
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
IMM-H004, a coumarin derivative, attenuated brain ischemia/reperfusion injuries and subsequent inflammation in spontaneously hypertensive rats through inhibition of VCAM-1 †
Peng-Fei Yanga,
Xiu-Yun Songa,
Ting Zengb,
Qi-Di Aib,
Dan-Dan Liuc,
Wei Zuoa,
Shuai Zhanga,
Cong-Yuan Xiaa,
Xin Hec and
Nai-Hong Chen *ab
*ab
aDepartment of State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica & Neuroscience Center, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China. E-mail: chennh@imm.ac.cn; Fax: +86-010-63165177; Tel: +86-010-63165177
bCollege of Pharmacy, Hunan University of Chinese Medicine, Changsha, Hunan, China
cTianjin University of Traditional Chinese Medicine, Tianjin, China
First published on 23rd May 2017
Abstract
Strokes are the leading cause of death and disability all over the world, however, there are few satisfactory therapies. IMM-H004 citrate (004), 7-hydroxy-5-methoxy-4-methyl-3-(4-methylpiperazin-1-yl)-coumarin, is a coumarin derivative which has potential therapeutic effects in brain ischemia with ambiguous mechanisms. We aim to study the anti-brain ischemia effect and mechanism of 004 in spontaneously hypertensive (SHR) rats. Adult male Wistar-Kyoto (WKY) rats and SHR rats were subjected to 24 h reperfusion after transient middle cerebral artery occlusion (tMCAO) for 1 h and were intravenously injected with 004 or Edaravone at the time of reperfusion. Behavioral scores, magnetic resonance imaging (MRI) and TTC staining were used to test the therapeutic effect of 004. To study the mechanism, the infiltration of leukocytes, the activation of microglia, blood viscosity, the expression of VCAM-1, MMPs and proteins involved in the MAPK/NF-κB pathway were researched. The results indicated that tMCAO/R induced serious injury, while 004 significantly alleviated the infarct volume and improved neurological deficits. 004 improved inflammatory processes, such as the enhancement of blood viscosity, expression of VCAM-1 and MMP2, release of TNF-α, IL-1β, IL-6 and IL-23, phosphorylation of JNK and p38 and translocation of NF-κB, which play crucial roles in brain I/R in SHR rats. An in vitro study also proved that 004 regulated JNK and NF-κB pathways and decreased the expression of VCAM-1, which eventually led to the suppression of neuroinflammation.
1. Introduction
Strokes are one of the leading cause of death and disability and are a great burden to the whole world.1,2 Since more than 85% of all strokes are caused by cerebral ischemia,3 researchers have put a great deal of effort into finding treatments for ischemic cerebrovascular diseases. Brain ischemia/reperfusion (I/R) triggers a complicated cascade of events, including abnormal energy supplement, disturbance of ion metabolism, oxidative stress, vascular damage and activation of the immune system. These pathological processes eventually cause damage to neuronal cells and brain tissue. A number of compounds are also designed based on these etiologic factors, but unfortunately most of them are effective in experiments, but invalid in clinical trials. The only Food and Drug Administration (FDA)-approved drug for clinical application is the recombinant tissue plasminogen activator (rtPA), which has a short therapeutic window and causes several side effects, such as allergic/vascular edema, systemic hemorrhage, myocardial ischemia and a high risk of hemorrhage transformation, and can be used only for patients with systolic pressure < 180 and diastolic pressure < 110. Nevertheless, the greatest risk factor for strokes is high blood pressure (BP), a reduction in risk by 41% for strokes was achieved for every 10 mmHg lower BP achieved.4 Of the patients with first-ever strokes, 70.3% were diagnosed with hypertension. Among which, 15.9% were unaware of their hypertension at the time of the stroke onset.5 Thus, there is an urgent need to develop new drugs to treat stroke patients with hypertension.Coumarin and its derivatives, an important class of organic heterocyclic compounds which are widely distributed in plants, have been demonstrated to exert various neuroprotective effects in previous studies. IMM-H004 [7-hydroxy-5-methoxy-4-methyl-3-(4-methylpiperazin-1-yl)-coumarin] (004, Fig. 1), a novel coumarin compound that is our own intellectual property, has been demonstrated to have neuroprotective effects in brain I/R models in our previous studies,6,7 but the effects in brain I/R models with hypertension are still unexplored and the relevant mechanism was also ambiguous. Since hypertension is the most common complication in a stroke, it is of great value to test the protective effects of 004 in a spontaneously hypertensive (SHR) rat brain I/R model.
 | ||
| Fig. 1 The chemical structure of IMM-H004 citrate. | ||
In strokes, the initial inflammatory response causes the activation of microglia and inflammatory cells, which is the organism’s attempt to alleviate brain injury. However, a mass amount of lysosomal enzymes and oxidative bursts that are secreted by the over-activation of inflammatory cells gradually lead to endothelial cell junction dysfunction, destroyed blood–brain barrier integrity, exacerbated neuron death and finally results in permanent brain injury which is called “secondary brain injury”.8 Besides, inflammation is believed to play an important role in the severe outcome of strokes in hypertensive patients.9 Thus, we aim to investigate the effect of 004 in brain I/R models in SHR rats, and clarify the effect of 004 on the inflammation in this model.
2. Experimental procedures
2.1 Animals
Adult male Wistar-Kyoto (WKY) rats and SHR rats (270 ± 10 g, 3–4 months) were purchased from Vital River Laboratories (Beijing, China). WKY rats were randomly assigned to 3 groups: sham, I/R and I/R + 004 (10 mg kg−1), while SHR rats were randomly assigned to 6 groups: sham, I/R, I/R + 004 (2.5, 5, 10 mg kg−1) and I/R + Edaravone (10 mg kg−1, Xiansheng, China). Rats were randomly assigned to each group and were given vehicles or drugs intravenously at the time of reperfusion after ischemia for 1 h and were kept for 24 h or 7 days. Ischemia was conducted by middle cerebral artery occlusion (MCAO) as previously described.10 Body temperature was maintained at 37.5 ± 0.5 °C until the rats recovered from anesthesia. All experiments were double-blind. A total of 236 rats were used, 33 rats died, 3 SHR rats were excluded because of low BP and the data of 7 rats were deleted because of data error in blood sample analysis. The total mortality rate was 14%. The effect of MCAO was confirmed by computed tomography angiography (CTA) (Additional file 2†). All animal experiments were approved by the Institutional Animal Care and Use Committee of the Chinese Academy of Medical Sciences and were performed in accordance with the standards of the National Institutes of Health guide for the care and use of Laboratory animals.2.2 Blood pressure measurement
The systolic blood pressure (SBP) of the animals was measured using the blood pressure (BP) measurement apparatus Softron BP-98A (Softron, Japan). The animals were kept in a warm place for 5 min and SBP was measured by a tail-cuff technique following the guidelines. For hypertension confirmation, SBP values were determined 3 times during 18:00–19:00 as the BP curve served as a reverse-dipper pattern in SHR rats.11 In order to understand the influence of 004 on BP, SBP values were measured after treatment with 004 for 2 h because the effects of the reagent on SBP appeared 1 to 2 hours after drug administration in most cases.12,132.3 Evaluation of neurological deficits
Neurological deficits were blindly scored at different time points after I/R using methods such as Zea Longa’s test,14 the grid test,15 the hanging wire test16 and the Garcia and Bederson (G&B) scores17 to comprehensively analyze the neurological function. The details are shown in Table 1.| Test type | Purpose | Scores | Unit | Criterion | |
|---|---|---|---|---|---|
| Mildest | Severest | ||||
| Zea Longa’s test | Classic assessment for overall condition | 0 | 4 | Point | 0: no neurological deficit; 1: failure to extend left forepaw fully; 2: circling to the left; 3: falling to the left; 4: did not walk spontaneously and had a depressed level of consciousness. |
| Grid test | Evaluating motor ability | 0 | 10 | Pace | Animals placed on an elevated (1 m) grid surface platform with square openings of 9 × 9 cm were encouraged to traverse the grid surface for 1 minute. The foot faults where animals inaccurately placed a limb through one of the openings in the grid were counted. The number of foot faults made per meter in 1 minute was calculated. |
| Hanging wire | Checking grasping ability | 120 | 0 | Second | A stainless steel bar (50 cm in length) resting on two vertical supports and elevated 60 cm above a flat surface was used in this test. Rats were placed on the bar midway between the supports and were observed for 120 s. The amount of time spent hanging was recorded. |
| G&B scores | Evaluating reaction and response ability | 18 | 3 | Point | Spontaneous activity, symmetry of movements and symmetry of forelimbs were evaluated as 0–3; climbing the wall of the wire cage, the reaction to touch on either side of the trunk and the response to vibrissae touch were evaluated as 1–3. The total scores were recorded. |
2.4 Magnetic resonance imaging (MRI) of brain lesions
After I/R at different time points rats were anaesthetized for MRI (PharmaScan 70/16 US, Bruker, Germany). The MRI protocol was a T2_TurboRARE with TR/TE = 4300/35 ms, 4 averages, 40 × 40 field-of-view, size 256 × 256, and 0.5 mm thickness.2.5 The measurement of brain infarct and edema volumes
2,3,5-Triphenyltetrazolium-chloride (TTC, Sigma, USA) staining was performed to determine the infarct and edema volumes after 24 h by using Image J. While the infarct and edema volumes after 0 hours, 3 hours, 6 hours, 12 hours, 1 day, 2 days, 4 days and 7 days after I/R were determined by MRI using the Image-Pro Plus 6.0 software. The infarct volumes were calculated by multiplying the white area of all slices and the edema volumes were measured by multiplying the enhanced area caused by I/R.2.6 Immunohistochemistry
After the brains were collected from the rats in each group, a series of 5 μm-thick sections were prepared. The sections were submitted to antigen retrieval and incubated with a primary antibody against ionized calcium-binding adapter molecule 1 (Iba1, Wako, Japan) or myeloperoxidase (MPO, Abcam, UK) overnight. Then the sections were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody and visualized by 3,3′-diaminobenzidine (DAB, ZSGB-Bio, China).2.7 Immunofluorescence staining
Sections with a thickness of 5 μm were incubated with primary antibodies for chemokine-like factor 1 (CKLF1), Iba1, NeuN, the glial fibrillary acidic protein (GFAP) or the vascular cell adhesion molecule (VCAM)-1 for 10 h and were then incubated with Alexa 488, 546 or 647-conjugated donkey secondary antibodies (life technologies, USA) to combine with the primary antibodies. Images were acquired in the ischemic regions with a fluorescence microscope (Eclipse TE2000e, Japan).2.8 Hemorheological tests
Blood samples were collected from the abdominal aorta of the rats in each group. Hematocrit was measured by Sysmex XE-2100 (Sysmex, Japan). The automatic blood rheometer Succeeder SA-6000 (Succeeder, China) was used for measuring whole blood viscosity (WBV), plasma viscosity (PV), whole blood relative viscosity at low (WBRV-L) and high (WBRV-H) shear rate, erythrocyte aggregation index (EAI) and erythrocyte rigidity index (ERI).2.9 ELISA assay
After blood collection, the ischemic cortex tissues were quickly dissected, washed, homogenized and centrifuged at 3000 g for 10 min and then the levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-23 were measured using the corresponding kits (Jiancheng, China) and strictly following the manufacturer’s instructions.2.10 Cell culture
Human brain microvascular endothelial cells (HBMEC) were obtained from National Infrastructure of Cell Line Resource (Beijing, China) and maintained in RPMI-1640 medium at 37 °C. We set up an oxygen glucose deprivation (OGD) model by using glucose-free RPMI-1640 and putting the culture flask into a modular incubator chamber (Billups-rothenberg, USA) that was gassed with 95% N2/5% CO2. The concentration of O2 (Measured by O2 Quickstick, Nuvair, USA) decreased to 0.03% and was considered as the hypoxia state. Then cells were reoxygenated (R) in glucose-containing RPMI-1640 in normal culture conditions.2.11 Western blotting
Brain tissues of the ischemic cortex were dissected from each rat, then washed and disrupted to obtain the tissue homogenates. Cell lysates were obtained by homogenization in lysis buffer. 25 μg of protein samples were separated on SDS-PAGE gels, then transferred to PVDF membranes (Millipore, USA). After incubation with the following primary antibodies overnight, anti-pIKKαβ, anti-IKKα, anti-IKKβ, anti-p-NF-κB, anti-NF-κB, anti-MMP2, anti-MMP9, anti-β-actin, anti-PCNA (all 1:500, Santa Cruz, USA), and anti-VCAM-1 (1:500, Abcam, UK), the membranes were incubated with HRP-conjugated secondary antibodies (1:5000, KPL, USA). The predicted bands were detected using chemical luminescence (PPLYGEN, China) in a chemiluminescence detection system (ImageQuant LAS4000mini, Sweden).2.12 Statistical analysis
Data were expressed as mean ± standard deviation (SD) and analyzed using the GraphPad Prism 5 software. One-way analysis of variance (ANOVA) analysis was used to analyze the differences between multiple groups within the same species. Student’s t-test was used to analyze the differences between two groups.3. Results
3.1 004 attenuates I/R-induced neurological deficits, brain edema and infarction in SHR and WKY rats
24 h was believed to be an appropriate time point to evaluate the effect of compounds in the brain I/R model18 so the effect of 004 was judged at 24 h as a preliminary test. Zea Longa’s test, the grid test, the hanging wire test and the Garcia and Bederson scores test (Fig. 2A) were performed so that the motor coordination, the motor ability, the grasping ability and the reaction and response ability of the animals could be judged. Rats in the I/R group displayed prominent deficits, while the 004 group showed an obvious dose-dependent improvement and the 10 mg kg−1 group displayed the best protective effect. The Edaravone (10 mg kg−1) group also exhibited significantly improved neurological performance compared to brain I/R SHR rats.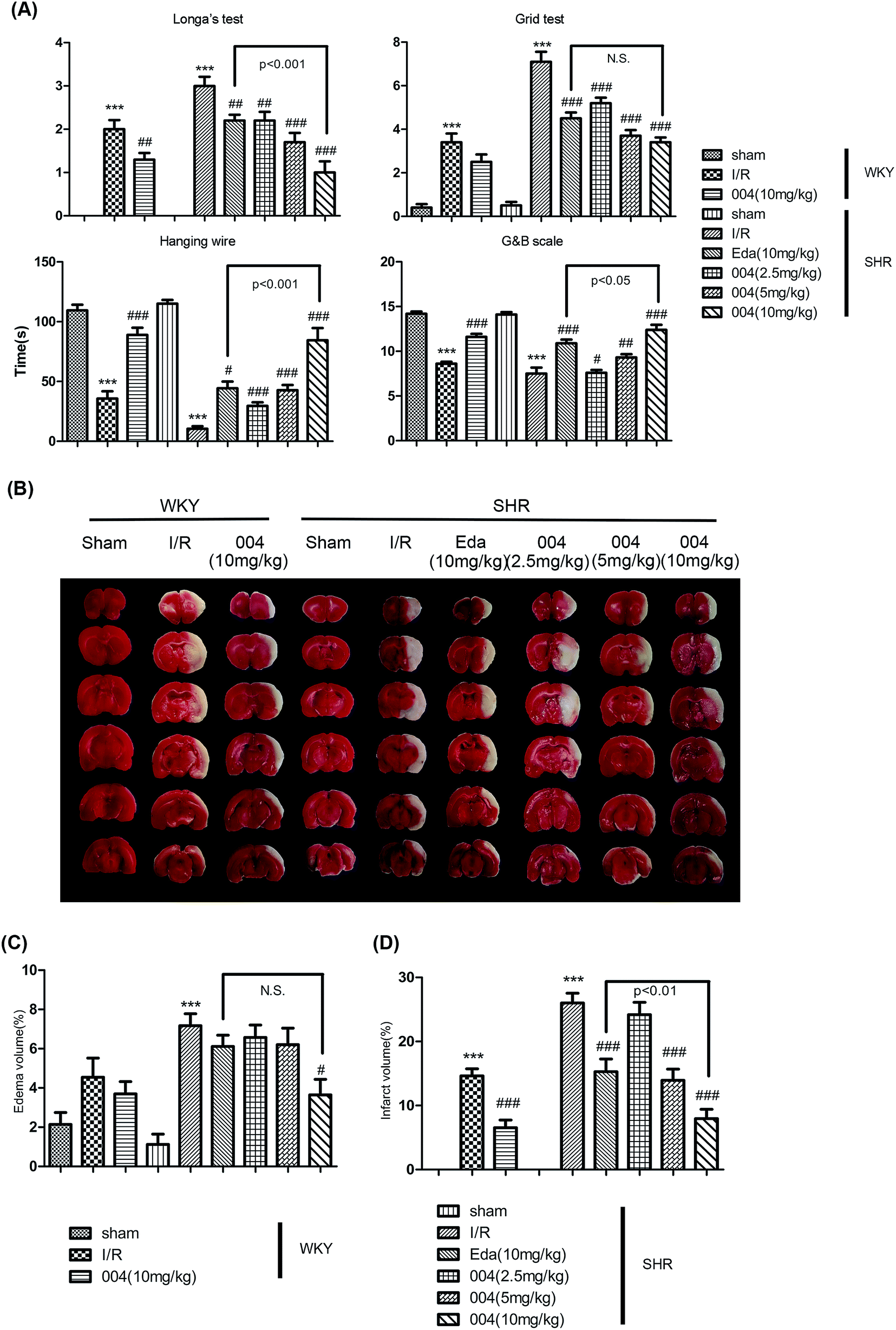 | ||
| Fig. 2 The effect of 004 on the behavioral and pathological changes induced by I/R in WKY and SHR rats at 24 h after reperfusion. (A) 004 improved the behavioral injury in Zea Longa’s test, the grid test, the hanging wire test, the Garcia and Bederson scores test. (B) Representative pictures for the effect of 004 on brain I/R injury using TTC staining. (C and D) 004 decreased the infarct area and edema volume in WKY and SHR rats. In A, C and D, n = 10 in each group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. I/R. | ||
TTC staining in Fig. 2B, as well as the graphs in Fig. 2C and Fig. 2D, showed that both 004 and Edaravone reduced brain edema (Fig. 2C) and cerebral infarction (Fig. 2D) significantly after reperfusion for 24 h. The effect of 004 was better than Edaravone in ameliorating the infarct volume. Although 004(10 mg kg−1) decreased the edema volume significantly and Edaravone showed no obvious effect, there were no significant differences between 004 and Edaravone in decreasing the edema volume.
In order to investigate the protective effect of 004 after a long period, we tested the effect of 004 until 7 days after reperfusion in SHR rats. The results of survival rates and neurological testing showed that 004 reduced the death rate (Fig. 3A) and alleviated behavioral deficits (Fig. 3B). There were slight differences in the survival rate between 004 and Edaravone treated groups (91.7% vs. 83.3%). We speculated that the phenomenon might be related to the results of the edema volume analysis, in which 004 and Edaravone showed no significant differences in ameliorating the edema volume, as clinically significant brain edema is a cause of substantial mortality.19 The results of the MRI also proved that 004 relieved brain lesions as the enhanced infarct volume and the edema volume caused by brain I/R dramatically decreased (Fig. 3C–E), especially at 24 h and 48 h. Nevertheless, the change of I/R injury by 004 was not due to BP alteration (Fig. 3F).
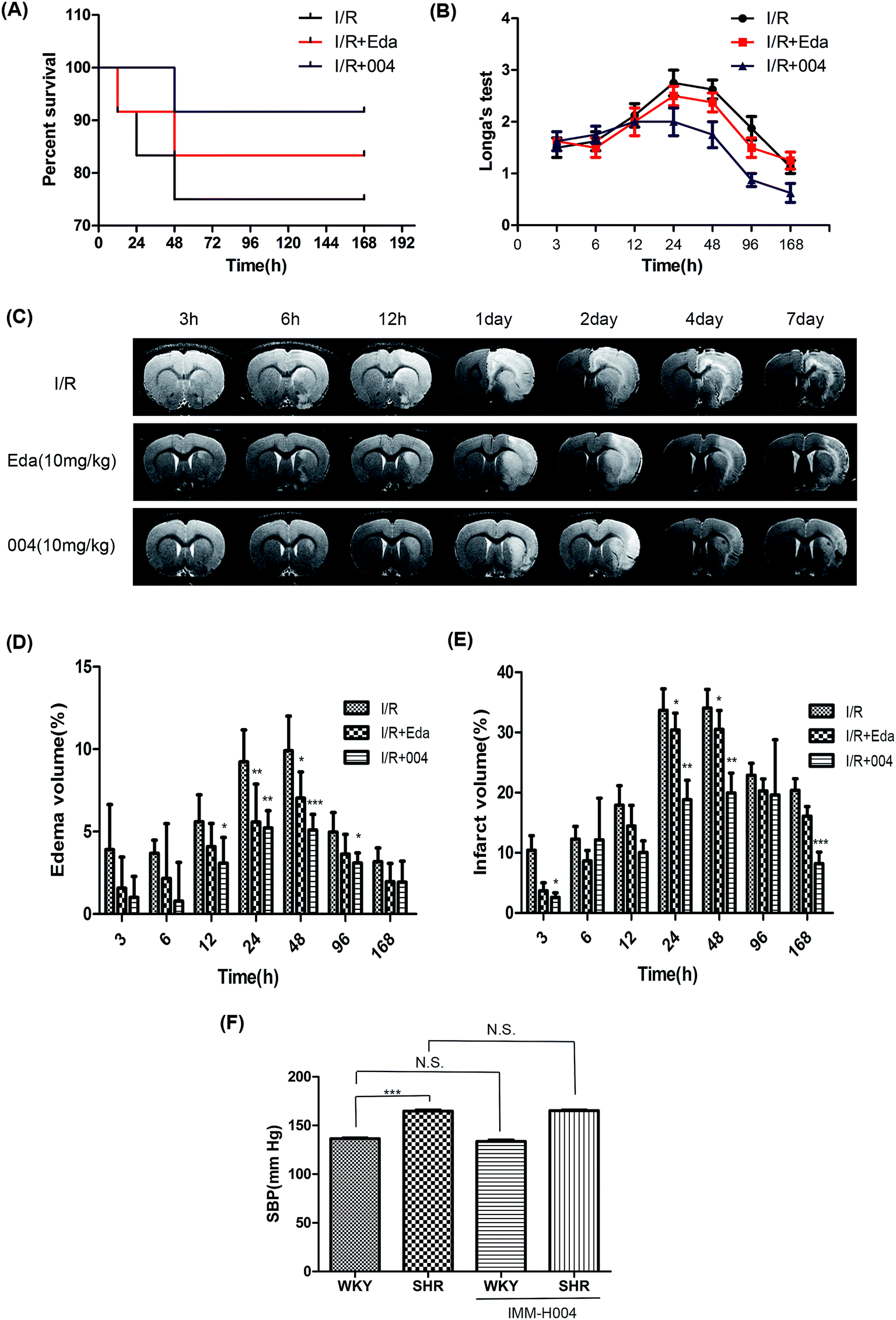 | ||
| Fig. 3 The effect of 004 on the survival rate and the behavioral and pathological changes induced by I/R in SHR rats were observed for 7 days. (A) 004 decreased the death rate in SHR rats after observation for 7 days. n = 12. (B) 004 relieved the neurological deficits in SHR rats after observation for 7 days. n = 8. (C) Representative pictures for the effect of 004 on brain I/R injury using magnetic resonance imaging (MRI). (D and E) 004 decreased the edema volume and infarct volume at different time points in SHR rats. In D and E, n = 6 in each group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. I/R. (F) Changes in SBP caused by 004 in WKY and SHR rats. In F, n = 15. Values expressed as mean ± standard deviation (SD). | ||
3.2 004 ameliorated inflammatory cell infiltration and microglia activation in SHR rats which is more severe than in WKY rats
I/R induced neuroinflammation is a progressive event and occurs from hours to days.20 Major inflammatory markers peaked at 24 h after I/R,21–23 so we chose 24 h as an appropriate time point to study the effect of 004 on inflammation, which might negatively influence the outcome of stroke. As MPO was seen as a marker of myeloid cells (mainly neutrophils and monocytes), we measured the number of MPO + cells in the damaged region to examine leukocyte infiltration (Fig. 4A) after I/R for 24 h. The results showed that the number of MPO + cells greatly increased in the damage region of I/R rats when compared to sham rats, while 004 notably reduced the number of MPO + cells (Fig. 4B). | ||
| Fig. 4 004 ameliorated inflammatory cell infiltration and microglia activation. (A and B) 004 decreased myeloperoxidase (MPO)+ cell numbers in the ischemic area of the cortex. (C and D) 004 decreased activated microglia numbers in the ischemic area of the cortex. (E) There were more ionized calcium-binding adapter molecule 1 (Iba1)+ cells per field in the brains of SHR than in WKY rats. n = 6 in each group. Scalebar = 200 μm. ***P < 0.001 vs. Sham, ###P < 0.001 vs. I/R. @P < 0.05 vs. WKY sham. Values expressed as mean ± SD. | ||
Iba1 was considered as a marker of microglia and activated microglia were identified as showing (1) thicker Iba1 immunohistochemical staining, (2) enlarged and/or irregular cell bodies and (3) complete or partial loss of thin, elongated processes.24 We observed that the markedly increased number of activated microglia (red arrow, Fig. 4C) in I/R groups was attenuated by 004 (Fig. 4D). Although the activated microglia showed no difference, there are many more Iba1+ cells (resting microglia, yellow arrow, Fig. 4E) in the brain of SHR sham rats than in WKY sham rats.
3.3 004 alleviated abnormal hemorheological indices changes caused by I/R in SHR and WKY rats
When acute inflammation occurs, the blood viscosity increases, so the leukocytes in the blood will easily accumulate along and stick to the vascular endothelium and eventually migrate through the vascular wall into the site of injury. Therefore, we monitored the hemorheological indices, which are also available clinically. The WBV at low (1.0 s−1), middle (50 s−1) or high (180 s−1) shear rates (Fig. 5A–C) and the WBRV-L or WBRV-H shear rates (Fig. 5D and E) gave similar results, that is, blood viscosity was enhanced after I/R, while 004 and Edaravone showed decreased blood viscosity. As the PV, hematocrit and fibrinogen concentration of the animals in each group did not show significant differences (Fig. 5F–H), WBRV-L and WBRV-H were mainly determined by EAI and ERI because both of them were thought to be more obviously correlated with the severity of hypertension when there is no obvious change in the aforementioned parameters.25 Results showed that the enhanced EAI and ERI in I/R groups were alleviated by Edaravone or 004 administration (Fig. 5I and J). It is known that oxidative stress is involved in the enhancement of EAI and ERI,26 so the effect of 004 may be derived from its antioxidant capacity which was proved in an earlier study.27 We also measured the coagulation function, which presented no useful information to our study (Additional file 1†).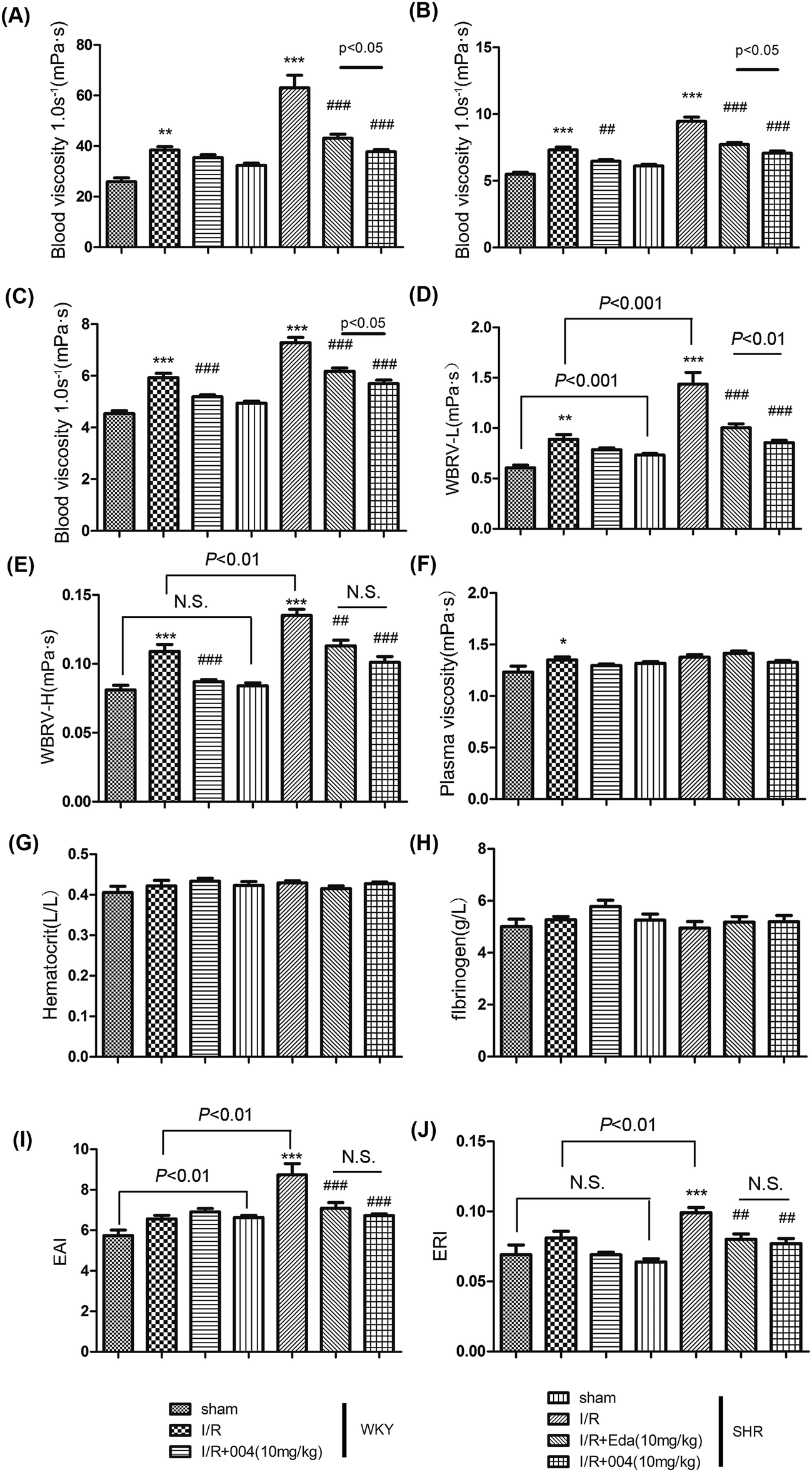 | ||
| Fig. 5 004 alleviated abnormal hemorheological indices changes induced by I/R in WKY and SHR rats. (A, B and C) 004 alleviated abnormal blood viscosity at 1.0, 50 and 180 s−1 shear rates. (D and E) 004 depressed the whole blood relative viscosity at low-(WBRV-L) or high-(WBRV-H) shear rates. (F, G and H) There were no changes in plasma viscosity (PV), hematocrit and fibrinogen in each group. (I and J) 004 decreased the erythrocyte aggregation index (EAI) and the erythrocyte rigidity index (ERI). n = 10 in each group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham, ##P < 0.01, ###P < 0.001 vs. I/R. Values expressed as mean ± SD. | ||
3.4 004 lowered the enhanced expression of VCAM-1 and MMP2 in I/R treated WKY and SHR rats
VCAM-1 is important in attaching circulating leukocytes to endothelium and in helping leukocytes migrate through vessels. In our study, the ameliorating effect on VCAM-1 by 004 was observed (Fig. 6A). We observed a similar outcome in the immunofluorescence images of SHR rats (Fig. 6B). | ||
| Fig. 6 004 decreased the vascular cell adhesion molecule (VCAM)-1 and matrix metalloproteinase (MMP)2 expression in SHR rats. (A) 004 administration decreased VCAM-1 expression. (B) VCAM-1 was attenuated by 004 near the positions of brain microvessels using immunofluorescence staining in SHR rats. Scalebar = 200 μm (C, D and E) 004 administration decreased MMP2 expression, but not MMP9 expression, although there is a similar trend. n = 4. **P < 0.01, ***P < 0.001 vs. Sham, #P < 0.05, ###P < 0.001 vs. I/R. Values expressed as mean ± SD. | ||
Matrix metalloproteinase (MMP) 2 and MMP9 are two key gelatinases in degradating type IV collagen, which is considered the major constituent of basal lamina. The breakdown of basal lamina is believed to be a critical process in inflammation. The expression of MMP2 increased after I/R, while the up-regulated state was attenuated by 004 administration (Fig. 6C and D). The results of MMP2 showed similar trends, although there were no significant differences between the groups (Fig. 6E).
3.5 004 suppressed the release of inflammatory factors in WKY and SHR rats through modulating the MAPKs and NF-κB pathways
The release of inflammatory factors promotes the chemotaxis of inflammatory cells in prolonged inflammation. As shown in Fig. 7A, the results revealed that there was an increase of TNF-α, IL-1β, IL-6 and IL-23 in I/R groups, while an obvious suppression effect occurred in the groups treated with 004.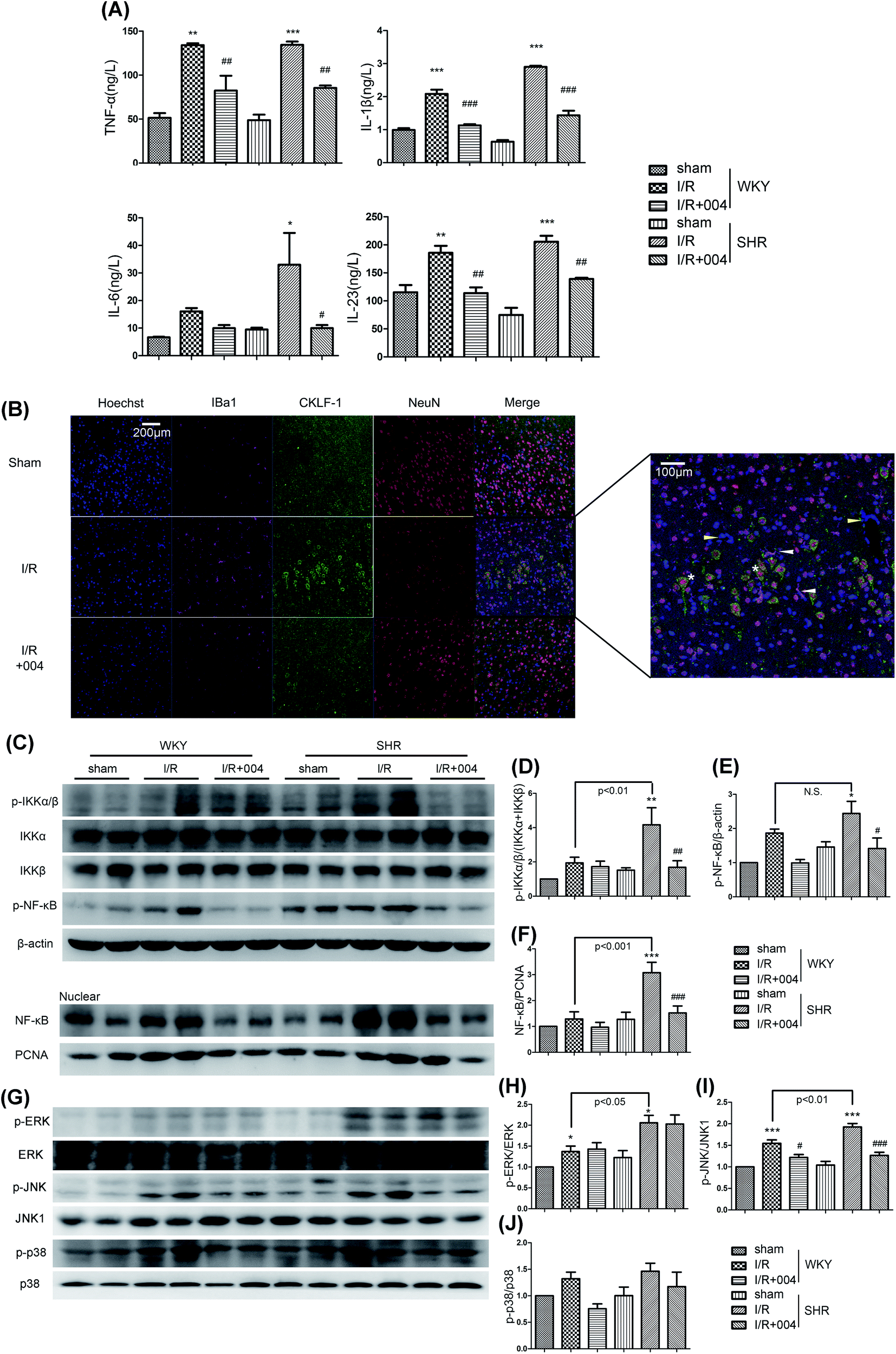 | ||
| Fig. 7 004 depressed neuroinflammation via decreasing JNK, p38 MAPK and NF-κB activity in SHR rats. (A) 004 abolished the release of TNF-α, IL-1β, IL-6 and IL-23. (B) The release of chemokine-like factor 1 (CKLF1) by neurons is attenuated by 004. Scalebar = 200 or 100 μm. (C–F) 004 suppressed the translocation of NF-κB. (G–J) 004 suppressed the phosphorylation of JNK and p38, but not ERK. n = 4. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. I/R. Values expressed as mean ± SD. | ||
CKLF1, a newly discovered novel cytokine, was also observed in our study. The expression of CKLF1 was high in the brains of fetuses and changed to be low in adults,28 but was up-regulated in the occurrence of cerebral I/R.29 However, the source of CKLF1 is still obscure. As shown in Fig. 7B, the obvious expression of CKLF1 was found to be restrained in the 004 treated SHR group. Of note, there was co-localization of CKLF1 and neuron cells (NeuN+, asterisk), but not microglia (Iba1+, white arrow), vascular endothelial cells (yellow arrow) or astrocytes (GFAP+, Additional file 1†), which indicated that the neurons secrete CKLF-1 in I/R brains for the first time.
Furthermore, we investigated whether 004 blocked the mitogen-activated protein kinase (MAPK) family and NF-κB signal pathways. As shown in Fig. 7C, the expression of p-IKKα/β (Fig. 7D), p-NF-κB (Fig. 7E) and the translocation of NF-κB to the nucleus (Fig. 7F) was greatly elevated in I/R rats, which were considerably restrained in the 004 group. Besides, up-regulated expression of p-JNK (Fig. 7I) and p-p38 (Fig. 7J) induced by I/R was also restrained in the 004 group, however, 004 showed no effect on the increased p-ERK expression (Fig. 7H).
3.6 The effects and mechanism of 004 on VCAM-1 expression in HBMEC cells
We examined the relationship between 004, MAPKs and NF-κB in modulating VCAM-1 expression in HBMEC cells. At first, we examined the effect of oxygen glucose deprivation/reoxygenation (OGD/R) with or without IMM-H004 on HBMEC cell viability to find out the proper OGD/R procedure to avoid severe damage to the cells. The results indicated that although an obvious decrease was found after OGD2h/R24h, the viability ratio was still as high as 92.09% (Fig. 8A). The viability of HBMEC cells showed no significant difference until reperfusion for 12 h following 2 h OGD (Fig. 8A). The maximum expression of VCAM-1 was observed in the OGD2h/R24h group when compared to groups treated with OGD for different time periods followed by 24 h reperfusion, and at OGD2h/R4h there was obvious up-regulation of VCAM-1 which lasted for 24 h (Fig. 8B), So we evaluated the expression of VCAM-1 using the model of OGD2h/R4h treated HBMEC cells.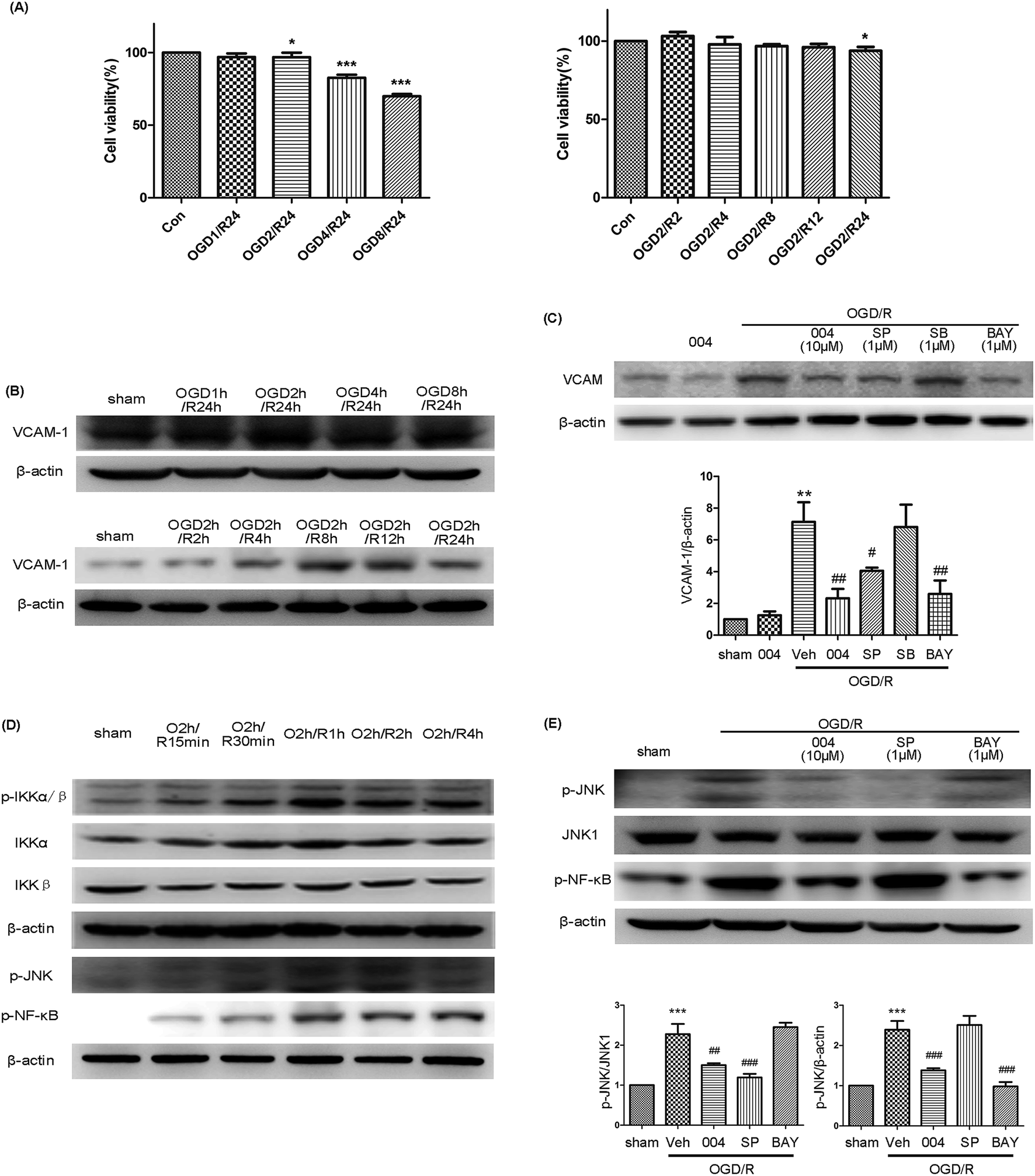 | ||
| Fig. 8 004 decreased VCAM-1 expression via inhibiting the phosphorylation of JNK and NF-κB. (A) The effect of OGD/R with or without IMM-H004 on HBMEC cell viability. *P < 0.05, ***P < 0.001 vs. Con. (B) The expression of VCAM-1 induced by OGD/R at various times. (C) 004, SP600125 and BAY11-7082 abolished VCAM-1 expression induced by OGD/R. (D) The maximum expression of p-JNK, p-IKKα/β and p-NF-κB was O2h/R1h. (E) 004 abolished the phosphorylation of JNK and NF-κB, but JNK and NF-κB pathways are independent of each other. n = 3 in each group. SP600125, SB203580, and BAY11-7082 are JNK, p38 and NF-κB inhibitors, respectively. **P < 0.01, ***P < 0.001 vs. Sham, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. I/R. Values expressed as mean ± SD. | ||
In this model, increased VCAM-1 expression after OGD/R was observed to be suppressed by 004, SP600125 (an inhibitor of JNK) and BAY11-7082 (an inhibitor of NF-κB), but SB203580 (an inhibitor of p38) showed no effect (Fig. 8C), demonstrating that JNK and NF-κB participated in the up-regulation of VCAM-1. However, the phosphorylation of JNK, IKKα/β and NF-κB occurred after reoxygenation at 1 h and then began to decline (Fig. 8D), so there may be underlying mechanisms mediating the delayed expression of VCAM-1. In order to test whether JNK and NF-κB pathways interact with each other or if the two pathways are independent, BAY11-7082 and SP600125 were administered. The expression of p-JNK and p-NF-κB was abolished in their corresponding inhibitor treated OGD2h/R1h groups or the 004 treated group when compared to the OGD/R group, however, the phosphorylation of JNK or NF-κB was not changed in the BAY11-7082 or SP600125 treated OGD/R groups respectively (Fig. 8E), which meant that JNK and NF-κB participated in the expression of VCAM-1 in HBMEC cells independently and 004 inhibited both of the two influential factors and therefore relieved VCAM-1 expression in HBMEC cells.
Actually, we are curious about the result where OGD2h/R24h showed only a minor detrimental effect on HBMEC cells as we observed serious injury when OGD2h/R24h was conducted in primary neuron cells (data not shown). But the results were consistent with a previous study, in which the cell viability was as high as 86% after OGD for 3 h.30 In another research study, the OGD time was set at 6 h in order to make the OGD/R-induced damage obvious.31 The results showed that endothelial cells are more tolerant to OGD injury compared to neurons which undergo more severe damage after identical OGD/R treatment. For example, only 42.5% of neuron cells remained viable at 24 h after OGD for 3 h.32 The reason for this phenomenon may come from the production of NO in endothelial cells. Vascular endothelial cells could even assist neurons in preventing hypoxic injury by the production of NO in the direct neuron-brain microvessel endothelial cell (-BMEC) co-culture system.33
4. Discussion
Hypertension is not only the main cause of a stroke, but also deteriorates the outcome and causes poor prognosis. However, the reason remains an enigma. Although there is a close relationship between hypertension and ischemic strokes in many clinical trials, lowering BP may not always lead to a favorable outcome in stroke patients. Actually, in 1977, scientists even tried to favorably alter stroke outcomes by maintaining hypertension because hypertension always means better perfusion, but the outcome was inevitably sad.34 Besides, although hypertension means an increased pressure on the vessel wall, the majority (∼80%) of hypertension strokes are thrombotic but not haemorrhagic,35 so we can conclude that it is the complications rather than hypertension itself that cause and influence stroke outcomes.Inflammation, which is thought to play a pivotal role in the progression of acute brain injury after I/R, has aroused our attention. Hypertension is always closely related to a plethora of inflammatory processes. Increased BP activates brain-resident astrocytes and microglia and upregulates adhesion molecule (AM) expression. Besides, hypertension correlates to monocyte activation and increased plasma levels of pro-inflammatory cytokines in the periphery.9 As 004 showed ROS reduction effects and inflammatory pathway inhibition in our previous study, we observed the inflammation inhibiting effect in this study. The results showed that neutrophil infiltration, microglia activation, blood viscosity, the expression of VCAM-1 and MMP2, inflammatory factors and pathways were enhanced in SHR I/R rats, while 004 suppressed these pathological changes by inhibiting inflammation mainly through down regulation of VCAM-1 (Fig. 9) and eventually improved neurological defects and alleviated infarction in a time- and dose-dependent manner in SHR rats.
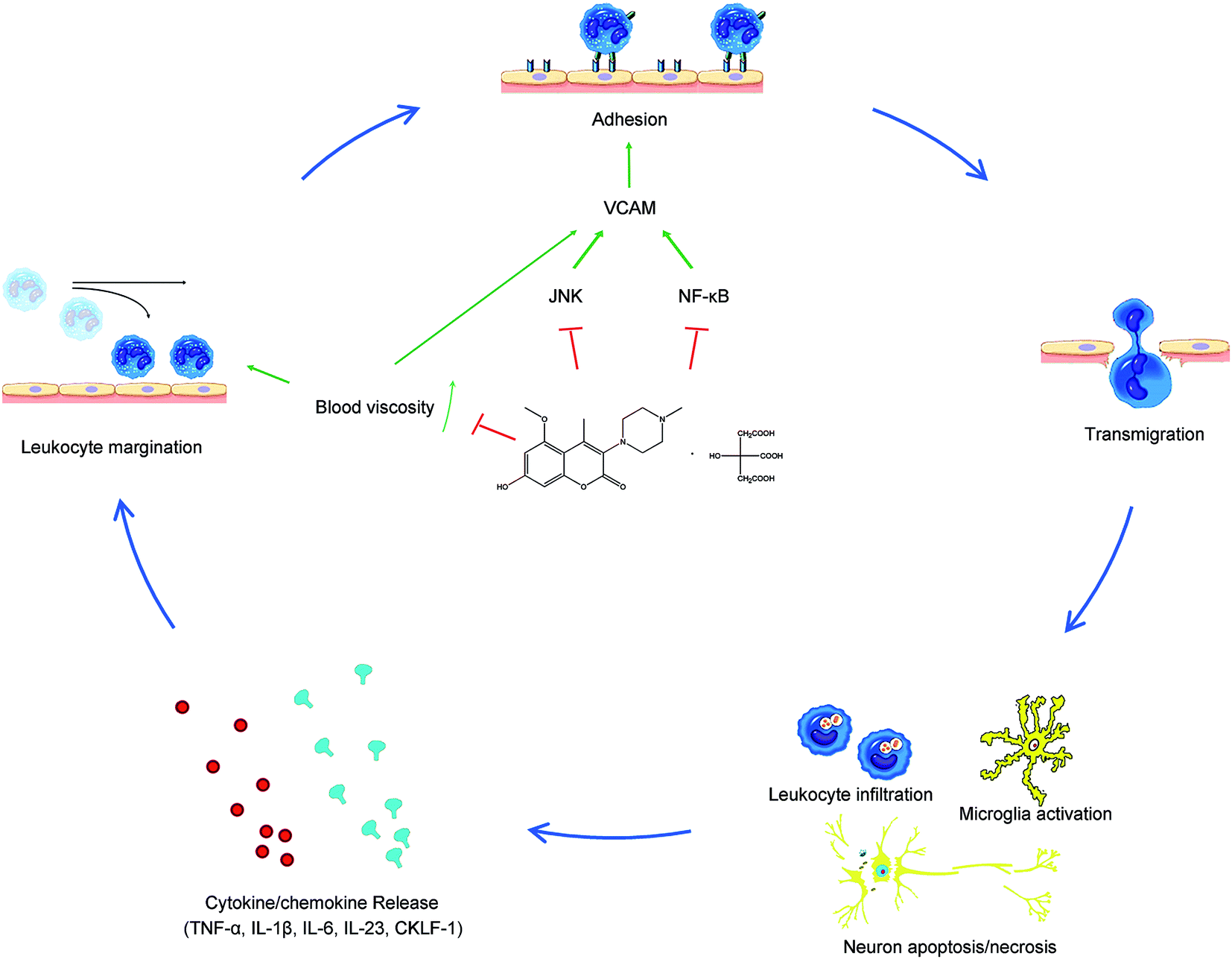 | ||
| Fig. 9 The mechanism of 004 in ameliorating inflammation caused by brain I/R. | ||
There are 3 questions we want to answer in our study.
The first is the question as to why hypertension gives rise to elevated inflammatory reaction. The relationship between hypertension and severe inflammation is known, but the reason is controversial. In our study, we emphasized the importance of hemorheology in hypertension. Although the science of hemorheology has evolved over 400 years, the importance of hemorheological variables in strokes have been known only for the last 20 years.36 The relationship that associates enhanced blood viscosity with hypertension was clarified as “chicken and eggs”:35,37 an increase in blood viscosity could result in hypertension by enhancing the vascular resistance in the periphery38 and evaluated BP is required to compensate physiologically for the increased viscosity.39 The changes in hemorheology often result from the dysfunction of the sympathetic nervous system or the renin-angiotensin-aldosterone system.37 Although there is a close correlation between hypertension and high blood viscosity, only a few studies consider the role of blood viscosity in strokes. There is also an interesting observation that satisfactory BP reduction with various kinds of antihypertensive drugs may not lead to an identical reduction in stroke patients, so it is inferred that this observation may partly be due to the unsatisfactory effects of some drugs in hemorheology.40 Furthermore, epidemiological studies have found an increased risk of cardiovascular disease in patients with higher mean blood viscosity levels, but it is not relevant to whether they belong to hypertensive or normotensive individuals.41 So hemorheological change might play a more important role in stroke patients.
However, the effect of the change in blood viscosity was neglected in most studies, while we believed that blood viscosity might serve as a potential breakthrough point for analyzing the mechanism. In our study, the results showed that elevated WBRV-L was observed to be much higher in SHR rats when compared to WKY rats and both WBRV-L and WBRV-H were markedly enhanced in SHR I/R rats compared to WKY I/R rats.
In normal blood flow, erythrocytes are confined to a central axis of the flow and displace the leukocytes toward the vessel wall. In the case of inflammation, as vasodilation and extravasation of plasma fluid and proteins happens consecutively, blood flow slows and hemodynamic changes occur (wall shear stress decreases) and a large number of leukocytes drift to a peripheral position along the endothelial surface. This process is called “margination”.42 Leukocytes must first become marginated to collide with the endothelial cells, and blood viscosity was thought to be one of the most important influencing factors in this process. A study showed that both enhanced erythrocyte aggregation and increased hematocrit led to elevated blood viscosity which correlated with more slow-flowing leukocytes near the wall and eventually facilitated leukocyte adhesion.43 As the wall shear rate decreased, which elevated blood viscosity, leukocytes were increasingly distributed toward the wall in blood flowing in glass capillaries.44
After margination, the leukocytes then adhere firmly and insert pseudopods into the junctions between endothelial cells, and escape into the extravascular space, which is called “adhesion”.42 In this stage, the immune cells were activated to express integrins like Very Late Antigen (VLA)-4 and lymphocyte function-associated antigen (LFA)-1. Endothelial cells were also activated to express ligands for integrins, especially VCAM-1 (ligand for VLA-4) and ICAM-1 (ligand for LFA-1), which are called adhesion molecules. Integrins and adhesion molecules showed a high-affinity state, so leukocytes stop rolling and spread out on the endothelial surface to prepare for infiltration. In this process, endothelial cells play important roles and blood viscosity could alter the adhesion of leukocytes to endothelial cells, as the physiological status of endothelial cells could be changed by hemorheological alteration in the blood. Vascular endothelial cells do not only act as components of blood vessels, but also act as sensors of mechanical stress and conductors of mechanical signals in the blood flow. It was found that pathological shear stress stimulates expression of VCAM-1.45 Because blood is particulate and has non-Newtonian nature, decreasing shear rate leads to an increase in viscosity43, therefore, at low shear stress in experiments in blood, expression of VCAM-1 was enhanced and inflammation was induced.46–48 Besides, these interactions between leukocytes and endothelial cells are both governed by local hydrodynamics such as fluid velocity and viscosity, as increased viscosity and decreased velocity means it is hard to separate the combination.49 In one study, the efficiency of integrin-dependent adhesion was highest (∼0.2) at high viscosity and it decreased to approximately zero at low viscosity.50 All of these results indicate that high blood viscosity facilitated the margination of leukocytes and adhesion of leukocytes to endothelial cells and eventually affected the occurrence of inflammation.48 So, we may conclude that 004 could alleviate VCAM-1 up-regulation by decreasing the enhanced blood viscosity so that inflammation could be suppressed (Fig. 9). Interestingly, we also found out that although the activated microglia numbers seemed the same, there were more resting microglia in the brains of SHR rats than those of WKY rats. That would also partly explain why SHR rats exhibited a stronger effect in activating microglia and thus underwent more serious injury induced by I/R compared to WKY rats.
The second question is whether MAPKs and NF-κB were involved in the up-regulation of VCAM-1 in HBMECs and whether they took part in this progress independently or were influenced by each other. It was found that MAPKs and NF-κB were deeply involved in VCAM-1 expression under different stimulation, such as gastrin-releasing peptide (GRP), lipopolysaccharides (LPS), thyroid stimulating hormone (TSH) and tumor necrosis factor α (TNF-α), in different endothelial cells. For example, the expression of VCAM-1 was observed in GRP treated human umbilical vein endothelial cells (HUVECs), while the inhibition of ERK1/2, p38 and NF-κB significantly reduced the expression of VCAM-1 and subsequent adhesion of monocytes to the endothelium.51 In another study using TSH as a stimulator, the enhancement of VCAM-1 was accompanied by ERK1/2 activation in HUVECs.52 It was also observed that TNF-α induced activation of JNK and p38 promoted endothelial activation resulting in enhanced VCAM-1 expression in an in vitro experiment using HUVECs, while inhibition of p38, JNK or NF-κB blocked the expression of VCAM-1.53 In another commonly used endothelial cell type, human aortic endothelial cells (HAECs), a similar phenomenon was observed. TNF-α was found to increase VCAM-1 expression via JNK, ERK1/2, p38 MAPK and NF-κB in HAECs.54 Activation of p38 MAPK is believed to be at least partially involved in LPS-induced VCAM-1 expression in HAECs.55 The triglyceride-rich lipoproteins mediated sensitization to inflammatory stimuli such as TNFα stimulated VCAM-1 expression, which was dependent upon p38 activity because the sensitization was abrogated by pharmacological blockade of p38.56 Many experiments indicated that NF-κB worked as a VCAM-1 promoter in both HUVECs and HAECs.57–63 It was also found that MAPKs and NF-κB participated in VCAM-1 expression in other cell types. For example, p38 was involved in the expression of VCAM-1, which was induced by adding inflammatory cytokines to cardiac cells.64 ERK and JNK took part in the synthesis of VCAM-1 in human bone marrow mesenchymal stem cells.65 All MAPKs facilitate the expression of VCAM-1 in human rheumatoid arthritis synovial fibroblasts.66 However, contrary results were also obtained in some experiments. For example, a study indicated that JNK might serve as a suppressor of VCAM-1 expression in HAECs.67 Another report proved that salusin-α could selectively decrease the expression of the VCAM-1 protein independently of p38 and JNK.68 The discrepancies between the reports implied that the divergent pathways that induce VCAM-1 production depend on the type of stimulus, cell type or experimental procedure.
Previous results regarding the relationship between MAPKs and NF-κB pathways were also obscure. Some studies showed that they were independent from each other.66 While some experiments indicated that JNK and p38 induce the activation and translocation of NF-κB,69,70 other reports proved that NF-κB facilitates the phosphorylation of MAPKs.70 In order to answer the question, we designed an OGD/R model in vitro using HBMEC cells and examined the role of MAPKs and NF-κB in regulating the expression of VCAM-1.
We observed for the first time that both JNK and NF-κB were involved in modulating the expression of VCAM-1, and they influenced the expression of VCAM-1 independently in OGD/R treated HBMEC cells. Although there was little discussion about the role of MAPKs and NF-κB in modulating VCAM-1 expression in HBMEC cells previously, we found that the results of an earlier study were similar to ours, although the cell types were not identical. In that study, TNF-α induced VCAM-1 expression in HAECs via JNK/p38 and NF-κB signaling pathways, while reduction of VCAM-1 expression in response to TNF-α treatment was affected by JNK and p38 inhibitors, not by an ERK inhibitor.71
The last question is which cell type released CKLF-1 in the cortical ischemia area of I/R rats. CKLF-1 is a newly discovered novel cytokine which is used as a functional ligand for the chemokine (C–C motif) receptor 4 (CCR4). The expression of CKLF1 was high in the brains of fetuses and changed to be low in adults,28 but it was up-regulated in the occurrence of cerebral I/R and the neutralization was associated with reduced damage by decreasing the production of inflammatory mediators.72 However, the source of CKLF1 is still unclear. We observed that it is neuron cells, rather than microglia, astrocyte or vascular endothelial cells, that secrete CKLF-1 in the process of brain I/R for the first time (Fig. 7B). However, the results need to be further confirmed by other experiments.
There has been a problem puzzling pharmaceutical scientists for a long time, that is, why there are so many “popstar” medicines that are effective for strokes in preclinical experiments, but are invalid in clinical trials. It may be that the internal environment of a human body is too complicated to be completely simulated. As there are so many stroke patients that suffer from high BP at the same time, it is of great value to use hypertensive instead of normal animals to evaluate the efficacy of a potential compound.
However, pathologic features should not be limited to only hypertension in stroke patients, others such as ageing, diet and hormone abnormality, all need to be discussed with medicinal judgment. Besides, the animal model of MCAO could also not accurately imitate the actual obstruction of a blood vessel. Therefore, additional experiments need to be designed to fully reveal the effects of 004. As a type of compound which shows protective effects in a variety of animal brain I/R models, 004 deserves further study. Of course, more effort is needed and there is a long way to go.
5. Conclusion
In this study, we used hypertensive SHR rats to study the effect of 004 in treating brain I/R injury and the results showed that 004 could efficiently ameliorate the neurological defects and infarct volume in a time and dose dependent manner. It was observed that 004 alleviated neuroinflammation, such as the infiltration of leukocytes, the activation of microglia and increased blood viscosity, and the alleviating effect was achieved through down-regulating the expression of VCAM-1 by inhibiting the JNK/NF-κB pathway.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81274122, 81373997, 81260650, U1402221, 81603315, 81603316), the National Mega-project for Innovative Drugs (2012ZX09301002-004), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) (IRT1007), the Beijing Natural Science Foundation (7131013, 7142115, 7161011), the Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study (BZ0150), the Key Research and Development Project of Hunan Province (2015SK2029-1) and the Scientific Research Foundation of the Higher Education Institutions of Hunan Province (15K091). The authors would like to thank Zixian Guo from the Xinxiang Medical College for his help in video editing.References
- R. W. Flynn, R. S. MacWalter and A. S. Doney, Neuropharmacology, 2008, 55, 250–256 CrossRef CAS PubMed.
- S. Zhang, X. Y. Song, C. Y. Xia, Q. D. Ai, J. Chen, S. F. Chu, W. B. He and N. H. Chen, RSC Adv., 2016, 6, 93815–93825 RSC.
- W. Rosamond, K. Flegal, G. Friday, K. Furie, A. Go, K. Greenlund, N. Haase, M. Ho, V. Howard, B. Kissela, S. Kittner, D. Lloyd-Jones, M. McDermott, J. Meigs, C. Moy, G. Nichol, C. J. O’Donnell, V. Roger, J. Rumsfeld, P. Sorlie, J. Steinberger, T. Thom, S. Wasserthiel-Smoller and Y. Hong, Circulation, 2007, 115, e69–171 CrossRef PubMed.
- J. Lv, B. Neal, P. Ehteshami, T. Ninomiya, M. Woodward, A. Rodgers, H. Wang, S. MacMahon, F. Turnbull, G. Hillis, J. Chalmers and V. Perkovic, PLoS Med., 2012, 9, e1001293 Search PubMed.
- Q. Cao, P. Pei, J. Zhang, J. Naylor, X. Fan, B. Cai, Q. Dai, W. Sun, R. Ye, R. Shi, K. Liu, Y. Jiang, W. Liu, F. Yang, W. Zhu, Y. Xiong, X. Liu and G. Xu, BMC Public Health, 2016, 16, 170 CrossRef PubMed.
- S. F. Chu, Z. Zhang, W. Zhang, M. J. Zhang, Y. Gao, N. Han, W. Zuo, H. Y. Huang and N. H. Chen, Mol. Neurobiol., 2017, 54, 524–540 CrossRef CAS PubMed.
- W. Zuo, W. Zhang, N. Han and N. H. Chen, CNS Neurosci. Ther., 2015, 21, 280–288 CrossRef CAS PubMed.
- S. A. Bhat, R. Goel, R. Shukla and K. Hanif, Brain, Behav., Immun., 2017, 59, 173–189 CrossRef CAS PubMed.
- K. Moller, C. Posel, A. Kranz, I. Schulz, J. Scheibe, N. Didwischus, J. Boltze, G. Weise and D. C. Wagner, Front. Cell. Neurosci., 2015, 9, 461 Search PubMed.
- W. Zuo, S. Zhang, C. Y. Xia, X. F. Guo, W. B. He and N. H. Chen, Neuropharmacology, 2014, 86, 103–115 CrossRef CAS PubMed.
- M. Munakata, Y. Imai, N. Minami, S. Sasaki, T. Ichijyo, M. Yoshizawa, H. Sekino, K. Abe and K. Yoshinaga, Tohoku J. Exp. Med., 1990, 161, 55–64 CrossRef CAS PubMed.
- A. Garcia-Tejedor, L. Sanchez-Rivera, M. Castello-Ruiz, I. Recio, J. B. Salom and P. Manzanares, J. Agric. Food Chem., 2014, 62, 1609–1616 CrossRef CAS PubMed.
- R. Balti, A. Bougatef, A. Sila, D. Guillochon, P. Dhulster and N. Nedjar-Arroume, Food Chem., 2015, 170, 519–525 CrossRef CAS PubMed.
- E. Z. Longa, P. R. Weinstein, S. Carlson and R. Cummins, Stroke, 1989, 20, 84–91 CrossRef CAS PubMed.
- S. Dang, X. Liu, P. Fu, W. Gong, F. Yan, P. Han, Y. Ding, X. Ji and Y. Luo, Neurol. Res., 2011, 33, 520–528 CrossRef PubMed.
- W. Zuo, J. Chen, S. Zhang, J. Tang, H. Liu, D. Zhang and N. Chen, Eur. J. Neurosci., 2014, 39, 2107–2118 CrossRef PubMed.
- J. H. Garcia, S. Wagner, K. F. Liu and X. J. Hu, Stroke, 1995, 26, 627–634 CrossRef CAS PubMed.
- J. Wang, C. Li, T. Chen, Y. Fang, X. Shi, T. Pang, L. Zhang and H. Liao, Neuropharmacology, 2016, 105, 398–410 CrossRef CAS PubMed.
- C. Ayata and A. H. Ropper, J. Clin. Neurosci., 2002, 9, 113–124 CrossRef PubMed.
- R. Meller, Neuroscientist, 2009, 15, 243–260 CrossRef CAS PubMed.
- B. Anuncibay-Soto, D. Perez-Rodriguez, M. Santos-Galdiano, E. Font, M. Regueiro-Purrinos and A. Fernandez-Lopez, J. Neurochem., 2016, 138, 295–306 CrossRef CAS PubMed.
- J. Lehmann, W. Hartig, A. Seidel, C. Fuldner, C. Hobohm, J. Grosche, M. Krueger and D. Michalski, Neuroscience, 2014, 279, 139–154 CrossRef CAS PubMed.
- Y. Gan, Q. Liu, W. Wu, J. X. Yin, X. F. Bai, R. Shen, Y. Wang, J. Chen, A. La Cava, J. Poursine-Laurent, W. Yokoyama and F. D. Shi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2704–2709 CrossRef CAS PubMed.
- C. Drake, H. Boutin, M. S. Jones, A. Denes, B. W. McColl, J. R. Selvarajah, S. Hulme, R. F. Georgiou, R. Hinz, A. Gerhard, A. Vail, C. Prenant, P. Julyan, R. Maroy, G. Brown, A. Smigova, K. Herholz, M. Kassiou, D. Crossman, S. Francis, S. D. Proctor, J. C. Russell, S. J. Hopkins, P. J. Tyrrell, N. J. Rothwell and S. M. Allan, Brain, Behav., Immun., 2011, 25, 1113–1122 CrossRef CAS PubMed.
- G. X. Fu, M. Ji, L. Z. Han, C. C. Xu, F. F. Pan, T. J. Hu and Y. Zhong, Zeitschrift fur Gerontologie und Geriatrie., 2017, 50, 233–238 CrossRef PubMed.
- V. Kucukatay, M. Bor-Kucukatay, G. Gundogdu, G. Erken, T. O. Ozcan, F. D. Miloglu and Y. Kadioglu, Folia Biol., 2012, 58, 157–165 CAS.
- X. Y. Song, J. F. Hu, M. N. Sun, Z. P. Li, Z. X. Zhu, L. K. Song, Y. H. Yuan, G. Liu and N. H. Chen, Brain Res. Bull., 2014, 106, 30–38 CrossRef CAS PubMed.
- Z. Z. Wang, G. Li, X. Y. Chen, M. Zhao, Y. H. Yuan, X. L. Wang and N. H. Chen, Brain Res., 2010, 1308, 24–34 CrossRef CAS PubMed.
- Y. Tan, Y. Wang, L. Li, J. Xia, S. Peng and Y. He, PLoS One, 2015, 10, e0125073 Search PubMed.
- W. Li, Z. Chen, M. Yan, P. He, Z. Chen and H. Dai, J. Neurochem., 2016, 136, 651–659 CrossRef CAS PubMed.
- T. Urbanek, W. Kuczmik, A. Basta-Kaim and B. Gabryel, Brain Res., 2014, 1553, 1–11 CrossRef CAS PubMed.
- G. Q. He, W. M. Xu, J. F. Li, S. S. Li, B. Liu, X. D. Tan and C. Q. Li, Mol. Brain, 2015, 8, 88 CrossRef PubMed.
- K. W. Wu, Z. W. Kou, J. L. Mo, X. X. Deng and F. Y. Sun, Neuroscience, 2016, 334, 275–282 CrossRef CAS PubMed.
- J. D. Michenfelder and J. H. Milde, Stroke, 1977, 8, 87–91 CrossRef CAS PubMed.
- G. Y. Lip, A. D. Blann, I. S. Farooqi, J. Zarifis, G. Sagar and D. G. Beevers, Blood Coagulation Fibrinolysis, 2001, 12, 307–315 CrossRef CAS PubMed.
- G. Feher, K. Koltai, G. Kesmarky, B. Horvath, K. Toth, S. Komoly and L. Szapary, Phytomedicine, 2009, 16, 111–117 CrossRef CAS PubMed.
- C. G. Spencer and G. Y. Lip, J. Hum. Hypertens., 2000, 14, 291–293 CAS.
- G. D. Sloop, J. J. Weidman and J. A. St Cyr, Ther. Adv. Cardiovasc. Dis., 2015, 9, 403–411 CrossRef CAS PubMed.
- K. S. Houschyar, R. Ludtke, G. J. Dobos, U. Kalus, M. Broecker-Preuss, T. Rampp, B. Brinkhaus and A. Michalsen, BMC Med., 2012, 10, 54 CrossRef CAS PubMed.
- C. G. Spencer, D. G. Beevers and G. Y. Lip, J. Hum. Hypertens., 2001, 15(1), S39–S42 Search PubMed.
- W. Koenig, M. Sund, B. Filipiak, A. Doring, H. Lowel and E. Ernst, Arterioscler., Thromb., Vasc. Biol., 1998, 18, 768–772 CrossRef CAS.
- V. Kumar, A. K. Abbas and J. C. Aster, Robbins & Cotran Pathologic Basis of Disease, Elsevier, Netherland, 2014 Search PubMed.
- K. B. Abbitt and G. B. Nash, Am. J. Physiol.: Heart Circ. Physiol., 2003, 285, H229–H240 CrossRef CAS PubMed.
- U. Nobis, A. R. Pries, G. R. Cokelet and P. Gaehtgens, Microvasc. Res., 1985, 29, 295–304 CrossRef CAS PubMed.
- P. Sucosky, K. Balachandran, A. Elhammali, H. Jo and A. P. Yoganathan, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 254–260 CrossRef CAS PubMed.
- B. Li, J. Zhang, Z. Wang and S. Chen, PloS One, 2016, 11, e0149694 Search PubMed.
- T. Marin, B. Gongol, Z. Chen, B. Woo, S. Subramaniam, S. Chien and J. Y. Shyy, Free Radical Biol. Med., 2013, 64, 61–68 CrossRef CAS PubMed.
- L. Luong, H. Duckles, T. Schenkel, M. Mahmoud, J. L. Tremoleda, M. Wylezinska-Arridge, M. Ali, N. P. Bowden, M. C. Villa-Uriol, K. van der Heiden, R. Xing, F. J. Gijsen, J. Wentzel, A. Lawrie, S. Feng, N. Arnold, W. Gsell, A. Lungu, R. Hose, T. Spencer, I. Halliday, V. Ridger and P. C. Evans, Thromb. Haemostasis, 2016, 116, 181–190 CrossRef PubMed.
- R. K. Jain, L. L. Munn, D. Fukumura and R. J. Melder, Methods Mol. Med., 1999, 18, 553–575 CAS.
- S. Neelamegham, A. D. Taylor, A. R. Burns, C. W. Smith and S. I. Simon, Blood, 1998, 92, 1626–1638 CAS.
- M. K. Kim, H. J. Park, Y. Kim, H. J. Kim, S. K. Bae and M. K. Bae, Biochem. Biophys. Res. Commun., 2017, 485, 542–549 CrossRef CAS PubMed.
- Y. Yan, F. Jiang, Y. Lai, H. Wang, A. Liu, C. Wang, Y. Zhang, W. Teng and Z. Shan, Int. J. Mol. Sci., 2016, 17(9), E1484 CrossRef PubMed.
- M. Huang, S. Zeng, Y. Zou, M. Shi, Q. Qiu, Y. Xiao, G. Chen, X. Yang, L. Liang and H. Xu, Br. J. Pharmacol., 2017, 174, 101–115 CrossRef CAS PubMed.
- W. Olejarz, D. Bryk, D. Zapolska-Downar, M. Malecki, A. Stachurska and D. Sitkiewicz, Eur. J. Clin. Invest., 2014, 44, 54–64 CrossRef CAS PubMed.
- B. Liu, L. Cheng, D. Liu, J. Wang, X. Zhang, R. Shu and J. Liang, J. Periodontol., 2012, 83, 955–962 CrossRef CAS PubMed.
- H. J. Ting, J. P. Stice, U. Y. Schaff, D. Y. Hui, J. C. Rutledge, A. A. Knowlton, A. G. Passerini and S. I. Simon, Circ. Res., 2007, 100, 381–390 CrossRef CAS PubMed.
- B. S. Kong, S. J. Im, Y. J. Lee, Y. H. Cho, Y. R. Do, J. W. Byun, C. R. Ku and E. J. Lee, PloS One, 2016, 11, e0149394 Search PubMed.
- H. B. Bai, Y. Wang, Y. H. Zhang and Y. Zhang, Acta Physiol. Sin., 2016, 68, 41–49 Search PubMed.
- T. M. Wang, C. J. Chen, T. S. Lee, H. Y. Chao, W. H. Wu, S. C. Hsieh, H. H. Sheu and A. N. Chiang, J. Nutr. Biochem., 2011, 22, 187–194 CrossRef CAS PubMed.
- F. Addabbo, C. Nacci, L. De Benedictis, V. Leo, M. Tarquinio, M. J. Quon and M. Montagnani, Am. J. Physiol.: Endocrinol. Metab., 2011, 301, E1143–E1154 CrossRef CAS PubMed.
- Y. S. Cho, C. H. Kim, H. N. Kim, T. S. Ha and H. Y. Ahn, Pharmazie, 2014, 69, 818–822 CAS.
- C. J. Liang, S. H. Wang, Y. H. Chen, S. S. Chang, T. L. Hwang, Y. L. Leu, Y. C. Tseng, C. Y. Li and Y. L. Chen, Free Radical Biol. Med., 2011, 51, 1337–1346 CrossRef CAS PubMed.
- D. Ross, H. Zhou and D. Siegel, Chem.-Biol. Interact., 2011, 192, 145–149 CrossRef CAS PubMed.
- R. Kacimi, J. S. Karliner, F. Koudssi and C. S. Long, Circ. Res., 1998, 82, 576–586 CrossRef CAS PubMed.
- Z. Y. Lu, W. C. Chen, Y. H. Li, L. Li, H. Zhang, Y. Pang, Z. F. Xiao, H. W. Xiao and Y. Xiao, Mol. Med. Rep., 2016, 14, 643–648 CAS.
- S. F. Luo, R. Y. Fang, H. L. Hsieh, P. L. Chi, C. C. Lin, L. D. Hsiao, C. C. Wu, J. S. Wang and C. M. Yang, Semin. Arthritis Rheum., 2010, 62, 105–116 CrossRef CAS PubMed.
- B. Liu, J. Wang, L. Cheng and J. Liang, Mol. Med. Rep., 2013, 8, 1594–1600 CAS.
- C. H. Zhou, J. Pan, H. Huang, Y. Zhu, M. Zhang, L. Liu and Y. Wu, PloS One, 2014, 9, e107555 Search PubMed.
- Y. Pan, Y. Wang, L. Cai, Y. Cai, J. Hu, C. Yu, J. Li, Z. Feng, S. Yang, X. Li and G. Liang, Br. J. Pharmacol., 2012, 166, 1169–1182 CrossRef CAS PubMed.
- R. Gorina, M. Font-Nieves, L. Marquez-Kisinousky, T. Santalucia and A. M. Planas, Glia, 2011, 59, 242–255 CrossRef PubMed.
- C. J. Liang, C. W. Lee, H. C. Sung, Y. H. Chen, S. H. Wang, P. J. Wu, Y. C. Chiang, J. S. Tsai, C. C. Wu, C. Y. Li and Y. L. Chen, Am. J. Chin. Med., 2014, 42, 619–637 CrossRef CAS PubMed.
- L. L. Kong, Z. Y. Wang, N. Han, X. M. Zhuang, Z. Z. Wang, H. Li and N. H. Chen, J. Neuroinflammation, 2014, 11, 112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02154b |
| This journal is © The Royal Society of Chemistry 2017 |