
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSintering of multiple Cu–Ag core–shell nanoparticles and properties of nanoparticle-sintered structures†
Jiaqi Wang and
Seungha Shin*
and
Seungha Shin*
Department of Mechanical, Aerospace and Biomedical Engineering, The University of Tennessee, Knoxville, Tennessee 37996-2210, USA. E-mail: sshin@utk.edu
First published on 18th April 2017
Abstract
Cu–Ag core–shell (CS) nanoparticles (NP) have been synthesized to replace pure Ag NP paste in order to lower the cost while maintaining excellent thermal and electrical conductivities for electronic applications. In this study, a multiple-CS-NP sintering model with molecular dynamics is employed to investigate the NP size and temperature dependency of the sintering process, as well as mechanical and thermodynamic properties of the sintered structures. Porosity and multiple particle effects are included, which allow for more accurate analysis than the conventional two- or three-NP sintering model. We unravelled the sintering mechanism at room temperature, and the interplay of liquid and solid surface diffusion during sintering at higher temperatures. Interfacial atoms have a higher mobility than surface atoms and contribute to a higher densification in the multiple-CS-NP model. A more densified structure yields higher Young's modulus, yield strength and Poisson's ratio, while lowering isothermal compressibility. The coefficient of thermal expansion and specific heat capacity exhibit grain-size and porosity independence. This multiple-CS-NP model provides a theoretical basis for determining NP configuration and sintering conditions for desirable properties.
Introduction
The miniaturization of electronic devices increasingly motivates the development of nanotechnology in synthesis, assembly, and the joining of metallic nanoparticles (NP).1–7 Among them, Ag NP have been widely produced and employed in the fabrication of flexible and low-cost electronic devices through sintering, where high thermal and electrical conductivities are required.8–12 However, the cost of Ag has increased significantly over the last few years and is not projected to display a reduction trend in the near future,13 which has limited the wide industrial applications of Ag NP. Thus, Cu–Ag core–shell (CS) NP have been synthesized as a potential alternative to pure Ag NP;14–16 this allows for the tremendous reduction of production costs, enhanced protection of the Cu core from oxidation, and thereby maintaining the desirable thermal and electrical properties for electronic applications.Joining of the CS NP has been widely used as a bottom-up nanotechnology to provide permanent unions or connections to form functional nanodevices.4 The main challenge to nanojoining lies in the formation of a robust junction between NPs with excellent mechanical, thermal, and electrical performances. Understanding the underlying sintering mechanisms of NPs can enhance the performance of the sintered structure through the manipulation of temperature, pressure, heating rate, NP size or relative crystallographic orientation. Numerous studies have been conducted on the sintering process of NPs both computationally and experimentally.17–22 While most studies have focused on the monometallic two- or three-NP sintering model, the sintering process of multiple bimetallic Cu–Ag CS NPs is still relatively unexplored. The two- or three-NP sintering model is valid for loosely packed NP sintering in the gas phase or on substrates, but they overlook important factors that affect sintering dynamics and the properties of sintered structures.23 For instance, agglomeration and pore effects, which significantly contribute to the sintering rate and porosity of final sintered structures, are neglected in the two-NP model.24 Consequently, the sintering mechanisms and properties of sintered structures of multiple NPs are different from a two- or three-NP model. Therefore, a model containing pores to simulate the real case is essential for elucidating the sintering mechanism of the porous structure and studying the porosity dependency of sintered structure properties.
As an initial attempt, we developed a multiple-CS-NP sintering model with molecular dynamics (MD) simulation to gain insight into the nanoscale sintering process by monitoring the atomic movement.17,25,26 Our model takes into account the effects of NP size and temperature in this research; we plan a further extension of this model to reveal the effects of pressure, size distribution and crystallographic orientation on the sintering process. The sintering simulations using the monometallic multiple-NP model have been confirmed to be very effective for reproducing the sintering process in porous anodes.17,27 With this multiple-CS-NP model, the sintering dynamics of the CS porous structure at one atmospheric pressure but various temperatures, as well as mechanical and thermodynamic properties of sintered structures, are investigated effectively. Following this introduction, the methodology of simulation and analysis is displayed. In the Results and discussion section, the sintering dynamics and sintered structure properties are analysed and compared between (1) different-sized multiple-CS-NP models, (2) two-CS-NP and multiple-CS-NP sintering model, and (3) the bimetallic Cu–Ag CS NP model and monometallic pure Ag NP model. Implications and future work are elucidated in our conclusion.
Methodology
Modelling of multiple CS NPs
Differing from the two-CS-NP sintering model,28,29 the multiple-CS-NP model possesses more degrees of freedom. Although particle packing arrangement can affect the sintering rate due to different numbers of contact points,24,30 the sintering rate is not a main focus of this research and it is formidable and unnecessary to test all possible arrangements. Therefore, among various packing arrangements [e.g., simple cubic (SC), body-centred cubic (BCC), face-centred cubic (FCC), hexagonal close-packed (HCP), etc.], we employed the SC arrangement, within which the NPs are facing each other with identical crystallographic orientation. For instance, we set the NPs facing each other with the [100] orientation in this research, then no other orientations such as [111] were induced in this SC arrangement. NPs were built within a simulation box with periodic boundary conditions in all three dimensions. Compared to the two-CS-NP sintering model under non-periodic boundary conditions,29 this model is more realistic, since the agglomeration and porosity effect can be directly addressed.17,24,27 To simplify the simulated system, the effects of core size, and size distribution of NPs were excluded; i.e., all CS NPs in a simulation cell had the same radius and ratio of shell-thickness to core-radius.The NP is denoted as AgxCuy, where x and y represent the overall NP and core radii in terms of aAg, where aAg (= 4.079 Å) is the lattice constant of Ag. Four kinds of NP are employed; (1) NP1-Ag5Cu0, (2) NP2-Ag5Cu2.5, (3) NP3-Ag8Cu4, and (4) NP4-Ag11Cu5.5. The NP1-Ag5Cu0 is pure Ag NP with a radius of 5aAg, and the latter three are Cu–Ag CS NPs with rcs of 5aAg, 8aAg and 11aAg and rc of 2.5aAg, 4aAg, and 5.5aAg. Therefore, the ratio of core radius to shell thickness remained as unity in CS NPs [i.e., rc: (rcs − rc) = 1].
Fig. 1a and b show the initial sintering configuration of multiple NP2-Ag5Cu2.5. Eight NPs are included in each simulation box: one (= 8 × 1/8) at eight corners, three (= 6 × 1/2) at the centres of the six faces, three (= 12 × 1/4) at the centres of the twelve edges, and one at the centre of the simulation box. The NPs are separated by a distance of aAg to prevent atoms from overlapping. This distance is still within the cut-off radius of the interaction potential so that the sintering can be initiated by attractive forces among the atoms. For analysis of the surface and shell diffusion during sintering, the NP is divided into three regimes: Cu core, Ag shell, and Ag surface (Fig. 1c); their atomic distances (r) from the centre of mass are in the range of 0 < r < rc, rc < r < rcs, and (rcs − aAg) < r < rcs, respectively.
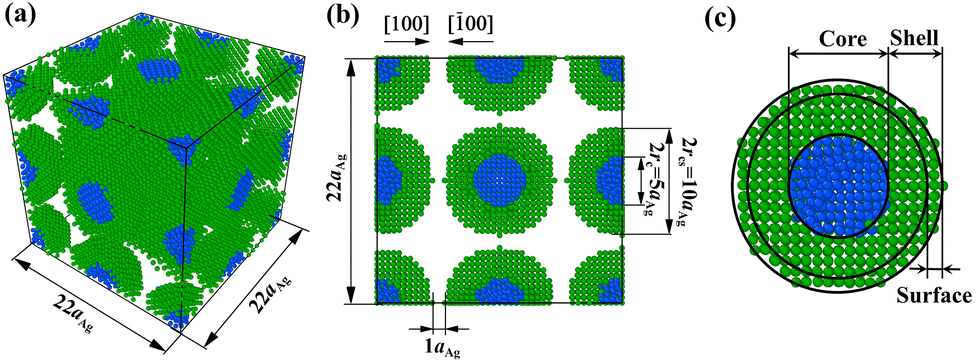 | ||
| Fig. 1 (a) 3-D view of the initial configuration for sintering simulations of multiple NP2-Ag5Cu2.5. (b) Cross-sectional image of (a). The initial sintering arrangements of other CS NPs are identical to NP2-Ag5Cu2.5. (c) One CS NP2-Ag5Cu2.5 extracted from (a), which is divided into three regimes (core, shell, and surface), as illustrated. The Ag shell atoms are coloured with green, while the Cu core atoms are coloured with blue. | ||
Simulation methodology
In this research, all the simulations were performed utilizing the LAMMPS code,31 and the Extreme Science and Engineering Discovery Environment (XSEDE) resources32 were employed for partial simulations. The embedded atom method (EAM) potential33 was applied for describing the interactions between Cu and Ag atoms.34 This potential has been proven to accurately calculate the cohesive energy, lattice parameters, elastic constant, phase diagram and high-temperature properties of Cu and Ag. A timestep of 1 fs was chosen for all simulations, and Newton's equation of motion was integrated with the Verlet algorithm.35Melting simulations were first performed to determine the melting temperature (Tm) as well as surface-premelting temperature (Tsm) of different-sized NPs. The simulation methodology was validated by comparing the obtained Tm with other reported values.36 Based on the resulting Tsm and Tm, we selected temperatures for sintering simulations, in which we (1) investigated the temperature and size effect on the sintering dynamics, and (2) obtained final structures sintered at different temperatures as simulation subjects for the subsequent studies of mechanical and thermodynamic properties.
Melting simulations
For melting simulations, the system was equilibrated at 300 K for 50 ps, simulations were then continued at various temperatures ranging from 300 K to 1300 K (employing increments of 100 K from 300 K to 900 K, and increments of 20 K from 900 K to 1300 K for a closer observation near Tm) for 50 ps at each temperature, while recording the system-averaged potential energy (Ep) and atom trajectories of the CS NP every 0.5 ps. Both the equilibration and data production phases were conducted in NVT canonical ensemble (constant number of atoms, volume, and temperature) with non-periodic boundary conditions. Tm was determined by the Ep curve; a sharp change in Ep indicated a phase transition.37–40 Atom trajectories were processed to obtain the Lindemann index (δLI), which was employed to determine the Tsm. Also, the Tm was determined by δLI to verify the Tm determined by Ep. For a system consisting of N atoms, the system-averaged δLI is given by41,42
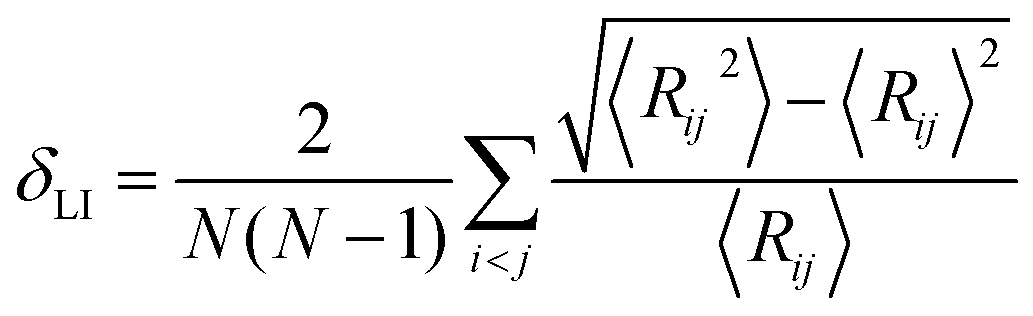 | (1) |
Sintering, diffusivity and activation energy
Sintering of four different multiple-CS-NP systems (NP1-Ag5Cu0, NP2-Ag5Cu2.5, NP3-Ag8Cu4, and NP4-Ag11Cu5.5) was simulated for 800 ps. All the initial sintering processes such as neck formation, growth, and densification were observed in the simulations. We employed the isothermal-heating method in sintering simulations;44 i.e., the sintering system was kept at 300 K for 800 ps using the 300 K relaxed structure, and then kept at 400 K for another 800 ps with the same structure, until the temperature reached 1300 K (with increments of 100 K). By applying the isothermal-heating method, temperature gradients inside the NP were eliminated, thus ignoring heat transfer effects. NpT isobaric–isothermal ensemble (constant number of atoms, constant pressure and temperature) was employed with Nosé–Hoover thermostat and barostat to control the system temperature and pressure (constant pressure of 1 atm at all temperatures). Sintering simulations were preceded by energy minimization on the initial multiple-CS-NP configuration with steepest decent algorithm.45 Atom trajectory, Ep, densification (ξ, the ratio of reduction in volume of simulation cell to initial volume during sintering process), and mean square displacement (〈d2〉) of surface and shell atoms were exported every 0.2 ps for further analysis. The 〈d2〉 was correlated with self-diffusivity (Dself) by Einstein's relation46 given by
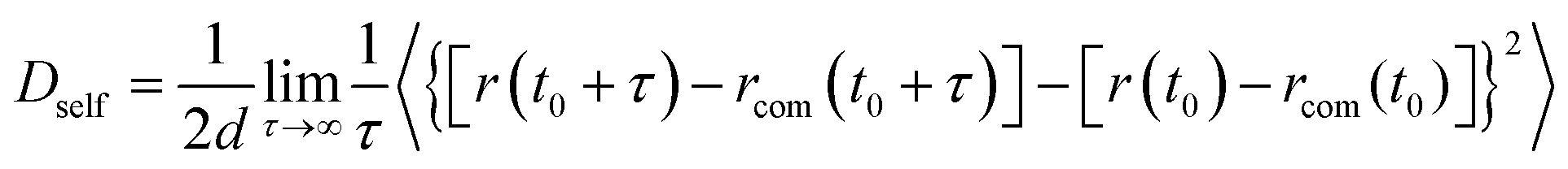 | (2) |
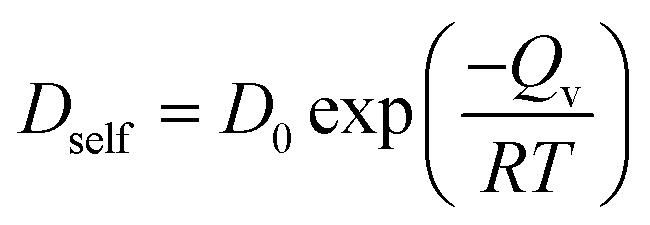 | (3) |
Mechanical properties
Before running simulations for mechanical properties, the final sintered structures (sintering products at 800 ps) were annealed from the sintering temperature (Tsinter) to room temperature (Troom = 300 K) within 100 ps, and the annealed structures were further relaxed at Troom for another 100 ps to obtain a fully equilibrated structure. Strain was then applied by uniformly extending the x dimension of the MD cell with a constant axial strain rate of 2 × 10−2 ps−1; i.e., extending the box length in the x dimension by 2% of its original length every picosecond, followed by rescaling the new x coordinates of the atoms to fit within the new dimension.Strain (ε), stress (σ), and x, y, and z dimensions of the simulation cell were recorded every 0.2 ps during the tensile test for mechanical properties, including Young's modulus (E100 = dσ/dε in the elastic regime), yield strength (σYS), and Poisson's ratio (ν100 = −εyy/εxx = −εzz/εxx). Since each NP was initially placed facing other NPs with the [100] direction and did not rotate during sintering, the E100, σYS and ν100 obtained below Tm were regarded as those of the {100} faces in the final structures. The E100 and σYS were extracted from the strain–stress plots, while ν100 was obtained by calculating the strain ratio in the y (or z) direction and x direction (strain direction).
Thermodynamic properties
We performed MD simulations on calculating the isothermal compressibility (βT), volumetric coefficient of thermal expansion (αp), and constant-volume specific heat capacity (cv), using the equations shown below:48–50
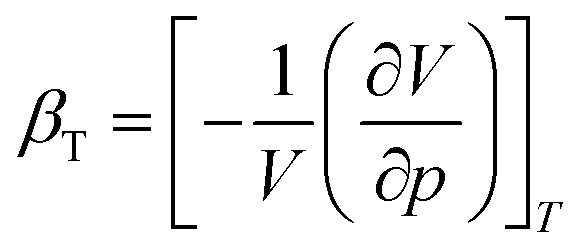 | (4) |
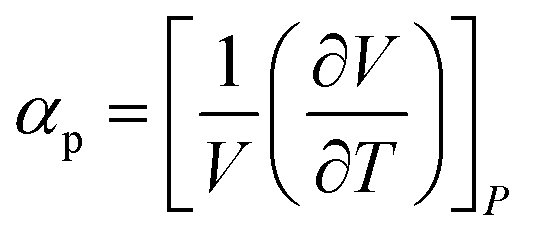 | (5) |
 | (6) |
In eqn (4) and (5), V is the volume of the system, kB is the Boltzmann constant (= 8.614 × 10−5 eV K−1) in eqn (6), m is the mass of the NP-sintered structure, and Ep,tot is the total potential energy of the system. The multiple-NP-sintered structures at different temperatures were equilibrated prior to the simulations for βT, similar to that in obtaining mechanical properties. Then, the multiple-NP-sintered structures were further simulated for 100 ps at Troom under five different pressures (81, 121, 161, 201, and 241 atm, respectively) within the NpT ensemble. As pressure increases, the NP-sintered structure is more compressed, thus decreasing the volume. The compressed volume was obtained by averaging the volume during the last 50 ps. The ratio of volume change to original volume (−ΔV/V0, where V0 is the original volume at 1 atm) was plotted with respect to pressure change (Δp), and βT was calculated using the slope of the plot obtained by eqn (4). In simulations for αp calculation, the NP-sintered structure was first relaxed at a temperature 200 K lower than the Tsinter for 200 ps, then the temperature was increased by a step of 20 K and the system maintained at each increased temperature for 100 ps. The volume was averaged during the last 50 ps as well. The αp is the slope of the (−ΔV/V0) − ΔT plot as in eqn (5). Since the cv is dependent on T below the Debye temperature (215 K for Ag and 315 K for Cu, respectively),51 we evaluated the cv of all sintered structures at 300 K using eqn (6) after the equilibration to exclude the temperature dependency.
Results and discussion
Thermal stability of different-sized NPs
Sintering dynamics are affected by diffusion behaviours of single NP, which depend on the NP shape, size, temperature, etc. For example, thermal diffusion of triangular nanoplates can be activated at Troom, allowing the nanoplates to self-sinter and form nanobelts with a growth-oriented capping agent,52 while the diffusion of the Cu–Ag CS spherical NP is not initiated at Troom.29 Thus, thermal stability of NPs is first studied under the heating process by determining the critical temperatures such as Tm and Tsm according to the two aforementioned criteria: Ep (Fig. 2a) and δLI (Fig. 2b). Steep jumps are observed in both curves at 960 K, 1180 K, and 1220 K for NP2-Ag5Cu2.5, NP3-Ag8Cu4 and NP4-Ag11Cu5.5, respectively. The obtained Tm's for different sizes of Cu–Ag CS NPs coincide well with the reported computational values.36 For further verification and comparison, we display the cross-sectional images of NP3-Ag8Cu4 in Fig. 2a. As temperature increases, the NP have a less crystallized structure, and the structure at the determined Tm is completely amorphous, also indicating the phase transition from solid to liquid.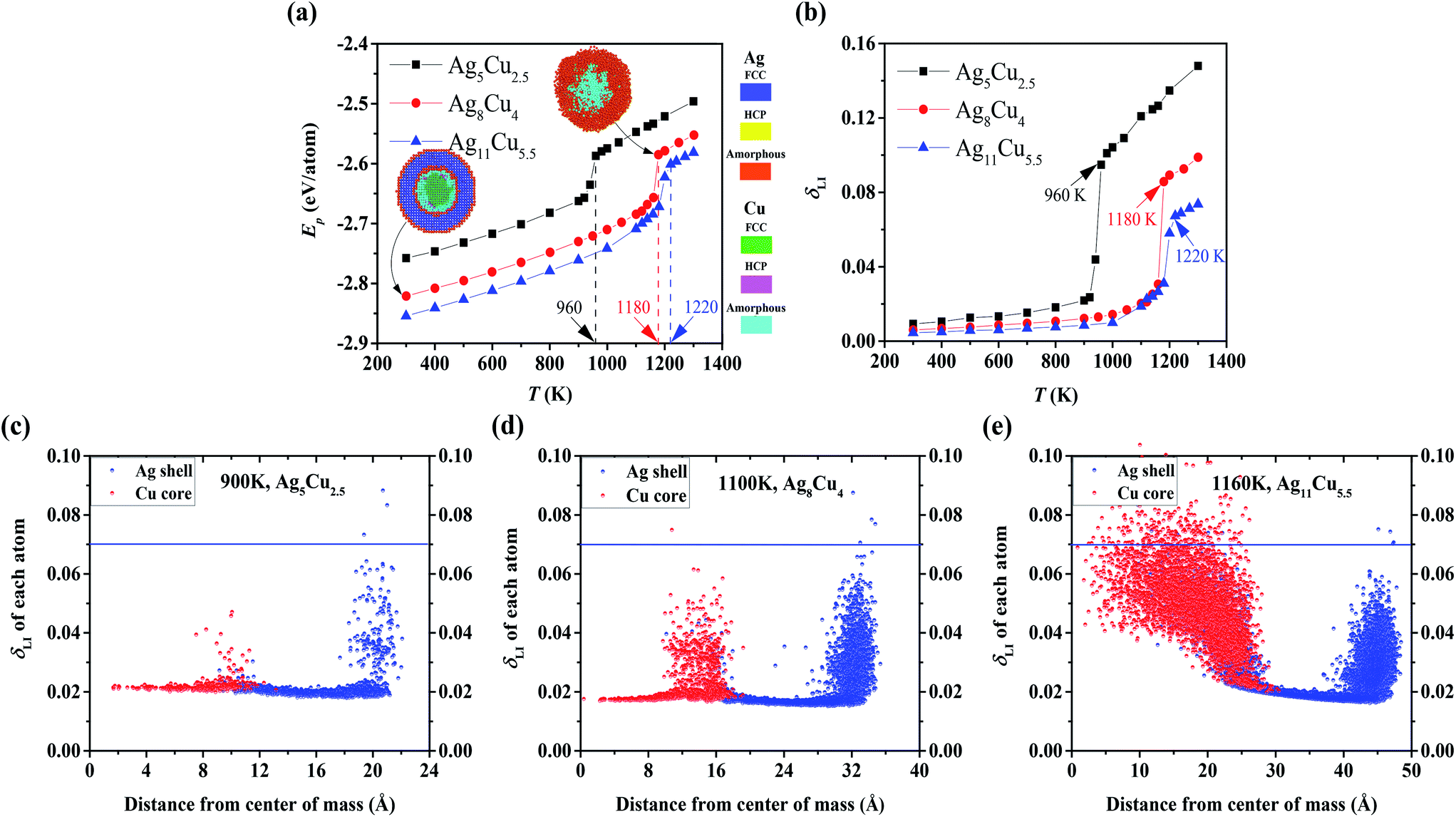 | ||
| Fig. 2 (a) Potential energy (Ep) of different-sized single CS NP during the heating process. (b) Lindeman index (δLI) of different-sized single CS NP during the heating process. The melting temperatures (Tm) determined by both the Ep and δLI coincide well with each other. Radial distribution of δLI in (c) NP2-Ag5Cu2.5, (d) NP3-Ag8Cu4, and (e) NP4-Ag11Cu5.5 at surface premelting temperature (Tsm). | ||
The Lindemann atom, which has δLI larger than 0.07, on the Ag surface indicates that the surface is premelted, and it appears at temperatures of 900 K in NP2-Ag5Cu2.5 (Fig. 2c), 1100 K in NP3-Ag8Cu4 (Fig. 2d), and 1160 K in NP4-Ag11Cu5.5 (Fig. 2e), which are determined as Tsm. Since the pure Ag NP, i.e., NP1-Ag5Cu0, has the identical size to NP2-Ag5Cu2.5, the NP1-Ag5Cu0 should have the same Tm and Tsm as the NP2-Ag5Cu2.5, which are 960 K and 900 K, respectively. As NP size increases, the portion of surface atoms with a lower coordination number decreases, which induces the increase in Tsm, the decrease in the surface energy, and thus the surface atom mobility. In addition to size effects, the Cu/Ag interface enhances the mobility of interfacial Cu and Ag atoms, which can make a difference in the sintering dynamics as compared to pure Ag NP sintering. Detailed physical analysis is demonstrated in the section on the comparison between the sintering of Cu–Ag CS and Pure Ag NP. Table 1 summarizes the geometrical details and the corresponding Tm and Tsm for each NP.
| NP type | rcs | rc | # Cu atoms | # Ag atoms | Tm (K) | Tsm (K) |
|---|---|---|---|---|---|---|
| NP1-Ag5Cu0 | 5aAg | 0 | 0 | 2120 | 960 | 900 |
| NP2-Ag5Cu2.5 | 5aAg | 2.5aAg | 369 | 1874 | 960 | 900 |
| NP3-Ag8Cu4 | 8aAg | 4aAg | 1505 | 7505 | 1180 | 1100 |
| NP4-Ag11Cu5.5 | 11aAg | 5.5aAg | 4093 | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 401 401 |
1220 | 1160 |
Sintering dynamics of the multiple-CS-NP model
As the size of the NP decreases, the fraction of surface-layer atoms, which are more sensitive to the surrounding temperature,42,53,54 increases. Thus, a smaller NP is more thermodynamically active as confirmed by their lower Tsm (Table 1). However, if the NP size is too small, the quantum-confinement effect should be considered,55 which cannot be properly addressed in MD. Hence, for a clear observation of the temperature-effect on sintering dynamics while avoiding the quantum-confinement effect, the multiple-CS-NP model of Ag5Cu2.5 (the smallest NP in this research) is selected.Ep is analysed not only for detecting the sintering mechanism, but also for evaluating the stability of the final sintered structures. As shown in Fig. 3a, the Ep of Ag shell atoms (obtained by averaging the Ep during last 50 ps) in the final structures sintered at different temperatures increases linearly as temperature increases from 300 K to 500 K, characterized as the low-temperature sintering without pore elimination (Fig. S1a–c†). The surface diffusion mechanism loses its dominance at low temperatures (300–500 K); instead, other mechanisms, such as plastic deformation involving dislocation or twinning, contribute to the densification.24 Therefore, we employ 300 K as a low-temperature case for the multiple-CS-NP model. There is an obvious decrease in Ep of the Ag shell in structures sintered at a temperature between 600 K (pore elimination temperature, Tpe) and 800 K. This decrease is induced by the annihilation of free surface, due to pore elimination after sintering (Fig. S1d–f†). The Ep decrease indicates that a more stable structure is achieved in this temperature range (Tpe < T < Tsm), compared to the low-temperature cases. At 900 K (Tsm), the pores are eliminated with a higher speed since surface-premelting occurs in multiple NP2-Ag5Cu2.5 (Fig. S1g†). The whole NP system is melted at 1000 K (the Tm is 960 K for NP2-Ag5Cu2.5) and the final sintered structure is shown as Fig. S1h.† The melting is also indicated by a steep jump in Ep from 900 K to 1000 K (Fig. 3a). However, unlike the Ag shell, the Ep of the Cu core shows a linear increase both before and after melting, indicating that the Cu cores do not participate in the sintering at temperatures below Tm. Based on the above analysis, we only selected 300, 600, 900, and 1000 K to efficiently analyse the temperature effect on the sintering dynamics of the multiple-CS-NP model with NP2-Ag5Cu2.5 in detail.
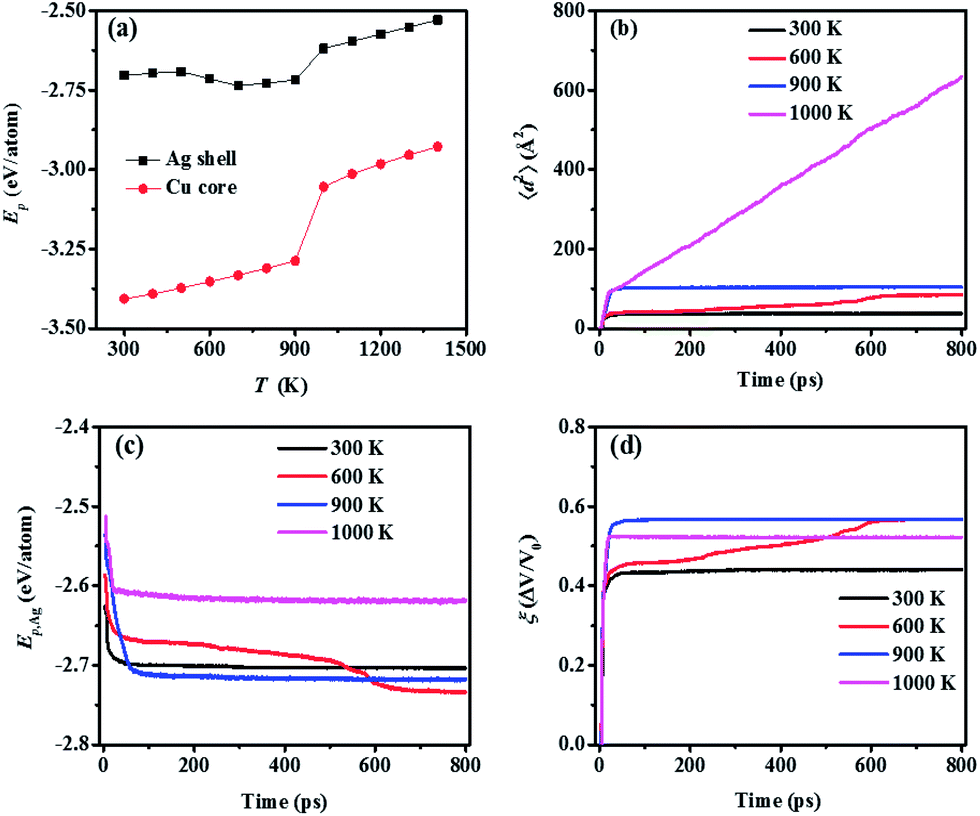 | ||
| Fig. 3 (a) Averaged potential energy (Ep) of the Ag shell and Cu core over the last 50 ps (total simulation time: 800 ps) with respect to temperature. (b) Mean square displacement (〈d2〉) of the surface atoms (atoms located within the depth of aAg) in multiple NP2-Ag5Cu2.5. (c) Potential energy of the Ag shell in NP Ag5Cu2.5 (Ep,Ag) at four representative temperatures (i.e., 300, 600, 900 and 1000 K). (d) Densification (ξ) of the sintered structure of multiple NP2-Ag5Cu2.5. The trends of the 〈d2〉, Ep,Ag, and ξ coincide well with each other. | ||
At each selected temperature, mean square displacement (〈d2〉) of surface atoms, potential energy of Ag shell (Ep,Ag), as well as densification (ξ) of the sintering system were monitored during sintering, and plotted to characterize the sintering dynamics as in Fig. 3b–d. Regardless of temperature, the agglomeration of NPs involving neck formation and fast broadening was achieved within 20 ps as evidenced by the steep slopes of the curves in Fig. 3b–d. This indicates that the initial migration of atoms is not dominated by the thermal energy of the system. Instead, the attractive forces existing between the atoms lead to initial contact only if the distance between the NPs is less than the cut-off radius of the interaction potential. The finding of the temperature-independent initial stage in multiple-CS-NP sintering coincides well with our previous two-CS-NP sintering model, as well as many other MD simulations of sintering.25,44,56–58
The sintering process after the initial agglomeration shows a distinct T-dependence. At 300 K, 〈d2〉 of the surface atoms remains constant due to insufficient kinetic energy (Ek) for diffusion, leading to the equilibrium of Ep and ξ, while at 600 K, Ep and ξ do not reach the equilibrium. Continuous densification of the multiple NPs occurs at 600 K with a moderate speed, which facilitates the observation of various sintering mechanisms. A detailed illustration of the sintering process at 600 K is shown in Fig. 4, during which the local order of each atom is identified by common neighbour analysis59–61 and categorized as (1) FCC, (2) HCP and (3) amorphous (all other local orders, including BCC).
 | ||
| Fig. 4 Cross-sectional images of the sintering process of porous multiple NP2-Ag5Cu2.5 at 600 K. Blue: Ag FCC; yellow: Ag HCP; red: Ag amorphous; green: Cu FCC; magenta: Cu HCP; cyan: Cu amorphous. | ||
We monitored the sintering behaviour at 600 K from 5 ps, at which the NPs make their initial contact (Fig. 4a). The surface and partial inner Ag atoms of the centre NP (NP C, as indicated in Fig. 4) deviate from original FCC lattice sites to form several high-energy surface layers. As NPs approach one another, surface energy is minimized, necks are created and rapidly broadened within 5 ps, and the curvatures of the pores are diminished (Fig. 4b). Due to the pore-induced curvature and packing arrangement, elastic collision behaviour (collision and then bouncing back), which is observed in the two-CS-NP sintering model, is not detected in this multiple-CS-NP model. Although the neck formation and broadening stages reduce the curvature, the strong propensity for further reducing the curvature and thus the surface energy leads to further diffusion. In addition, because of the symmetry of the multiple-CS-NP system, the NPs located on the left and right of NP-C (NP-L and NP-R, respectively) can collide elastically with NP-C from both sides at the same time, which cancels the elastic collision behaviour. From 10 ps to 100 ps, amorphized atoms in the neck region rearrange themselves, leading to recrystallization and thus contributing to further Ep reduction. This detected amorphization–recrystallization coincides with both the CS two-NP and pure two-NP sintering model, such as nickel62 and copper.25 After 100 ps, pores are gradually diminished with a moderate Ep reduction rate until 600 ps, at which several stacking faults (double HCP layers) are formed. This multiple-CS-NP sintered structure reaches quasi-equilibrium after 700 ps, and stable stacking faults are left within the structure, which is potentially eliminated by the annealing process. The reduction of Ep at 600 K in Fig. 3c, acting as a driving force for the sintering,19,63 contributes to densification comparable to that at 900 K, indicating that sintering at a lower Tpe can yield the same densification as high T. Note that the sintered structure at 600 K, even at 900 K, is not a fully densified structure, i.e., some pores are still left in the structure.
Based on the 〈d2〉 value (Fig. 3b), solid surface diffusion is observed only at 600 K after the initial neck formation and growth stage. A dominant mechanism at Troom is not the solid surface diffusion, but the plastic deformation, including stacking deformation and twin boundary formation during the neck formation and growth. At Tsm (900 K), pores in the multiple-CS-NP system are quickly eliminated due to a higher mobility of premelted surface atoms. This irreversible pore elimination locates the system in a quasi-equilibrium low-energy state; thus, the premelted atoms recrystallize without diffusion, due to insufficient kinetic energy (Ek).
Surface and bulk diffusion of atoms dominate the liquid phase sintering at Tm (1000 K). The sintering process is dominated by the inter-diffusion of melted core and shell atoms, similar to that of the two-CS-NP sintering model at Tm.29 The sintering product is an alloy structure with well mixed Cu and Ag atoms. The ξ of the final structure at 1000 K (Fig. 3d) is smaller (i.e., larger volume) than that at 900 K, and even smaller than that at 600 K, due to the thermal expansion of the alloy.
In order to validate the reliability of our results and investigate the size effect, we performed sintering simulations with identical sintering conditions, but with different NP sizes, i.e., the multiple-CS-NP sintering model with NP3-Ag8Cu4 and NP4-Ag11Cu5.5, 〈d2〉 and the corresponding final sintered structures of these two multiple-CS-NP sintering models at each temperature are shown in Fig. S2.† At Troom, it is commonly observed that the plastic deformation precedes others in the sintering process, especially at the initial stages. In the multiple-CS-NP system of NP3-Ag8Cu4, the solid surface diffusion dominates sintering at a temperature (900 K is tested for multiple-CS-NP Ag8Cu4) lower than its Tsm (Fig. S2a†). Since pores are eliminated at Tsm and a stable structure is formed (Fig. S2c†), the liquid surface diffusion at Tsm is deactivated. However, for the multiple-CS-NP model with Ag11Cu5.5, pores are not eliminated even at Tsm (1160 K, Fig. S2d†). As a result, solid surface diffusion dominates the sintering both at a lower temperature (1100 K is tested for multiple-CS-NP Ag11Cu5.5) and Tsm (Fig. S2b†).
Comparison between the sintering of Cu–Ag CS and pure Ag NPs
Sintering simulations on multiple NP1-Ag5Cu0 under the same conditions (size, temperature and pressure, etc.) as multiple NP2-Ag5Cu2.5 demonstrate that the CS NP is a more suitable candidate for higher ξ as shown in Fig. 5a–c. Since the CS and pure Ag NPs have the same overall radius, the higher ξ of CS NP is not induced by the higher mobility of surface atoms. 〈d2〉 of the surface atoms in both the CS and pure Ag NPs presents direct evidence for the similar mobility of surface atoms at Troom (black and green lines, Fig. 5d). However, the mobility of the overall shell atoms in the multiple-CS-NP model is over 60% higher than that of the pure Ag NP (blue and red lines, Fig. 5d–f), where this increased shell mobility is attributed to the lattice mismatch and interfacial restructuring between Ag and Cu atoms;64 thus, the mobility of Ag atoms in the vicinity of the Cu–Ag interface is much higher than surface atoms. As the temperature increases to 600 K, pores are not eliminated (Fig. S3†) in pure Ag NP sintering, due to a lack of sufficient solid surface diffusion (smaller 〈d2〉 of pure Ag NP than the CS NP is demonstrated in Fig. 5e). Pore elimination at 600 K in the multiple-CS-NP model suggests that the activated interfacial Ag atoms facilitate the densification. At 900 K, even though surface premelting occurs in CS NP, the 〈d2〉 of the surface atoms is smaller than the pure Ag (black and green lines, Fig. 5f). This supports the conclusion that the surface diffusion is not a dominant mechanism at Tsm in small CS NP (rcs < 8aAg), while it contributes to the densification of pure Ag NP at 900 K.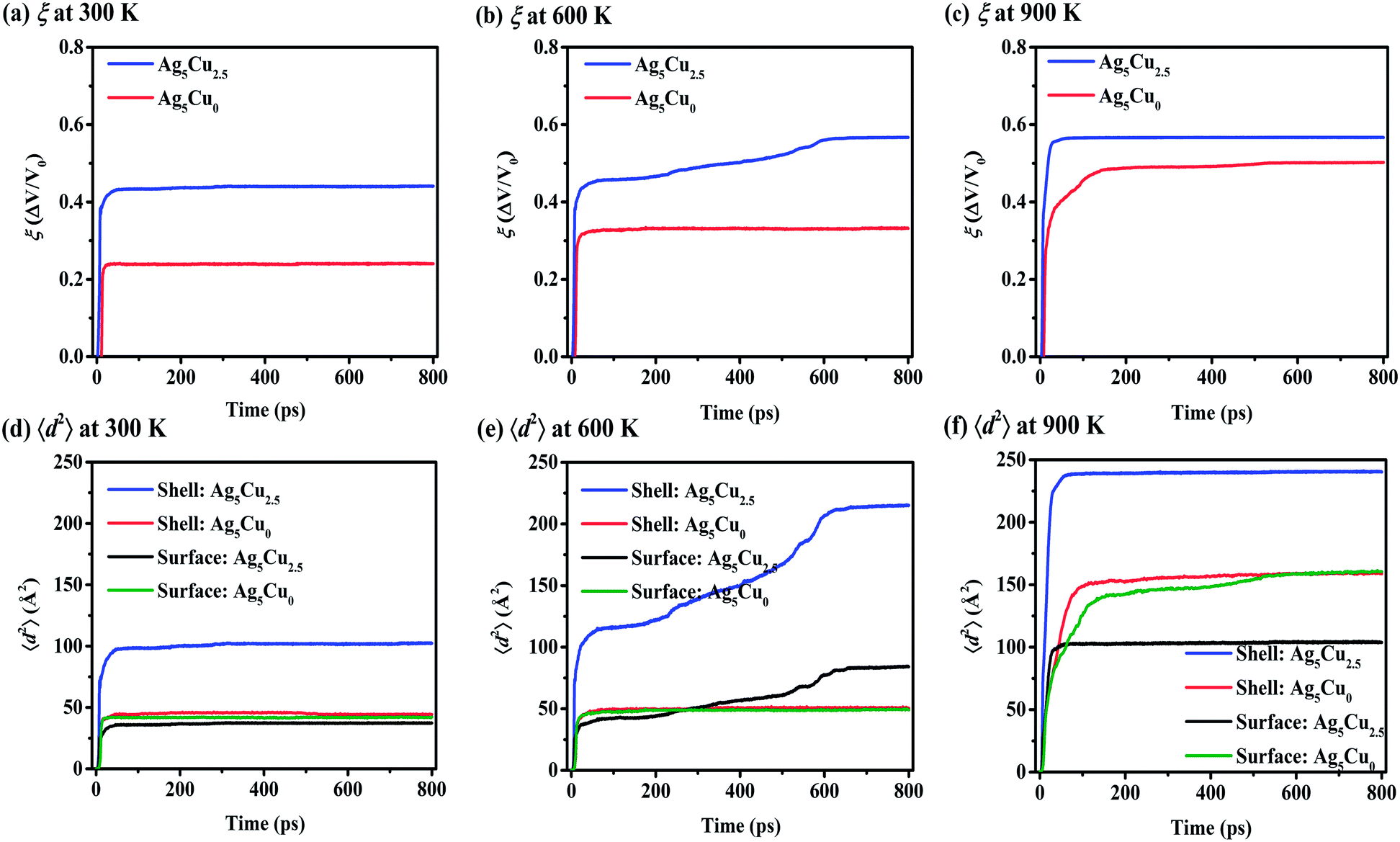 | ||
| Fig. 5 (a–c) Densification (ξ) and (d–f) mean square displacement (〈d2〉) during multiple-NP sintering with Ag5Cu2.5 and Ag5Cu0 at 300 K, 600 K, and 900 K. At each temperature, the ξ of CS NP2-Ag5Cu2.5 is higher than that of pure Ag NP1-Ag5Cu0 due to the high mobility of interfacial Ag atoms in the CS NP model. | ||
Self-diffusivity and activation energy
Self-diffusivity (Dself) characterizes atom mobility during sintering, and activation energy (Qv) is a good measure of sinterability within porous materials.23 Dself and Qv for surface and shell diffusion in the multiple-CS-NP sintering model with Ag5Cu2.5 are calculated in this research. Accurate diffusivity can be obtained only if the atoms travel a sufficient distance to make the 〈d2〉 have a linear, infinite time behaviour, which is required by the Einstein's relation. However, below Tm, the atoms vibrate around the equilibrium positions once the sintering system reaches the minimum-energy state; i.e., diffusion ends before it reaches linearity. Thus, 〈d2〉 is recorded during the diffusion period (between the initial contact of multiple NPs and the system minimum-energy state) for calculation of Dself and Qv (starting and ending points of the diffusion period are indicated by spherical dots in Fig. S4a and b†). In Fig. 6, the Qv of the shell and surface diffusion are 0.42 eV and 0.46 eV, respectively, which are in a fairly good agreement with the reported Qv for the silver {100} surface diffusion (0.4 eV).65 However, these values are smaller than the Qv of the grain boundary (84.4 kJ mol−1 = 0.87 eV)66 and lattice self-diffusion (192.1 kJ mol−1 = 1.99 eV)67 in Ag. These discrepancies can be attributed to (1) a higher surface mobility induced by the ultrafine size, and (2) a higher mobility of interfacial Ag atoms in CS structures. Note that the Qv of the overall shell is lower than that of the surface; i.e., the shell diffusion is easier to be activated, compared to the surface only. These results also coincide with our previous findings that the interfacial atoms of the Ag shell have a higher mobility than surface atoms and lead to a much higher densification in Cu–Ag CS NP, as compared to same-sized pure Ag NPs. Smaller Dself and higher Qv are expected for larger NPs due to the size effect, although significant size effect has not been reported on surface diffusivity for Ag NP in the size range of 10–40 nm.68 More detailed examination of the size dependency of the activation energy will be conducted with the MD approach in the near future.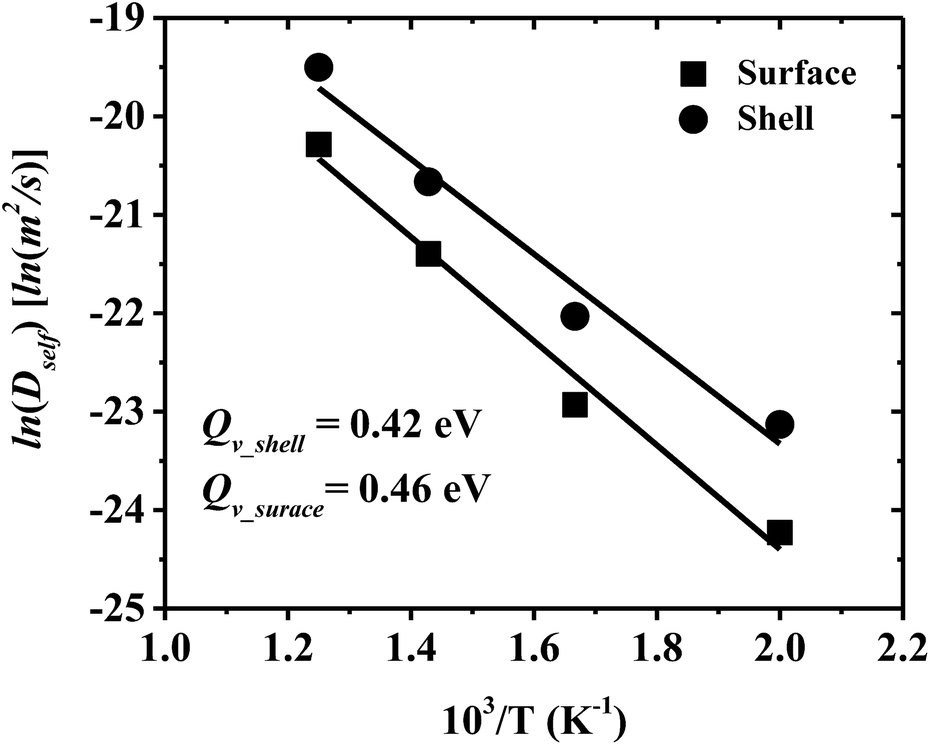 | ||
| Fig. 6 Arrhenius plot of self-diffusivity of shell and surface in the multiple-CS-NP sintering model Ag5Cu2.5. The solid line is the Arrhenius equation fitting. The activation energies obtained for the shell and surface are 0.42 eV and 0.46 eV, respectively. | ||
Mechanical properties
Sintering-temperature- and NP-size-dependent mechanical properties, including Young's modulus (E100), yield strength (σYS), and Poisson's ratio (ν100) are discussed in this section. Table 2 summarizes these properties of final structures sintered at four representative temperatures; (i) T1 = Troom, (ii) Troom < T2 < Tsm, (iii) T3 = Tsm, and (iv) T4 > Tm (i.e., 300, 600, 900, and 1000 K for both NP1-Ag5Cu0 and NP2-Ag5Cu2.5; 300, 900, 1100, and 1200 K for NP3-Ag8Cu4; 300, 1100, 1160 and 1300 K for NP4-Ag11Cu5.5). During the tensile test, isotropic shrinkage in the y and z directions is observed, demonstrating the identical Poisson's ratio in the final sintered structures. The strain–stress plots are shown in Fig. S5.†| Tsinter (K) | Young's modulus (E100, GPa) | Yield strength (σYS, GPa) | Poisson's ratio (ν100) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NP1 | NP2 | NP3 | NP4 | NP1 | NP2 | NP3 | NP4 | NP1 | NP2 | NP3 | NP4 | |
| 300 | 17.04 | 21.33 | 14.11 | 11.24 | 1.18 | 1.83 | 1.30 | 1.04 | 0.12 | 0.30 | 0.26 | 0.24 |
| 600 | 21.72 | 33.64 | — | — | 1.63 | 2.55 | — | — | 0.23 | 0.37 | — | — |
| 900 | 33.15 | 38.51 | 22.12 | — | 2.62 | 2.84 | 2.04 | — | 0.38 | 0.39 | 0.29 | — |
| 1000 | 27.72 | 33.22 | — | — | 1.63 | 1.51 | — | — | 0.40 | 0.41 | — | — |
| 1100 | — | — | 40.34 | 21.43 | — | — | 3.73 | 1.71 | — | — | 0.39 | 0.28 |
| 1160 | — | — | — | 23.37 | — | — | — | 2.13 | — | — | — | 0.31 |
| 1200 | — | — | 28.13 | — | — | — | 1.87 | — | — | — | 0.41 | — |
| 1300 | — | — | — | 34.05 | — | — | — | 1.68 | — | — | — | 0.41 |
Through scrutinized comparison and analysis, we have the following findings:
(1) Below Tm, all three properties (E100, σYS, and ν100) of the sintered structures by multiple NP1-Ag5Cu0 are smaller than the counterparts sintered by multiple NP2-Ag5Cu2.5. NP1 has a smaller elastic resistance, and its maximum force, which NP can bear for recovering the original shape, is also smaller; however, the resistance in the orthogonal directions of the strain are larger. Note that all of these properties also increase with sintering temperature, regardless of the NP size. This suggests that the pore narrowing and elimination in the final sintered product enhance its resistance to elastic deformation in the elongation direction, and the maximum force, while decreasing the resistance of deformation in the shrinkage directions.
(2) In general, all properties tend to decrease as the nanoscale grain size increases below Tsm. E100 of nanocrystalline materials increases with decreasing the grain size, which contradicts the previous study.69 However, the porosity of the larger grain sized NP-sintered structure is also higher than the smaller grain-sized structure (even though the sintering temperature of the larger grain sized structure is higher), and the porosity has a substantially greater effect than grain size (larger porosity induces smaller E100).69 Therefore, a more dominant porosity effect leads to a smaller E100 in larger grain sized structures. The grain-size dependency of σYS and ν100 coincides well with previous experimental and theoretical results.70,71
(3) For the sintered structure above Tm, E100 and σYS are smaller than those at Tsm, except for the E100 of sintered NP4-Ag11Cu5.5. Pores in the sintered structure of multiple NP4-Ag11Cu5.5 are not eliminated at Tsm, leading to a smaller E100, compared to that at the temperature (1300 K) above Tm. Quenching has been executed before the tensile test with rates of 7 × 1012 K s−1, 9 × 1012 K s−1, and 1013 K s−1 for sintered multiple NP1-Ag5Cu0 and NP2-Ag5Cu2.5, NP3-Ag8Cu4, and NP4-Ag11Cu5.5, respectively, all of which are a few orders of magnitude higher than the critical quenching rate for glass formation (105 to 106 K s−1).72,73 The Ep evolutions during quenching (Fig. S6†) also indicate that all three sintered structures form metallic glass, without an abrupt decrease in the Ep curve.74,75 The formation of the metallic glass yields lower E100 and σYS, compared to those of nanostructured porous materials. Contrarily, ν100 increases with temperature, regardless of whether the quenched structure is crystallized or metallic glass, which is different from the decrease of E100 and σYS at temperatures above Tm. A uniform ν100 of 0.41 is obtained for all metallic glass structures, manifesting the identical resistance in shrinkage direction.
Thermodynamic properties
Thermodynamic properties (i.e., βT, αp, and cv) calculated from our simulations are summarized in Table 3. The response of the volumetric change to pressure is more significant in a sintered structure with a larger porosity to yield a higher isothermal compressibility (βT). At Troom, all four sintered structures possess large porosities, which induce large βT (0.099, 0.053, 0.068, and 0.095 GPa−1 for NP1, NP2, NP3, and NP4 sintered structures, respectively), while βT decreases with increasing Tsinter due to pore narrowing and elimination. Differing from sintered NP3 and NP4, βT of the molten sintered structures of NP1 and NP2 are 0.018 GPa−1and 0.015 GPa−1, both of which are larger than those at Tsm of 900 K (0.017 GPa−1and 0.010 GPa−1, respectively). The sintered structures by multiple NP3's and NP4's still contain pores at Tsm (Fig. S2d†), but not at Tm. Thus, the βT at Tsm is larger than for the molten sintered structures of NP3 and NP4. On the other hand, pores in NP1 and NP2 are completely eliminated above Tsm, so we attribute the larger βT above Tsm to its lower crystallinity instead of porosity, which results in higher entropy and greater compressibility. This analysis concludes that porosity dominates the decreasing trend of βT in the NP-sintered structure below Tsm, while the dominant factor switches to crystallinity once pores are completely eliminated.| Tsinter (K) | Isothermal compressibility (βT, GPa−1) | Coefficient of thermal expansion [(αp × 105), K−1] | Specific heat capacity (cv, kJ kg−1 K−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NP1 | NP2 | NP3 | NP4 | NP1 | NP2 | NP3 | NP4 | NP1 | NP2 | NP3 | NP4 | |
| 300 | 0.099 | 0.053 | 0.068 | 0.095 | 5.50 | 6.43 | 6.69 | 6.48 | 0.051 | 0.134 | 0.122 | 0.122 |
| 600 | 0.068 | 0.014 | — | — | 6.07 | 6.50 | — | — | 0.055 | 0.122 | — | — |
| 900 | 0.017 | 0.010 | 0.041 | — | 7.40 | 7.26 | 5.90 | — | 0.056 | 0.111 | 0.150 | — |
| 1000 | 0.018 | 0.015 | — | — | 15.40 | 12.03 | — | — | 0.052 | 0.149 | — | — |
| 1100 | — | 0.009 | 0.049 | — | 7.14 | 6.848 | — | 0.128 | 0.167 | |||
| 1160 | — | — | 0.038 | — | — | 6.14 | — | — | 0.170 | |||
| 1200 | — | 0.008 | — | — | 12.02 | — | — | 0.132 | — | |||
| 1300 | — | — | 0.009 | — | — | 11.89 | — | — | 0.128 | |||
αp exhibits porosity and grain-size independence, i.e., no obvious difference is observed for all NP-sintered structures below Tsm. A uniform αp (∼1.2 × 10−4 K−1) is achieved for all amorphous Cu–Ag alloy structure, which is ∼75% higher than the αp of the crystallized structure. Thus, the higher entropy of the less-crystallized melted structure induces not only more compressibility, but also more expansivity. Similar to αp, no obvious dependency on porosity, grain size and crystallinity is detected for cv. Values ranging from 0.111 to 0.170 kJ kg−1 K−1 are obtained for all CS NP-sintered structures, while a much smaller averaged cv for the pure Ag NP sintered structure is calculated as 0.054 kJ kg−1 K−1, induced by the larger atomic mass of Ag. The weak bonding existing between the Cu and Ag atoms may also contribute to higher cv in CS sintered structures.
Conclusions
We report the sintering differences and similarities between (1) the different-sized multiple-CS-NP model, (2) the multiple Cu–Ag CS NPs and two Cu–Ag CS NPs models, and (3) the multiple Cu–Ag CS NPs and multiple Ag NPs models. The interplay among porosity, grain size, and crystallinity on the mechanical and thermodynamic properties of NP-sintered structures are also unravelled. The main conclusions drawn are as follows:1. Differing from the Cu–Ag two-CS-NP sintering model, this research on the sintering of the multiple-CS-NP model exhibits an accurate description of agglomeration and pore elimination. Simultaneous interactions with multiple particles accelerate the sintering process in simulations.
2. For smaller CS NPs (rc < 8aAg), solid surface diffusion dominates sintering at an intermediate temperature (Troom < T < Tsm), while plastic deformation plays a significant role at the Troom; liquid surface diffusion is deactivated after recrystallization at Tsm. Solid surface diffusion can induce continuous pore narrowing and elimination at Tsm in larger NPs (rc > 11aAg).
3. Activated interfacial atoms induced by lattice mismatch and interfacial interaction contribute to a higher densification, and thus a higher bonding strength in multiple CS NPs sintered structures, compared to the pure Ag NP sintered structures.
4. E100, σYS, and ν100 of the NP-sintered structure under Tsm are positively correlated with grain size, but negatively correlated with porosity. In general, the metallic glass structure yields a lower E100 and σYS, but identical ν100, compared with nanoporous crystallized structures. In terms of thermodynamics properties, the βT also has a negative dependency on porosity while the αp and cv are independent of porosity and grain size.
This research illustrates a more realistic sintering scheme for the multiple-CS-NP model at various critical temperatures than the two-CS-NP sintering model. The size and temperature dependency of the properties of the final sintered structures are investigated to provide a theoretical basis and roadmap for selecting suitable NP size and temperature to meet specific property requirements. These findings corroborate our previous research on the sintering dynamics of the Cu–Ag two-CS-NP model. Further simulations for the effects of relative crystallographic orientation and size distribution should be performed to gain an integrative insight into Cu–Ag CS NP sintering.
Acknowledgements
This work utilized the resources of Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant number ACI-1053575. We are thankful for the fruitful insight and discussions with Dr Anming Hu. J.W. appreciates the great efforts of Drew C. Marable and Ali Yousefzadi Nobakht in professional assistance to significantly improve the quality of this manuscript. J.W. also acknowledges helpful discussions with Dr Delong Ma, Denzel Bridges, Yongchao Yu, Shutong Wang, Chaoli Ma, Suhong Zhang, and Zhiming Liu, regarding the topic of scientific writing.References
- S. Ding, Y. Tian, Z. Jiang and X. He, AIP Adv., 2015, 5, 057120 CrossRef.
- F. Gao, S. Mukherjee, Q. Cui and Z. Gu, J. Phys. Chem. C, 2009, 113, 9546–9552 CAS.
- K. N. Thakkar, S. S. Mhatre and R. Y. Parikh, Nanomedicine, 2010, 6, 257–262 CrossRef CAS PubMed.
- P. Peng, A. Hu, A. P. Gerlich, G. Zou, L. Liu and Y. N. Zhou, ACS Appl. Mater. Interfaces, 2015, 7, 12597–12618 CAS.
- L. Lin, L. Liu, P. Peng, G. Zou, W. W. Duley and Y. N. Zhou, Nanotechnology, 2016, 27, 125201 CrossRef PubMed.
- B. Buesser, A. J. Grohn and S. E. Pratsinis, J. Phys. Chem. C, 2011, 115, 11030–11035 CAS.
- R. Strobel and S. E. Pratsinis, J. Mater. Chem., 2007, 17, 4743–4756 RSC.
- S. Magdassi, M. Grouchko, O. Berezin and A. Kamyshny, ACS Nano, 2010, 4, 1943–1948 CrossRef CAS PubMed.
- T. H. J. VanOsch, J. Perelaer, A. W. M. DeLaat and U. S. Schubert, Adv. Mater., 2008, 20, 343–345 CrossRef.
- S. Sivaramakrishnan, P. J. Chia, Y. C. Yeo, L. L. Chua and P. K. Ho, Nat. Mater., 2007, 6, 149–155 CrossRef CAS PubMed.
- D. Kim, S. Jeong, B. K. Park and J. Moon, Appl. Phys. Lett., 2006, 89, 264101 CrossRef.
- W. Xie, Y. Zheng, J. Kuang, Z. Wang, S. Yi and Y. Deng, Russ. J. Phys. Chem. A, 2016, 90, 848–855 CrossRef CAS.
- H. T. Hai, H. Takamura and J. Koike, J. Alloys Compd., 2013, 564, 71–77 CrossRef CAS.
- M. Tsuji, S. Hikino, Y. Sano and M. Horigome, Chem. Lett., 2009, 38, 518–519 CrossRef CAS.
- M. Miyakawa, N. Hiyoshi, M. Nishioka, H. Koda, K. Sato, A. Miyazawa and T. M. Suzuki, Nanoscale, 2014, 6, 8720–8725 RSC.
- J. Sarkar, M. Bhattacharyya, R. Kumar, N. Mandal and M. Mallik, Adv. Sci. Lett., 2016, 22, 193–196 CrossRef.
- J. Xu, R. Sakanoi, Y. Higuchi, N. Ozawa, K. Sato, T. Hashida and M. Kubo, J. Phys. Chem. C, 2013, 117, 9663–9672 CAS.
- A. Hu, J. Y. Guo, H. Alarifi, G. Patane, Y. Zhou, G. Compagnini and C. X. Xu, Appl. Phys. Lett., 2010, 97, 153117 CrossRef.
- E. Marzbanrad, A. Hu, B. Zhao and Y. Zhou, J. Phys. Chem. C, 2013, 117, 16665–16676 CAS.
- S. Jiang, Y. Zhang, Y. Gan, Z. Chen and H. Peng, J. Phys. D: Appl. Phys., 2013, 46, 335302 CrossRef.
- P. Grammatikopoulos, C. Cassidy, V. Singh, M. Benelmekki and M. Sowwan, J. Mater. Sci., 2013, 49, 3890–3897 CrossRef.
- B. J. Henz, T. Hawa and M. Zachariah, Mol. Simul., 2009, 35, 804–811 CrossRef CAS.
- K. Nakao, T. Ishimoto and M. Koyama, J. Phys. Chem. C, 2014, 118, 15766–15772 CAS.
- B. Cheng and A. H. W. Ngan, Comput. Mater. Sci., 2013, 74, 1–11 CrossRef CAS.
- P. Zeng, S. Zajac, P. C. Clapp and J. A. Rifkin, Mater. Sci. Eng., A, 1998, 252, 301–306 CrossRef.
- J. S. Raut, R. B. Bhagat and K. A. Fichthorn, Nanostruct. Mater., 1998, 10, 837–851 CrossRef CAS.
- J. Xu, S. Bai, Y. Higuchi, N. Ozawa, K. Sato, T. Hashida and M. Kubo, J. Mater. Chem. A, 2015, 3, 21518–21527 CAS.
- J. Wang and S. Shin, J. Nanopart. Res., 2017, 19, 53 CrossRef.
- J. Wang, S. Shin and A. Hu, J. Phys. Chem. C, 2016, 120, 17791–17800 CAS.
- R. M. German, Sintering Theory and Practice, Wiley-Interscience, New York, 1996 Search PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- J. Towns, T. Cockerill, M. Dahan, I. Foster, K. Gaither, A. Grimshaw, V. Hazlewood, S. Lathrop, D. Lifka, G. D. Peterson, R. Roskies, J. R. Scott and N. Wilkins-Diehr, Comput. Sci. Eng., 2014, 16, 62–74 CrossRef.
- M. S. Daw and M. I. Baskes, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 6443–6453 CrossRef CAS.
- P. L. Williams, Y. Mishin and J. C. Hamilton, Modell. Simul. Mater. Sci. Eng., 2006, 14, 817–833 CrossRef CAS.
- L. Verlet, Phys. Rev., 1967, 159, 98–103 CrossRef CAS.
- S. Li, W. Qi, H. Peng and J. Wu, Comput. Mater. Sci., 2015, 99, 125–132 CrossRef CAS.
- P. Song and D. Wen, J. Phys. Chem. C, 2010, 114, 8688–8696 CAS.
- R. Huang, Y. Wen, Z. Zhu and S. Sun, J. Phys. Chem. C, 2012, 116, 11837–11841 CAS.
- Z. Yang, X. Yang, Z. Xu and S. Liu, Phys. Chem. Chem. Phys., 2009, 11, 6249–6255 RSC.
- J. Tang and J. Yang, J. Nanopart. Res., 2015, 17, 299 CrossRef.
- Z. Yang, X. Yang and Z. Xu, J. Phys. Chem. C, 2008, 112, 4937–4947 CAS.
- S. Alavi and D. L. Thompson, J. Phys. Chem. A, 2006, 110, 1518–1523 CrossRef CAS PubMed.
- Y. Tamura and N. Arai, Mol. Simul., 2015, 41, 905–912 CrossRef CAS.
- P. Song and D. Wen, J. Nanopart. Res., 2009, 12, 823–829 CrossRef.
- K. B. Wiberg, J. Am. Chem. Soc., 1965, 87, 1070–1078 CrossRef CAS.
- D. Keffer and P. Adhangale, Chem. Eng. J., 2004, 100, 51–69 CrossRef CAS.
- R. Xiong, K. Odbadrakh, A. Michalkova, J. P. Luna, T. Petrova, D. J. Keffer, D. M. Nicholson, M. A. Fuentes-Cabrera, J. P. Lewis and J. Leszczynski, Sens. Actuators, B, 2010, 148, 459–468 CrossRef CAS.
- A. Mashreghi, Comput. Mater. Sci., 2012, 62, 60–64 CrossRef CAS.
- M. H. Ghatee and K. Shekoohi, Fluid Phase Equilib., 2013, 355, 114–122 CrossRef CAS.
- V. M. Dadarlat and C. B. Post, J. Phys. Chem. B, 2001, 105, 715–724 CrossRef CAS.
- M. Kaviany, Heat Transfer Physics, Cambridge University Press, New York, 2014 Search PubMed.
- R. Z. Li, A. Hu, D. Bridges, T. Zhang, K. D. Oakes, R. Peng, U. Tumuluri, Z. Wu and Z. Feng, Nanoscale, 2015, 7, 7368–7377 RSC.
- G. Guenther and O. Guillon, J. Mater. Sci., 2014, 49, 7915–7932 CrossRef CAS.
- Y. Shibuta and T. Suzuki, Chem. Phys. Lett., 2007, 445, 265–270 CrossRef CAS.
- J. G. Hou, B. Wang, J. Yang, K. Wang, W. Lu, Z. Li, H. Wang, D. M. Chen and Q. Zhu, Phys. Rev. Lett., 2003, 90, 246803 CrossRef CAS PubMed.
- A. Dutta, Rev. Adv. Mater. Sci., 2014, 39, 25–33 Search PubMed.
- H. Zhu, Philos. Mag. Lett., 1996, 73, 27–33 CrossRef CAS.
- S. Arcidiacono, N. R. Bieri, D. Poulikakos and C. P. Grigoropoulos, Int. J. Multiphase Flow, 2004, 30, 979–994 CrossRef CAS.
- G. J. Ackland and A. P. Jones, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 054104 CrossRef.
- W. Lechner and C. Dellago, J. Chem. Phys., 2008, 129, 114707 CrossRef PubMed.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2012, 20, 045021 CrossRef.
- M. Tavakol, M. Mahnama and R. Naghdabadi, Mater. Manuf. Processes, 2015, 30, 1397–1402 CrossRef CAS.
- Z. Z. Fang and H. Wang, Int. Mater. Rev., 2013, 53, 326–352 CrossRef.
- S. Shin, M. Kaviany, T. Desai and R. Bonner, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081302 CrossRef.
- G. Antczak and G. Ehrlich, Surface Diffusion: Metals, Metal Atoms, and Clusters, Cambridge University Press, New York, 2010 Search PubMed.
- E. A. Oliber, C. Cugno, M. Moreno, M. Esquivel, N. Haberkorn, J. E. Fiscina and C. J. R. G. Olivera, Matéria, 2003, 8, 350–357 Search PubMed.
- R. E. Hoffman and D. Turnbull, J. Appl. Phys., 1951, 22, 634–639 CrossRef CAS.
- M. A. Asoro, P. J. Ferreira and D. Kovar, Acta Mater., 2014, 81, 173–183 CrossRef CAS.
- H. S. Kim and M. B. Bush, Nanostruct. Mater., 1999, 11, 361–367 CrossRef CAS.
- N. Wang, Z. Wang, K. T. Aust and U. Erb, Acta Metall. Mater., 1995, 43, 519–528 CrossRef CAS.
- T.-Y. Kim, J. E. Dolbow and E. Fried, Int. J. Solids Struct., 2012, 49, 3942–3952 CrossRef.
- Q. Li, Mater. Lett., 2007, 61, 3323–3328 CrossRef CAS.
- D. V. Louzguine-Luzgin, T. Saito, J. Saida and A. Inoue, J. Mater. Res., 2011, 23, 515–522 CrossRef.
- X. Cheng, J. Zhang, H. Zhang and F. Zhao, J. Appl. Phys., 2013, 114, 084310 CrossRef.
- J. Wang, P. D. Hodgson, J. Zhang, W. Yan and C. Yang, J. Mater. Process. Technol., 2009, 209, 4601–4606 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Final morphology of sintered multiple-CS-NP model Ag5Cu2.5 at different temperatures (Fig. S1); mean square displacement and cross-sectional images of multiple-CS-NP model with Ag8Cu4 and Ag11Cu5.5 (Fig. S2); final sintered structure of multiple Ag5Cu0 NPs at 600 K (Fig. S3); mean square displacement of the surface and shell atoms during the sintering of multiple Ag5Cu2.5 NPs (Fig. S4); stress–strain plots for four sintered structures (Fig. S5); potential energy evolution during the quenching process of the sintered structures (Fig. S6); isothermal compressibility of four sintered structures (Fig. S7); coefficient of thermal expansion of four sintered structures (Fig. S8). See DOI: 10.1039/c7ra02611k |
| This journal is © The Royal Society of Chemistry 2017 |