
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Artificial tubular connections between cells based on synthetic lipid nanotubes
Alexander Kozintsev and
Kaori Sugihara *
*
Department of Physical Chemistry, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: kaori.sugihara@unige.ch
First published on 10th April 2017
Abstract
Tunneling nanotubes (TNTs) have become a major topic of interest as a form of intercellular communication due to their recent discovery. However, research on this subject has often suffered from a lack of controllability in the generation of the nanotubular connections. In this work, we demonstrate a simplified approach to selectively create a direct nanotubular connection between eukaryotic cells by manually manipulating self-assembling lipid nanotubes (LNTs) from inverted hexagonal-phase lipid blocks. The technique requires minimal instrumentation for creating the LNT connection between cells compared to conventional approaches. Based on the diffusion of fluorescent lipids from LNTs into cell membranes (D = 0.032 ± 0.003 μm2 s−1), the probability of observing membrane fusion between LNTs and cell membranes was estimated as 30%. Among these cell–LNT junctions the resulting structure is open-ended roughly 75% of the time, as evidenced from observations of the diffusion of a water-soluble dye between two cells connected with this nanotubular structure.
Introduction
Cell–cell communication is a fundamental process for the development and maintenance of multicellular organisms. Synapses, gap junctions, and exosomes are the well-known communication tools for cells,1 which have been studied extensively. Tunneling nanotubes (TNTs) are another means for intercellular communication discovered in 2004.2 They are tubular structures that connect cell bodies, which permit the direct intercellular transfer of diverse components between cells over a long distance (up to 100's of micrometers). Their functions include (i) the transport of proteins, vesicles, HIV viruses3 and prions4 between cells, (ii) the diffusion of membrane proteins and mitochondria through plasma membranes,5 and (iii) the propagation of electrical signals.6 Research over the last decade has revealed that TNTs are universal communication tools for many types of cells that have diverse structures and functions. Currently, TNTs are being studied with either in vivo or in vitro cell models. However, such traditional biological approaches often suffer from the complexities and a lack of controllability in the connection.To have more control over their network structures, the formation of artificial nanotube connections between cells has been demonstrated by several research groups. Optical tweezers have been used to pull a part of a cell membrane for creating a tubular structure as an extension of the cell membrane. This tube was attached to another gap-junction over-expressed cell, allowing for an electrical connection between two cells through gap junctions.7 A technique based on a micromanipulator combined with voltage pulse application previously used to fabricate networks between giant unilamellar vesicles (GUV) has also been applied to form tubular connections between cells.8 These artificial TNTs offer a well-controlled model system for studying the functions of TNTs.
In this work, we demonstrate a simplified approach to create a direct tubular connection between cells. This approach requires neither the over-expression of gap junctions in cells nor voltage pulse application. Previously we have found that the main component of bacterial cell membranes, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), self-assembles into single-wall tubes with an outer diameter of 19.1 ± 4.5 nm.9 DOPE is a zwitterionic conically shaped lipid that organizes into the inverted hexagonal phase (HII) in aqueous solution at room temperature.10–12 The HII lipid blocks can adsorb on either glass or plastic surfaces coated by cationic polyelectrolytes such as polyethylenimine (PEI) and polylysine (PLL). Upon solution flow, the lipid blocks move while a part of the block is attached to the substrate, thus generating lipid nanotubes (LNTs),9 somewhat similarly to the lipid nanotubes fabricated by microfluidic systems.13 These pre-assembled LNTs or HII lipid blocks can be used as a lipid reservoir to pull new LNTs by applying a point load with a micromanipulator, which enables the free-drawing of single LNT patterns.14 The extreme simplicity of our technique originates from the fundamental nature of DOPE lipids that prefer the HII phase. When GUVs are pushed against these LNTs, their membranes fuse. Between two GUVs connected with a LNT, a transport of water-soluble dye has been monitored, which suggests that the LNT–GUV junction is open-ended.14 This ability of DOPE to fuse with other lipid objects may be linked to its well-known fusogenic characteristic in transfection.15 We will apply this single LNT patterning method for creating a tubular connection between cells. The technique requires only a micromanipulator and a fluorescence microscope for controlling the LNT positioning and the connection to the cells.
Materials and methods
Preparation of lipid pre-deposited cell culture dishes
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti Polar Lipids, #850725) and 0.5% 1,2-dioleoyl-sn-glycero-3-3-phosphoethanolamine-N-(lissamine Rhodamine B sulfonyl) (Liss Rhodamine-PE, Avanti Polar Lipids, #810150) were dissolved in chloroform at a lipid concentration of 25 mg mL−1. Droplets (∼5 μL) of lipid solution were deposited directly onto 35 mm (40.28 mm O.D. × 6.17 mm) plastic cell culture dishes (Corning, #353001), dried with nitrogen gas, and then stored in darkness under low vacuum for at least 4 h. Thirty minutes prior to experiments, 2 mL of CO2-independent DMEM (Gibco, #18045-054) was added to the plates.Cell culture
HeLa cells were grown to 80% confluency in 25 mL tissue culture flasks (SIGMA, #Z707481) using high glucose DMEM with Glutamax™ (Gibco, #61965-059), supplemented with 10% fetal bovine serum (Gibco, #10270-106) and 1% penicillin streptomycin (Gibco, #15140-122). Cells were incubated in a 37 °C incubator in an atmosphere of 5% CO2.Cell plate preparation
HeLa cells were seeded at a density of ∼1.0 × 106 onto poly-D lysine coated glass-bottom dishes (MatTek, #P35GC-1.0-14-C) using medium consisting of high glucose DMEM with Glutamax™ supplemented with 10% fetal bovine serum and 1% penicillin streptomycin. Cells were incubated in the incubator for at least 4 h. Thirty minutes prior to imaging and manipulation the medium was washed twice with and swapped for CO2-independent DMEM supplemented with 1% L-glutamine (Gibco, #25030-081) and 1% penicillin streptomycin.Caged-dye loading to cells
HeLa cells were loaded with CMNB-caged fluorescein dye using the Influx™ Pinocytic Cell-Loading Reagent (Molecular Probes, #I14402) kit based on the osmotic lysis of pinocytic vesicles.16 Briefly, prior to seeding cells were incubated first in hypertonic medium solution containing the CMNB-caged fluorescein to induce pinocytic vesicle uptake and then transferred to a hypotonic medium to induce osmotic lysis of the vesicles; after loading, cells were seeded in 35 mm glass bottom plates as described previously in cell plate preparation.Micromanipulator setup
A manual micromanipulator (MM-33 by Marzhauser Wetzlar GmbH & Co. KG) along with a magnetic stand (Holex, #441210) was purchased from Science Products GmbH. Glass microtips were fabricated from borosilicate glass rods (Harvard Apparatus, #30-0085) using a micropipette puller (Sutter Instrument Co, #P-2000).Epifluorescent microscope setup
All fluorescent images were taken with Nikon Eclipse Ti-E (Nikon) equipped with a DS-Qi1 camera (Nikon), a metal–halide fluorescence lamp (Nikon) with long-distance objective lenses 20× and 40× (CFI S Plan Fluor ELWD, Nikon) and a TRITC fluorescent filter (543/593). The contrast and the brightness were adjusted and the images were presented with false colors for figures.Confocal laser scanning microscopy setup
Individual confocal scanning images as well as movies were taken using a CLSM (confocal A1, Nikon AG) with a 60× (CFI PLAN APO LBDA 60XH, Nikon) and 40× oil objectives (CFI PLAN FLUOR 40X Oil A, Nikon). The microscope, which has an integrated perfect focus system, resides on an active vibration isolation platform (Accurion, i4 series) and is housed in an acoustic enclosure (JPK Instruments). For imaging of fluorescein, 488 nm laser excitation was paired with an FITC A1 filter (525/50). For imaging of Liss-Rhod-PE, 561 nm laser excitation was paired with a Rhodamine Red A1 filter (595/50). The contrast and the brightness were adjusted and the images were presented with false colors for figures. The resolution in the z direction was set at 114.9 μm, which corresponded to pinhole sizes of 3.5 AU when scanning with the 561 nm laser (monitoring of Liss-Rhod-PE) and 4.0 AU when scanning with the 488 nm laser (monitoring of fluorescein). The large z-range was set in order to capture all fluorescence from the HeLa cell.Lipid diffusion experiment
Experimental setup began with cell plate preparation and preparation of lipid pre-deposited plates as described in preceding techniques. The previously described micromanipulator setup was used to coat the fabricated microtips with small lipid blocks by dragging the tips through large lipid blocks in the lipid pre-deposited plate. The lipid-coated microtip was then introduced into the cell plate and was carefully pushed into cell membranes in order to generate lipid nanotubes, as illustrated in Fig. 1A. Images of cells successfully connected to LNTs were taken at different timepoints using the epifluorescent and confocal laser scanning microscope setups.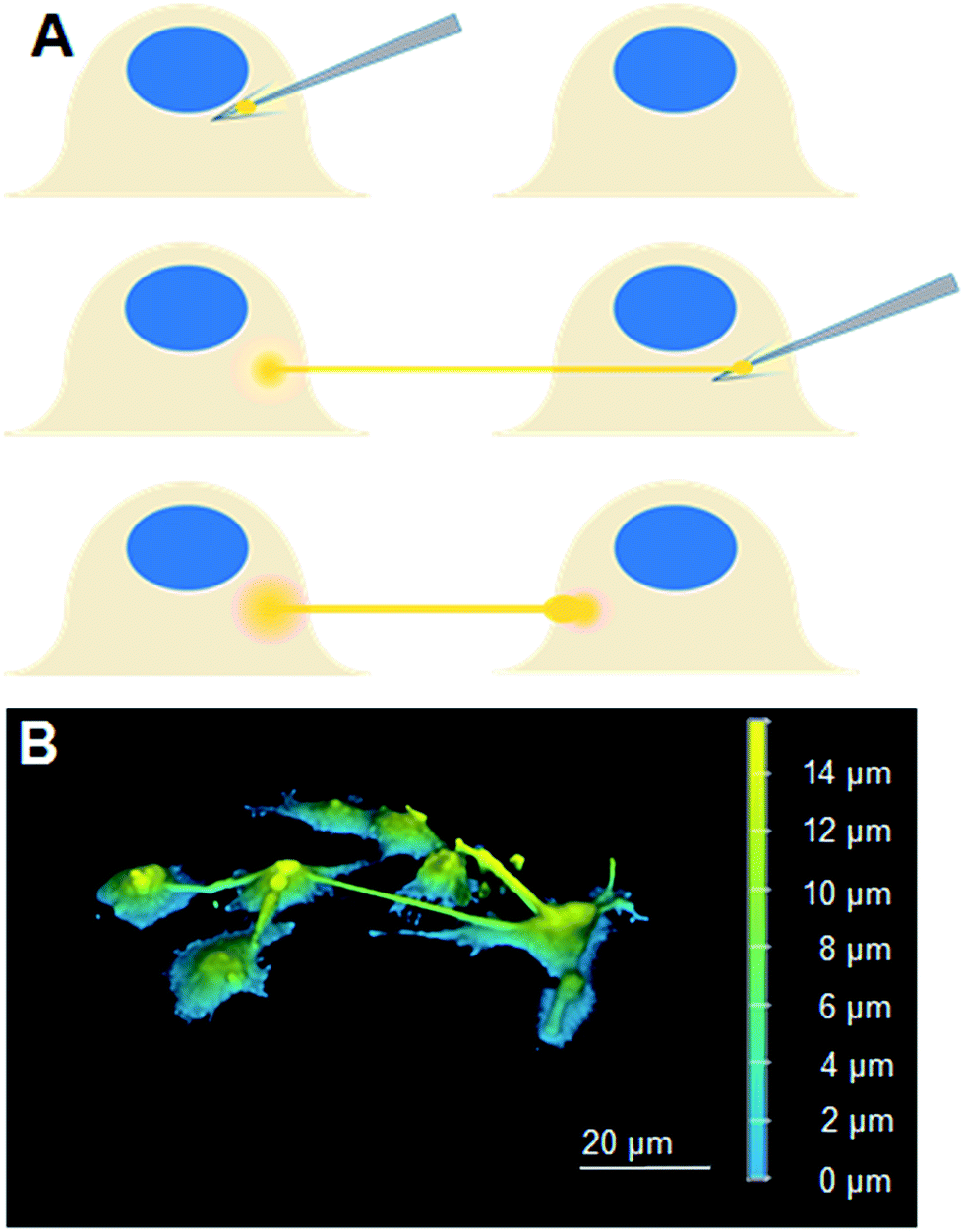 | ||
| Fig. 1 (A) A scheme describing our approach to fabricate LNT–cell network. (B) A 3D reconstituted confocal laser scanning microscopy image of Liss-Rhod-PE stained LNT–cell network. | ||
Analysis of lipid diffusion into cell membrane
Qualitative analysis of Liss-Rhod-PE diffusion was performed in a total of N = 23 cells. Quantitatively, fluorescence intensity within the LNT-attached cells at a known distance r from the LNT–cell junction was measured using ImageJ software and the average values were plotted over time (see the inset scheme in Fig. 2). Note that the background fluorescence intensity was subtracted at each frame. The diffusion coefficient of fluorescent lipids was estimated by fitting the obtained plot with the following two-dimensional diffusion model with fixed r using Matlab software:
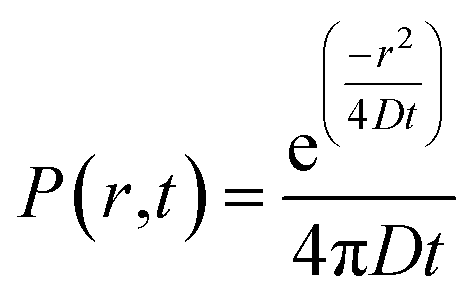 | (1) |
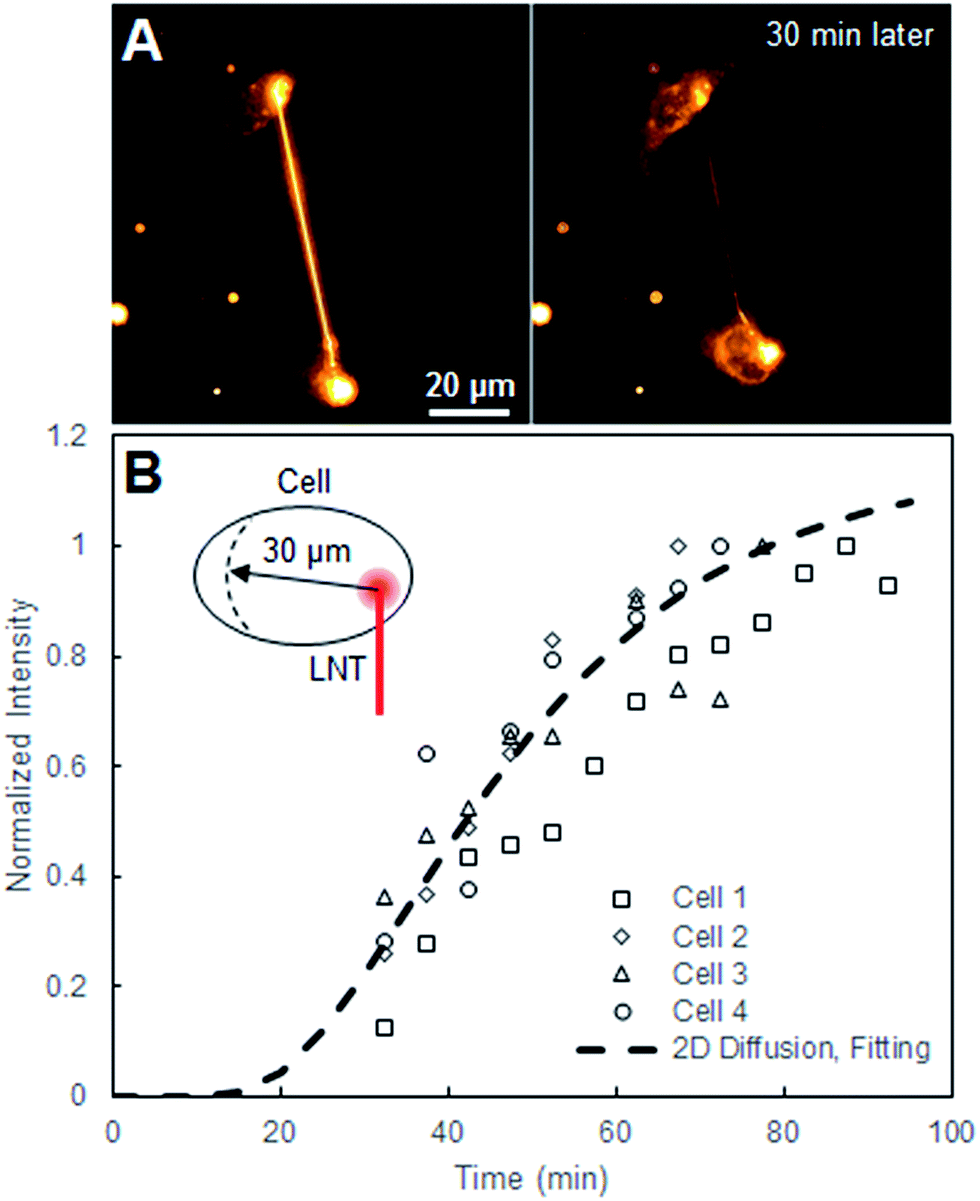 | ||
| Fig. 2 (A) Fluorescence images of two cells connected with a Liss-Rhod-PE-stained LNT right after the connection and 30 min later. (B) Normalized intensity plot at the position r = 30 μm, where r = 0 μm is defined as the LNT–cell junction, over time from four cells alongside the fitted 2D diffusion curve (eqn (1)) for r = 30 μm. | ||
The equation describes the probability of finding a diffusing molecule at time t and a radius r from its initial point source, where D is the effective diffusion coefficient. The intensity data was related to the probability function by introducing a scaling coefficient for normalizing the intensity values during the fitting. Among 8 cells we analyzed, r was selected at 30 μm for four cells and the fitting curves are shown in Fig. 2. For the other four cells, r was chosen such that the position of the nucleus and the change in the cell size and shape interfered with the analysis the least.
Fluorescein diffusion experiment
For fluorescein diffusion through LNTs, experimental setup began with caged-dye loading into cells followed by seeding them in a cell plate, and preparation of lipid pre-deposited plates as described in preceding techniques. LNT connections between cells were fabricated as described previously. For fluorescein diffusion through TNTs, experimental setup began with caged-dye loading into cells and their seeding in a plate, followed by qualitative identification of cell pairs connected by natural TNTs. Diffusion experiments were performed by taking either single (FITC) or dual channel (FITC and Rhod Red) confocal laser scanning microscopy images of the sample before and after uncaging fluorescein in donor cells. For both LNT and TNT experiments, the region of interest (ROI) and pinhole size (114.9 μm) of the confocal laser scanning microscope were chosen large enough to contain entire cells within the detection volume. This enables the total fluorescence signals within the cells to be captured. Intensity measurements were done using ImageJ software by selecting a region containing only the cell of interest and calculating the difference between the average intensity of this region and the background. In total N = 37 fluorescein diffusion experiments were performed.Theoretical fluorescein diffusion model
We study the diffusion of free fluorescein from an uncaged fluorescein-donor cell (Cell 1) to a receptor cell (Cell 2) through a nanotube in the following two steps.
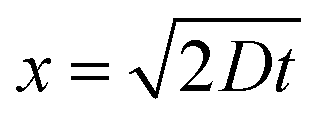 | (2) |
Setting x = L, the length of the nanotube, we can solve the equation for the time t, which is effectively the duration of time required for the dye molecules to reach the boundary with Cell 2:
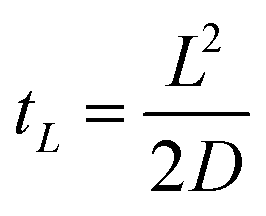 | (3) |
Using the known diffusion coefficient D of fluorescein in water at 298 K, D = 425 μm2 s−1,17 we can estimate the magnitude of tL for our system, which ends up being at maximum a few seconds. Therefore, tL can be neglected with respect to the timescales of our experiments and we may treat the diffusion as beginning to occur instantaneously.
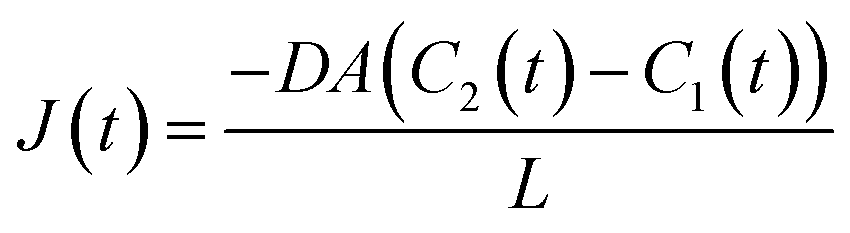 | (4) |
| N1(t) + N2(t) = NTot ∴ C1(t)V1 + C2(t)V2 = CTotVTot = C1(0)V1 | (5) |
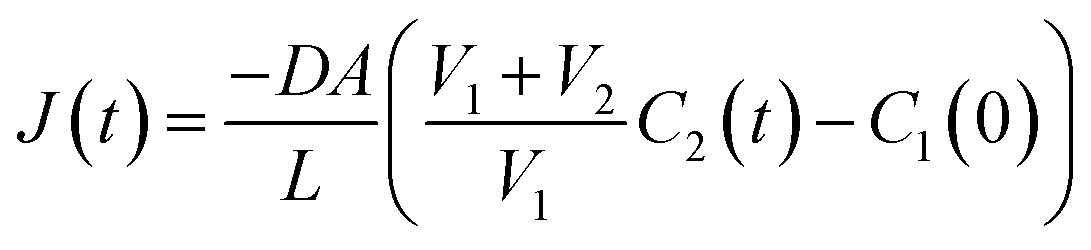 | (6) |
The flux, J(t), describes the rate of diffusion through the nanotube boundary with Cell 2 as a function of time. We can relate the flux to the rate of change in concentration of Cell 2, dC2(t)/dt, by dividing by its volume V2.
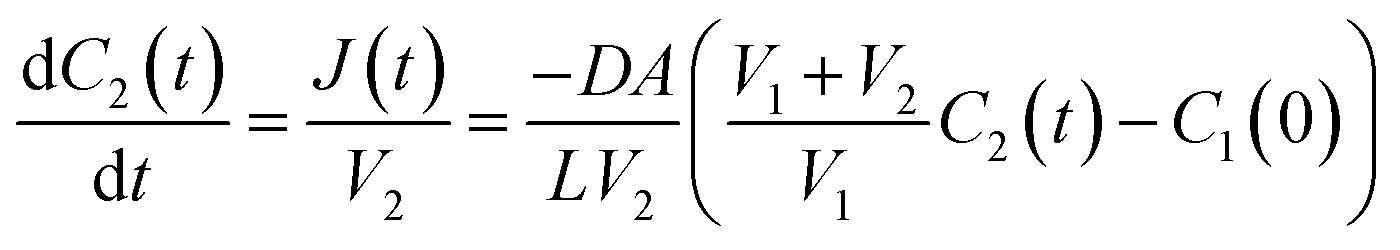 | (7) |
Separating this differential equation in terms of dC2(t) and dt and taking an integral with boundary condition C2(0) = 0 yields the following solution:
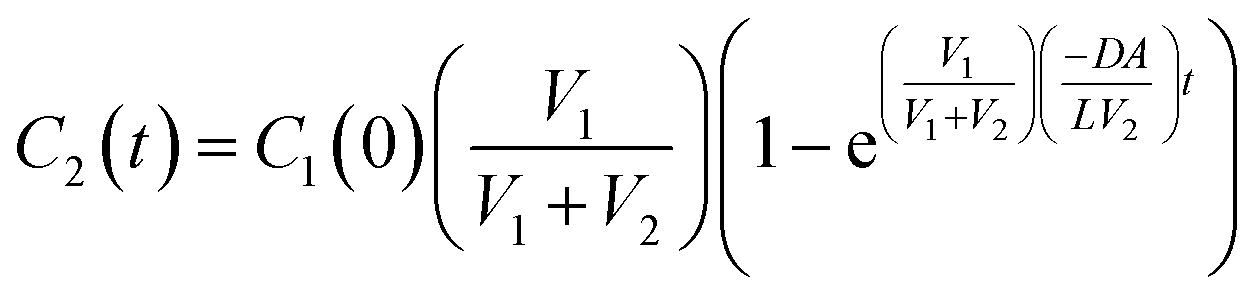 | (8) |
This function C2(t) describes the time evolution of the fluorescein concentration in the receptor cell, which we can directly relate to our experimental data. In Fig. 3, C2(t)/C1(0) (concentration of fluorescein in Cell 2 normalized by the initial concentration at Cell 1) was plotted for the LNT-connected cells using nanotube radii r of 10, 40, and 70 nm using the following parameters: A = πr2, V1 = 2000 μm3, V2 = 2000 μm3, L = 64.8 μm, and D = 425 μm2 s−1. In Fig. 4, C2(t)/C1(0) was plotted for the TNT system using radii r of 50, 100, and 150 nm using the following parameters: A = πr2, V1 = 2000 μm3, V2 = 2000 μm3, L = 20.1 μm, D = 425 μm2 s−1. Volumes of the donor and receptor cells in Fig. 3 and 4 were estimated using the reported average HeLa cell volume18 of 2000 μm3. The lengths of the natural tunneling and artificial lipid nanotubes were calculated using ImageJ and a known μm per pixel scale.
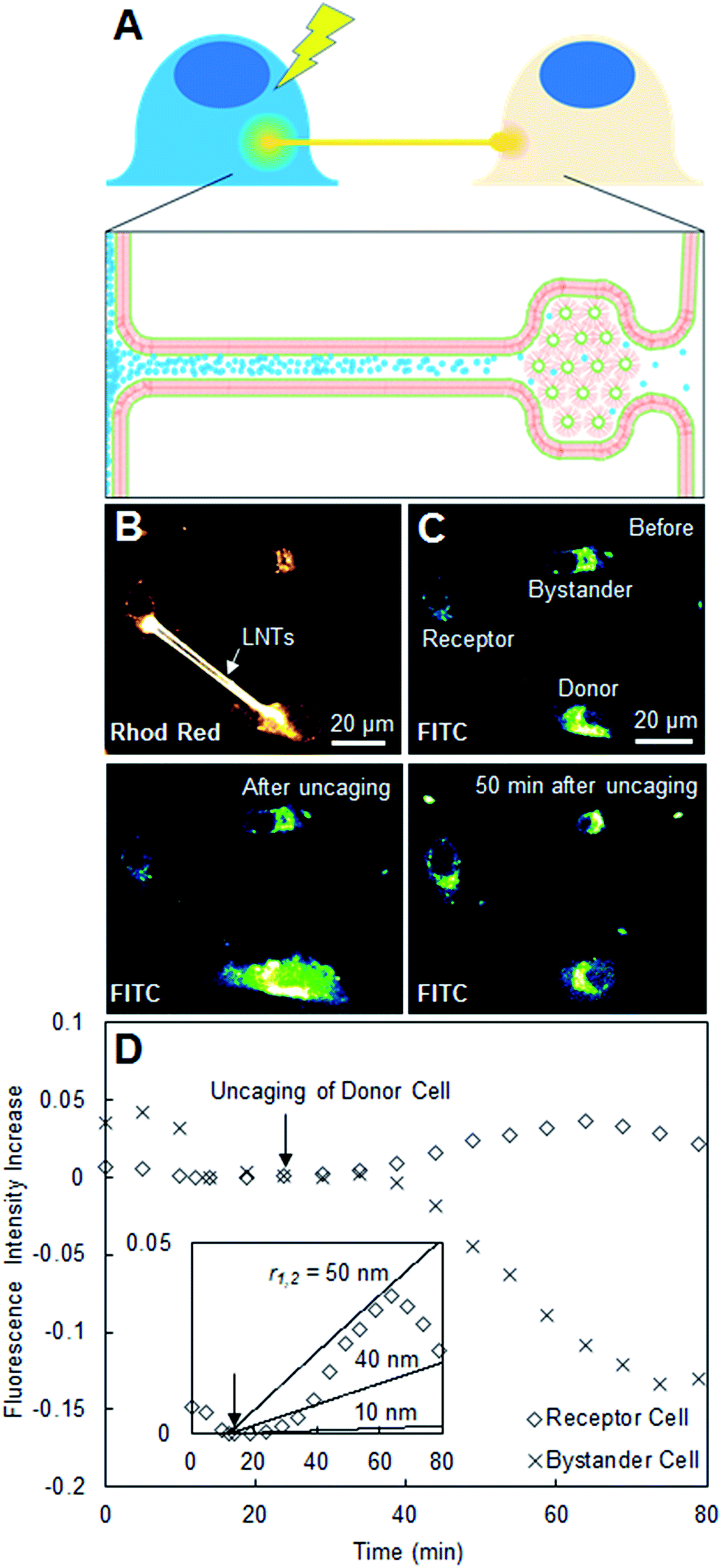 | ||
| Fig. 3 (A) Scheme describing the diffusion of uncaged fluorescein between two connected cells. (B) Confocal laser scanning microscopy image in the Rhod Red channel of three cells of interest loaded with caged fluorescein, two of which are connected with Liss-Rhod-PE-stained LNTs. (C) Confocal laser scanning microscopy image of the same cells in the FITC channel before, immediately after, and 50 min after uncaging in the selected donor cell. (D) Plot of observed fluorescence intensity increase of the receptor and bystander cells over time including inset with the theoretical estimation at nanotube radii r of 10, 40 and 70 nm (eqn (8)). | ||
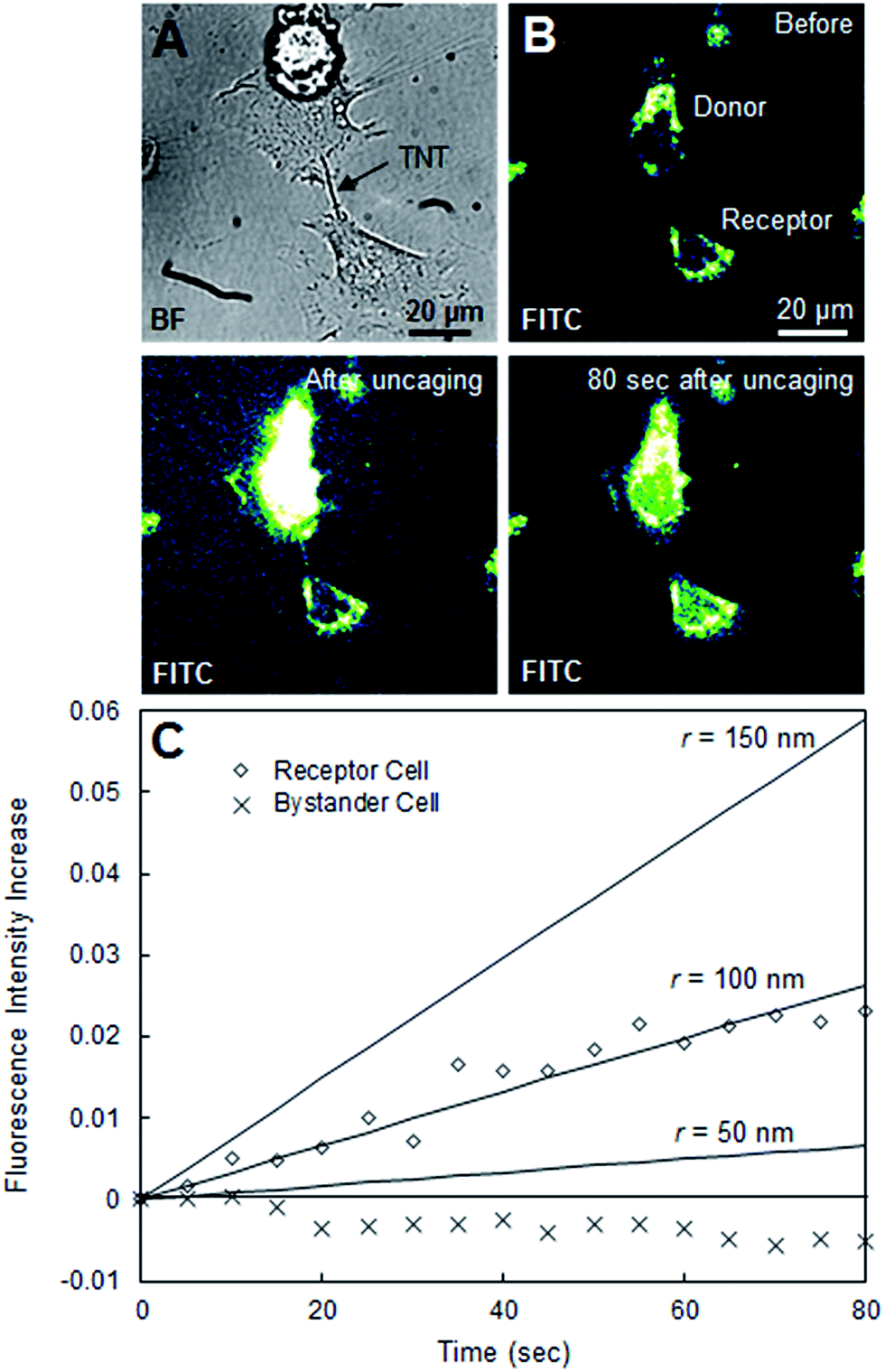 | ||
| Fig. 4 (A) Bright-field image of two HeLa cells connected by a naturally-occurring TNT (B) confocal laser scanning microscopy images of the same cells in the FITC channel before, immediately after, and 80 s after uncaging in the selected donor cell. (C) Plot of observed fluorescence intensity increase of the receptor cell over time alongside the theoretical estimation at several TNT-radii. | ||
Sample preparation for electron microscopy
Lipid blocks were deposited directly on poly-D lysine-coated glass-bottom dishes as described previously, and co-incubated with cells according to the described cell plate preparation. Nanotubes were generated from the lipid block using the micromanipulator and connected to immediately adjacent cells. Separately, an aqueous fixation solution containing 0.67% osmium tetroxide (Electron Microscopy Sciences, CAS #20816-12-0), 0.84% glutaraldehyde (Electron Microscopy Sciences, CAS #111-30-8), and 0.1 M cacodylate buffer was prepared and stored on ice. Following LNT sample plate preparation, 1 mL of medium was carefully withdrawn and 1 mL of fixation solution was added directly to the dish, which was then immediately placed on ice and left to fix for 40 min. After fixation, all of the medium was withdrawn and the sample was carefully rinsed 5× with buffer (10 mM HEPES with 150 mM NaCl, pH 7.4). Next, the sample was dehydrated through a 30%, 50%, 70%, 80%, 95%, and 100% ethanol/water gradient, allowing 20–25 min for each step; after the final step, the 100% ethanol was withdrawn and the sample was allowed to air-dry overnight. Prior to imaging the sample was removed from the dish by carefully detaching the glass coverslip bottom and mounting it on a SEM sample holder using two-sided carbon tape. Finally, the sample was coated with ∼2 nm of gold using a fine sputter-coater (Jeol, JFC-1200) in order to create an electrically conductive surface.Scanning electron microscopy
Electron microscopy images were taken using a low-vacuum scanning electron microscope (Jeol, JSM-6510LV). The microscope was operated using an acceleration voltage of 20 kV, a spot size of 35 nm, and a working distance of 10 mm. The images in Fig. 5A–D were taken as 80 second scans at magnifications of 2200, 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 23000, and 950, respectively. Brightness, contrast, astigmatism, and lens modulation were adjusted manually prior to taking the final scans of the sample.
000, 23000, and 950, respectively. Brightness, contrast, astigmatism, and lens modulation were adjusted manually prior to taking the final scans of the sample.
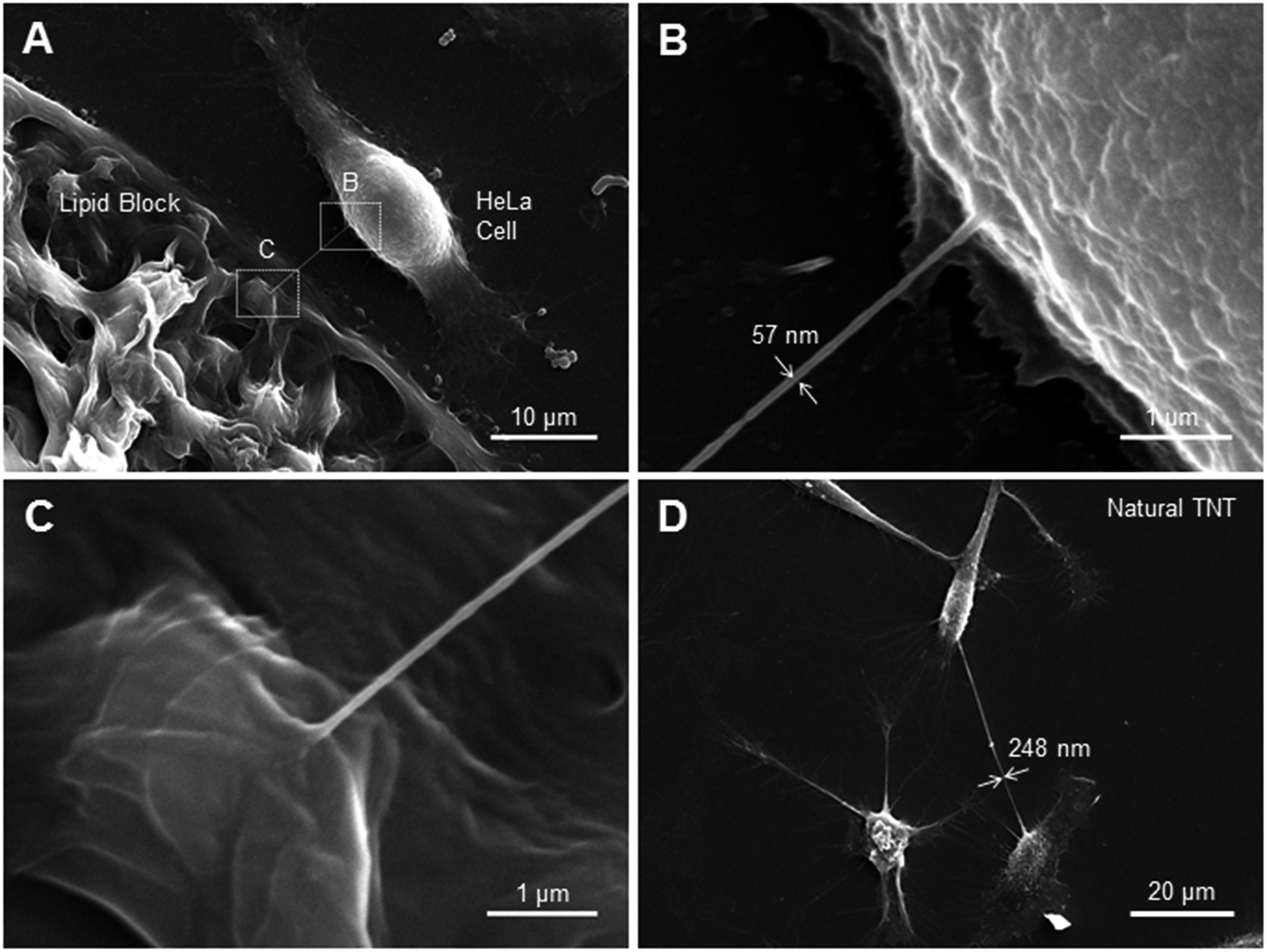 | ||
| Fig. 5 (A) Scanning electron microscopy (SEM) image of a HeLa cell connected to a lipid block by a LNT. (B) Higher magnification SEM image of the cell–LNT junction. (C) Higher magnification SEM image of the LNT-lipid block junction. (D) SEM image of natural tunneling nanotube between two HeLa cells. | ||
Results and discussion
The scheme in Fig. 1A describes our approach to create tubular connections between cells. First, a glass microtip connected to a micromanipulator is coated with small lipid blocks by dragging it through a large lipid block prepared in a separate plate. Once sufficiently coated, the microtip is introduced into the cell culture plate and carefully pushed into a cell membrane to form a connection between the lipids and the cell membrane. The micropipette is subsequently carefully retracted, drawing out a LNT. The newly-generated LNT is further pushed into another nearby cell until it adheres, fabricating a LNT–cell network. The simplicity of the LNT formation in this system comes from the nature of the lipid blocks in HII phase. They act as a lipid reservoir with a fixed surface tension for efficiently extracting tubes.14 This procedure works extremely reproducibly, enabling fabrication of arbitrary LNT–cell network structures (Fig. 1B). HeLa cells were selected for the experiment because of their demonstrated small pericellular matrix,19 which increases the chance of direct lipid–lipid contact between the LNT and the lipid part of the cell membranes for membrane fusion. Furthermore, natural tunneling nanotubes between HeLa cells have been observed,20 though it has not yet been reported if these nanotubes are open-ended.To study the structure of the LNT–cell junction, diffusion of fluorescence-tagged lipids from LNTs to cell membranes was monitored. Fig. 2A shows fluorescence images of two non-labeled cells that were connected with a LNT stained with Lissamine Rhodamine B PE lipids (Liss-Rhod-PE) immediately, as well as 30 min, after connection. The fluorescence from Liss-Rhod-PE in the LNT diffused into the cell membranes and equilibrated throughout the entire system (the membranes of the two cells and the LNT) over time. This indicates that the bilayer of the LNT and the cell membranes from these two cells are laterally connected, presenting the first implication for the membrane fusion. The result is qualitatively similar to what has been previously observed with GUVs connected with a LNT.14 Note that serum was depleted from the cell culture medium when LNTs were attached to cells to increase the chance of the direct lipid–lipid contact between cell membranes and LNTs. In presence of serum, it was previously observed that the LNT–cell interaction is protein-mediated via focal contact.21 We found that after connection with a lipid nanotube, approximately 30% of cells (N = 23) presented qualitatively the same results, while other cells displayed no observable diffusion. This suggests that 30% is the fusion rate between LNTs and HeLa cell membranes under these conditions. In order to analyze the diffusion behavior quantitatively, the increase in the fluorescence intensity at position r = 30 μm inside cells, where the LNT–cell junction was defined as r = 0 μm (see the inset in Fig. 2B), was plotted over time in Fig. 2B. By fitting these curves with a two-dimensional diffusion model (see materials and methods), we roughly estimated the diffusion coefficient of the fluorescent lipids in the cells. The extracted effective diffusion coefficient from analysis of the 8 cells was D = 0.032 ± 0.003 μm2 s−1. Though on the same order of magnitude, the estimated value is five times smaller than the reported diffusion coefficient for Cy3-DOPE in HeLa cell membranes, D = 0.17 μm2 s−1.22 This discrepancy is possibly due to either an underestimation of the radius in our experiments on account of the natural wrinkles in cell membrane, or a slowing of diffusion at the LNT–cell membrane junction due to the membrane curvature.23 Our observed value of D is substantially smaller than, for example, three-dimensional isotropic liposome diffusion in water (D = 2.2 μm2 s−1, estimated using the Stokes–Einstein equation with a particle size of 100 nm). Had the majority of fluorescent lipids been internalized by the cell as vesicles, this might have yielded a false positive due to random dispersion of the vesicles within the cytoplasm. Nevertheless, the sizeable difference in diffusion coefficients refutes this, further supporting our conclusion that the nanotube indeed fuses to the cell membrane.
To study if the LNT–cell junction is open-ended, we monitored the transport of a water-soluble yet membrane-impermeable dye, fluorescein, between two LNT-connected cells. CMNB-caged fluorescein is introduced inside cells via induced pinocytic vesicle fusion and internal osmotic lysis prior to plating. After connecting two cells with a pair of LNTs, we uncaged the CMNB-caged fluorescein in one of the cells and monitored the change in fluorescence intensity of the connected cell (propagation of uncaged fluorescein) over time (Fig. 3A). Fig. 3B shows a Rhod Red channel (bilayer staining) confocal laser scanning microscopy image of three cells, where two are connected with LNTs (labeled in Fig. 3C as donor and receptor) while one is not (bystander). Fig. 3C shows snapshots of the same cells in the second channel monitoring for fluorescein (FITC) before, immediately after, and 50 min after uncaging the CMNB-caged fluorescein. The image after uncaging clearly shows that fluorescein was successfully released in the donor cell, while in the image 50 min after uncaging the receptor cell shows an observable increase in fluorescence intensity. The increase in the fluorescence intensity of the receptor cell (in the FITC channel) was plotted against time in Fig. 3D together with that of the bystander cell for comparison. The receptor cell presented a noticeable increase in the fluorescence intensity over time after uncaging, compared to that of the bystander cell, which typically decreases over time due to photobleaching. This suggests that the increase in the fluorescence intensity of the receptor cell is due to the transport of uncaged fluorescein via LNTs rather than other causes such as release and uptake of uncaged fluorescein via exosomes. The result also suggests that the LNT–cell junction is open-ended for these two cells. The inset in Fig. 3D is the same fluorescence data from Cell 2 in a different scale plotted with theoretical predictions. These predictions were calculated using a simplified diffusion model (eqn (8)) at several nanotube-radii starting with 10 nm, as expected for LNTs.9 We found that the observed diffusion was approximately 25× that expected for an LNT with r = 10 nm. This suggests that the observed fluorescein diffusion is either due to the contribution of two LNTs and/or the diameter of the LNT slightly enlarged after it was connected to the cell membranes. We observed the transport of fluorescein in LNT-connected systems with a probability of 5% (N = 37). The low success rate implies the low probability of the formation of a successfully fused, open-ended structure at the LNT–cell interface. This rate is reasonable when considering the 30% probability of successful nanotube–cell membrane fusion as characterized before in Fig. 2. To observe the transport of fluorescein, both nanotube–cell membrane junctions must be open-ended for two connected cells, which occurs with a 9% probability (30% for the donor cell × 30% for the receptor cell). The experimental success rate of the observed fluorescein diffusion (5%) is slightly less than 9%, which could be because the lipid blocks which are used to generate the connections sometimes retain the inverse hexagonal phase, effectively blocking the junction (Fig. 3A). An additional factor in the success rate of caged-dye experiments was the viability of the donor cells, and this too could have played a part in observing diffusion. A cell which had been loaded with too much caged fluorescein could experience a significant change in internal osmotic pressure upon uncaging (one caged fluorescein would become two CMNB molecules and a fluorescein), potentially leading to cell death and permeation of dye from the cell before the diffusion takes place.
During our experiments, we sometimes encountered HeLa cells connected by natural tunneling nanotubes, such as the pair shown in Fig. 4A. To test the credibility of our simplified model, we performed the same uncaging experiments and analysis with these natural TNTs. Fig. 4B shows snapshots from an FITC confocal laser scanning microscopy movie of two TNT-connected, CMNB-caged fluorescein-loaded cells before, immediately after, and 80 s after uncaging in the donor cell. We observed a significant fluorescence increase in the receptor cell on a time scale of a couple of minutes after uncaging (Fig. 4B). Though this is significantly faster than the transport of fluorescein between synthetic LNT–connected cells, this result confirms that natural TNT connections in HeLa cells, like our LNT connection, can indeed be open-ended and allow for intercellular transport. In Fig. 4C, the time evolution of normalized fluorescence intensity in the receptor cell was plotted. Theoretical estimations of fluorescence were also plotted for radii of 25, 50, and 100 nm as benchmarks. This is the same diffusion model as the one we used for the synthetic LNT–connected cells, where the only changed parameter is the length of the tube L derived from the microscopy image. Based on the correlation of the experimental data with the benchmarks the effective inner radius of our HeLa TNT can be estimated as around 100 nm. Although there were no reported radius values specific to HeLa cell TNTs, in general the radius of natural TNTs has been reported as ranging from 25 to 250 nm.24 Therefore, our simplified model could explain the time scale of the transport through a natural TNT with a radius within reported values, supporting the validity of our analysis.
In order to visualize the junction of the cell–LNT connection with high resolution, as well as to estimate LNT and TNT radii, we performed scanning electron microscopy (SEM) imaging. SEM overcomes the diffraction limit of standard light microscopy techniques and should allow for the visualization of nanostructures such as LNTs. Unfortunately, the fixation and dehydration steps required for SEM sample preparation can destroy such structures. In order to increase a chance for finding intact lipid tubes in specimen, we needed a high throughput way to connect cells with nanotubes. We achieved this by incubating deposited lipid blocks directly with HeLa cells, and drawing many nanotubes between the block and adjacent cells. A lipid block-cell LNT connection formed using this technique is depicted in Fig. 5A. Fig. 5B and C show zoomed-in SEM images of the cell–LNT junction and the lipid block-LNT junction respectively. These images imply that the LNT and the cell membrane are laterally connected by membrane fusion in agreement with the fluorescein diffusion experiment (Fig. 3), although we cannot exclude possible artifacts from fixation. The diameter of the LNT appears to be ∼57 nm in the SEM image (Fig. 5B), which also roughly agrees with value estimated from the fluorescein diffusion experiment (Fig. 3D). This value is slightly larger than the reported diameter of DOPE-based LNT (∼20 nm).9 As discussed previously, this could be due to the mixture of cell membrane components into the LNT upon membrane fusion, which modulated the surface tension and average bending modulus of LNTs. We also examined several natural tunneling nanotubes between HeLa cells by SEM, one of which is shown in Fig. 5D. They typically appear to have a diameter of 200–250 nm, which is larger than the nanotube we observed between the cell and lipid block in Fig. 5A. This is also in agreement with the value obtained from the fluorescein diffusion experiment (Fig. 4C).
Conclusion
The approach demonstrated in this work allows for the arbitrary generation of nanotubular connections between eukaryotic cells using lipid nanotubes. In HeLa cells, the generated nanotubes fuse to the cell membrane with an experimental success rate of approximately 30% and allow for the diffusion (D = 0.032 ± 0.003 μm2 s−1) of lipid components through the nanotube–cell interface. Furthermore, the transport of water-soluble dye between two cells connected with this nanotubular structure was demonstrated. The observed time scale of the transport (0.04% increase of fluorescence intensity in the receptor cell after 60 min) is in agreement with that derived from our theoretical diffusion model, where the analysis indicated that the radius of the synthetic LNT became slightly larger when it was connected to cells. Analysis of SEM images of the LNT–cell connection also suggested an enlarged nanotube structure. This widening of the LNTs can be explained by the lipid exchange between the cell membrane and the nanotube, which alters the balance between surface tension and average bending modulus, which determine the diameter. Overall, our statistical analysis concluded that the probability of observing membrane fusion between LNT and cell membranes is 30%, among which roughly 75% of the time the resulting structure is open-ended. The presented work is a proof of concept for the facile fabrication of artificial LNT–cell network and the demonstration of passive exchange of free molecules between cells. The approach demonstrated in this work can be used to fabricate controlled cellular networks on substrates as a model system for TNTs and as a single cell molecular delivery system in the future. Furthermore, the good correlation between the diameters estimated from the high-resolution SEM images and the ones from the theoretical model implicates a possible use of the presented theoretical approach to estimate the radii of nanotubular connections between cells.Acknowledgements
We acknowledge the Swiss National Centre for Competence in Research (NCCR) in Chemical Biology for providing access to their cell culture facilities, and in particular Daniel Abegg and Dominic Hoch for their support. We also acknowledge the Bioimaging Center at the University of Geneva for access to a scanning electron microscope as well as preparatory equipment and facilities, and in particular Jérôme Bosset for training and support. Part of the research leading to these results has received funding from Novartis Foundation for medical-biological research (#15A010) and Swiss National Science Foundation (#200021_159567). The master's thesis containing this research was organized and supported by the Swiss National Centre for Competence in Research (NCCR) in Chemical Biology.References
- M. Mittelbrunn and F. Sanchez-Madrid, Nat. Rev. Mol. Cell Biol., 2012, 13, 328–335 CAS.
- A. Rustom, R. Saffrich, I. Markovic, P. Walther and H. H. Gerdes, Science, 2004, 303, 1007–1010 CrossRef CAS PubMed.
- M. Hashimoto, F. Bhuyan, M. Hiyoshi, O. Noyori, H. Nasser, M. Miyazaki, T. Saito, Y. Kondoh, H. Osada, S. Kimura, K. Hase, H. Ohno and S. Suzu, J. Immunol., 2016, 196, 1832–1841 CrossRef CAS PubMed.
- S. Abounit, J. W. Wu, K. Duff, G. S. Victoria and C. Zurzolo, Prion, 2016, 10, 344–351 CrossRef CAS PubMed.
- A. Osteikoetxea-Molnar, E. Szabo-Meleg, E. A. Toth, A. Oszvald, E. Izsepi, M. Kremlitzka, B. Biri, L. Nyitray, T. Bozo, P. Nemeth, M. Kellermayer, M. Nyitrai and J. Matko, Cell. Mol. Life Sci., 2016, 73, 4531–4545 CrossRef CAS PubMed.
- X. Wang and H. H. Gerdes, BBA, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2082–2086 CrossRef CAS PubMed.
- P. Pascoal, D. Kosanic, M. Gjoni and H. Vogel, Lab Chip, 2010, 10, 2235–2241 RSC.
- H. Zhang, S. Xu, G. D. M. Jeffries, O. Orwar and A. Jesorka, Nano Commun Netw., 2013, 4, 197–204 CrossRef.
- K. Sugihara, M. Chami, I. Derenyi, J. Voros and T. Zambelli, ACS Nano, 2012, 6, 6626–6632 CrossRef CAS PubMed.
- R. Koynova and M. Caffrey, Chem. Phys. Lipids, 1994, 69, 1–34 CrossRef CAS PubMed.
- R. P. Rand and N. L. Fuller, Biophys. J., 1994, 66, 2127–2138 CrossRef CAS PubMed.
- E. Y. Shalaev and P. L. Steponkus, BBA, Biochimica et Biophysica Acta, Biomembranes, 1999, 1419, 229–247 CrossRef CAS.
- J. West, A. Manz and P. S. Dittrich, Lab Chip, 2008, 8, 1852–1855 RSC.
- K. Sugihara, A. Rustom and J. P. Spatz, Soft Matter, 2015, 11, 2029–2035 RSC.
- H. Farhood, N. Serbina and L. Huang, Biochim. Biophys. Acta, 1995, 1235, 289–295 CrossRef.
- C. Y. Okada and M. Rechsteiner, Cell, 1982, 29, 33–41 CrossRef CAS PubMed.
- C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Talanta, 2002, 56, 365–373 CrossRef CAS PubMed.
- L. Zhao, C. D. Kroenke, J. Song, D. Piwnica-Worms, J. J. H. Ackerman and J. J. Neil, NMR Biomed., 2008, 21, 159–164 CrossRef CAS PubMed.
- R. Zhou, H. Y. Zhou, B. Xiong, Y. He and E. S. Yeung, J. Am. Chem. Soc., 2012, 134, 13404–13409 CrossRef CAS PubMed.
- C. Schiller, K. N. Diakopoulos, I. Rohwedder, E. Kremmer, C. von Toerne, M. Ueffing, U. H. Weidle, H. Ohno and E. H. Weiss, J. Cell Sci., 2013, 126, 767–777 CrossRef CAS PubMed.
- K. Sugihara, M. Delai, R. Mahnna, J. Kusch, D. Poulikakos, J. Voros, T. Zambelli and A. Ferrari, Integr. Biol., 2013, 5, 423–430 RSC.
- T. K. Fujiwara, K. Iwasawa, Z. Kalay, T. A. Tsunoyama, Y. Watanabe, Y. M. Umemura, H. Murakoshi, K. G. N. Suzuki, Y. L. Nemoto, N. Morone and A. Kusumi, Mol. Biol. Cell, 2016, 27, 1101–1119 CrossRef CAS PubMed.
- N. S. Gov, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2006, 73, 041918 CrossRef PubMed.
- M. W. Austefjord, H. H. Gerdes and X. Wang, Commun. Integr. Biol., 2014, 7, e27934 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |