
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interplay between the σ-tetrel bond and σ-halogen bond in PhSiF3⋯4-iodopyridine⋯N-base†
Huili Xu,
Jianbo Cheng*,
Xin Yang,
Zhenbo Liu,
Xiao Bo and
Qingzhong Li *
*
The Laboratory of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Yantai University, Yantai 264005, People's Republic of China. E-mail: liqingzhong1990@sina.com; Fax: +86 535 6902063; Tel: +86 535 6902063
First published on 18th April 2017
Abstract
The ternary complexes of PhSiF3⋯4-iodopyridine⋯N-base (N-base = HCN, NH3, NHNH2, and NH2CH3), PhTF3⋯4-iodopyridine⋯NH3 (T = C and Ge), PhSiY3⋯4-iodopyridine⋯NH3 (Y = H and Cl), PhSiF3⋯4-bromopyridine⋯NH3 and the respective binary complexes have been investigated. 4-Halopyridine in these ternary complexes plays a dual role of both a Lewis acid with the σ-hole on the halogen atom in the halogen bond and a Lewis base with the nitrogen atom in the tetrel bond. The interplay between both interactions in the ternary complexes has been analyzed in terms of the binding distance, binding energy, charge transfer, electron density and electrostatic potentials. A synergistic effect is found for the tetrel and halogen bonds in most of the ternary complexes, while a diminutive effect is present for the hydrogen and halogen bonds in PhCF3⋯4-iodopyridine⋯NH3. The magnitude of cooperative energy depends on the strength of both interactions. Interestingly, PhSiCl3⋯4-iodopyridine has a stronger tetrel bond than PhSiH3⋯4-iodopyridine, inconsistent with the size of the σ-hole on the Si atom. In addition, the tetrel bond exhibits a partially covalent interaction nature.
1. Introduction
Noncovalent interactions have a special importance in supramolecular chemistry, crystal engineering, and biological systems.1,2 It is well known that hydrogen bonds are the main driving force in maintaining the stability of complex structures.3,4 Recently, other interactions such as halogen bonds have been widely recognized because of their similarities in geometries and applications with hydrogen bonds. Halogen bonds also play an important role in controlling molecular recognition in biological systems and determining molecular orientation within crystals.5–9 The origin of the Lewis acid in halogen bonding is mainly attributed to a region of positive electrostatic potential (σ-hole),10–15 being the consequence of the diminished electron density on the extension of a covalent bond to the halogen atom. The applications of halogen bonds are dependent on their strength, which becomes stronger in the order F ≪ Cl < Br < I. Thus iodine-containing molecules are often used to form a halogen bond in crystal materials and solution.16–20 In these investigations, the I⋯N halogen bond has drawn much attention with experimental20–22 and theoretical23–25 methods. For example, tetraiodoethynyl resorcinarene cavitands are formed using halogen bonded supramolecular assemblies with the nitrogen in pyridine.21 In constructing crystal materials, more than one interaction is present, thus cooperative effect occurs, which is one of important properties of noncovalent interactions. It was demonstrated that halogen bonds exhibit cooperative effects with themselves or other interactions,26–35 through which the strength of halogen bonds is modulated.Like the halogen atom in halogen bonding, a group 14 atom also acts as a Lewis acid to interact with a Lewis base and the corresponding interaction has been coined as tetrel bonding recently.36 Actually, this interaction had been explored37–39 before this term was proposed in 2013. Now tetrel bonding has been attracting more attention due to its roles in crystal materials40–42 and chemical reactions.43–45 However, tetrel bonding attracted less interest than hydrogen and halogen bonds; even so, there are relatively many studies that focus on the cooperativity involving tetrel bonds.46–61 Esrafili et al. performed a comparative investigation on the cooperative effects between tetrel bond and other σ-hole bond interactions in complexes YH3M⋯NCX⋯NH3 (M = C, Si; Y = F, CN and X = Cl, SH, PH2).56 Ab initio calculations were performed to study the cooperativity between the π-hole tetrel bond and σ-hole halogen bond in complexes XCN⋯F2CO⋯YCN (X = H, F, Cl, Br; Y = F, Cl, Br).60 Zeng and co-authors61 discovered a pseudo π-hole in cyclopropane and its derivatives M3H6 (M = C, Si, Ge, Sn, Pb), and found that this π-hole is enlarged due to the coexistence of a π-hole tetrel bond and a σ-hole halogen bond in complexes M3H6⋯(NCF)n (n = 1, 2, 3). The results showed that the strength of tetrel bond can be affected by the coexistence with another interaction.
In the present paper, we study the complexes PhSiF3⋯4-iodopyridine⋯N-base (N-base = HCN, NH3, NHNH2, and NH2CH3) to investigate the synergistic effect between σ-hole tetrel bond and σ-hole halogen bond and to compare the effect of different N-bases on this synergistic effect. 4-Iodopyridine is a drug intermediate and it is denoted as PyI in the following sections for simplicity. Phenyltrifluorosilane (PhSiF3) turned out to be an original and effective reagent and synthon in organoelemental and organic synthesis.62 Comparison is made for the complexes of PhCF3⋯PyI⋯NH3, PhGeF3⋯PyI⋯NH3, PhSiH3⋯PyI⋯NH3, PhSiCl3⋯PyI⋯NH3, and PhSiF3⋯PyBr⋯NH3. The synergistic effect between both interactions is characterized in views of binding distances, interaction energies, cooperative energy, and electron density. Natural bond orbital (NBO) and molecular electrostatic potential (MEP) analyses are used to unveil the mechanism of such cooperativity.
2. Computational methods
The geometries of the complexes and monomers were optimized at the RI-MP2/aug-cc-pVTZ63 level of theory using the TURBOMOLE 6.5 software.64 For iodine, the aug-cc-pVTZ-PP65 basis set with pseudopotentials was used to accelerate the calculations and account for relativistic effects. It has been demonstrated that the RI-MP2 method is very convenient for studying a variety of noncovalent interactions in large systems.66,67 Frequency calculations were also performed at the same level to affirm that the corresponding structures are minima on the potential energy surfaces. The binding energies were calculated with supermolecular method and corrected for the basis set superposition error (BSSE) using the Boys–Bernardi counterpoise technique.68MEPs were computed on the 0.001 au contour of electronic density at the MP2/aug-cc-pVTZ level using the Wave Function Analysis-Surface Analysis Suite (WFA-SAS) program.69 The topological analysis of the electron density at bond critical point (BCP) was performed by means of the AIM2000 program.70 NBO analysis71 was carried out at the HF/aug-cc-pVTZ level using NBO 5.0 program.72 Non-covalent interaction (NCI) index73 was plotted with the Multiwfn program.74
3. Results and discussion
3.1 MEPs and geometries
Fig. 1 shows the MEP maps of PhSiF3 and 4-iodopyridine (PyI). Four σ-holes (red region) are found on the tetrahedral surfaces of the Si atom in PhSiF3, locating along the C–Si and F–Si bond ends, respectively. We pay our attention to the σ-hole at the C–Si end, having a positive MEP of 134.98 kJ mol−1 (Table 1). This value amounts to 15.85 and 138.43 kJ mol−1 in PhCF3 and PhGeF3, respectively. Clearly, the σ-hole at the C–T end enlarges in the sequence C ≪ Si < Ge, due to the smaller electronegativity and larger polarizability of the heavier T atom. When –SiF3 is respectively replaced by –SiH3 and –SiCl3, the corresponding value is 82.59 and 55.84 kJ mol−1, which is smaller than that in PhSiF3 owing to the smaller electronegativity of H and Cl atoms. Interestingly, the σ-hole at the C–Si end in PhSiCl3 has a smaller MEP than that in PhSiH3, which is inconsistent with the relative electronegativity of both H and Cl elements. We ascribe it to a strong orbital interaction from the lone pair on the Cl atom to the C–Si* anti-bonding orbital in PhSiCl3 (Fig. S1†). There are three Lp(Cl) → σ*(C–Si) orbital interactions in PhSiCl3 and the sum of their perturbation energies is ∼116 kJ mol−1. Similarly, a σ-hole is found at the outer of C–I bond in PyI and it has a greater MEP than that in PyBr. On the other hand, the N atom of PyI is surrounded by a negative MEP, and its value is almost equal to that in PyBr. That is, the halogen substituent in pyridine has a slight effect on the basicity of its N atom. Therefore, the N atom of PyX (X = Br and I) acts as a Lewis base to form a tetrel bond with the tetrel atom of PhTF3 (T = C, Si, Ge) and the X atom acts as a Lewis acid to form a halogen bond with the N-base. The structures of the corresponding complexes are shown in Fig. 2.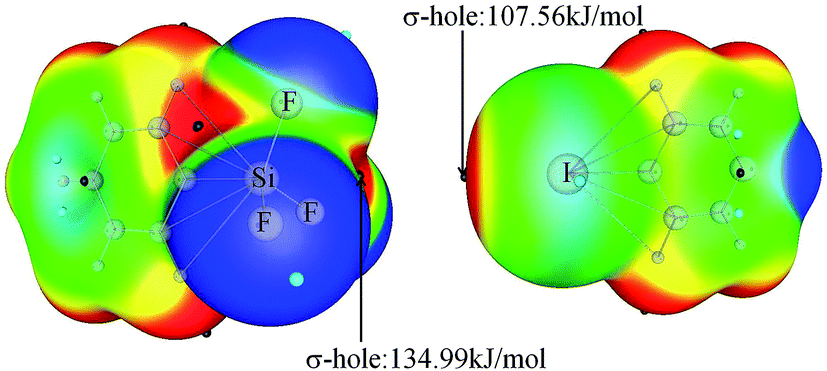 | ||
| Fig. 1 MEP maps of PhSiF3 (left) and 4-iodopyridine (right). Color ranges in kJ mol−1, are: red, greater than 47; yellow, between 47 and 13; green, between 13 and −26, blue, less than −26. | ||
| Molecules | Vmax,T | Complexes | Vmax,X | Complexes | Vmin,N |
|---|---|---|---|---|---|
| a Note: Vmax,X is 75.29 and 107.56 kJ mol−1 in PyBr and PyI, respectively. Vmin,N is −141.67 and −142.02 kJ mol−1 in PyBr and PyI, respectively. | |||||
| PhCF3 | 15.85 | PhCF3⋯PyI | 96.43 | PyI⋯NH3 | −161.80 |
| PhSiF3 | 134.98 | PhSiF3⋯PyI | 145.39 | PyI⋯NCH | −161.89 |
| PhGeF3 | 138.43 | PhGeF3⋯PyI | 150.69 | PyI⋯NHCH2 | −161.86 |
| PhSiH3 | 82.59 | PhSiH3⋯PyI | 119.52 | PyI⋯NH2CH3 | −162.86 |
| PhSiCl3 | 55.84 | PhSiCl3⋯PyI | 150.15 | PyBr⋯NH3 | −157.84 |
| — | — | PhSiF3⋯PyBr | 113.01 | — | — |
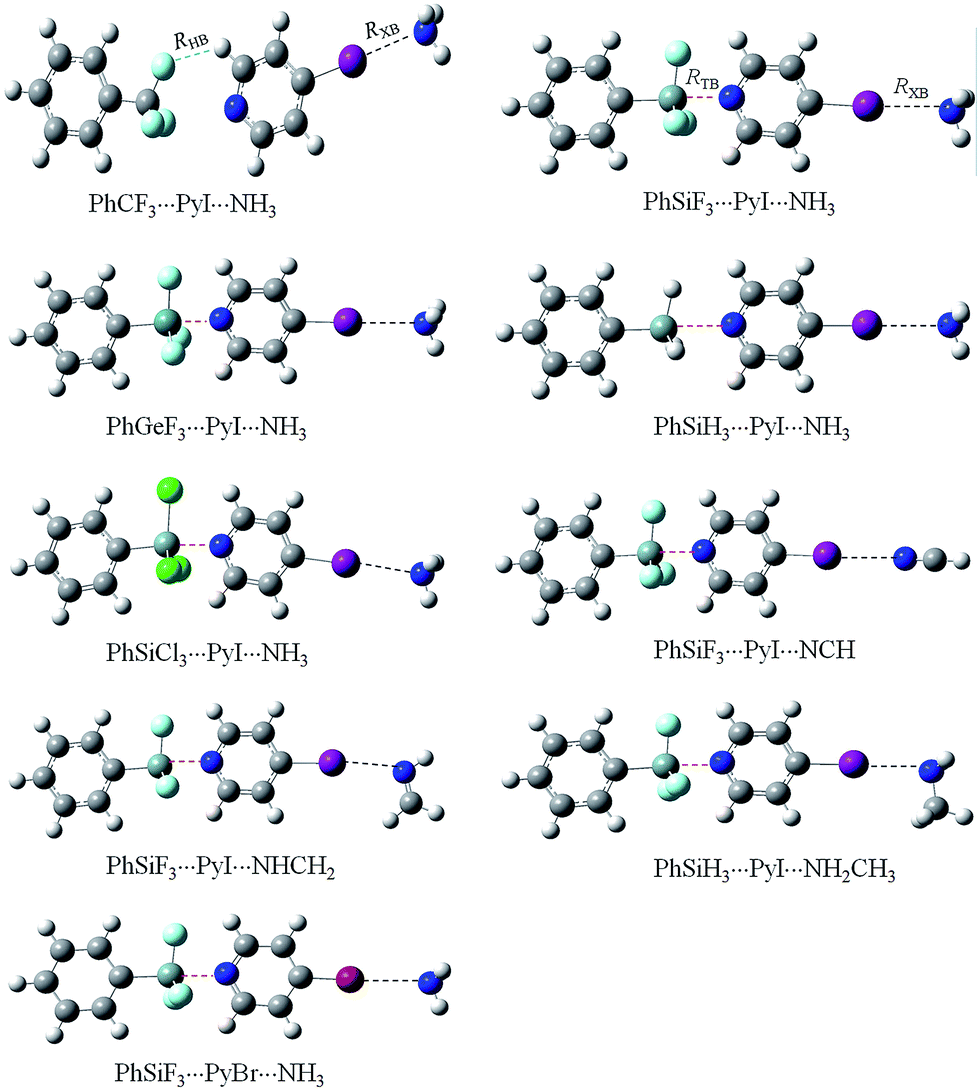 | ||
| Fig. 2 Optimized structures of the ternary complexes. | ||
Table 2 presents the binding distances in the dyads and triads. One can see that the binding distances of both tetrel and halogen bonds are shorter in the trials except PhCF3⋯PyI⋯NH3 when the complex varies from the dyad to the triad. In PhCF3⋯PyI⋯NH3, the two interatomic separations are longer in the triad with respect to those in the dyad. Moreover, the shortening of the halogen bonding distance is much larger than that of tetrel bonding distance in most triads. For the tetrel bond, the longer the binding distance in the dyad, the greater the shortening of the binding distance in the triad. This is also hold true for the binding distance of halogen bond in the complexes of PhSiF3⋯PyI⋯N-base. The shortening of halogen bonding distance in PyI⋯NH3 is larger for the shorter tetrel bonding distance. The largest shortening of halogen bonding distance is 0.684 Å in PhGeF3⋯PyI⋯NH3 and PhSiCl3⋯PyI⋯NH3.
| Triads | RTB/HB | ΔRTB/HB | RXB | ΔRXB |
|---|---|---|---|---|
| a Note: data in parentheses are from the respective dyads. | ||||
| PhCF3⋯PyI⋯NH3 | 2.4271(2.4181) | 0.0096 | 3.0506(3.0463) | 0.0043 |
| PhSiF3⋯PyI⋯NH3 | 2.0980(2.1278) | −0.0298 | 2.9852 | −0.0611 |
| PhGeF3⋯PyI⋯NH3 | 2.0642(2.0787) | −0.0145 | 2.9779 | −0.0684 |
| PhSiH3⋯PyI⋯NH3 | 2.5442(2.6189) | −0.0747 | 3.0157 | −0.0306 |
| PhSiCl3⋯PyI⋯NH3 | 2.1058(2.1413) | −0.0355 | 2.9779 | −0.0684 |
| PhSiF3⋯PyI⋯NCH | 2.1013 | −0.0265 | 3.0587(3.1142) | −0.0555 |
| PhSiF3⋯PyI⋯NHCH2 | 2.0981 | −0.0297 | 2.9151(2.9749) | −0.0598 |
| PhSiF3⋯PyI⋯NH2CH3 | 2.0958 | −0.0320 | 2.8804(2.9381) | −0.0577 |
| PhSiF3⋯PyBr⋯NH3 | 2.1011(2.1271) | −0.0260 | 3.0174(3.0589) | −0.0415 |
3.2 Binding energies
Table 3 presents the total binding energy in the triads as well as the binding energies of tetrel and halogen bonds in the binary and the ternary complexes. The total binding energy is changed in a wide range from 16.95 kJ mol−1 in PhCF3⋯PyI⋯NH3 to 148.38 kJ mol−1 in PhGeF3⋯PyI⋯NH3. The binding energy of halogen bond is related with the nature of the halogen donor and the N-base. It is expected that PyI forms a stronger halogen bond with NH3 than PyBr, which is consistent with the magnitude of σ-hole on the halogen atom. The halogen bond is stronger in the order HCN(sp) < NH3(sp3) < NHCH2(sp2) < NH2CH3(sp3). Obviously, the basicity of N-base is related with the electronegativity of N element. In addition, the methyl group in the electron donor plays an electron-donating role in the formation of halogen bond.48 The strength of tetrel bond is mainly dependent on the nature of tetrel atom and its substituents, but it shows a slight dependence on the halogen substitution in pyridine. The binding energy of tetrel bond is almost equal in both PhSiF3⋯PyBr and PhSiF3⋯PyI, consistent with the negative MEP on the N atom of PyX (X = Br and I). PhGeF3 forms a stronger tetrel bond than PhSiF3 and their binding energies have a difference of about 25 kJ mol−1. However, the positive MEP on the Ge atom in PhGeF3 is larger only by 3.45 kJ mol−1 than that on the Si atom in PhSiF3. This indicates that not only electrostatic interaction but also polarization interaction is responsible for the stability of tetrel bonded complexes. The binding energy of tetrel bond is more negative in PhSiF3⋯PyI than that in PhSiH3⋯PyI, having a consistent change with the positive MEP on the Si atom in both molecules. PhSiCl3 forms a weaker tetrel bond with PyI than PhSiF3 but a stronger tetrel bond than PhSiH3. The former is consistent with the change of σ-hole on the Si atom, but the latter is reverse to the change of σ-hole on the Si atom. This inconsistency is also attributed to the larger polarization in PhSiCl3⋯PyI than in PhSiH3⋯PyI. The larger polarization in PhSiCl3⋯PyI can be visualized by comparing the electron density shift on the silicon atom in PhSiCl3⋯PyI and PhSiH3⋯PyI. As shown in Fig. 3, the red area on the silicon atom in PhSiCl3⋯PyI is larger than that in PhSiH3⋯PyI. This indicates that the silicon atom in PhSiCl3⋯PyI suffers larger polarization in the formation of tetrel bond. Similarly, the role of polarization in halogen bonds has been unveiled in CF3Cl⋯OH2 complex by Clark et al.75 Halogen bond is weaker than tetrel bond in most dyads but is stronger than hydrogen bond in PhCF3⋯PyI. The binding energy of tetrel bond is larger than 90 kJ mol−1 in PhTY3⋯PyX (T = Si and Ge; Y = F and Cl; X = Br and I). Thus this tetrel bond is very strong, resulting in a prominent deformation of –TY3 group. This deformation has been considered in calculating the binding energy of tetrel bond by using the energy of the monomer in the complexes. The magnitude of –TY3 deformation can be estimated with the change of angle C–T–Y in the complex relative to the monomer (Table S1†). This angle is larger than 10° in PhTY3⋯PyX (T = Si and Ge; Y = F and Cl; X = Br and I). Thus the deformation energy has the larger contribution to the strong tetrel bond in the above complexes. The prominent deformation of –TY3 group is mainly caused by the formation of a partially covalent tetrel bond.| Triads | ΔEtotal | ΔETB/HB | ΔEXB | Ecoop |
|---|---|---|---|---|
| a Note: data in parentheses are from the respective dyads. The interaction energies of tetrel and halogen bonds in the triad are calculated with formula of ΔEA–BC = EA–BC(T) − EA(T) − EBC(T), where EA–BC(T) is the energy of A–BC corrected for BSSE with counterpoise = 2, EA(T) the energy of A in the triad, and EBC(T) the energy of BC in the triad. | ||||
| PhCF3⋯PyI⋯NH3 | −16.95 | −1.42(−2.42) | −14.54(−15.48) | 0.68(4.0%) |
| PhSiF3⋯PyI⋯NH3 | −126.27 | −111.20(−96.94) | −20.25 | −13.51(10.7%) |
| PhGeF3⋯PyI⋯NH3 | −148.38 | −133.42(−121.84) | −20.79 | −10.81(7.3%) |
| PhSiH3⋯PyI⋯NH3 | −34.95 | −19.72(−16.39) | −17.29 | −3.03(8.7%) |
| PhSiCl3⋯PyI⋯NH3 | −122.27 | −107.21(−92.85) | −20.92 | −13.81(11.3%) |
| PhSiF3⋯PyI⋯NCH | −119.94 | −109.42 | −14.96(−10.92) | −2.72(2.3%) |
| PhSiF3⋯PyI⋯NHCH2 | −128.22 | −111.23 | −22.18(−17.29) | −13.64(10.6%) |
| PhSiF3⋯PyI⋯NH2CH3 | −132.04 | −112.53 | −25.25(−19.95) | −14.85(11.2%) |
| PhSiF3⋯PyBr⋯NH3 | −118.11 | −108.85(−96.90) | −13.49(−9.43) | −11.42(9.7%) |
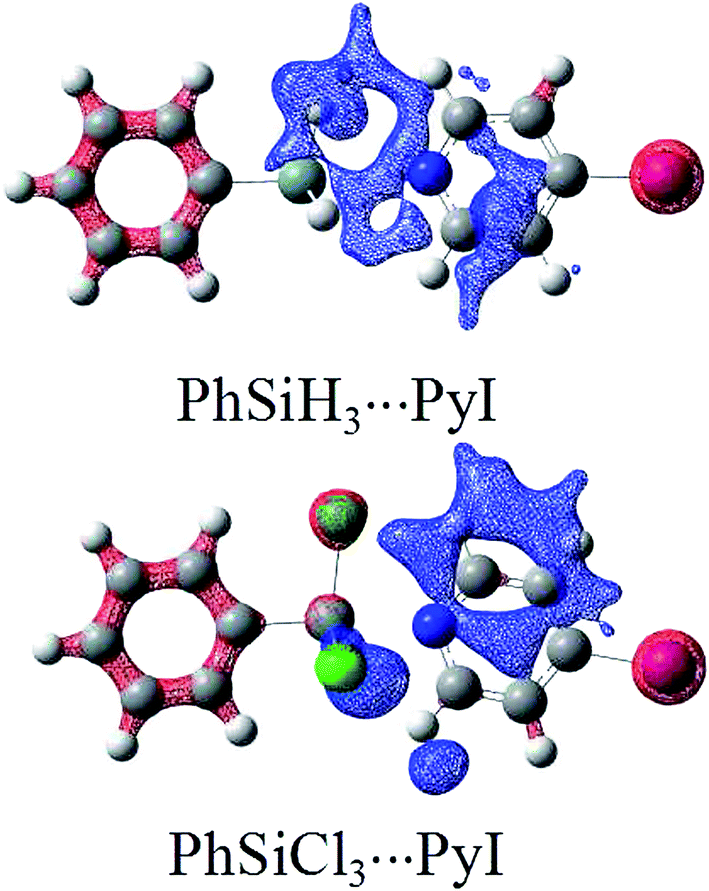 | ||
| Fig. 3 Electron density shifts in PhSiH3⋯PyI and PhSiCl3⋯PyI. Contours are shown at the 0.25 au level. Red/blue regions indicate increased/decreased density. | ||
In most triads, the binding energies of tetrel and halogen bonds are more negative than those in the dyads, indicating that both tetrel and halogen bonds are stronger in the triads. Thus there exists positive cooperativity between tetrel and halogen bonds in the triads except PhCF3⋯PyI⋯NH3. However, the binding energies of both interactions in PhCF3⋯PyI⋯NH3 are less negative, showing that they are weaker in this triad. As a result, negative cooperativity is found between hydrogen and halogen bonds in PhCF3⋯PyI⋯NH3. Table S2† lists the increased/decreased percentage of binding energies of both interactions in the triads relative to those in the dyads. It is found that this percentage is pertinent to the relative strength of both interactions. Clearly, the weaker interaction has a larger increased/decreased percentage than the stronger one. Specially, the hydrogen bond has a larger weakening than the halogen bond in PhCF3⋯PyI⋯NH3; the tetrel bond has a larger enhancement than the halogen bond in PhSiH3⋯PyI⋯NH3, while the halogen bond has a larger enhancement than the tetrel bond in other triads.
According to the electrostatic nature of tetrel and halogen bonds, the change of binding energy in the triads can be understood with the change of the negative MEP on the N atom of PyX in PyX⋯N-base and the positive MEP on the X atom of PyX in PhTY3⋯PyX (Table 1). The former is more negative in PyX⋯N-base and the latter is more positive in PhTY3⋯PyX (T = Si and Ge). This indicates that the N atom of PyX in PyX⋯N-base is a stronger Lewis base and the X atom of PyX in PhTY3⋯PyX (T = Si and Ge) is a stronger Lewis acid. Consequently, the former forms a stronger tetrel bond and the latter forms a stronger halogen bond. However, the positive MEP on the iodine atom of PyI is smaller in PhCF3⋯PyI, thus it is a weaker Lewis acid and forms a weaker halogen bond in PhCF3⋯PyI⋯NH3. The positive MEP on the H atom adjoined to the N atom of pyridine reduces from 86.90 kJ mol−1 in 4-iodopyridine to 66.36 kJ mol−1 in PyI⋯NH3, thus a weaker hydrogen bond is obtained in PhCF3⋯PyI⋯NH3.
This cooperative effect can also be better estimated with cooperative energy (Ecoop), calculated with the formulas of Ecoop = ΔEtotal − ΔETB(D) − ΔEXB(D), where ΔEtotal is the total binding energy of triad, ΔETB(D) and ΔEXB(D) are the binding energies of tetrel bond and halogen bond in the optimized dyads, respectively. It is found from Table 3 that this term is negative in most triads but positive in PhCF3⋯PyI⋯NH3, confirming the positive cooperativity in the former and the negative one in the latter. The cooperative energy amounts to about 2.3–11.3% of the total binding energy. The smallest percentage is found in PhSiF3⋯PyI⋯NCH and the largest percentage is found in PhSiCl3⋯PyI⋯NH3 and PhSiF3⋯PyI⋯NH2CH3. The contribution of cooperative energy to the total binding energy is small if one of two interactions is weak. This percentage in most triads is larger than that reported in hydrogen bonds (less than 6%).76
3.3 AIM analyses
Fig. 4 shows the molecular maps of the dyads. A H⋯F BCP and two N⋯F BCPs are found in PhCF3⋯PyI, and the former confirms the existence of C–H⋯F hydrogen bond. The tetrel bond in PhTY3⋯PyX (T = Si and Ge) is characterized with a T⋯N BCP. In addition, a H⋯F/Cl BCP is also found in the strong tetrel bonded complexes. This H⋯F/Cl BCP corresponds to a C–H⋯F/Cl hydrogen bond. However, such BCP is not present in PhSiH3⋯PyI. Thus the C–H⋯Cl hydrogen bond has some contribution to the larger binding energy in PhSiCl3⋯PyI relative to that in PhSiH3⋯PyI. A X⋯N BCP is found in the halogen bonded complexes.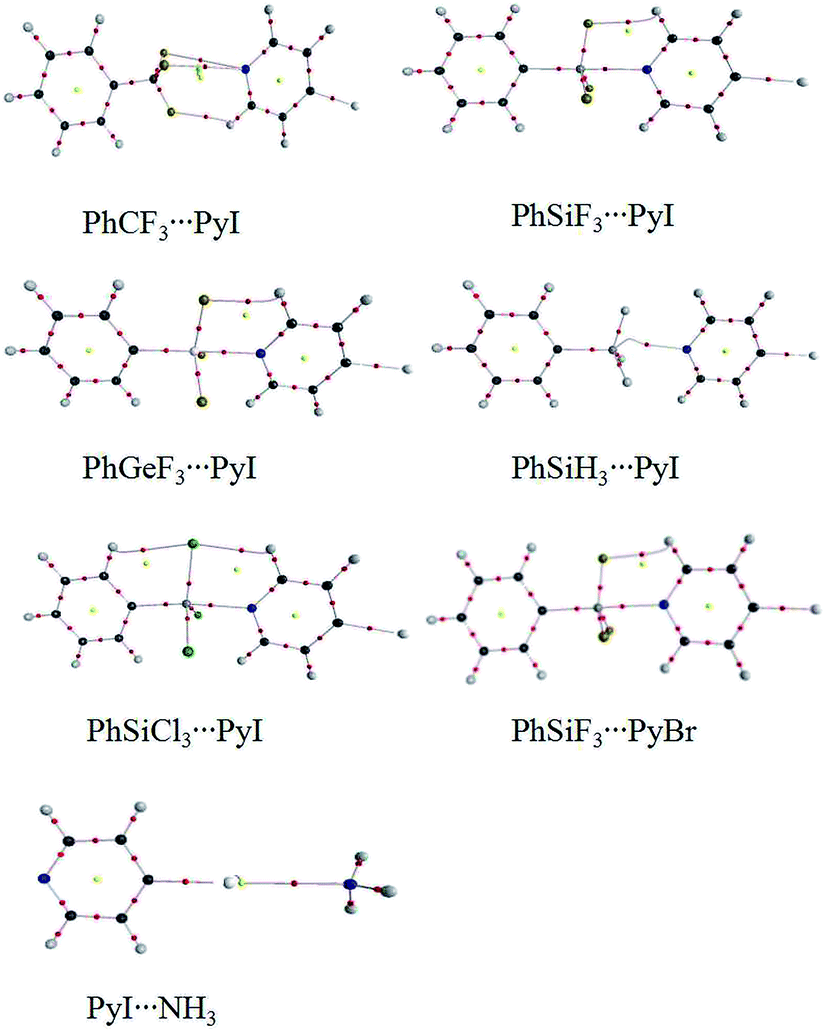 | ||
| Fig. 4 Molecular graph of the binary complexes. | ||
The electron density, Laplacian, and energy density at these BCPs are collected in Table 4. The electron density at the X⋯N BCP is smaller than 0.024 au and that at the Si/Ge⋯N BCP is larger than 0.021 au. The electron density at the H⋯F BCP is very small, consistent with the weak C–H⋯F interaction. The Laplacian at the T⋯N BCP is a large positive value in PhTY3⋯PyX (T = Si and Ge) and its energy density is negative. This shows that the tetrel bond is a partially covalent interaction with a big strength.77 The positive energy density at the X⋯N BCP supports the conclusion that halogen bond is electrostatic in nature. The electron density at the X⋯N BCP is increased in the sequence HCN < NH3 < NHCH2< NH2CH3, showing that the electron density can be used to measure the strength of halogen bond. This conclusion holds true for the tetrel bond. In Fig. 5, we plotted the relationship between the electron density at the same BCP and the corresponding atom separation for the tetrel and halogen bonds. An exponential relationship is found for the Si⋯N tetrel bond but a linear relationship for the I⋯N halogen bond. Table S3† presents the change of electron density (Δρ) at the triads relative to the dyads. The value of Δρ is positive in most triads with an exception of PhCF3⋯PyI⋯NH3. The increase/decrease of electron density confirms the change of both interactions in the triads.
| Triads | ρTB | ∇2ρTB | HTB | ρXB | ∇2ρXB | HXB |
|---|---|---|---|---|---|---|
| a Note: The corresponding values are from the H⋯F BCP in PhCF3⋯PyI⋯NH3. The corresponding values at the N⋯F BCP are 0.0039, 0.0159 and 0.0008 au in PhCF3⋯PyI⋯NH3. | ||||||
| PhCF3⋯PyI⋯NH3 | 0.0073 | 0.0355 | 0.0019 | 0.0163 | 0.0544 | 0.0016 |
| PhSiF3⋯PyI⋯NH3 | 0.0557 | 0.2167 | −0.0169 | 0.0185 | 0.0605 | 0.0014 |
| PhGeF3⋯PyI⋯NH3 | 0.0844 | 0.2419 | −0.0342 | 0.0188 | 0.0612 | 0.0014 |
| PhSiH3⋯PyI⋯NH3 | 0.0243 | 0.0565 | −0.0024 | 0.0174 | 0.0577 | 0.0015 |
| PhSiCl3⋯PyI⋯NH3 | 0.0596 | 0.1863 | −0.0211 | 0.0188 | 0.0612 | 0.0014 |
| PhSiF3⋯PyI⋯NCH | 0.0553 | 0.2141 | −0.0167 | 0.0135 | 0.0541 | 0.0022 |
| PhSiF3⋯PyI⋯NHCH2 | 0.0557 | 0.2163 | −0.0169 | 0.0204 | 0.0687 | 0.0013 |
| PhSiF3⋯PyI⋯NH2CH3 | 0.0561 | 0.2180 | −0.0171 | 0.0234 | 0.0717 | 0.0006 |
| PhSiF3⋯PyBr⋯NH3 | 0.0552 | 0.2144 | −0.0167 | 0.0139 | 0.0518 | 0.0020 |
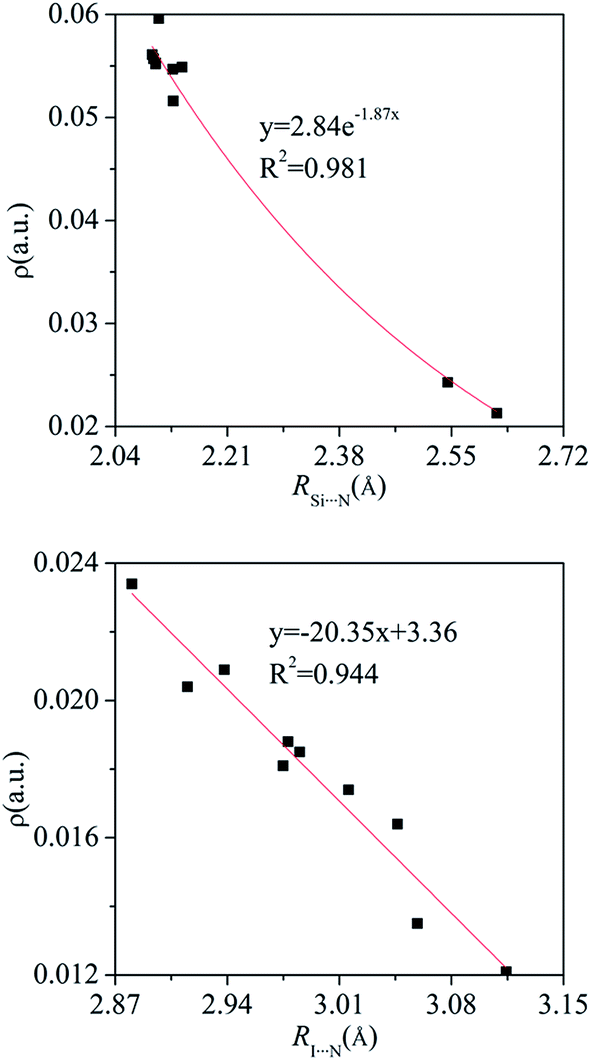 | ||
| Fig. 5 Electron density (ρ) at the same BCP versus the corresponding atom separation (R) for the Si⋯N tetrel bond and I⋯N halogen bond in the binary and ternary complexes. | ||
The cooperativity between both interactions in the ternary complexes can be visualized by the NCI analysis. The ternary complexes of PhCF3⋯PyI⋯NH3 and PhSiF3⋯PyI⋯NH3 are respectively chosen as an example to obtain visualization of the negative and positive cooperativity. Fig. 6 shows the plot of the reduced density gradient versus sign(λ2)ρ in these complexes. The F⋯H hydrogen bond in PhCF3⋯PyI and I⋯N halogen bond in PyI⋯NH3 are characterized by the pink and green spikes, respectively. Although their shifts are not obviously observed in PhCF3⋯PyI⋯NH3, a tiny shift to the lower electron density can be found by a careful comparison, confirming the negative cooperativity in PhCF3⋯PyI⋯NH3. The tetrel bond in PhSiF3⋯PyI and halogen bond in PyI⋯NH3 are also represented by the pink and green spikes. In PhSiF3⋯PyI⋯NH3, the corresponding spikes move to the higher electron density, thus the positive cooperativity is present in this ternary complex.
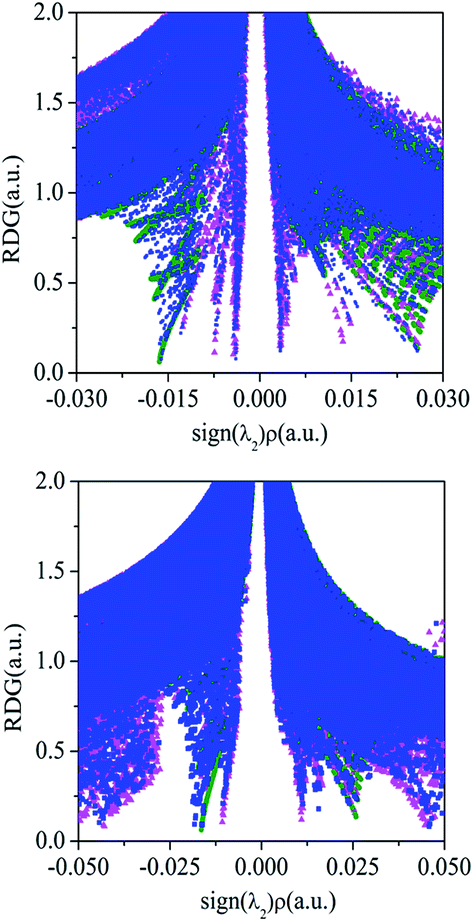 | ||
| Fig. 6 Reduced density gradient (RDG) versus sign(λ2)ρ in the ternary complexes (blue) of PhCF3⋯PyI⋯NH3 (up) and PhSiF3⋯PyI⋯NH3 (down) as well as the corresponding hydrogen/tetrel (pink) and halogen (green) bonded binary complexes. | ||
3.4 NBO analyses
The interactions in these complexes were analyzed in views of orbital interactions and charge transfer. There are two orbital interactions of Lp(N) → and Lp(N) →
and Lp(N) → 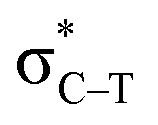 in the tetrel bonded complexes of PhTF3⋯PyX (T = Si and Ge), where Lp(N) denotes the lone pair orbital on N atom,
in the tetrel bonded complexes of PhTF3⋯PyX (T = Si and Ge), where Lp(N) denotes the lone pair orbital on N atom,  the empty p orbital on T atom, and
the empty p orbital on T atom, and 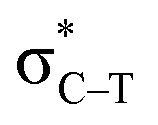 the C–T antibonding orbital. Lp(N) →
the C–T antibonding orbital. Lp(N) → 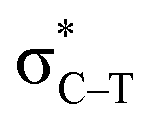 is still present in PhSiY3⋯PyI (Y = H and Cl), but Lp(N) →
is still present in PhSiY3⋯PyI (Y = H and Cl), but Lp(N) →  is replaced by Lp(N) →
is replaced by Lp(N) → 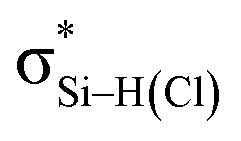 . Only Lp(F) →
. Only Lp(F) → 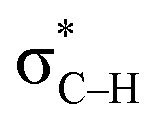 /Lp(N) →
/Lp(N) → 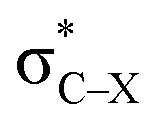 is found in the hydrogen/halogen bond. These orbital interactions are estimated with second-order perturbation energies (Table 5). In PhTF3⋯PyX (T = Si and Ge), Lp(N) →
is found in the hydrogen/halogen bond. These orbital interactions are estimated with second-order perturbation energies (Table 5). In PhTF3⋯PyX (T = Si and Ge), Lp(N) →  is far stronger than Lp(N) →
is far stronger than Lp(N) → 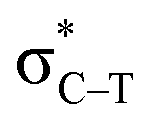 , which is a feature of strong tetrel bonds. For the weak tetrel bond of PhSiH3⋯PyI, Lp(N) →
, which is a feature of strong tetrel bonds. For the weak tetrel bond of PhSiH3⋯PyI, Lp(N) → 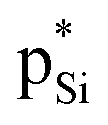 is not present and Lp(N) →
is not present and Lp(N) → 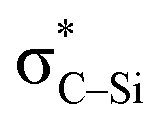 is stronger than Lp(N) →
is stronger than Lp(N) → 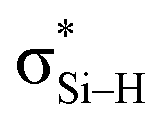 , where the sum of perturbation energy of three Lp(N) →
, where the sum of perturbation energy of three Lp(N) → 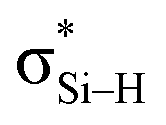 is listed in Table 5. In PhSiCl3⋯PyI, Lp(N) →
is listed in Table 5. In PhSiCl3⋯PyI, Lp(N) → 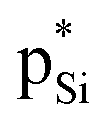 is also not present, but Lp(N) →
is also not present, but Lp(N) → 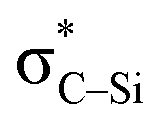 is weaker than Lp(N) →
is weaker than Lp(N) → 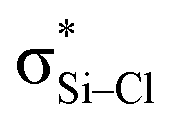 . The three strong Lp(N) →
. The three strong Lp(N) → 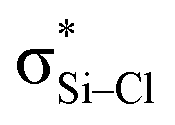 orbital interactions are partly responsible for the strong tetrel bond in PhSiCl3⋯PyI. Lp(F) →
orbital interactions are partly responsible for the strong tetrel bond in PhSiCl3⋯PyI. Lp(F) → 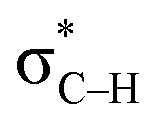 is very weak in PhCF3⋯PyI, corresponding to the weak C–H⋯F hydrogen bond. Lp(N) →
is very weak in PhCF3⋯PyI, corresponding to the weak C–H⋯F hydrogen bond. Lp(N) → 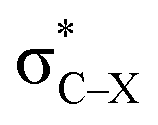 has an inconsistent change with the interaction energy of halogen bond, indicating the less importance of orbital interaction in the formation of halogen bond. The changes of second-order perturbation energies in the triads relative to the dyads are given in Table S4.† The second-order perturbation energies of the above orbital interactions are increased in most triads except PhCF3⋯PyI⋯NH3, where they are almost not changed. The strengthening of Lp(N) →
has an inconsistent change with the interaction energy of halogen bond, indicating the less importance of orbital interaction in the formation of halogen bond. The changes of second-order perturbation energies in the triads relative to the dyads are given in Table S4.† The second-order perturbation energies of the above orbital interactions are increased in most triads except PhCF3⋯PyI⋯NH3, where they are almost not changed. The strengthening of Lp(N) →  /Lp(N) →
/Lp(N) → 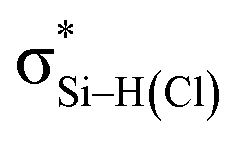 is larger than that of Lp(N) →
is larger than that of Lp(N) → 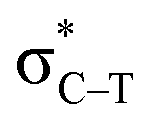 and Lp(N) →
and Lp(N) → 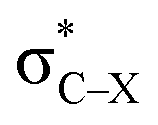 in PhTF3⋯PyX⋯N-base/PhSiY3⋯PyI⋯NH3 (Y = H and Cl). That is, the orbital interaction has some contribution to the cooperativity between tetrel and halogen bonds. However, the orbital interaction has a slight contribution to the interplay between hydrogen and halogen bonds in PhCF3⋯PyI⋯NH3.
in PhTF3⋯PyX⋯N-base/PhSiY3⋯PyI⋯NH3 (Y = H and Cl). That is, the orbital interaction has some contribution to the cooperativity between tetrel and halogen bonds. However, the orbital interaction has a slight contribution to the interplay between hydrogen and halogen bonds in PhCF3⋯PyI⋯NH3.
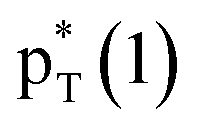 /Lp(N) →
/Lp(N) → 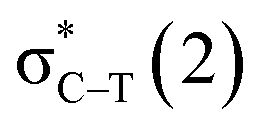 in the tetrel bond and Lp(N) →
in the tetrel bond and Lp(N) → 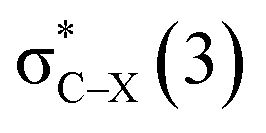 in the halogen bond in the triadsa
in the halogen bond in the triadsa
| Triads | E(2)1 | E(2)2 | E(2)3 |
|---|---|---|---|
a Note: E(2)1 corresponds to Lp(F) → 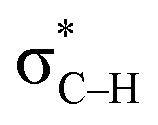 in PhCF3⋯PyI⋯NH3 and Lp(N) → in PhCF3⋯PyI⋯NH3 and Lp(N) → 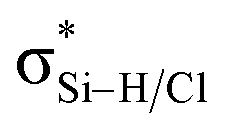 in PhSiH3⋯PyI⋯NH3/PhSiCl3⋯PyI⋯NH3, respectively. Data in parentheses are from the respective dyads. in PhSiH3⋯PyI⋯NH3/PhSiCl3⋯PyI⋯NH3, respectively. Data in parentheses are from the respective dyads. |
|||
| PhCF3⋯PyI⋯NH3 | 0.92(0.92) | — | 29.18(29.59) |
| PhSiF3⋯PyI⋯NH3 | 617.89(599.99) | 17.60(16.18) | 36.53 |
| PhGeF3⋯PyI⋯NH3 | 803.06(762.93) | 8.53(8.36) | 37.41 |
| PhSiH3⋯PyI⋯NH3 | 54.01(41.26) | 46.06(38.62) | 32.85 |
| PhSiCl3⋯PyI⋯NH3 | 331.85(304.97) | 83.52(80.51) | 37.62 |
| PhSiF3⋯PyI⋯NCH | 613.08(561.96) | 17.26(16.18) | 17.43(14.04) |
| PhSiF3⋯PyI⋯NHCH2 | 619.94(599.99) | 17.39(16.18) | 34.53(26.79) |
| PhSiF3⋯PyI⋯NH2CH3 | 624.58(561.96) | 17.47(16.18) | 45.19(36.16) |
| PhSiF3⋯PyBr⋯NH3 | 612.12(560.75) | 17.31(16.30) | 17.97(15.30) |
Table 6 presents the charge transfer (CT) in the ternary complexes and its change (ΔCT) relative to the corresponding binary complexes. One can see that CT is much larger in the tetrel bond than that in the halogen bond but it is much smaller in the hydrogen bond than that in the halogen bond. Generally, the bigger CT corresponds to a stronger interaction although there is not a good relationship between CT and binding energy. There are some exceptions. For example, CT is larger in PhSiCl3⋯PyI than that in PhSiF3⋯PyI, which disagrees with the change of binding energy. The larger CT in PhSiCl3⋯PyI is partly attributed to the greater polarization of Cl atom. ΔCT is positive for the tetrel and halogen bonds but negative for the hydrogen and halogen bonds in the triads, indicating that charge transfer is also important in strengthening both interactions.
| Triads | CTTB/HB | CTXB | ΔCTTB/HB | ΔCTXB |
|---|---|---|---|---|
| a Note: data in parentheses are CT in the respective dyads. | ||||
| PhCF3⋯PyI⋯NH3 | 0.0015(0.0019) | 0.0182(0.0186) | −0.0004 | −0.0004 |
| PhSiF3⋯PyI⋯NH3 | 0.1358(0.1243) | 0.0234(0.0186) | 0.0115 | 0.0048 |
| PhGeF3⋯PyI⋯NH3 | 0.1737(0.1647) | 0.0241(0.0186) | 0.0090 | 0.0055 |
| PhSiH3⋯PyI⋯NH3 | 0.0443(0.0349) | 0.0207(0.0186) | 0.0094 | 0.0021 |
| PhSiCl3⋯PyI⋯NH3 | 0.1511(0.1372) | 0.0241(0.0186) | 0.0139 | 0.0055 |
| PhSiF3⋯PyI⋯NCH | 0.1350(0.1234) | 0.0061(0.0044) | 0.0107 | 0.0017 |
| PhSiF3⋯PyI⋯NHCH2 | 0.1366(0.1234) | 0.0195(0.0145) | 0.0123 | 0.0050 |
| PhSiF3⋯PyI⋯NH2CH3 | 0.1375(0.1234) | 0.0297(0.0234) | 0.0132 | 0.0063 |
| PhSiF3⋯PyBr⋯NH3 | 0.1348(0.1245) | 0.0108(0.0089) | 0.0103 | 0.0019 |
4. Conclusions
Ab initio calculations have been performed to study the σ-tetrel bond and σ-halogen bond in the ternary complexes. PhTF3 (T = Si and Ge) forms a stronger tetrel bond with 4-iodopyridine, while PhCF3 forms a weaker hydrogen bond with 4-iodopyridine. 4-Iodopyridine forms a halogen bond with a series of nitrogen bases. The Si atom of PhSiCl3 has the small σ-hole than that of PhSiH3, but the former forms a stronger tetrel bond than the latter. The tetrel bond exhibits a nature of partially covalent interaction although it is dominated by electrostatic interaction. In most ternary complexes, both tetrel and halogen bonds are strengthened with positive cooperativity. However, both hydrogen and halogen bonds in PhCF3⋯4-iodopyridine⋯NH3 are weakened with negative cooperativity. The cooperativity between tetrel and halogen bonds is attributed to the electrostatic and orbital interactions. The cooperative energy grows up in the order HCN < NH3 < NHCH2 < NH2CH3, depending on the strength of halogen bond. The title complexes are involved with molecules usually used in crystal materials, thus these results are significant for the applications of tetrel and halogen bonds in these fields.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21573188).References
- B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818 CrossRef CAS PubMed.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–168 CrossRef.
- Q. Shao and Y. Q. Gao, J. Chem. Theory Comput., 2010, 6, 3750–3760 CrossRef CAS.
- A. J. Parker, J. Stewart, K. J. Donald and C. A. Parish, J. Am. Chem. Soc., 2012, 134, 5165–5172 CrossRef CAS PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- S. Triguero, R. Llusar, V. Polo and M. Fourmigúe, Cryst. Growth Des., 2008, 8, 2241–2247 CAS.
- P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461–1464 CrossRef CAS PubMed.
- K. Ueda, M. Oguni and T. Asaji, Cryst. Growth Des., 2014, 14, 6189–6196 CAS.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137, 499–507 CrossRef CAS PubMed.
- P. Politzer, P. Lane, M. C. Concha, Y. G. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2008, 14, 659–665 CrossRef CAS PubMed.
- K. E. Riley, J. S. Murray, P. Politzer, M. C. Concha and P. Hobza, J. Chem. Theory Comput., 2009, 5, 155–163 CrossRef CAS PubMed.
- K. E. Riley, J. S. Murray, J. Fanfrlík, J. Řezáč, R. J. Solá, M. C. Concha, F. M. Ramos and P. Politzer, J. Mol. Model., 2011, 17, 3309–3318 CrossRef CAS PubMed.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541–548 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, Theor. Chem. Acc., 2012, 131, 1114 CrossRef.
- A. C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdélyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- Q. N. Zheng, X. H. Liu, T. Chen, H. J. Yan, T. Cook, D. Wang, P. J. Stang and L. J. Wan, J. Am. Chem. Soc., 2015, 137, 6128–6131 CrossRef CAS PubMed.
- E. Cariati, G. Cavallo, A. Forni, G. Leem, P. Metrangolo, F. Meyer, T. Pilati, G. Resnati, S. Righetto, G. Terraneo and E. Tordin, Cryst. Growth Des., 2011, 11, 5642–5648 CAS.
- F. C. Pigge, P. P. Kapadia and D. C. Swenson, CrystEngComm, 2013, 15, 4386–4391 RSC.
- L. C. Roper, C. Präsang, V. N. Kozhevnikov, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, Cryst. Growth Des., 2010, 10, 3710–3720 CAS.
- L. Turunen, N. K. Beyeh, F. Pan, A. Valkonen and K. Rissanen, Chem. Commun., 2014, 50, 15920–15923 RSC.
- E. M. Karlsen and J. Spanget-Larsen, Chem. Phys. Lett., 2009, 473, 227–232 CrossRef CAS.
- A. Bauzá, D. Quiñonero, A. Frontera and P. M. Deyà, Phys. Chem. Chem. Phys., 2011, 13, 20371–20379 RSC.
- S. Tsuzuki, A. Wakisaka, T. Ono and T. Sonoda, Chem.–Eur. J., 2012, 18, 951–960 CrossRef CAS PubMed.
- A. C. C. Carlsson, M. Uhrbom, A. Karim, U. Brath, J. Gräfenstein and M. Erdélyi, CrystEngComm, 2013, 15, 3087–3092 RSC.
- Q. Z. Li, Q. Q. Lin, W. Z. Li, J. B. Cheng, B. A. Gong and J. Z. Sun, ChemPhysChem, 2008, 9, 2265–2269 CrossRef CAS PubMed.
- M. Gao, Q. Z. Li, J. B. Cheng, W. Z. Li and H. B. Li, RSC Adv., 2015, 5, 105160–105168 RSC.
- Q. Z. Li, R. Li, X. F. Liu, W. Z. Li and J. B. Cheng, ChemPhysChem, 2012, 13, 1205–1212 CrossRef CAS PubMed.
- Q. Z. Li, H. Qi, R. Li, X. F. Liu, W. Z. Li and J. B. Cheng, Phys. Chem. Chem. Phys., 2012, 14, 3025–3030 RSC.
- M. Gao, J. B. Cheng, X. Yang, W. Z. Li, B. Xiao and Q. Z. Li, J. Chem. Phys., 2015, 143, 054308 CrossRef PubMed.
- Y. L. Zeng, W. J. Wu, X. Y. Li, S. J. Zheng and L. P. Meng, ChemPhysChem, 2013, 14, 1591–1600 CrossRef CAS PubMed.
- Y. X. Lu, Y. T. Liu, H. Y. Li, X. Zhu, H. L. Liu and W. L. Zhu, ChemPhysChem, 2012, 13, 2154–2161 CrossRef CAS PubMed.
- C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, ChemPhysChem, 2011, 12, 2742–2750 CrossRef CAS PubMed.
- H. Y. Li, Y. X. Lu, Y. T. Liu, X. Zhu, H. L. Liu and W. L. Zhu, Phys. Chem. Chem. Phys., 2012, 14, 9948–9955 RSC.
- I. Alkorta, J. Elguero, O. Mó, M. Yáñez and J. E. Del Bene, Phys. Chem. Chem. Phys., 2015, 17, 2259–2267 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed.
- R. S. Ruoff, T. Emilsson, A. I. Jaman, T. C. Germann and H. S. Gutowsky, J. Chem. Phys., 1992, 96, 3441–3446 CrossRef CAS.
- N. W. Mitzel, A. J. Blake and D. W. H. Rankin, J. Am. Chem. Soc., 1997, 119, 4143–4148 CrossRef CAS.
- I. Alkorta, I. Rozas and J. Elguero, J. Phys. Chem. A, 2001, 105, 743–749 CrossRef CAS.
- M. S. Gargari, V. Stilinović, A. Bauzá, A. Frontera, P. McArdle, D. V. Derveer, S. W. Ng and G. Mahmoudi, Chem.–Eur. J., 2015, 21, 17951–17958 CrossRef PubMed.
- G. Mahmoudi, A. Bauzá and A. Frontera, Dalton Trans., 2016, 45, 4965–4969 RSC.
- A. Bauzá, A. Frontera and T. J. Mooibroek, Phys. Chem. Chem. Phys., 2016, 18, 1693–1698 RSC.
- J. Mikosch, S. Trippel, C. Eichhorn, R. Otto, U. Lourderaj, J. X. Zhang, W. L. Hase, M. Weidemüller and R. Wester, Science, 2008, 319, 183–186 CrossRef CAS PubMed.
- S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824–1834 RSC.
- M. X. Liu, Q. Z. Li, J. B. Cheng, W. Z. Li and H. B. Li, J. Chem. Phys., 2016, 145, 224310 CrossRef PubMed.
- Q. Z. Li, X. Guo, X. Yang, W. Z. Li, J. B. Cheng and H. B. Li, Phys. Chem. Chem. Phys., 2014, 16, 11617–11625 RSC.
- X. Guo, Y. W. Liu, Q. Z. Li, W. Z. Li and J. B. Cheng, Chem. Phys. Lett., 2015, 620, 7–12 CrossRef CAS.
- M. X. Liu, Q. Z. Li, W. Z. Li and J. B. Cheng, J. Mol. Graphics Modell., 2016, 65, 35–42 CrossRef CAS PubMed.
- M. X. Liu, L. Yang, Q. Z. Li, W. Z. Li, J. B. Cheng, B. Xiao and X. F. Yu, J. Mol. Model., 2016, 22, 192 CrossRef PubMed.
- Q. J. Tang and Q. Z. Li, Comput. Theor. Chem., 2014, 1050, 51–57 CrossRef CAS.
- S. A. C. McDowell, Chem. Phys. Lett., 2014, 598, 1–4 CrossRef CAS.
- M. D. Esrafili, N. Mohammadirad and M. Solimannejad, Chem. Phys. Lett., 2015, 628, 16–20 CrossRef CAS.
- M. Solimannejad, M. Orojloo and S. Amani, J. Mol. Model., 2015, 21, 183 CrossRef PubMed.
- S. Yourdkhani, T. Korona and N. L. Hadipour, J. Comput. Chem., 2015, 36, 2412–2428 CrossRef CAS PubMed.
- M. Marín-Luna, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2016, 120, 648–656 CrossRef PubMed.
- M. D. Esrafili, R. Nurazar and F. Mohammadian-Sabet, Mol. Phys., 2015, 113, 3703–3711 CrossRef CAS.
- M. D. Esrafili and F. Mohammadian-Sabet, Mol. Phys., 2016, 114, 1528–1538 CrossRef CAS.
- M. D. Esrafili and F. Mohammadian-Sabet, Mol. Phys., 2016, 114, 83–91 CrossRef CAS.
- Z. Rezaei, M. Solimannejad and M. D. Esrafili, Comput. Theor. Chem., 2015, 1074, 101–106 CrossRef CAS.
- M. Vatanparast, E. Parvini and A. Bahadori, Mol. Phys., 2016, 114, 1478–1484 CrossRef CAS.
- W. Li, Y. Zeng, X. Li, Z. Sun and L. Meng, Phys. Chem. Chem. Phys., 2016, 18, 24672–24680 RSC.
- M. G. Voronkov, O. M. Trofimova, E. A. Grebneva, N. F. Chernov and K. A. Abzaeva, Russ. J. Gen. Chem., 2011, 81, 2391–2411 CrossRef CAS.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- R. Ahlrichs, M. Bär, M. Häcer, H. Horn and C. Komel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877–13883 CrossRef CAS PubMed.
- A. Frontera, D. Quiñonero, C. Garau, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2005, 109, 4632–4637 CrossRef PubMed.
- D. Quiñonero, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2006, 110, 5144–5148 CrossRef PubMed.
- S. B. Boys and F. Bernardy, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- F. A. Bulat, A. Toro-Labbe, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS PubMed.
- R. F. W. Bader, AIM2000 Program, v. 2.0, McMaster University, Hamilton, Canada, 2000 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.0, 2001 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 3, 580–592 CrossRef PubMed.
- T. Clark, J. S. Murray and P. Politzer, Aust. J. Chem., 2014, 67, 451–456 CrossRef CAS.
- R. Rivelino and S. Canuto, J. Phys. Chem. A, 2001, 105, 11260–11265 CrossRef CAS.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835–12841 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02068f |
| This journal is © The Royal Society of Chemistry 2017 |