183.7, 152.9, 147.6, 143.1, 138.9 (×2), 137.9, 134.3, 131.1, 129.7 (×2), 120.4 (×2), 107.9 (×2), 61.0, 56.2 (×2) ppm; MS (ESI): [M + H]+ = 374.1, [M + Na]+ = 396.1.
2-(2-Bromophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8h). Compound 8h was obtained from 7h as a light yellow solid (0.30 g, 73%); mp 168–172 °C; 1H NMR (600 MHz, CDCl3) δ 8.48 (s, 1H), 7.81 (d, J = 1.1 Hz, 1H), 7.79 (s, 2H), 7.64 (dd, J = 7.7 Hz, 1.6 Hz, 1H), 7.51 (m, 1H), 7.42 (td, J = 7.7 Hz, 1.6 Hz, 1H), 3.95 (s, 3H), 3.95 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.9, 161.4, 153.0, 147.9, 143.2, 138.8, 134.7, 134.2, 131.2 (×2), 128.3, 128.0, 118.5, 108.1 (×2), 61.0, 56.4 (×2) ppm; MS (ESI): [M + H]+ = 418.0, [M + Na]+ = 440.1.
2-(3-Bromophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8i). Compound 8i was obtained from 7i as a light yellow solid (0.32 g, 76%); mp 156–158 °C; 1H NMR (600 MHz, CDCl3) δ 8.48 (s, 1H), 7.79 (s, 2H), 7.70 (dd, J = 7.5 Hz, 2.0 Hz, 1H), 7.62 (dd, J = 7.5 Hz, 1.6 Hz, 1H), 7.47 (m, 2H), 3.95 (s, 3H), 3.95 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.9, 153.0, 148.0, 143.1, 138.8, 137.6, 131.2, 131.2 (×2), 130.8, 129.1, 127.7, 127.6, 108.0 (×2), 61.0, 56.3 (×2) ppm; MS (ESI): [M + H]+ = 418.0, [M + Na]+ = 440.0.
2-(4-Bromophenyl)-4-(3,4,5-trimethoxybenzoyl)-2H-1,2,3-triazol (8j). Compound 8j was obtained from 7j as a light yellow solid (0.33 g, 79%); mp 150–153 °C; 1H NMR (600 MHz, CDCl3) δ 8.38 (s, 1H), 8.00 (d, J = 8.0 Hz, 2H), 7.70 (s, 2H), 7.63 (d, J = 8.0 Hz, 2H), 3.96 (s, 3H), 3.94 (s, 6H) ppm; 13C NMR (100 MHz, CDCl3) δ 183.6, 152.9 (×2), 147.6, 143.1, 138.9, 138.3, 132.6 (×2), 131.1, 122.2, 120.6 (×2), 107.9 (×2), 60.9, 56.2 (×2) ppm; MS (ESI): [M + H]+ = 418.0, [M + Na]+ = 440.0.
General synthetic procedures for compounds (9a–j)
An excess of hydroxylamine hydrochloride (15 mmol) was added to a solution of compound 8a–j (1 mmol) and sodium acetate (15 mmol) in absolute ethanol (5 mL). After reacting at 80 °C for 2 h, the reaction was entirely finished (TLC control). Then the mixture was added to water (20 mL) and extracted with EtOAc (20 mL × 3). The combined organic layers were washed with water, dried over Na2SO4, and the solvent was then evaporated under reduced pressure. The residue was purified by column chromatography (petroleum ether/AcOEt = 2:1 as eluent) on silica gel to obtain pure product.
(Z + E)-(2-Phenyl-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9a). Compound 9a was obtained from 8a as a white solid (0.33 g, 94%) with a 76:24 mixture of Z and E isomers; mp 132–135 °C; 1H NMR (400 MHz, CDCl3) (Z isomer): δ 9.23 (s, 1H), 8.57 (s, 1H), 8.10 (d, J = 8.0 Hz, 2H), 7.49 (dd, J = 7.6 Hz, 8.0 Hz, 2H), 7.38 (t, J = 7.6 Hz, 1H), 6.98 (s, 1H), 3.92 (s, 3H), 3.88 (s, 6H) ppm; (E isomer): δ 8.05 (d, J = 7.9 Hz, 2H), 7.96 (s, 1H), 7.45 (dd, J = 7.9 Hz, 2H), 7.35 (m, 1H), 6.86 (s, 1H), 3.94 (s, 3H), 3.86 (s, 6H) ppm; 1H NMR (600 MHz, d6-DMSO) (Z isomer): δ 12.26 (s, 1H), 8.67 (s, 1H), 8.03 (d, J = 7.9 Hz, 2H), 7.59 (t, J = 7.6 Hz, 2H), 7.45 (t, J = 7.1 Hz, 1H), 6.99 (s, 1H), 3.78 (s, 3H), 3.73 (s, 6H) ppm; MS (ESI): [M + H]+ = 355.1, [M + Na]+ = 377.1.
(Z + E)-(2-(o-Tolyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9b). Compound 9b was obtained from 8b as a white solid (0.33 g, 89%) with a 92:8 mixture of Z and E isomers; mp 307–310 °C; 1H NMR (400 MHz, CDCl3) (Z isomer) δ 10.30 (s, 0.6H), 8.60 (s, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.35 (m, 3H), 6.95 (s, 2H), 3.90 (s, 3H), 3.86 (s, 6H), 2.40 (s, 3H) ppm; (E isomer) δ 8.04 (s, 1H), 7.58 (m, 1H), 7.28 (m, 3H), 6.85 (s, 2H), 3.92 (s, 3H), 3.84 (s, 6H), 2.40 (s, 3H) ppm; MS (ESI): [M + H]+ = 369.2.
(Z + E)-(2-(p-Tolyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9c). Compound 9c was obtained from 8c as a white solid (0.34 g, 92%) with a 82:18 mixture of Z and E isomers; mp 142–145 °C; 1H NMR (400 MHz, CDCl3) (Z isomer) δ 8.39 (s, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.77 (s, 2H), 7.33 (d, J = 8.4 Hz, 2H), 3.98 (s, 3H), 3.97 (s, 6H), 2.44 (s, 3H) ppm; (E isomer) δ 8.17 (s, 1H), 7.95 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.2 Hz, 2H), 6.68 (s, 2H), 3.93 (s, 3H), 3.81 (s, 6H), 2.42 (s, 3H) ppm; MS (ESI): [M + H]+ = 369.1, [M + Na]+ = 391.1.
(Z + E)-(2-(4-Methoxyphenyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9d). Compound 9d was obtained from 8d as a white solid (0.33 g, 87%) with a 89:11 mixture of Z and E isomers; mp 178–180 °C; 1H NMR (400 MHz, CDCl3) (Z isomer) δ 9.80 (s, 1H); 8.57 (s, 1H); 8.08 (d, J = 7.7 Hz, 2H); 7.46 (d, J = 7.7 Hz, 2H); 6.97 (s, 2H); 3.93 (s, 3H); 3.88 (s, 6H), 3.48 (s, 3H) ppm; MS (ESI): [M + H]+ = 385.1, [M + Na]+ = 407.1.
(Z + E)-(2-(4-Fluorophenyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9e). Compound 9e was obtained from 8e as a white solid (0.31 g, 84%) with a 78:22 mixture of Z and E isomers; mp 139–147 °C; 1H NMR (600 MHz, CDCl3) (Z isomer) δ 8.40 (s, 1H), 8.14 (m, 2H), 7.73 (s, 2H), 7.23 (m, 2H), 6.66 (s, 1H), 3.98 (s, 3H), 3.97 (s, 6H) ppm; (E isomer) δ 8.19 (s, 1H), 8.06 (m, 2H), 7.73 (s, 2H), 7.20 (m, 2H), 6.66 (s, 2H), 3.93 (s, 3H), 3.81 (s, 6H). ppm; MS (ESI): [M + H]+ = 373.1.
(Z + E)-(2-(2-Chlorophenyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9f). Compound 9f was obtained from 8f as a white solid (0.33 g, 86%) with a 84:16 mixture of Z and E isomers; mp 215–218 °C; 1H NMR (600 MHz, d6-DMSO) (Z isomer) δ 12.29 (s, 0.2H), 8.75 (s, 1H), 8.14 (m, J = 9.8 Hz, 2H), 7.74 (m, J = 8.0 Hz, 1H), 7.63 (s, 2H), 7.60 (m, 1H), 3.81 (s, 3H), 3.78 (s, 6H); (E isomer) δ 11.89 (s, 0.2H), 8.78 (s, 1H), 8.26 (s, 2H), 8.02 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 1H), 7.58 (m, 1H), 3.90 (s, 9H) ppm; MS (ESI): [M + H]+ = 389.1, [M + Na]+ = 411.1.
(Z + E)-(2-(4-Chlorophenyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9g). Compound 9g was obtained from 8g as a white solid (0.35 g, 91%) with a 88:12 mixture of Z and E isomers; mp 184–189 °C; 1H NMR (600 MHz, CDCl3) (Z isomer) δ 8.55 (s, 1H), 8.03 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 8.6 Hz, 2H), 6.94 (s, 2H), 3.91 (s, 3H), 3.86 (s, 6H) ppm; (E isomer) 8.41 (s, 1H), 8.09 (d, J = 8.8 Hz, 2H), 7.72 (s, 2H), 7.51 (d, J = 8.8 Hz, 2H), 3.98 (s, 3H), 3.96 (s, 6H) ppm; MS (ESI): [M + H]+ = 389.1, [M + Na]+ = 411.1.
(Z + E)-(2-(2-Bromophenyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9h). Compound 9h was obtained from 8h as a white solid (0.39 g, 90%) with a 71:29 mixture of Z and E isomers; mp 195–198 °C; 1H NMR (600 MHz, CDCl3) (Z isomer) δ 8.65 (s, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.56 (d, J = 6.0 Hz, 1H), 7.45 (d, J = 7.4 Hz, 1H), 7.36 (t, J = 7.2 Hz, 1H), 6.96 (s, 2H), 3.88 (s, 3H), 3.86 (s, 6H) ppm; (E isomer) δ 8.09 (s, 1H), 7.72 (s, 1H), 7.55 (s, 1H), 7.42 (d, J = 7.2 Hz, 1H), 7.32 (s, 1H), 6.87 (s, 2H), 3.90 (s, 3H), 3.85 (s, 6H) ppm; MS (ESI): [M + H]+ = 433.0, [M + Na]+ = 455.0.
(Z + E)-(2-(3-Bromophenyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9i). Compound 9i was obtained from 8i as a white solid (0.40 g, 92%) with a 82:18 mixture of Z and E isomers; mp 180–185 °C; 1H-NMR (600 MHz, CDCl3): (Z isomer) δ 9.23 (s, 0.6H), 8.63 (s, 1H), 7.61 (dd, J = 1.8 Hz, 7.57 Hz, 1H), 7.6 (dd, J = 1.6 Hz, 7.9 Hz, 1H), 7.43 (dd, J = 1.8 Hz, 7.9 Hz, 1H), 7.41 (dd, J = 1.6 Hz, 7.6 Hz, 1H), 6.96 (s, 2H), 3.88 (s, 3H), 3.87 (s, 6H) ppm; (E isomer) δ 8.49 (s, 1H), 7.79 (s, 2H), 7.70 (dd, J = 1.9 Hz, 7.5 Hz, 1H), 7.63 (d, J = 1.8 Hz, 1H), 7.47 (d, J = 1.9 Hz, 1H), 7.45 (d, J = 1.8 Hz, 1H), 3.95 (s, 3H), 3.94 (s, 6H) ppm; MS (ESI): [M + H]+ = 433.0, [M + Na]+ = 455.1.
(Z + E)-(2-(4-Bromophenyl)-2H-1,2,3-triazol-4-yl)(3,4,5-trimethoxyphenyl)methanone oxime (9j). Compound 9j was obtained from 8j as a white solid (0.40 g, 93%) with a 83:17 mixture of Z and E isomers; mp 201–205 °C; 1H NMR (600 MHz, d6-DMSO) (Z isomer) δ 12.26 (s, 1H), 8.66 (s, 1H), 8.03 (m, 2H), 7.42 (m, 2H), 6.97 (s, 2H), 3.78 (s, 6H), 3.76 (s, 3H) ppm; (E isomer) δ 11.83 (s, 1H), 8.27 (s, 1H), 7.95 (m, 1H), 7.47 (m, 1H), 6.86 (s, 1H), 3.73 (s, 3H), 3.72 (s, 6H) ppm; MS (ESI): [M + H]+ = 433.0.
Biology
Cell line and culture conditions. The human gastric adenocarcinoma SGC-7901 cells, lung adenocarcinoma A549 cells, fibrosarcoma HT-1080 cells and normal fibroblasts L929 cells were purchased from Shanghai Institute of Cell Resources Center of Life Science (Shanghai, China). All cells were cultured in RPMI-1640 medium (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, USA), streptomycin and penicillin at 37 °C in humidified atmosphere with 5% CO2.
Antiproliferative activity assay. The antiproliferative activity assay in vitro was carried out referring to the previously reported method.24 9a–j was used without separation. (Z)-9a and (E)-9a were separated from mixture and was used immediately.
Tubulin polymerization assay. The tubulin polymerization assay was carried out referring to the previously reported method.24 (Z)-9a was separated from mixture and was used immediately.
Immunofluorescence assay. The immunofluorescence assay was carried out referring to the previously reported method.29 (Z)-9a was separated from mixture and was used immediately. 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Sigma Chemical (St. Louis, MO, USA) and the primary α-tubulin antibody was used (Santa Cruz, CA, USA).
Cell cycle assay. The cell cycle assay was carried out referring to the previously reported method.29 (Z)-9a was separated from mixture and was used immediately.
Calculational assay. Geometry optimization of molecules was carried out at the B3LYP level of theory with the 6-31G(d) basis set on Gaussian 09 software package. The angles were calculated by methods of analytic geometry manually according to the coordinate data from Gaussian software. The molecular modelling studies were performed with Accelrys Discovery Studio 3.0.17,18 The crystal structures of tubulin complexed with DAMA-colchicine (PDB: 1SA0) was retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/pdb).27 The ligand 9a model was optimized by molecular dynamic force field, and docked into the binding site using the CDOCKER protocol with the default settings. The image of Fig. 2 and 7 was possessed by Discovery Studio 4.5 Visualizer.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (81673293, 81502932, 81602969) and State Key Laboratory of Natural Medicines and Active Substance (No. GTZK201603) for their generous financial support. This work was also supported by Program for Innovative Research Team of the Ministry of Education and Program for Liaoning Innovative Research Team in University.
References
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790 CrossRef CAS PubMed.
- E. A. Perez, Mol. Cancer Ther., 2009, 8, 2086 CrossRef CAS PubMed.
- Y. Lu, J. Chen, M. Xiao, W. Li and D. D. Miller, Pharm. Res., 2012, 29, 2943 CrossRef CAS PubMed.
- M. N. Islam and M. N. Iskander, Mini-Rev. Med. Chem., 2004, 4, 1077 CrossRef CAS PubMed.
- G. G. Dark, S. A. Hill, V. E. Prise, G. M. Tozer, G. R. Pettit and D. J. Chaplin, Cancer Res., 1997, 57, 1829 CAS.
- G. R. Pettit, B. Toki, D. L. Herald, P. V. Pinard, M. R. Boyd, E. Hamel and R. K. Pettit, J. Med. Chem., 1998, 41, 1688 CrossRef CAS PubMed.
- Y. Lu, C.-M. Li, Z. Wang, C. R. Ross II, J. Chen, J. T. Dalton, W. Li and D. D. Miller, J. Med. Chem., 2009, 52, 1701 CrossRef CAS PubMed.
- J. Chen, Z. Wang, C. M. Li, Y. Lu, P. K. Vaddady, B. Meibohm, J. T. Dalton, D. D. Miller and W. Li, J. Med. Chem., 2010, 53, 7414 CrossRef CAS PubMed.
- J. Chen, S. Ahn, J. Wang, Y. Lu, J. T. Dalton, D. D. Miller and W. Li, J. Med. Chem., 2012, 55, 7285 CrossRef CAS PubMed.
- Q. Guan, D. Feng, Z. Bai, Y. H. Cui, D. Zuo, M. Zhai, X. Jiang, W. Zhou, K. Bao, Y. Wu and W. Zhang, MedChemComm, 2015, 6, 1484 RSC.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed.
- V. D. Bock, D. Speijer, H. Hiemstra and J. H. V. Maarseveen, Org. Biomol. Chem., 2007, 5, 971 CAS.
- Y. C. Duan, Y. C. Ma, E. Zhang, X. J. Shi, M. M. Wang, X. W. Ye and H. M. Liu, Eur. J. Med. Chem., 2013, 62, 11 CrossRef CAS PubMed.
- N. R. Madadi, N. R. Penthala, K. Howk, A. Ketkar, R. L. Eoff, M. J. Borrelli and P. A. Crooks, Eur. J. Med. Chem., 2015, 103, 123 CrossRef CAS PubMed.
- A. D. Baecke, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, J. C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Wang, Y. Yin, Y. Zhang, S. Mi, M. Zhao, P. Lv, B. Wang and H. Zhu, Eur. J. Med. Chem., 2015, 93, 291 CrossRef CAS PubMed.
- H. Behbehani, H. M. Ibrahim and S. Makhseed, Heterocycles, 2009, 78, 3081 CrossRef CAS.
- L. Wu, S. Guo, X. Wang, Z. Guo, G. Yao, Q. Lin and M. Wu, Tetrahedron Lett., 2015, 56, 2145 CrossRef CAS.
- C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 171 CrossRef PubMed.
- A. Hoz, A. Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164 RSC.
- Z. Wen, J. Xu, Z. Wang, H. Qi, Q. Xu, Z. Bai, Q. Zhang, K. Bao, Y. Wu and W. Zhang, Eur. J. Med. Chem., 2015, 90, 184 CrossRef CAS PubMed.
- T. Yang, G. Chen, Z. Sang, Y. Liu, X. Yang, Y. Chang, H. Long, W. Ang, J. Tang, Z. Wang, G. Li, S. Yang, J. Zhang, Y. Wei and Y. Luo, J. Med. Chem., 2015, 58, 6389 CrossRef CAS PubMed.
- Y. Lu, C.-M. Li, Z. Wang, J. Chen, M. L. Mohler, W. Li, J. T. Dalton and D. D. Miller, J. Med. Chem., 2011, 54, 4678 CrossRef CAS PubMed.
- R. B. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198 CrossRef CAS PubMed.
- A. Massarotti, A. Coluccia, R. Silvestri, G. Sorba and A. Brancale, ChemMedChem, 2012, 7, 33 CrossRef CAS PubMed.
- Q. Xu, Y. Wang, J. Xu, M. Sun, H. Tian, D. Zuo, Q. Guan, K. Bao, Y. Wu and W. Zhang, ACS Med. Chem. Lett., 2016, 7, 1202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c7ra02720f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a