
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive dechlorination of endosulfan isomers and its metabolites by zero-valent metals: reaction mechanism and degradation products†
Swatantra P. Singh‡
 and
Purnendu Bose*
and
Purnendu Bose*
Department of Civil Engineering, Indian Institute of Technology, Kanpur-208016, India. E-mail: pbose@iitk.ac.in; Tel: +91-512-2597403
First published on 24th May 2017
Abstract
The widely used organochlorine pesticide endosulfan (ES) is extremely toxic to fishes, other aquatic species and mammals. Current evidence suggests that all known natural attenuation residues of ES, i.e., ES-metabolites, retain the original chlorinated skeleton of ES and are potential carcinogens. The objective of the present study was to identify the dechlorinated products formed during the reduction of ES-isomers, i.e., endosulfan-1 and endosulfan-2 and ES-metabolites, namely endosulfan sulfate (ES-S), endosulfan lactone (ES-L), endosulfan ether (ES-E) and endosulfan alcohol (ES-A), by various metallic surfaces. During dechlorination by nano zero-valent iron (NZVI), the mass spectra of the degradation products were consistent with the loss of one, two and three chlorine atoms from the parent molecules. During the interaction of ES-E with Mg0, the degradation products had mass spectra consistent with the loss of 1–6 chlorine atoms from the parent molecule. In all cases, dechlorination appears to occur through sequential electron transfer at the metallic surface. Tentative chemical structures for various degradation products were proposed. Synthesis of some of the observed degradation products was possible using ES-E as the starting molecule. 1H NMR, crystallographic (X-ray diffraction) and GC-MS analysis of the synthesized products provide final confirmation of the proposed chemical structures. A pathway for stepwise reductive dechlorination of ES-isomers and metabolites is proposed. Some of the degradation products identified in the present study being less chlorinated, may be less toxic and more amenable to subsequent biodegradation and ultimate mineralization in the natural environment.
1. Introduction
The organochlorine pesticide endosulfan (ES) has been widely used since the 1950s.1,2 Commercial grade ES is available as a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of the two isomers, endosulfan-1 (ES-1) and endosulfan-2 (ES-2). ES is an endocrine disrupter with a primary effect on the central nervous system (CNS). It is very toxic to fishes and other aquatic species3–5 and also reported to cause mammalian gonadal toxicity, genotoxicity, and neurotoxicity.6,7 ES-isomers were included in the list of POPs (Persistent Organic Pollutants) in the Stockholm convention of 2011.8 Natural attenuation of ES through hydrolysis, photo-degradation and biodegradation leads to the formation of degradation products, i.e., ES-metabolites (Fig. 1), such as, endosulfan sulfate (ES-S), endosulfan lactone (ES-L), endosulfan ether (ES-E) and endosulfan alcohol (ES-A).9–14 All known natural attenuation residues of ES retain the original chlorinated ring structure of ES and are toxic and potential carcinogens.15 There is no concrete evidence in literature that natural attenuation through chemical or aerobic/anaerobic biological processes can disrupt the chlorinated ring structure of ES, i.e., the ES skeleton. Studies using C14-labelled ES have shown that the rate of mineralization of ES by natural attenuation processes is negligibly small.9,16
:3 mixture of the two isomers, endosulfan-1 (ES-1) and endosulfan-2 (ES-2). ES is an endocrine disrupter with a primary effect on the central nervous system (CNS). It is very toxic to fishes and other aquatic species3–5 and also reported to cause mammalian gonadal toxicity, genotoxicity, and neurotoxicity.6,7 ES-isomers were included in the list of POPs (Persistent Organic Pollutants) in the Stockholm convention of 2011.8 Natural attenuation of ES through hydrolysis, photo-degradation and biodegradation leads to the formation of degradation products, i.e., ES-metabolites (Fig. 1), such as, endosulfan sulfate (ES-S), endosulfan lactone (ES-L), endosulfan ether (ES-E) and endosulfan alcohol (ES-A).9–14 All known natural attenuation residues of ES retain the original chlorinated ring structure of ES and are toxic and potential carcinogens.15 There is no concrete evidence in literature that natural attenuation through chemical or aerobic/anaerobic biological processes can disrupt the chlorinated ring structure of ES, i.e., the ES skeleton. Studies using C14-labelled ES have shown that the rate of mineralization of ES by natural attenuation processes is negligibly small.9,16
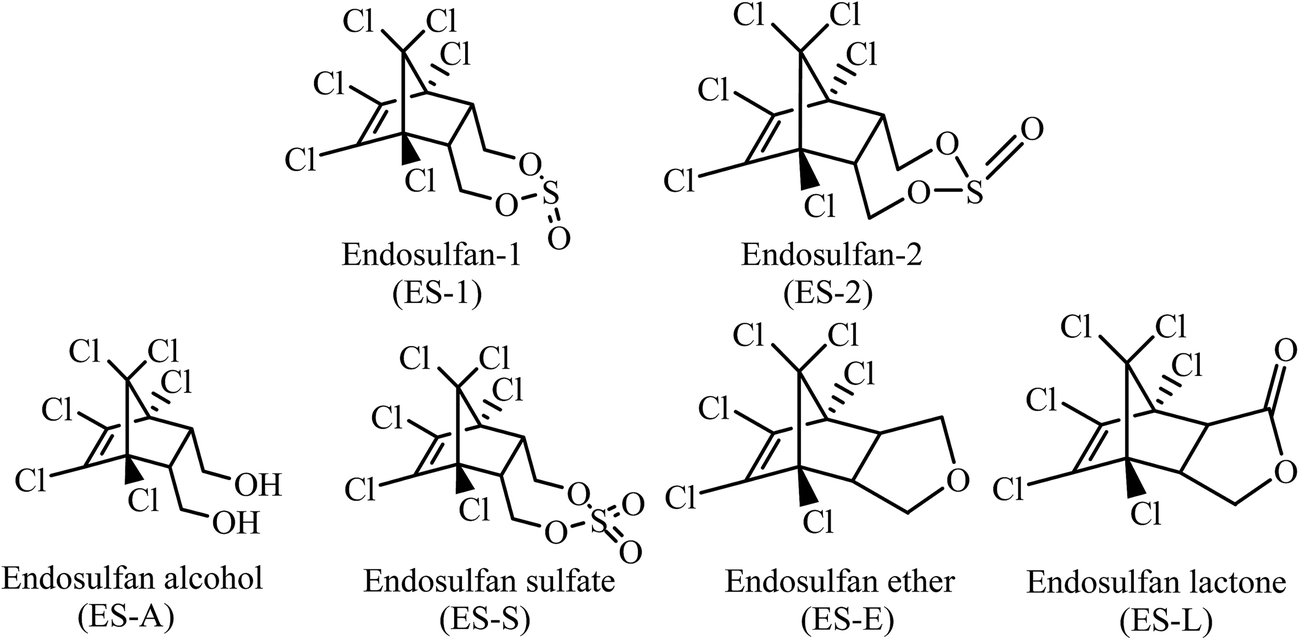 | ||
| Fig. 1 Chemical structures of ES-isomers and metabolites. | ||
Henschler17 did a comprehensive review of the toxicity of poly-chlorinated compounds and concluded that the toxicity of poly-chlorinated compounds was directly related to the number of chlorine atoms in the compounds.18 Hydrophobicity and stability of chlorinated compounds generally increases with chlorine atoms19,20 and this leads to higher accumulation and persistency of poly-chlorinated compounds in the environment. A computational study by Zahedi-Tabrizi and Farahati (2011) reported an increase in the strength of intermolecular hydrogen bonding with increase in the number of chlorine atoms in a compound.21 Biodegradation of poly-chlorinated compounds is difficult because of their stability and toxic nature.22,23 Furukawa et al. related the degree of chlorination in 31 isomers of poly-chlorinated biphenyl (PCBs) to their biodegradation potential and found an inverse relation between biodegradation potential and the number of chlorine atoms in the molecule.22,23
Based on the above information it may be hypothesized that dechlorination of ES-isomers and ES-metabolites produces degradation products, which owing to their lower chlorine content may be less toxic and more amenable to subsequent biodegradation and eventual mineralization. Hence, there is a need to study the methods for dechlorination of ES-isomers and ES-metabolites, identify the resultant degradation products and screen the identified degradation products for toxicity and biodegradation potential.
Zero-valent iron (ZVI) has been used as an effective agent for dechlorination of many toxic organic compounds.24–29 Nano-scale zero valent iron (NZVI) particles are generally more effective than micron-scale zero valent iron (MZVI) particles for degradation of pollutants.30–32 ZVI interaction with chlorinated organic compounds in reducing environment leads to dechlorination either via beta elimination or sequential elimination of chlorine molecules from the chlorinated compound.33 Many other metals, namely Pd0, Ni0, Mg0 etc. and bi-metallic alloys, such as Ni0–Fe0 or Pd0–Fe0 etc. particles have also been used for reductive dechlorination of chlorinated pollutants.34,35 Bimetallic nano-particles, i.e., NZVI doped with other trace metals have been used by many researchers for the degradation of chlorinated pollutants.36,37 The trace constituents, such as metals like palladium (Pd), platinum (Pt), silver (Ag) or nickel (Ni) may enhance the reactivity of NZVI particles. The presence of a noble metal controls the passivation of iron surface besides acting as a catalyst,26,34,36–41 thus increasing the effectiveness of reductive dechlorination of HOCs.
The main objective of present study was to suggest a mechanism for reductive dechlorination of ES-isomers and ES-metabolites through interaction with NZVI/Mg0. The main tasks involved monitoring of the evolution of degradation products during the interactions of ES-isomers and ES-metabolites with NZVI (Fe0) and Mg0; tentative identification of some of the degradation products using mass spectroscopy, proton NMR and X-ray crystallography and chemical synthesis and characterization of some of the degradation products were also carried out.
2. Materials & methods
2.1. Chemicals and glassware
Technical standards of ES-1, ES-2, ES-S, ES-A, ES-L, ES-E and 2,4,5,6-tetrachloro-m-xylene were obtained from Sigma-Aldrich, India. Technical grade ES were obtained from United Phosphorous Ltd., Ankleshwar, India. Thin layer chromatography in 7:3 hexane–acetone medium was performed to isolate ES-1 and ES-2 from technical grade ES. The purity of ES-1 and ES-2 thus isolated was ∼99% when checked against the standards using GC-MS.
Solvents used for sample preparation and extraction, i.e., ethyl acetate, n-hexane, acetonitrile and acetone (>99% purity, HPLC grade) were procured from Merck, India. Sodium hydroxide pellets (98%), anhydrous sodium sulfate (99% purity), ferric chloride heptahydrate (96% purity), sodium borohydride (97% purity), soluble starch (>99.5% purity), Mg0 metal powder (99%), glacial acetic acid (99.7% purity), and hydrochloric acid (AR Grade) were procured from Loba Chemicals, India; sodium sulfate anhydrous (99%) and zinc sulfate (99%) were obtained from Thomas Baker Chemical Limited, India. Triethyl amine (>98% purity), methane sulfonyl chloride (mesyl chloride) (>99% purity) and ZnCl2 (95% purity) were purchased from Fisher Scientific, India; Na metal cubes were procured form Sigma-Aldrich, India; NZVI was synthesized and characterized as described in earlier studies.42,43
Borosilicate glass vials (40 mL) equipped with screw caps and Teflon faced re-sealable septa were used for various experiments. GC vials of 2 mL capacity with 11 mm PTFE rubber lined aluminium seal were used for the storage of extracted samples prior to analysis. Both types of vials were procured from Wheaton Science, Millville, NJ, USA.
2.2. Experiments with NZVI
The chemical structures of ES-isomers and ES-metabolites used in this study are as shown in Fig. 1. Concentrated stock solutions of ES-1, ES-2, ES-S, ES-L, ES-E and ES-A were prepared in acetone. For experiments with each compound, 40 mL vials was filled with 0.1 g L−1 aqueous NZVI suspension and the required volume of stock solution was added to each vial. All such vials were capped tightly and put on a rotating shaker at 30 rpm for mixing; control vials with no NZVI were also prepared. All operations were carried out in a glove box and in an atmosphere of nitrogen. Vials were removed in duplicate (along with one control) at specified time intervals for analysis. Ambient temperature was approximately 31 ± 3 °C and the pH maintained between 8.5–8.9 in these vials.2.3. Synthesis of endosulfan ether
In addition to ES-1 and ES-2, ES-E was also required in substantial quantities during this study. ES-E was synthesized from technical grade ES by a two-step process as shown in Fig. 2. In the first step, 5 g of technical grade ES was dissolved in 50 mL of 1:1 acetone–water mixture. Next, 0.1 mol of NaOH dissolved in 10 mL of deionised water was added to the mixture followed by stirring for 12 hours at 40 °C. The resulting mixture was extracted twice with 100 mL aliquots of ethyl acetate. Ethyl acetate was evaporated at 60 °C under high vacuum, leaving a white residue. This residue was purified by column chromatography and eluted with ethyl acetate/hexane. The solvent was evaporated yielding 3.55 g of white powder, which was confirmed to be 97% pure ES-A by GC-ECD analysis. In the next step, 2.0 g ES-A powder was dissolved in 50 mL of chloroform containing 7.0 mmol of triethylamine and the solution was stirred for 1 hour at room temperature (27 ± 3 °C). Thereafter 7.0 mmol of mesyl chloride was added slowly to the solution, followed by further stirring for 16 hours at room temperature. The organic phase was washed using NaHCO3 solution and dried under high vacuum. The dried white crystalline solid obtained was further purified by column chromatography and extracted with mixture of ethyl acetate and hexane. The solvent was evaporated yielding 1.63 g of white powder, which was confirmed to be 98% pure ES-E by GC-ECD analysis in comparison with ES-E standard.
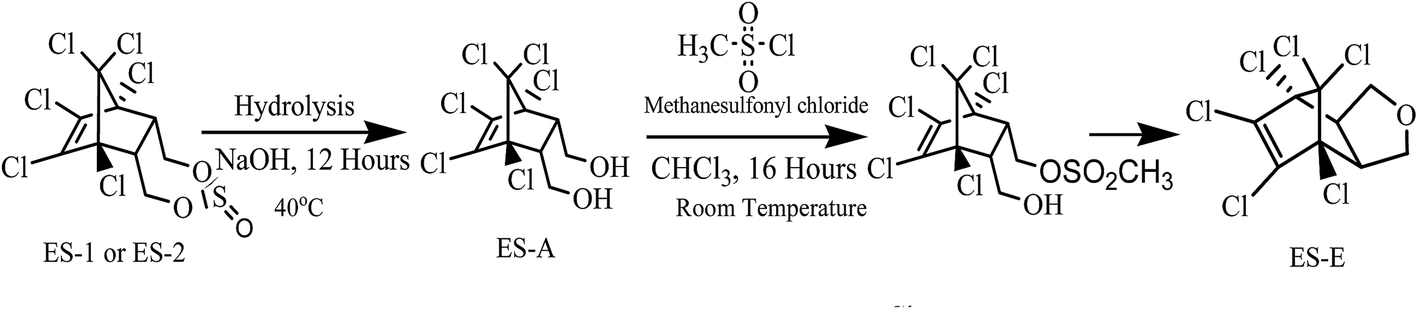 | ||
| Fig. 2 Synthesis of (a) ES-E and (b–d) dechlorinated degradation products of ES-E. | ||
2.4. Experiments with Mg0and Na0
Experiments involving interaction between ES-E and Mg0 were carried out in 40 mL vials as per established procedure;35 2 g Mg0 powder, 50 mg L−1 ZnCl2, 4 mL of acetone and 1 mg L−1 of ES-E were added to the vial and the solution volume made up to 20 mL with deionised water. 0.5 mL of glacial acetic acid was slowly added to the mixture with constant manual shaking. The vials were tightly capped and after 30 minutes, were put on the rotor at 30 rpm for 2 hours. Vials were removed in duplicate (along with one control) at various time intervals for analysis. Ambient temperature was approximately 31 ± 3 °C. For experiments involving interaction between ES-E and Na0, 1.5 g ES-E was dissolved in 50 mL of ethanol and refluxed with 0.4 mol of Na0 metal for three hours at 100 °C.2.5. Extraction methods
The procedure for the extraction of ES-isomers and ES-metabolites for GC and GC-MS analysis was adapted from Singh and Bose (2016).43 In brief, the aqueous samples were mixed with ethyl acetate (1:4 ratio) and 250 mg NaCl in tightly capped vials and mixed on a vortex mixer (Vertex Genie-2, Scientific Industries, USA) for 10 minutes. The contents were then centrifuged (BiofugeStratos, Heraeus, Germany) at 3000 × g for phase separation. After extraction, the samples were passed through anhydrous sodium sulfate to remove moisture. These were stored in 2 mL autosampler vials for analysis in GC-ECD and GC-MS. The percent recoveries of ES-1, ES-2, ES-S, ES-A, ES-A and ES-E by using the above extraction procedure were 98.6 ± 11.3%, 97.4 ± 6.6%, 94.2 ± 5.9%, 92.01 ± 7.3%, 93.1 ± 7.1% and 94.2 ± 4.8%, respectively. These recoveries were comparable to those obtained by the soxhlet extraction procedure (USEPA Method 3540).
2.6. Analytical methods
A Gas Chromatograph (GC) was used for analysis (Model Clarus 500, Perkin-Elmer, USA), equipped with an electron capture detector (ECD) and a capillary column (MXT-5) of length 30 m, internal diameter 0.25 mm and film thickness 0.25 μm. The internal standard used was 2,4,5,6-tetrachloro-m-xylene. Nitrogen was used as the carrier gas at 68.95 kPa (10 psi) and as the makeup gas at 30 mL min−1. Injector and detector temperatures were 250 °C and 375 °C, respectively. The oven temperature program was as follows: start at 120 °C, hold for 1 minute; increase to 170 °C @ 10 °C min−1, hold for 8 minutes; increase to 220 °C @ 18 °C min−1, hold for 8 minutes; increase to 240 °C @ 12 °C min−1 and hold for 5 minutes.GC-MS analysis was done by Ion trap GC-MS (Polaris Q, ThermoFinnigan, USA) using 30 m × 0.25 mm × 0.25 mm DB-5MS and DB-XLB capillary columns (Agilent Technologies, India). The career gas was helium at a flow rate of 1 mL min−1. Injector, ion source and auxiliary line temperatures were 250, 200 and 260 °C, respectively. Oven temperature program was as follows: start at 120 °C, increase to 180 °C @ 8 °C min−1, hold for 10 minutes; increase to 220 °C @ 10 °C min−1 and hold for 10 minutes. Ionization was carried out at 70 eV. The analysis for each sample was done in the full scan mode (50–500 m/z) as well as in the MS/MS mode. The GC-MS/MS details were adopted from Tiwari and Guha (2013).44 A different oven temperature program was used for lower dechlorinated by-products analysis. The temperature program was as follows: start at 70 °C, hold for 10 minutes; increase to 180 °C @ 8 °C min−1, hold for 5 minutes; increase to 220 °C @ 10 °C min−1 and hold for 12 minutes.
3. Results & discussion
3.1. Reductive dechlorination by NZVI
The evolution of degradation products during reductive dechlorination of ES-isomers, i.e., ES-1 and ES-2, and ES-metabolites, namely ES-S, ES-L, ES-E and ES-A, by NZVI was recorded by a time series of chromatograms corresponding to each experiment. The data presentation scheme is summarized in Table 1. In Table 1, the parent compounds are referred by abbreviations used earlier, i.e., ES-1, ES-2, etc. The degradation products are denoted as ES-1x, where the suffix ‘x’ corresponds to order of appearance of the degradation product peaks during the dechlorination process shown in the gas chromatogram (Fig. 3b). For example, ES-12 denotes the degradation product which appeared second during the interaction of ES-1 with NZVI. Only the following information is presented for each experiment; (a) chromatogram at the start of the experiment showing the peak corresponding to the parent molecule; (b) chromatogram at an intermediate time during the experiment, where the peaks corresponding to the parent molecule and all degradation products are simultaneously visible; (c) mass spectra of the parent compound and corresponding reference spectra from the NIST library; (d) mass spectra corresponding to each degradation product peak and the corresponding proposed chemical structure. The location information, i.e., figure/table numbers for the above data for each experiment are given in Table 1.| Expt. no. | Parent molecule (chemical structures in Fig. 1) | By-products | |
|---|---|---|---|
| Chromatogram | Mass spectra and proposed chemical structure | ||
| a #The parent molecule and the dechlorination products of ES-A were derivatized to obtain the corresponding ethers. Thus the elution times and the mass spectra of ES-E1, derivatized ES-A1* and ES-E2, and derivatized ES-A2* were identical. | |||
| 1. | ES-1 | ES-11, ES-12, ES-13 | Table SI1 |
| Chromatogram: Fig. 3a | Chromatogram: Fig. 1b | ||
| Mass spectra: Fig. 3c and d | |||
| 2. | ES-2 | ES-21, ES-22 | Table SI2 |
| Chromatogram: Fig. SI1a | Chromatogram: Fig. SI1b | ||
| Mass spectra: Fig. SI1c and 2d | |||
| 3. | ES-S | ES-S1, ES-S2, ES-S3 | Table SI3 |
| Chromatogram: Fig. SI2a | Chromatogram: Fig. SI2b | ||
| Mass spectra: Fig. SI2c and d | |||
| 4. | ES-L | ES-L1, ES-L2 | Table SI4 |
| Chromatogram: Fig. SI3a | Chromatogram: Fig. SI3b | ||
| Mass spectra: Fig. SI3c and d | |||
| 5. | ES-E | ES-E1, ES-E2, ES-E3, ES-E4 | Table SI5 |
| Chromatogram: Fig. SI4a | Chromatogram: Fig. SI4b | ||
| Mass spectra: Fig. SI4c and d | |||
| 6. | ES-A# | ES-A1*, ES-A2* | |
| Derivatization: Fig. SI5a | Chromatogram: Fig. SI5 | ||
| Chromatogram: Fig. SI5b | |||
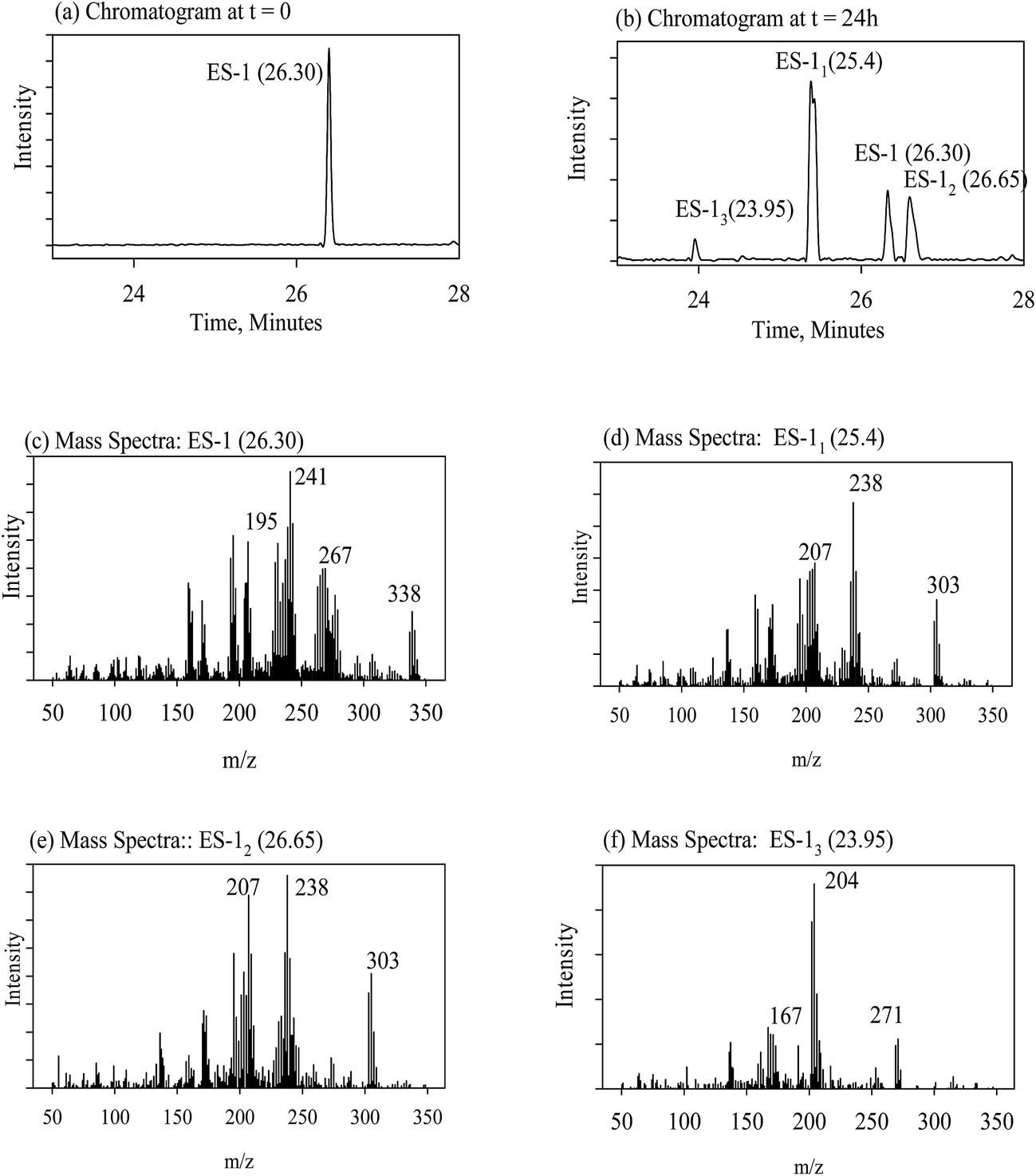 | ||
| Fig. 3 Reductive dechlorination of ES-1 by NZVI; (a) chromatogram at, t = 0; (b) chromatogram at, t = 24 h; (c) mass spectra of ES-1; (d) mass spectra of ES-11; (e) mass spectra of ES-12; (f) mass spectra of ES-13. | ||
3.2. Cyclization of endosulfan alcohol
The analysis of ES-A and its dechlorinated by-products was difficult in GC-MS due to the presence of hydroxyl group. However, ES-A and its dechlorination products could be converted to their corresponding ethers by the procedure shown in Fig. 2. Thus, the peak elution times and mass spectra of ES-A and its dechlorination products as shown in Fig. SI5† were identical to those obtained in case of ES-E (Fig. SI4†).The variation in pH during NZVI experiments was between 7.5–8.8, and at a higher pH, hydrolysis of ES-1 and ES-2 to ES-A is reported.13,14,45 In the present experiments, ES-1, ES-2 and its dechlorination products disappeared from the chromatograph after 48 hours. However, ES-A and its dechlorination products could still be detected in these solutions after conversion of ES-A and its dechlorination products to their corresponding ethers. Moreover, these dechlorination products were identical to those detected during dechlorination of ES-E. So the dechlorination products obtained from the dechlorination experiments of ES-1, ES-2, ES-A and ES-E were identical, which supports the hypothesis of a similar dechlorination sequence for ES skeleton, irrespective of the particular compound being dechlorinated. Considering the central role of ES-E as described above and the fact that ES-E does not hydrolyze, it was selected as the representative compound for further experiments.
3.3. Reductive dechlorination of ES-E by Mg0 and Na0
The degradation products formed during reductive dechlorination of ES-E by Mg0 were all observed in a chromatogram obtained from a sample extracted at an intermediate point during the experiment (Fig. 4b). Based on this chromatogram and the associated mass spectra, the following general observations were possible regarding formation of degradation products during the reductive dechlorination of ES-E by Mg0, (a) reductive dehalogenation resulted in the formation of six degradation products, ES-E1a, ES-E3a–ES-E7a (Fig. 4b); (b) mass spectra of ES-E1a, ES-E3a and ES-E4a (Table SI6†) were consistent with the loss of one, two and three chlorine atoms, respectively from the parent compound and were nearly identical to the mass spectra of ES-E1, ES-E3 and ES-E4 (Table SI5†) obtained earlier during reductive dechlorination of ES-E by NZVI; (c) mass spectra of ES-E5a, ES-E6a and ES-E7a were consistent with the loss of four, five and six chlorine atoms, respectively from the parent compound (Table SI6†).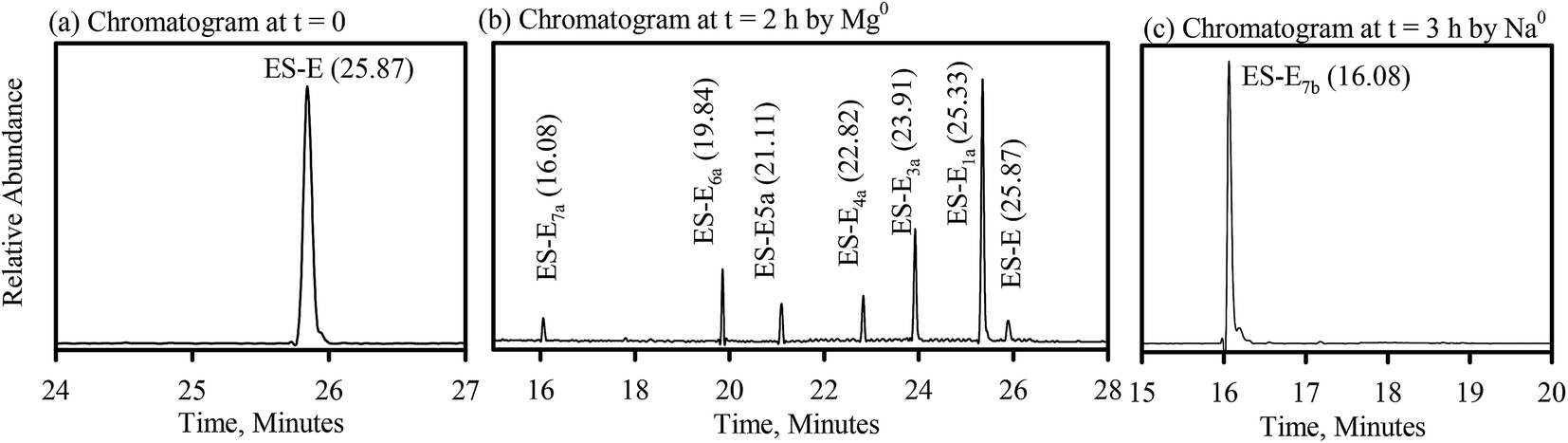 | ||
| Fig. 4 Reductive dechlorination of ES-E by Mg0; (a) chromatogram at, t = 0; (b) chromatogram at, t = 2 h by Mg0; (c) chromatograph at t = 3 h by Na0. | ||
Only one degradation product (ES-E7b), consistent with the loss of six chlorine atoms from the parent compound, was observed during reductive dechlorination of ES-E by Na0 (Fig. 4c) and confirmed by NIST library (NIST no. 105262) as endo-4-oxatricyclo[5.2.1.0(2.6)]dec-8-ene. The mass spectra of ES-E7b (Table SI7†) was nearly identical to the mass spectra of ES-E7a (Table SI6†) obtained earlier during reductive dechlorination of ES-E by Mg0.
3.4. Dechlorination mechanism
In all experiments involving reductive dechlorination of ES and its metabolites by NZVI, two distinct degradation products were initially observed. Both had mass spectra analogous to the loss of one chlorine atom from the parent compound (Tables SI1–5†). In some cases, other degradation products, having mass spectra consistent with the loss of two or three chlorine atoms from the parent compound, were also observed sequentially. The experiment involving reductive dechlorination of ES-E by Mg0 showed sequential formation of degradation products having mass spectra consistent with the loss of 1–6 chlorine atoms from the parent compound, i.e., ES-E1a, ES-E3a–ES-E7a. In this case, only one degradation product (ES-E1a) was observed with mass spectrum corresponding to the loss of a single chlorine atom. It is possible that a degradation product similar to ES-E2 (with NZVI) was also formed in this case, but was quickly further dechlorinated due to vigorous interaction with Mg0 and thus escaped observation.It is increasingly apparent that the mechanisms for reductive dechlorination in the above experiments with NZVI and Mg0 are essentially the same, i.e., dechlorination occurs sequentially, but may or may not proceed to the dechlorinated end product depending on the intensity of the reducing conditions employed. Reductive dechlorination appeared to be through a surface mediated single electron transfer mechanism. Such a mechanism is consistent with the observed experimental data and has also been proposed by several researchers for the reductive dechlorination of other organic pollutants.32,46–48
The reductive dechlorination of ES-E by Na0 showed only one degradation product having mass spectra consistent with the loss of all six chlorine atoms from the parent molecule. The dechlorination process in this case was very rapid and it is possible that the dechlorination mechanism probably involved simultaneous attack on all chlorinated sites by electron shuttles transporting electrons from the Na surface to the ES-E molecule. Such a mechanism is consistent with the observed rapid and near simultaneous dechlorination at all sites.
3.5. Chemical structure of degradation products
Considering the ES-skeleton shown in Fig. 5, it is apparent that Fe0/Mg0-mediated sequential reductive dechlorination can start with the removal of chlorine atoms at positions 1, 2 or 3. The formation of two mono-dechlorinated products with the same mass spectra (Fig. 3d and e), suggests that the initial attack takes place on position 1 and it can be assumed that dechlorination can start at positions 1a or 1b. Since the chemical environment of the newly attached hydrogen atom will be different depending on whether it is attached at position 1a or 1b, the resulting dechlorinated isomers are likely to have different physical properties and hence these are expected to appear as separate peaks in chromatograms. This was indeed observed in this study in experiments with NZVI; third, dechlorination commencing at positions 2a or 2b and 3a or 3b will result in the formation of enantiomers, which will not be seen as separate peaks in chromatograms; fourth, we may assume that the initial dechlorination occurs randomly at positions either 1, 2 or 3. However, in that case, the second dechlorination step will result in the formation of several degradation products consistent with the loss of two chlorine atoms from the parent molecule. However, in all the above experiments, only a single degradation product was observed in accordance with the loss of two chlorine atoms from the parent molecule.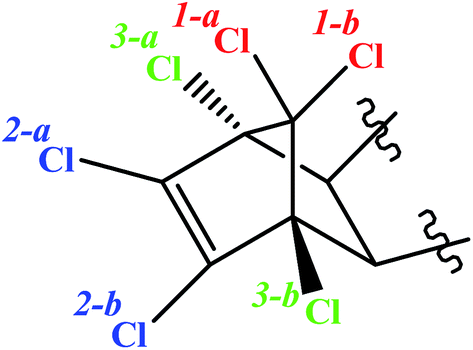 | ||
| Fig. 5 Structure of ES-skeleton. | ||
On the basis of the above reasoning, it is apparent that the second dechlorination step results in the loss of chlorine atoms at both positions 1a and 1b. Further, the third dechlorination step is likely to occur at position 2a or 2b for the following reasons; the carbon atom at site 3a or 3b is at a saturated bridge position of a bicyclic compound, rendering electron transfer to the attached chlorine atom very difficult; a study on electrochemical dechlorination of ES indicated that dechlorination at position 3a and 3b is not favored;49 analogy with a study on reductive dechlorination of 1,2-dichloroethene50 also indicates that dechlorination at position 2 is favored. As mentioned earlier, the two products formed due to the third dechlorination step are enantiomers and will hence appear as a single peak in the chromatogram. Fourth dechlorination step would results in the removal of chlorine atoms from both 2a and 2b positions. The fifth dechlorination step is expected to occurs at position 3a or 3b again resulting in the formation of two enantiomers and the final dechlorination step thereafter results in the formation of the fully dechlorinated product. The tentative chemical structures for the various degradation products observed during this study have been proposed in consonance with the dechlorination sequence as discussed above and the corresponding mass spectra (for degradation products of ES-1, ES-2, ES-S, ES-L, see Tables SI1–3 and 5,† respectively; for degradation products of ES-E, see Tables SI4, 6 and 7†). Although the exact pathway for all compounds (ES-isomer and metabolites) is matter of investigation with more experimental evidence.
3.6. Synthesis of dechlorinated by-products
Dechlorination of the ES-skeleton (Fig. 5) can commence at positions 1, 2 or 3. The preponderance of evidence discussed in the previous section suggests that dechlorination commences at position 1. Unequivocal determination of the first dechlorination position is critical, since the proposed chemical structures of the degradation products hinges on this determination. An unequivocal confirmation of the initial dehalogenation position requires the synthesis and subsequent analysis of the actual dechlorinated products. As noted earlier, there is a strong possibility that the degradation pathway of ES-isomers and ES-metabolites are similar. Further, considering the central role of ES-E as discussed earlier, it was decided that the degradation mechanism of ES-E will be studied in detail. Thus, some of the degradation products of ES-E formed through reductive dechlorination were synthesized.The first dechlorinated product synthesized corresponded to the loss of one chlorine atom from the parent molecule. The procedure for this synthesis is summarized in Fig. 6a. First, 2 g of ES-E was dissolved in 30 mL of acetone–water mixture (1:1) followed by the addition of 5.0 g of Mg0 metal powder and 5.0 mmol of ZnCl2. Then, 5 mL of glacial acetic acid was added dropwise with constant stirring. The mixture was stirred for another 2 hours for completion of the reaction at room temperature (27 ± 3 °C). The degradation products thus formed were extracted twice with 50 mL of ethyl acetate. The solvent in the extract was dried at 60 °C under high vacuum. The required product was purified by column chromatography and eluted with ethyl acetate/hexane. Evaporation of the solvent yielded 1.51 g of a white crystalline powder. The fact that this compound was consistent with the loss of one chlorine atom from the parent compound was confirmed by GC-MS and 1H-NMR (500 MHz, DMSO-d6, 25 °C, TMS).
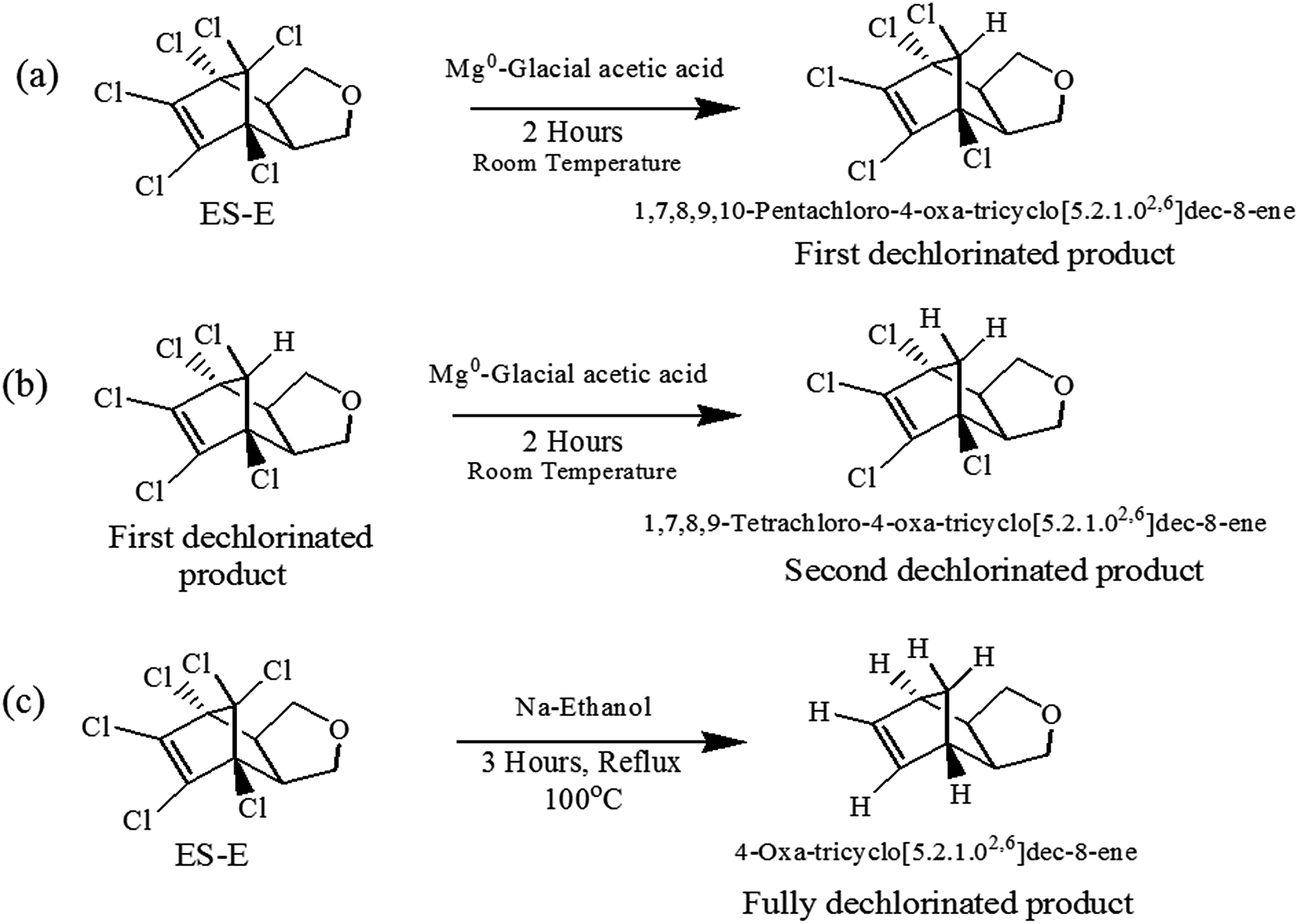 | ||
| Fig. 6 Synthesis of (a) first dechlorinated product, (b) second dechlorinated product and (c) fully dechlorinated product of ES-E. | ||
The second dechlorinated product synthesized corresponded to the loss of two chlorine atoms from the parent ES-E molecule. The starting material for this synthesis was 300 mg of the first dechlorinated product. The procedure for this synthesis is summarized in Fig. 6b. However, the quantities of the reagents used were modified proportionately to account for the lower amount of starting material used in this case. Purification of the product was done on TLC plates using mixture of acetone hexane as eluting solvent. The fact that this compound was consistent with the loss of two chlorine atoms from the parent molecule was confirmed by GC-MS analysis.
The third dechlorinated product synthesized corresponded to the loss of all six chlorine atoms from the parent molecule. The procedure of synthesis is summarized in Fig. 6c. 1.5 g ES-E was dissolved in 50 mL of ethanol and refluxed with 0.4 mol of Na0 metal for three hours at 100 °C. The cooled mixture was mixed with crushed ice and then extracted with 100 mL ethyl acetate. The solvent was evaporated at 45 °C under high vacuum.51 The resulting product was purified on TLC plates using mixture of acetone and hexane as eluting solvent. The fact that this compound was consistent with removal of all chlorine atoms from the parent molecule was confirmed by GC-MS and the analogy of its mass spectra with the corresponding mass spectra available in NIST library.
3.7. Experimental evidence for a plausible degradation pathway
The theoretical 1H NMR for singly dechlorinated ES-E was obtained assuming that dechlorination occurred from position 1, 2 or 3 as shown in Fig. SI6.† The actual 1H NMR of ES-E and the dechlorinated product ES-E1a are shown in Fig. SI7.† The 1H NMR of the degradation product ES-E1a provides the position for introduction of new proton at δ (PPM) 4.28, which is very close to the δ (PPM) 4.48 value obtained from the theoretical 1H NMR corresponding to the proton insertion at position 1.Further confirmation of position of dechlorination either from 1a or 1b was obtained via single crystal X-ray diffraction measurements mounted on a CCD Bruker SMART APEX diffractometer. The crystallographic figure has been generated using Diamond 3.2g (Fig. SI8†). The contribution of all hydrogen atoms has been incorporated in both empirical formulas and formula weights of the complexes. X-ray diffraction measurements confirmed that the first dechlorination takes place from 1b position (Fig. SI8†).
All evidence presented suggest that dechlorination mechanism for all ES and ES-metabolite molecules through interaction with NZVI/Mg0 surface was identical. Based on the above results, the most probable pathway for the dechlorination process was proposed. Dechlorination seems to occurs sequentially, yielding products 1a/1b, 2, 3a/3b, 4, 5a/5b and 6 (Fig. 7). Products 1a and 1b are isomers and hence observed as separate peaks in chromatograms. Products 3a/3b and 5a/5b are enantiomers and hence appeared as the overlapping peaks in chromatogram. Peaks corresponding to intermediate products 2, 4 and the dechlorinated product 6 were also observed. The proposed pathway (Fig. 7) is a tentative pathway and exact pathway is matter of future investigation, as 3rd, 4th and 5th dechlorination needs more experimental supports.
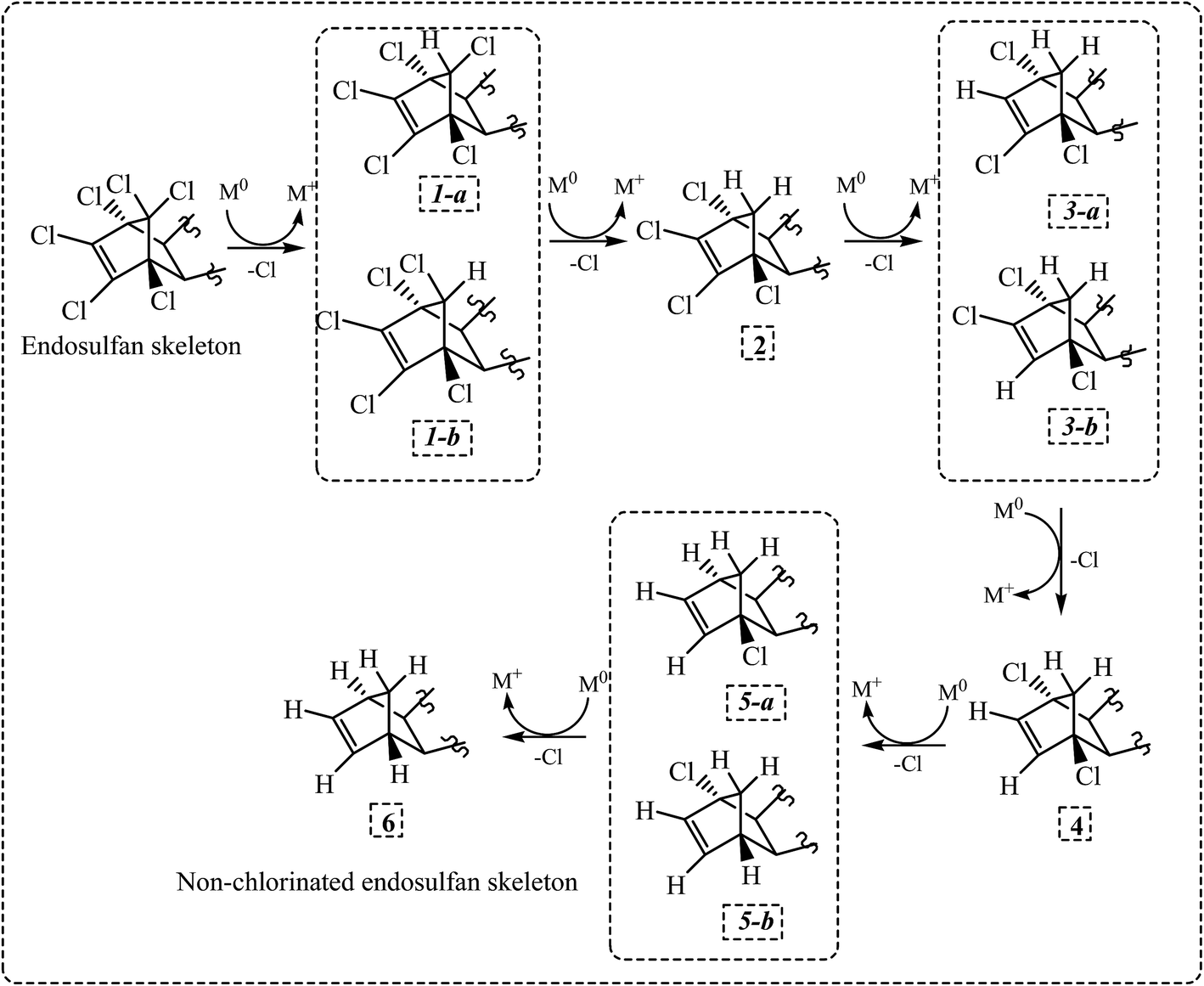 | ||
| Fig. 7 Proposed dechlorination pathway for ES-isomers and metabolites. | ||
The extent of dechlorination of the various ES-isomers and ES-metabolites appeared to depend on the intensity of the reducing conditions employed; hence, dechlorination was more complete during interaction with the Mg0 surface. The rate of dechlorination was however, different for different molecules depending on the nature of the side-chain attached to the ES skeleton shown in Fig. 5.
4. Conclusions
Reductive dechlorination of ES and ES-metabolites through interaction with NZVI and Mg0 was investigated. During dechlorination of ES-isomers and ES-metabolites by NZVI, two distinct degradation products, both having mass spectra consistent with the loss of one chlorine atom from the parent compound, were initially observed. Other degradation products were observed later and had mass spectra consistent with the loss of two or three chlorine atoms from the parent compound. During dechlorination of ES-E by Mg0, degradation products having mass spectra consistent with the loss of 1–6 chlorine atoms from the parent compound were observed sequentially. The dechlorination mechanism for all ES-isomers and ES-metabolite molecules through interaction with NZVI/Mg0 surface appeared to be the similar. It was proposed that the abstraction of chlorine atoms occurred sequentially from positions 1a/1b, 2a/2b and 3a/3b of the ES skeleton shown in Fig. 5. Chemical structures of various degradation products are proposed based on the above order of chlorine abstraction and mass spectra of the degradation products. Dechlorinated products were synthesized using ES-E as the parent molecule. 1H NMR, X-ray diffraction measurements, and GC-MS analysis of the synthesized products provide the final proof of initial chlorine abstraction from position 1b as shown in Fig. 5.The extent of dechlorination of the various ES-isomers and ES-metabolites appeared to be variable and appeared to depend on the nature of the side-chain attached to the ES skeleton. Further, it appears that the extent of dechlorination could be enhanced by increasing the intensity of the reducing conditions employed. While using Mg0 as a reducing agent may be difficult in natural environment, application of bimetallic nano-particles, i.e., Ni0–Fe0, or Pd0–Fe0 etc., may be an environmentally feasible option, which may result in more complete dechlorination of ES-isomers and ES-metabolites as compared to NZVI. Finally, the significance of the present study lies in the fact that partially or fully dechlorinated degradation products of ES and ES-metabolites may be less toxic and more amenable to subsequent biodegradation and ultimate mineralization in the natural environment. The toxicity study of these dechlorinated degradation products and their biodegradation potential requires further evaluation.
Acknowledgements
The authors thank Dr. Jitendra Kumar Bhatt and Dr. Narendra Mishra for their help in synthesis and solving crystal structure for new by-products.References
- Y. F. Li and R. W. Macdonald, Sci. Total Environ., 2005, 342, 87–106 CrossRef CAS PubMed.
- J. Weber, C. J. Halsall, D. Muir, C. Teixeira, J. Small, K. Solomon, M. Hermanson, H. Hung and T. Bidleman, Sci. Total Environ., 2010, 408, 2966–2984 CrossRef CAS PubMed.
- R. I. M. Sunderam, D. M. H. Cheng and G. B. Thompson, Environ. Toxicol. Chem., 1992, 11, 1469–1476 CrossRef CAS.
- L. Beyger, R. Orrego, H. Guchardi and D. Holdway, Ecotoxicol. Environ. Saf., 2012, 76, 71–78 CrossRef CAS PubMed.
- H. L. Henderson, J. Townsend and D. J. Tortonese, J. Endocrinol., 2008, 197, 343–350 CrossRef CAS PubMed.
- A. Caride, A. Lafuente and T. Cabaleiro, Toxicol. Lett., 2010, 197, 106–112 CrossRef CAS PubMed.
- O. Ozmen and F. Mor, Pestic. Biochem. Physiol., 2012, 102, 129–133 CrossRef CAS.
- C. J. Hapeman, L. L. McConnell, T. L. Potter, J. Harman-Fetcho, W. F. Schmidt, C. P. Rice, B. A. Schaffer and R. Curry, Atmos. Environ., 2013, 66, 131–140 CrossRef CAS.
- R. Martens, Appl. Environ. Microbiol., 1976, 31, 853–858 CAS.
- J. R. W. Miles and P. Moy, Bull. Environ. Contam. Toxicol., 1979, 23, 13–19 CrossRef CAS PubMed.
- T. F. Guerin, Environ. Pollut., 2001, 115, 219–230 CrossRef CAS PubMed.
- T. E. Archer, I. K. Nazer and D. G. Crosby, J. Agric. Food Chem., 1972, 20, 954–956 CrossRef CAS PubMed.
- S. M. Peterson and G. E. Batley, Environ. Pollut., 1993, 82, 143–152 CrossRef CAS PubMed.
- H. M. Shivaramaiah, F. Sanchez-Bayo, J. Al-Rifai and I. R. Kennedy, J. Environ. Sci. Health, Part B, 2005, 40, 711–720 CrossRef CAS PubMed.
- C. N. G. Bedor, R. J. L. Morais, L. S. Cavalcanti, J. V. Ferreira and A. C. Pavão, Sci. Total Environ., 2010, 408, 6281–6284 CrossRef CAS PubMed.
- R. Martens, Bull. Environ. Contam. Toxicol., 1977, 17, 438–446 CrossRef CAS PubMed.
- D. Henschler, Angew. Chem., Int. Ed. Engl., 1994, 33, 1920–1935 CrossRef.
- G. Bonse, D. Henschler and P. J. Gehring, Crit. Rev. Toxicol., 1976, 4, 395–409 CrossRef CAS PubMed.
- A. Sato and T. Nakajima, Arch. Environ. Health, 1979, 34, 69–75 CrossRef CAS PubMed.
- A. Bobra, W. Shiu and D. Mackay, in QSAR in Environmental Toxicology, ed. K. E. Kaiser, Springer Netherlands, 1984, ch. 2, pp. 3–16 Search PubMed.
- M. Zahedi-Tabrizi and R. Farahati, Comput. Theor. Chem., 2011, 977, 195–200 CrossRef CAS.
- K. Furukawa, K. Tonomura and A. Kamibayashi, Appl. Environ. Microbiol., 1978, 35, 223–227 CAS.
- K. Furukawa, N. Tomizuka and A. Kamibayashi, Appl. Environ. Microbiol., 1979, 38, 301–310 CAS.
- R. W. Gillham and S. F. O'Hannesin, Ground Water, 1994, 32, 958–967 CrossRef CAS.
- T. L. Johnson, M. M. Scherer and P. G. Tratnyek, Environ. Sci. Technol., 1996, 30, 2634–2640 CrossRef CAS.
- X.-q. Li and W.-x. Zhang, Langmuir, 2006, 22, 4638–4642 CrossRef CAS PubMed.
- R. A. Crane and T. B. Scott, J. Hazard. Mater., 2012, 211–212, 112–125 CrossRef CAS PubMed.
- S. P. Singh and P. Bose, RSC Adv., 2015, 5, 94418–94425 RSC.
- B. I. Kharisov, H. V. Rasika Dias, O. V. Kharissova, V. Manuel Jimenez-Perez, B. Olvera Perez and B. Munoz Flores, RSC Adv., 2012, 2, 9325–9358 RSC.
- J. Gotpagar, E. Grulke, T. Tsang and D. Bhattacharyya, Environ. Prog., 1997, 16, 137–143 CrossRef CAS.
- B. Deng, D. R. Burris and T. J. Campbell, Environ. Sci. Technol., 1999, 33, 2651–2656 CrossRef CAS.
- W. A. Arnold and A. L. Roberts, Environ. Sci. Technol., 2000, 34, 1794–1805 CrossRef CAS.
- S. M. Cook, Assessing the use and application of zero-valent iron nanoparticle technology for remediation at contaminated sites, Jackson State University, 2009 Search PubMed.
- W.-J. Liu, T.-T. Qian and H. Jiang, Chem. Eng. J., 2014, 236, 448–463 CrossRef CAS.
- A. Begum and S. K. Gautam, Water Res., 2011, 45, 2383–2391 CrossRef CAS PubMed.
- T. Hua, L. Jinjun, M. Zhen, L. Landong and H. Zhengping, Sep. Purif. Technol., 2009, 66, 84–89 CrossRef.
- T.-T. Lim, J. Feng and B.-W. Zhu, Water Res., 2007, 41, 875–883 CrossRef CAS PubMed.
- F. He and D. Zhao, Environ. Sci. Technol., 2005, 39, 3314–3320 CrossRef CAS PubMed.
- B. Schrick, J. L. Blough, A. D. Jones and T. E. Mallouk, Chem. Mater., 2002, 14, 5140–5147 CrossRef CAS.
- S. Luo, S. Yang, X. Wang and C. Sun, Chemosphere, 2010, 79, 672–678 CrossRef CAS PubMed.
- Y. Cho and S.-I. Choi, Chemosphere, 2010, 81, 940–945 CrossRef CAS PubMed.
- S. P. Singh, P. Bose, S. Guha, S. K. Gurjar and S. Bhalekar, Chemosphere, 2013, 92, 811–820 CrossRef CAS PubMed.
- S. P. Singh and P. Bose, J. Chem. Technol. Biotechnol., 2016, 91, 2313–2321 CrossRef CAS.
- M. Tiwari and S. Guha, Environ. Monit. Assess., 2013, 185, 8451–8463 CrossRef CAS PubMed.
- S. S. Walse, K. D. Shimizu and J. L. Ferry, Environ. Sci. Technol., 2002, 36, 4846–4853 CrossRef CAS PubMed.
- M. Elsner, S. B. Haderlein, T. Kellerhals, S. Luzi, L. Zwank, W. Angst and R. P. Schwarzenbach, Environ. Sci. Technol., 2004, 38, 2058–2066 CrossRef CAS PubMed.
- A. L. Roberts, L. A. Totten, W. A. Arnold, D. R. Burris and T. J. Campbell, Environ. Sci. Technol., 1996, 30, 2654–2659 CrossRef CAS.
- E. J. Bylaska, M. Dupuis and P. G. Tratnyek, J. Phys. Chem. A, 2008, 112, 3712–3721 CrossRef CAS PubMed.
- J. V. J. Gassmann and G. Adiwidjaja, Zeitschrift für Naturforschung B, 1996, 51, 417–420 CrossRef CAS.
- C. Nonnenberg, W. A. van der Donk and H. Zipse, J. Phys. Chem. A, 2002, 106, 8708–8715 CrossRef CAS.
- B. V. Lap and M. N. Paddon-Row, J. Org. Chem., 1979, 44, 4979–4981 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary data has 17 pages (S1–S17), 9 Figures (SI1–SI9), and 7 Tables (SI1–SI7). CCDC 1534755. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra02430d |
| ‡ Present address: Department of Desalination and Water Treatment, Zuckerberg Institute of Water Research, Ben-Gurion University of Negev, Sede-Boqer-89990, Israel. |
| This journal is © The Royal Society of Chemistry 2017 |