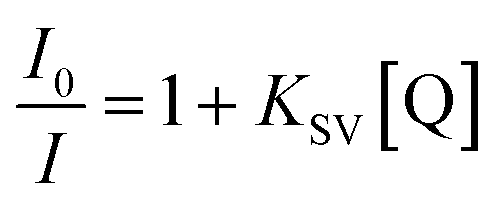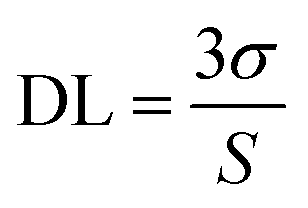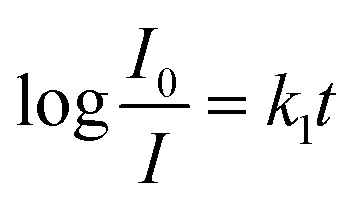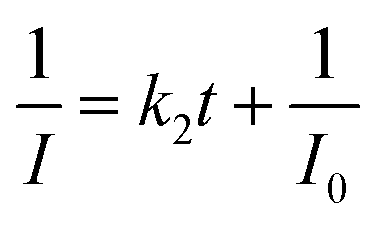DOI:
10.1039/C7RA02040F
(Paper)
RSC Adv., 2017,
7, 26849-26856
A fluorescent and colorimetric sensor based on a porphyrin doped polystyrene nanoporous fiber membrane for HCl gas detection†
Received
18th February 2017
, Accepted 28th April 2017
First published on 19th May 2017
Abstract
A facile fluorescent and colorimetric sensor capable of naked-eye detection of gaseous hydrogen chloride (HCl) was developed. This technology uses the optical probe 5,10,15,20-tetraphenylporphyrin immobilized in a polystyrene nanoporous fiber membrane (TPPH2-PS NFM) which was fabricated by electrospinning. The surface morphology and porous structures of the TPPH2-PS NFM were characterized and HCl gas sensing properties of the sensor was monitored by fluorescence spectrophotometry. Exposure to HCl gas causes apparent color change of the sensor from pink to yellow-green. The sample of THF-40 showed the maximum pseudo-second-order rate constant due to its largest specific surface area and pore volume. The detection limit was found to be 46 ppb and the response time was 5 s. The sensor exhibits excellent reversibility and prompt response and recovery which shed light on developing low-cost HCl gas sensing systems.
Introduction
Fluorescent and colorimetric sensors have emerged as a powerful new approach toward the detection of gaseous pollutants at ppm or ppb concentration levels.1–4 In contrast to other detecting approaches, fluorescent and colorimetric sensors present a number of advantages, such as instrumental simplicity, low-cost, high sensitivity, fast response, good reversibility, permit real-time analyses, etc.5 Monitoring of gaseous pollutants is very important because the quality of the atmospheric environment is directly relevant for human health and life. HCl gas is one of the toxic gases which is mainly derived from complete burning of halogenated polymers, incineration of plants and absorbing towers of semiconductor factories and is strictly regulated in many countries now.6–8 The threshold for tolerably safe exposure to HCl gas is 10 ppm and the exposure limit of 5 ppm has been identified as a work place hazard.9,10 For this reason, a reliably fluorescent and colorimetric sensor for HCl gas detection is strongly demanded.
Hitherto, the centerpiece of most fluorescent and colorimetric sensors is the fluorescent sensing materials.11–13 Porphyrin is a common organic dye and has attracted much interest as the fluorescent sensing materials for toxic gas detection due to its highly conjugated π-electron structure, distinctive fluorescence bands in the visible region as well as selectivity of the designated gas.14–16 Most of these porphyrin-based optochemical sensors mainly incorporate porphyrin into glasses, sol–gel, solid state films, etc.17–22 It is well known that the efficiency of these gas sensor is greatly dependent on the structure of the sensing materials.11 Due to its large specific surface area, high porosity and excellent accessibility for gaseous analytes, porous structure is highly attractive to the sensing materials. For example, F. W. Lin et al.23 fabricated honeycomb-patterned porous films for sensing of hazardous HCl gas. The well-ordered porous films sensor shows remarkable fluorescence quenching behavior toward HCl gas with quenching ratios of 72% higher than that of the corresponding dense film. Y.-Y. Lv et al.24 prepared a nanofibrous membrane of porphyrinated polyimide for the rapid detection of HCl gas. The fluorescence quenching of nanofibrous membrane is much significant than that of dense film due to its three dimensional and porous structures, and hence good gas accessibility.
Recently, electrospinning is one of the most promising approaches to fabricate nanofibers with nano- and micro-porous structure.25–27 Polystyrene (PS), a common thermoplastic polymer with low surface energy, has been widely used to fabricate porous nanofibers via directly electrospinning method.28,29 Owing to its low toxicity, cheaper cost and excellent photostability, PS has become the most popular smart material to envelop sensitive probes for oxygen measurement.30 However, to the best of our knowledge, there have been few studies regarding PS nanoporous fibers for the detection of HCl gas.13,31–33
In this work, we successfully demonstrated that TPPH2-PS NFM can be used for the naked-eye detection of HCl gas at ppm-level based on a simple color change. The PS nanoporous fiber membrane was prepared by electrospinning and TPPH2 was immobilized in the PS NFM uniformly. Furthermore, HCl gas sensing properties of the TPPH2-PS NFM sensor was monitored via distinct fluorescence variation.
Experimental
Materials
Polystyrene (PS) with molecular weight of Mw = 208![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 was provided by Wako, Japan. 5,10,15,20-Tetraphenylporphyrin (TPPH2) (reagent grade, 97%) was purchased from J&K Technology Co., China. Tetrahydrofuran (THF) and N,N-dimethylformamide (DMF) were obtained from Tianjin Fengchuan Chemical Reagent Co., Ltd., China.
000 g mol−1 was provided by Wako, Japan. 5,10,15,20-Tetraphenylporphyrin (TPPH2) (reagent grade, 97%) was purchased from J&K Technology Co., China. Tetrahydrofuran (THF) and N,N-dimethylformamide (DMF) were obtained from Tianjin Fengchuan Chemical Reagent Co., Ltd., China.
Preparation of the TPPH2-PS NFM
PS and TPPH2 were dissolved in THF:DMF mixtures to obtain the electrospinning solutions. The concentration of PS, TPPH2 contents and weight ratios of THF:DMF mixtures were listed in Table 1. All the chemical reagents were used without further purification. Then the spinning solution was placed in a syringe and was pumped through a metal capillary with an inner diameter of 0.8 mm by a syringe pump (WZL-506, Smiths Medical Instrument Zhejiang Co., Ltd., China). The solution feeding rate was controlled at 1.0 mL h−1. A high voltage power supply (DW-P303-1ACD8, Tianjin Dongwen High Voltage Co., China) was used to generate a potential difference of 20 kV between the capillary and an aluminum foil covered grounded metallic rotating roller rotated at 280 rpm. The work distance (WD) was kept at 20 cm. All the experiments were carried out at 25 °C and the relative humidity (RH) was 50 ± 2%. The spinning time was fixed in 5 h so that the thickness of TPPH2-PS NFMs was 0.11 ± 0.005 mm. After electrospinning the TPPH2-PS NFMs were dried in vacuum at 40 °C for 24 h.
Table 1 Formulas of the TPPH2-PS electrospinning solutions
| Sample |
PS concentration (wt%) |
TPPH2 content (mg mL−1) |
m(THF):m(DMF) |
TPPH2/PS (g g−1) |
| THF-0 |
25 |
2.0 |
0:100 |
0.0063 |
| THF-20 |
25 |
2.0 |
20:80 |
0.0064 |
| THF-40 |
25 |
2.0 |
40:60 |
0.0067 |
| THF-60 |
25 |
2.0 |
60:40 |
0.0066 |
| TPPH2-1.0 |
25 |
1.0 |
40:60 |
0.0033 |
| TPPH2-1.5 |
25 |
1.5 |
40:60 |
0.0050 |
| TPPH2-2.0 |
25 |
2.0 |
40:60 |
0.0067 |
| TPPH2-2.5 |
25 |
2.5 |
40:60 |
0.0083 |
Characterization
The morphology and cross-section of the electrospun TPPH2-PS NFMs was observed by field emission scanning electron microscope (FE-SEM) (S-4800, Hitachi, Japan) at an acceleration voltage of 5 kV and high vacuum mode after gold coating. The average fiber diameter of each sample was measured by image analysis software (Image Pro Plus 6.0). Fluorescent images of the samples were captured on fluorescence microscope (Nikon A1+, Nikon, Japan). The wavelength of excitation was 488 nm. N2 adsorption–desorption data were measured using a specific surface and micropore analyzer (Autosorb-iQ, Quantachrome, USA). The specific surface area of the TPPH2-PS NFM was obtained from the N2 adsorption data in the relative pressure range from 0.05 to 0.3 by using the Brunauer–Emmett–Teller (BET) method. The total pore volume, pore size and its distribution of the fiber membrane were calculated by the Barrett–Joyner–Halenda (BJH) method. The thermogravimetric (TG) analysis was performed using a thermal analyzer (STA 409 PC/PG, Netzsch, Germany), under nitrogen flow, in a temperature range from 20 °C to 800 °C and at a heating rate of 10 °C min−1. UV-vis absorbance spectra of the TPPH2-PS NFMs were measured by UV-Visible spectrophotometer (EVOLUTION 220, Thermo, USA) which is assembly attached to the ISA-220 integrating sphere. The Czerny–Turner monochromator (Thermo, USA) was used and the acquisition time was 2 min. Fluorescence emission spectra were conducted on fluorescence spectrophotometer (F-380, Gangdong Sci., China) with a solid assembly to have excitation and emission at 45° to the membrane surface. Samples were excited at 420 nm and emission was measured at wavelength of 600–760 nm. Water contact angle measurements were performed with contact angle instrument (JYSP-180, Jinshengxin, China) using the static sessile drop method with DROP image advanced software to determine static contact angles.
HCl gas sensing setup
Sensor samples were prepared by cutting the TPPH2-PS NFMs into round shape (the diameter is about 1 cm). HCl gas detection was carried out in a specially designed gas testing chamber comprising two high precision mass flow controllers (red-y, Voegtlin, Germany) which delivered a mixture of HCl gas and pure N2 to obtain required HCl gas concentration. The volume of the gas testing chamber was 1 L. The HCl gas was detected at 40 °C in 50 ± 2% RH. The fluorescence intensity at 645 nm of the sensor (I0 and I) before and after HCl gas exposure were recorded by the fluorescence spectrophotometer. After each exposure, the chamber was flushed with pure nitrogen to recover the TPPH2-PS NFM until the fluorescence intensity at 645 nm of the sensor approximate the original I0. Visible color variation of the sensor when exposed to HCl gas was recorded by a digital camera (IXUS 210, Canon, Japan) in the same condition to ensure the accuracy of these color pictures.
Results and discussion
Characterization of the TPPH2-PS NFM
Fig. 1 shows the FE-SEM images of TPPH2-PS NFMs from TPPH2-PS solutions of various solvent compositions via electrospinning method. Nonporous and bead-on-string structure was obtained from only DMF solvent due to the high boiling point of DMF (153 °C) which was incompletely evaporated before the fibers were collected at the collector (Fig. 1a). With the increase of the low boiling point of THF (66 °C), the amount of bead-on-string structure decreased and disappeared when the ratio of THF was up to 40%. It can be seen that the fiber comprised a grooved sheath and a solid core when a solvent mixture with 20% THF was used (insets of Fig. 1b). With the increasing THF percentage in the solvent mixtures, the nanopores presented on the fiber surfaces, and the porous structure appeared to be throughout the cross-sections of these fibers (insets of Fig. 1c). As the THF content further increased, the 3D interconnected nanopores inside the fibers became more uniform and dense as shown in insets of Fig. 1d. The morphology of the TPPH2-PS NFMs with different TPPH2 content is shown in Fig. S1.† The TPPH2 content has little impact on the fiber preparation and pore formation and the fiber diameter is in range of 800–3000 nm. Fluorescent images of the TPPH2-PS NFM are shown in Fig. 2. Clearly, the TPPH2-PS NFM shows uniform red fluorescence, while the pure PS NFM without TPPH2 doped shows none fluorescence. It confirms that TPPH2 is homogeneously dispersed in the surface and inside of the nanoporous PS fiber membrane.
 |
| | Fig. 1 FE-SEM images of the TPPH2-PS NFMs formed from various weight ratios of THF:DMF in solvent: (a) 0:100, (b) 20:80, (c) 40:60, (d) 60:40, respectively. The inset images were the cross-section of TPPH2-PS fibers. | |
 |
| | Fig. 2 Fluorescent images of the sample THF-40 with different TPPH2 content: (a) 0 mg mL−1, (b) 2.0 mg mL−1 (a1, b1-bright-field image, a2, b2-fluorescent image) (λex = 488 nm). | |
The main reasons for pores formation of the fibers have been proved to be the thermally-induced phase separation and vapor-induced phase separation by previous studies.29,34 During electrospinning, the solvent evaporated from the polymer fluid jets involves the flash vaporization at the fiber surface and the diffusion from core to surface. In the meantime, moisture in the air will penetrate into the fluid jet while the solvent will diffuse out from the core of the jet. This is accompanied by cooling the fluid jets, leading to thermodynamically unstable jets and phase separation into polymer-rich and solvent-rich domains.31 Eventually, the polymer-rich phase solidified into fiber matrix and the solvent-rich phase transformed into nanopores as the solvent diffused out. The difference in the porous structure of these fibers can be attributed to the competition between the rapid phase separation and solidification resulting from the mutual diffusion of the solvent within the jet and the surrounding moisture due to the different solvent compositions of THF and DMF utilized in electrospinning.
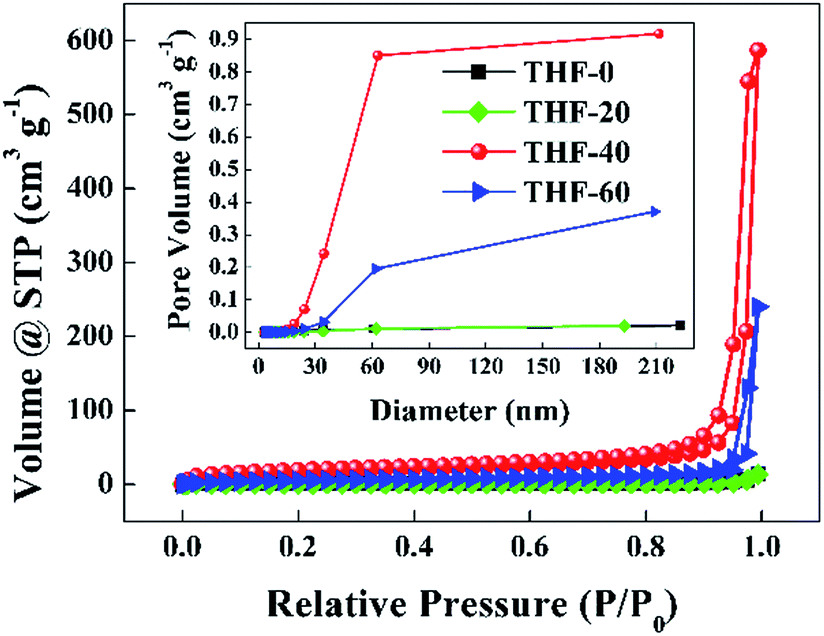
Fig. 3 presents the nitrogen adsorption–desorption isotherms and pore size distribution curves of the TPPH2-PS NFMs formed with various ratios of THF–DMF. The isotherms of THF-0 and THF-20 are in accordance with the type III isotherm which is characteristic of macropores, while the isotherms of THF-40 and THF-60 are akin to a type II isotherm with a hysteresis loop which is the characteristics of mesopores and macropores according to the International Union and Applied Chemistry (IUPAC) classification.34,35 The inset of Fig. 3 reveals that the pore size is in the range of 2–60 nm. Such porous structures are expected to contribute to higher specific surface area of these TPPH2-PS NFMs. The specific surface area and pore volume derived from BET measurements as well as average fiber diameter of the TPPH2-PS NFMs formed with various ratios of THF–DMF are show in Table 2. It is obvious that the varied solvent compositions exhibit the significant effects. THF-40 has the highest specific surface area of 58.63 m2 g−1, the biggest pore volume of 0.851 cm3 g−1 and the smallest average fiber diameter of 1609 nm. As the low boiling point of THF content increased, the average fiber diameter of THF-60 increased while the its pore volume decreased due to the faster speed on solidification of the electrospinning jet, result to lower specific surface area. THF-0 and THF-20 have the lowest specific surface area and pore volume because of their poor porous structure in spite of their fiber diameters are small. In addition, the weight loss of the TPPH2-PS NFM happens at approximately 400 °C, which confirm its good thermostability used as HCl gas sensor (Fig. S2†).
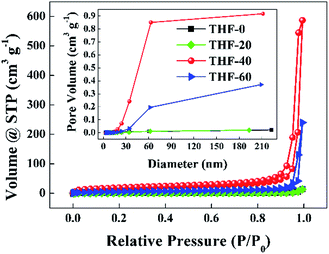 |
| | Fig. 3 Nitrogen adsorption–desorption isotherms and pore size distribution curves (inset) of the TPPH2-PS NFMs formed with various ratios of THF–DMF. | |
Table 2 The specific surface area, pore volume and average fiber diameter of the TPPH2-PS NFMs formed with various ratios of THF–DMF
| Sample |
Specific surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Average fiber diameter (nm) |
| THF-0 |
2.52 |
0.009 |
951 |
| THF-20 |
2.27 |
0.008 |
1933 |
| THF-40 |
58.63 |
0.851 |
1609 |
| THF-60 |
17.81 |
0.195 |
2807 |
HCl gas sensing properties of the TPPH2-PS NFM
The detection of HCl gas was carried out to study the sensing properties of the as-prepared TPPH2-PS NFM sensor. HCl gas of certain concentration was introduced to the test chamber where the sensor was placed. The sensor samples were prepared in the same size, which the diameter is about 1 cm and the thickness is about 0.11 mm. The HCl gas exposure test was carried out at 40 °C in 50 ± 2% RH. As shown in Fig. 4, the sensor presents a visible color variation from pink to yellow green with a concentration of HCl gas up to 1000 ppm. When repeatedly flushing the protonated TPPH2-PS NFM sensor with pure N2 three times for 1 min, the color of the sensor is completely reverted back to its original state. This vivid color variation can be ascribed to the reversible protonation reaction between H+ of HCl and the central nitrogen of the TPPH2 molecule.17,36 Initially, TPPH2 molecule has a planar structure with all phenyl group perpendicular to the plane of the porphyrin ring. After protonated with HCl gas, the planarity of the porphyrin ring is destroyed and change to saddle conformation resulting to the obvious color variation of the sensor.10,37 Considering the threshold for tolerably safe exposure to HCl gas (10 ppm) and the exposure limit in work place (5 ppm), the as-prepared TPPH2-PS NFM sensor could be used to briefly evaluate the concentration of HCl gas ranging from 1 ppm to 60 ppm without resorting to the use of expensive instruments which is necessary for developing low-cost HCl gas sensing systems.
 |
| | Fig. 4 Color pictures of the sample THF-40 exposed to different concentrations of HCl gas (CHCl) for 30 s at 40 °C in 50 ± 2% RH and refreshed by N2. | |
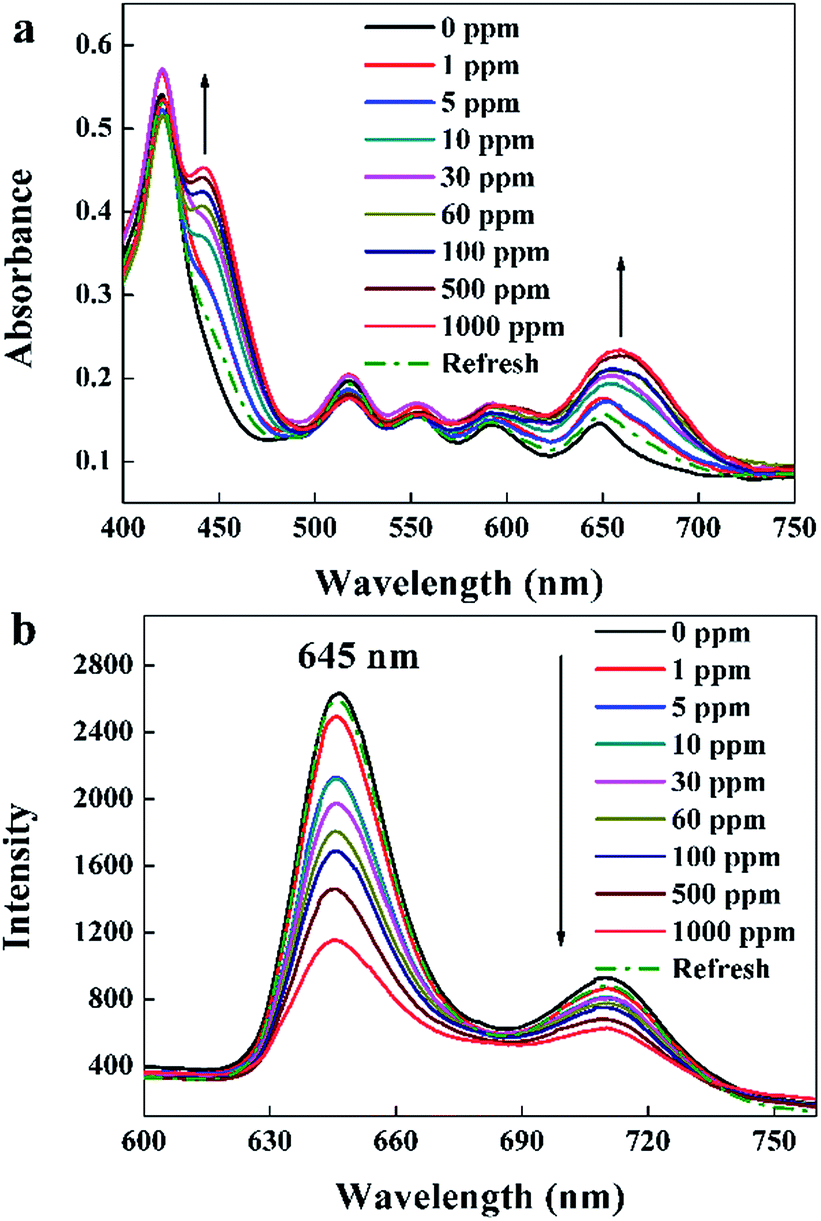
UV-visible absorption spectra of the TPPH2-PS NFM exposed to 0–1000 ppm HCl gas are shown in Fig. 5a. It displays a distinctive Soret band around 420 nm and four weaker Q bands at 518, 552, 593 and 650 nm before HCl gas exposure. The Soret band features a gradual bathochromic shift of 22 nm, from 420 to 442 nm upon the addition of HCl gas exposed to the TPPH2-PS NFM, which is consistent with the research result of Y.-Y. Lv et al.24 Also the Q band at 650 nm becomes stronger as the concentration of HCl gas increases. These spectral changes are attributed to the protonation reaction between H+ and the central nitrogen of the TPPH2, which causes the charge-transfer complex between H+ and TPPH2, and hence the visible color variation of the TPPH2-PS NFM.38 As shown in Fig. 5b, fluorescence quenching is also obvious and the intensities of emission spectra at 645 nm and 710 nm decrease with the increased concentration of HCl gas which is another proof of the protonation on TPPH2. When the sensor is refreshed by N2, the absorption and emission spectra of the TPPH2-PS NFM return to nearly the original states shown by the dash line of Fig. 5 which confirms the deprotonation of the TPPH2 resulting the color recovery of the sensor.
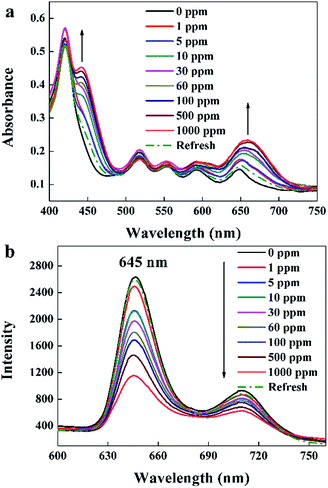 |
| | Fig. 5 UV-visible absorption spectra (a) and fluorescence spectra (b, λex = 420 nm) of the sample THF-40 exposure to different concentrations of HCl gas for 30 s at 40 °C in 50 ± 2% RH and refreshed by N2. | |
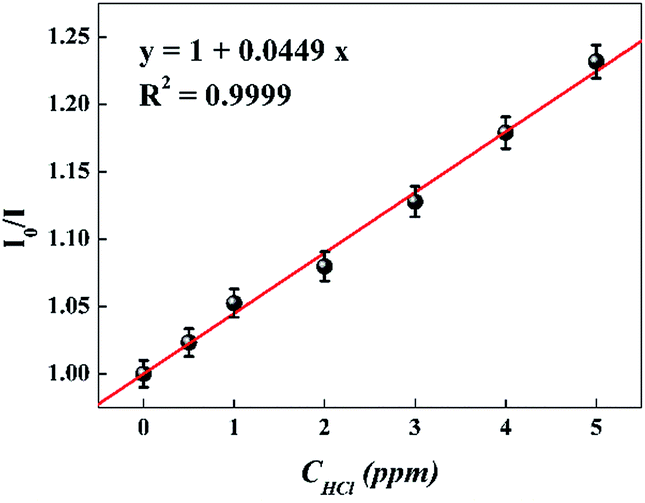
For further study of the sensing response of TPPH2-PS NFM sensor, fluorescence spectra of the sensor exposed to HCl gas with concentration of 0–3000 ppm were measured. The sensing response were calculated from specific value of the intensity at 645 nm (I0/I). Fig. S3† shows the sensing response of the sample THF-40 exposure to different concentration of HCl gas. The slope of the curve presents a linear increase at lower HCl concentration range in 0–5 ppm. At higher concentrations, the concentration dependence conform to non-linear due to the saturation effects.39 As shown in Fig. 6, a linear fitting is used to fit the value of I0/I at 645 nm against the concentration of HCl gas ranging from 0 ppm to 5 ppm, which can be described by the Stern–Volmer eqn (1).40
| |
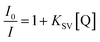 | (1) |
where
I0 and
I represented the fluorescence intensity at 645 nm of the sensor before and after exposure to HCl gas for 30 s at 40 °C, respectively. [Q] is the concentration of HCl gas and
KSV is the Stern–Volmer quenching constant. The detection limit (DL) of the sensor was computed as the
eqn (2).
39,41| |
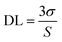 | (2) |
where
σ is the standard deviation of the blank signal (
n = 6) and
S is the slope of the linear calibration plot. The DL of the TPPH
2-PS NFM sensor toward HCl gas was found to be 46 ppb.
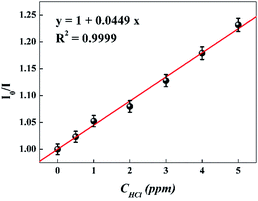 |
| | Fig. 6 Stern–Volmer plot of the sensor. I0 and I represented the fluorescence intensity of the sensor before and after exposure to HCl gas, respectively. (λex = 420 nm, λem = 645 nm). | |
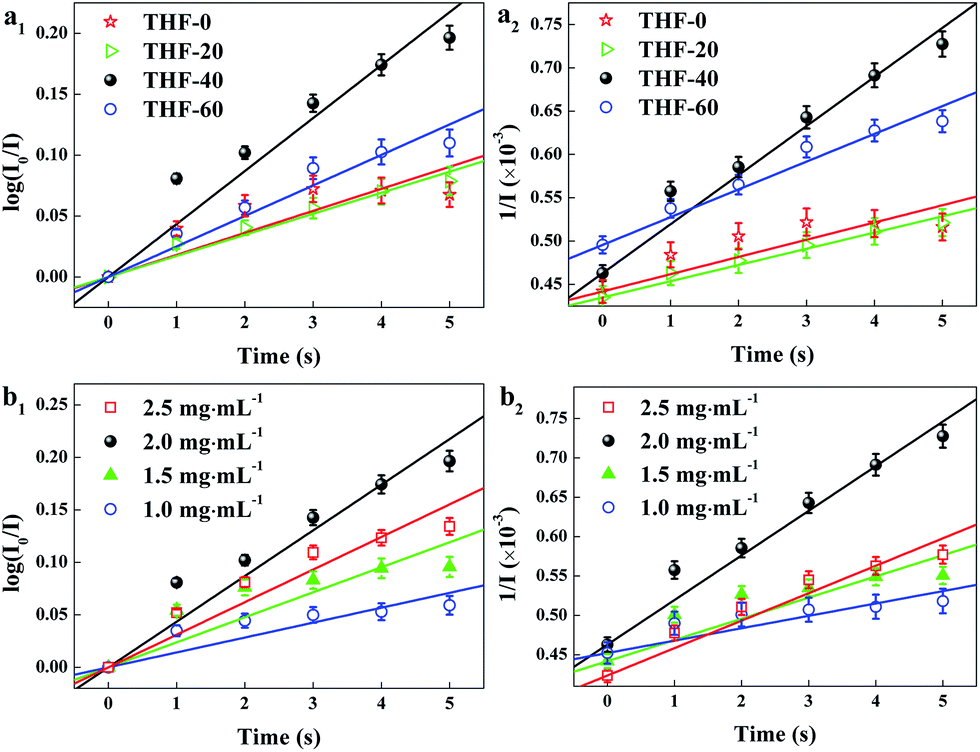
Fast response is a significant requirement to be considered for gas sensor. Fig. S4† shows the exposure time dependence of I0/I at 645 nm after the sensor exposed to 100 ppm HCl gas. The slope of all the curves presents a linear increase at the initial region and turns to equilibrium after 5 s. However, the response time of compact films, such as composite film42 and LS film,38 based gas sensors is in several minutes which indicates that the structure of these sensing films is the major factor influencing the response time of the gas sensor. Kinetic data were fitted based on the linear region of the curves shown in Fig. S4† using kinetic models to assess the HCl gas adsorption of the TPPH2-PS NFM with different structure and TPPH2 content. According to eqn (1), the linear equation of pseudo-first-order and pseudo-second-order kinetics can be deformed to eqn (3) and (4).43–46
| |
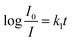 | (3) |
| |
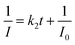 | (4) |
where
I0 and
I represented the fluorescence intensity at 645 nm of the sensor before and after exposure to 100 ppm HCl gas for
t s at 40 °C, respectively.
k1 and
k2 are the pseudo-first-order and pseudo-second-order rate constant. As illustrated in
Table 2, the porous structure of the TPPH
2-PS NFM is relevant to the solvent compositions. Therefore, kinetic fitting of the TPPH
2-PS NFMs with various ratios of THF–DMF was shown in
Fig. 7a to investigate the effect of porous structure on HCl gas adsorption. The kinetic parameters is shown in
Table 3.
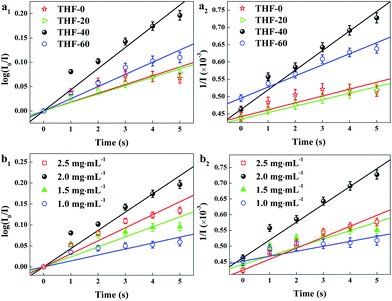 |
| | Fig. 7 Kinetic fitting of the TPPH2-PS NFMs with various ratios of THF–DMF (a) and different TPPH2 content (b) for the adsorption of 100 ppm HCl gas for 0–5 s. (a1, b1) was pseudo-first-order and (a2, b2) was pseudo-second-order kinetic fitting. | |
Table 3 Kinetic parameters of the effect of porous structure and TPPH2 content on HCl gas adsorption
| Sample |
Pseudo-first-order kinetics |
Pseudo-second-order kinetics |
| k1 (×10−2) |
R12 |
k2 (×10−5) |
R22 |
| THF-0 |
1.81 |
0.890 |
2.00 |
0.998 |
| THF-20 |
1.73 |
0.985 |
1.87 |
0.999 |
| THF-40 |
4.35 |
0.975 |
5.67 |
0.999 |
| THF-60 |
2.51 |
0.980 |
3.21 |
0.999 |
| TPPH2-1.0 |
1.42 |
0.911 |
1.57 |
0.999 |
| TPPH2-1.5 |
2.38 |
0.913 |
2.69 |
0.998 |
| TPPH2-2.0 |
4.35 |
0.975 |
5.67 |
0.999 |
| TPPH2-2.5 |
3.11 |
0.967 |
3.49 |
0.999 |
It is clearly indicate that the pseudo-second-order model fit the kinetic data well, with R2 over 0.99. The sample of THF-40 has the maximum k2 of 5.67 × 10−5 which confirm that high specific surface area and pore volume is suitable for gas adsorption and diffusion. Fig. 7b shows kinetic fitting of the TPPH2-PS NFMs with different TPPH2 content and the kinetic parameters of the effect of TPPH2 content on HCl gas adsorption is also shown in Table 3. With the increase of the TPPH2 content, the value of k2 increases and reaches the peak with TPPH2 content up to 2.0 mg mL−1. As the TPPH2 content further increased, the value of k2 decreases because a large proportion of the porphyrin results in some porphyrin crystals.42
Reversibility, photostability and humidity insensitivity of the sensor
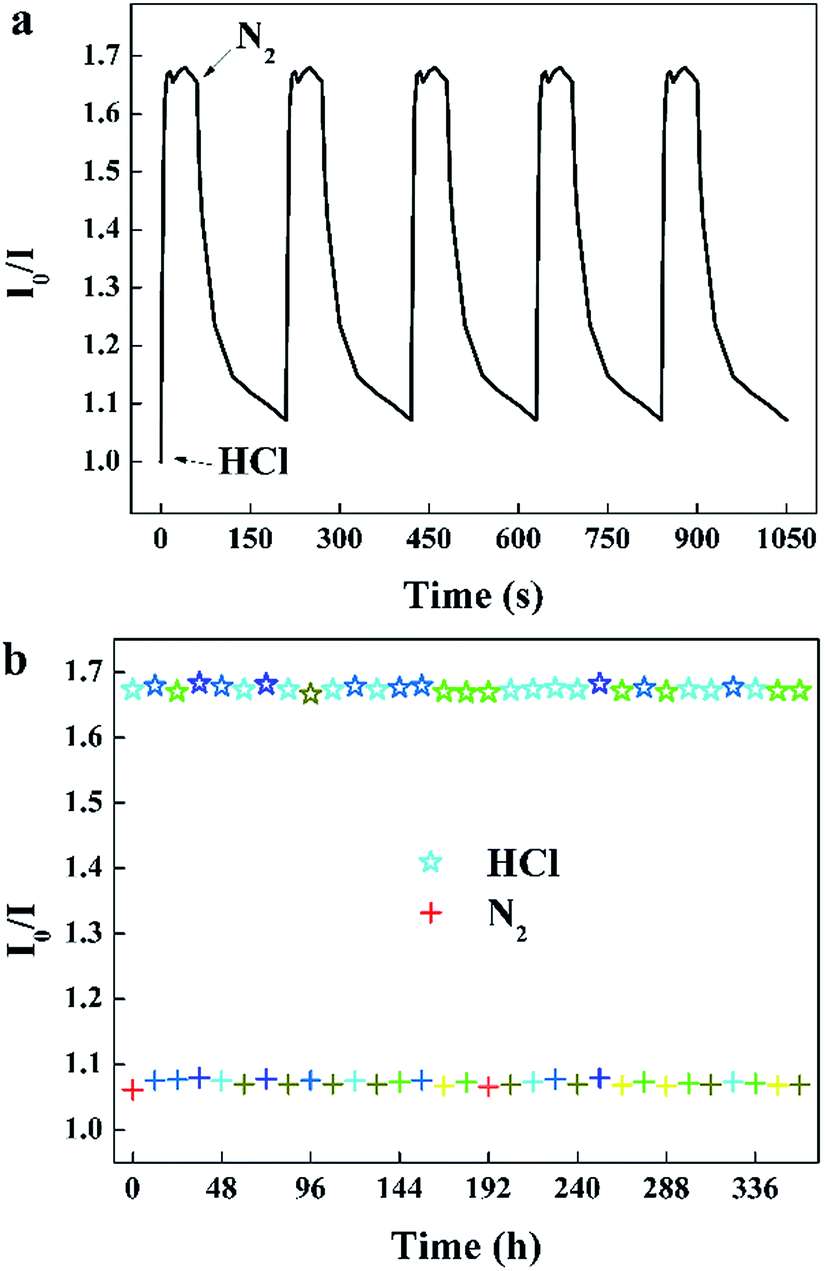
When the sensor is continuously monitored in rapidly alternated gaseous atmospheres, the specific value of I0/I at 645 nm rise and recover with the environment switched between 100 ppm HCl gas and pure N2 (Fig. 8a). The sensor exhibits excellent reversibility and prompt response and recovery. The response time taken as the time required to reach 90% of the steady-state value is approximately 5 s, and the recovery time taken as the time to recover the sensor to 10% of the span signal is approximately 90 s. The photostability of this sensor was measured by recording I0/I at 645 nm per 12 h exposed to 100 ppm HCl gas and N2.
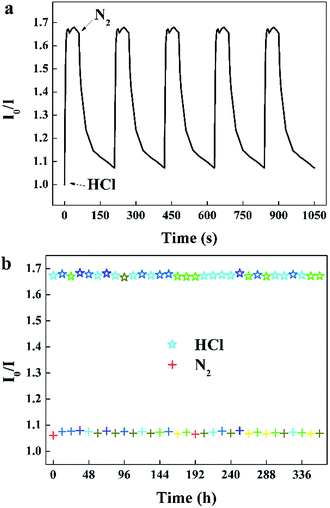 |
| | Fig. 8 (a) Reversibility of the sensor to alternating 100 ppm HCl gas and pure N2, (b) photostability of the sensor exposed to 100 ppm HCl gas and N2 for 30 s per 12 hours (λem = 645 nm). | |
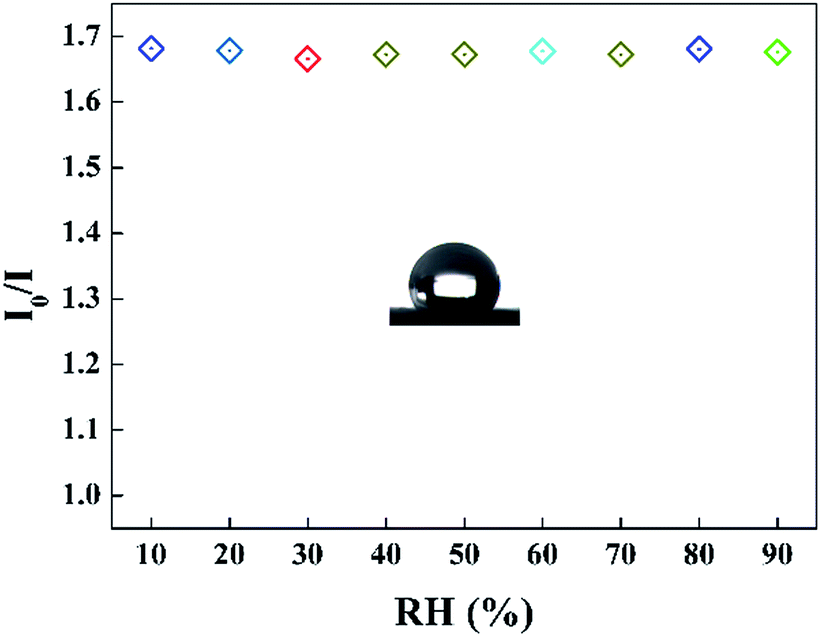
It can be seen that the sensing response of the sensor remains unchanged and the TPPH2-PLA NFM shows nearly no photobleaching (Fig. 8b). In this way, the sensor can be reused many times which is important in developing low-cost HCl gas sensing systems. As shown in Fig. 9, the RH conditions have nearly no effect on sensing response of the sensor exposed to 100 ppm HCl gas in 5 s due to the hydrophobicity of the TPPH2-PS NFM. The inset figure on the water contact angle of the TPPH2-PLA NFM in Fig. 9 confirms its excellent hydrophobicity.
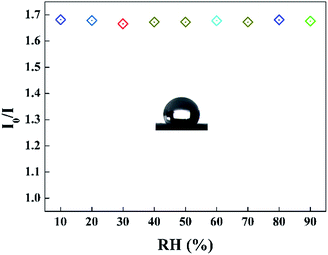 |
| | Fig. 9 Sensing response of the sensor in different RH conditions (λem = 645 nm) and the inset image presents the water contact angle of the TPPH2-PS NFM. | |
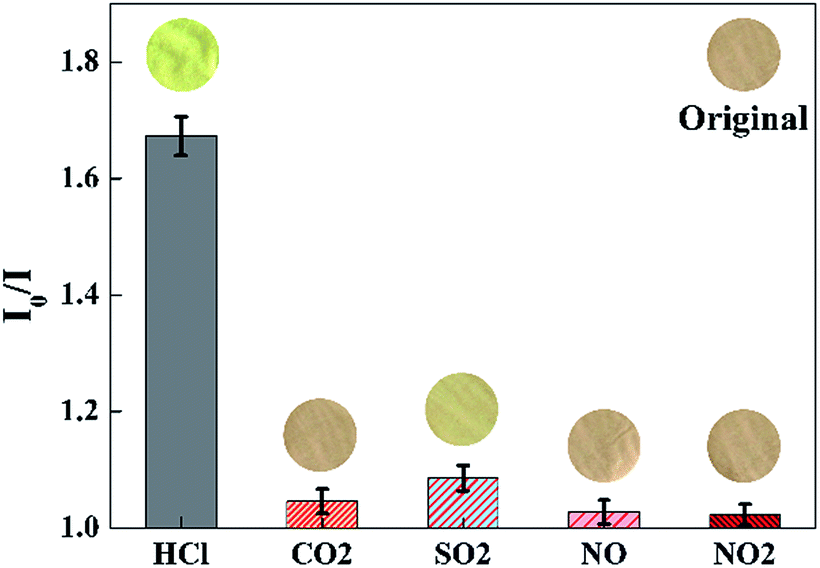
Selectivity of the sensor
To evaluate the selectivity of the proposed sensor, the sensing response (I0/I at 645 nm) is measured in the presence of various acid gases with the concentration of 100 ppm, such as gaseous CO2, SO2, NO, NO2, etc. As shown in Fig. 10, a significant increase of I0/I at 645 nm could be observed when the sensor was exposed to HCl gas. By contrast, the value of I0/I at 645 nm presented little increase when the sensor exposure to CO2, SO2, NO, NO2, respectively. As shown in the inset images of Fig. 10, the color of the sensor changed from pink to yellow green exposure to HCl gas, while remained unchanged exposure to other gaseous analytes of the test.
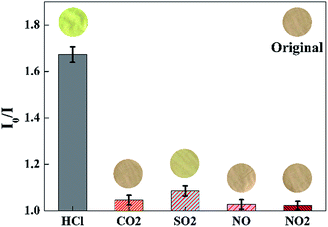 |
| | Fig. 10 Selectivity of the sensor exposed to other acid gases with the concentration of 100 ppm for 30 s at 40 °C in 50 ± 2% RH. Inset images were the color pictures of the sensor at each gaseous analytes. (λem = 645 nm). | |
Comparison with other HCl gas sensors based on porphyrin
The comparison of some reported porphyrin based sensors for detection of HCl gas was show in Table 4. Compared to other conventional optical sensors of HCl gas based on porphyrin, this TPPH2-PS NFM sensor exhibits high sensitivity and fast response due to the large specific surface area, controllable pore sizes, well-defined porosity of the three dimensional electrospun nanoporous fiber membrane and hence good accessibility for HCl gas. The sensor also presents good visual color change when exposure to HCl gas, which may be used for low-cost HCl sensor.
Table 4 Comparison with other optical sensors for detection of HCl gas based on porphyrin
| Material |
Method |
DL (ppb) |
Response time (s) |
Ref. |
| TPPH2-PS NFM |
Fluorescence/colorimetric |
46 |
5 |
This method |
| TMPyP/TiO2 composite thin film |
Absorbance |
100 |
16.8 ± 0.7 |
47 |
| Porphyrinated PI nanofibrous membrane |
Fluorescence/colorimetric |
5000 |
10 |
24 |
| MTMP deposited glass plate |
Absorbance/fluorescence |
30 |
>180 |
37 |
| TP(OH)4PH2 dispersed MTEOS/TEOS sol–gel film |
Reflectance |
500 |
— |
18 |
| Porphyrinated PI honeycomb film |
Fluorescence |
— |
10 |
23 |
Conclusions
A fluorescent and colorimetric HCl gas sensor based on TPPH2-PS NFM was facilely fabricated by electrospinning for the naked-eye detection of HCl gas. The sensor presented apparent color change from pink to yellow green when exposed to ppm-level of HCl gas at 40 °C. The sample of THF-40 showed very high sensitivity and the maximum pseudo-second-order rate constant of 5.67 × 10−5 due to its largest specific surface area and pore volume. The detection limit was found to be 46 ppb and the response time was 5 s. The sensor also showed excellent reversibility and prompt response and recovery. As this colorimetric sensing approach is simple, low-coast, sensitive and prompt, the sensor foresees a high potential for the development of visual HCl gas sensor working in smart textiles.
Acknowledgements
This work was supported by National Natural Science Foundation of China (51102178), National Key Technology Support Program (2015BAE01B03) and Innovation Fund for Technology of Tianjin (14ZXCXGX00776).
References
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- G. De Luca, G. Pollicino, A. Romeo and L. M. Scolaro, Chem. Mater., 2006, 18, 2005–2007 CrossRef CAS.
- M. K. LaGasse, J. M. Rankin, J. R. Askim and K. S. Suslick, Sens. Actuators, B, 2014, 197, 116–122 CrossRef CAS.
- N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710–713 CrossRef CAS PubMed.
- J. Sun, B. Ye, G. Xia, X. Zhao and H. Wang, Sens. Actuators, B, 2016, 233, 76–82 CrossRef CAS.
- P. Kalimuthu, A. Sivanesan and S. A. John, J. Chem. Sci., 2012, 124, 1315–1325 CrossRef CAS.
- P. Muthukumar and S. A. John, Sens. Actuators, B, 2011, 159, 238–244 CrossRef CAS.
- Y. Itagaki, S. Yamanaka and Y. Sadaoka, Sens. Lett., 2011, 9, 114–117 CrossRef CAS.
- K. Nakagawa, K. Kumon, C. Tsutsumi, K. Tabuchi, T. Kitagawa and Y. Sadaoka, Sens. Actuators, B, 2000, 65, 138–140 CrossRef CAS.
- K. Nakagawa, Y. Sadaoka, H. Supriyatno, A. Kubo, C. Tsutsumi and K. Tabuchi, Sens. Actuators, B, 2001, 76, 42–46 CrossRef CAS.
- X. C. Sun, C. Bruckner, M. P. Nieh and Y. Lei, J. Mater. Chem. A, 2014, 2, 14613–14621 CAS.
- B. P. Regmi, W. I. S. Galpothdeniya, N. Siraj, M. H. Webb, N. C. Speller and I. M. Warner, Sens. Actuators, B, 2015, 209, 172–179 CrossRef CAS.
- C. Wolf, M. Tscherner and S. Kostler, Sens. Actuators, B, 2015, 209, 1064–1069 CrossRef CAS.
- L. Maqueira, A. C. Valdés, A. Iribarren and C. P. de Melo, J. Porphyrins Phthalocyanines, 2013, 17, 283–288 CrossRef CAS.
- P. Castillero, J. R. Sanchez-Valencia, M. Cano, J. M. Pedrosa, J. Roales, A. Barranco and A. R. Gonzalez-Elipe, ACS Appl. Mater. Interfaces, 2010, 2, 712–721 CAS.
- A. D. F. Dunbar, T. H. Richardson, J. Hutchinson and C. A. Hunter, Sens. Actuators, B, 2008, 128, 468–481 CrossRef CAS.
- Y. Itagaki, K. Deki, S.-I. Nakashima and Y. Sadaoka, Sens. Actuators, B, 2005, 108, 393–397 CrossRef CAS.
- Y. Itagaki, K. Deki, S.-I. Nakashima and Y. Sadaoka, Sens. Actuators, B, 2006, 117, 302–307 CrossRef CAS.
- J. Spadavecchia, G. Ciccarella, P. Siciliano, S. Capone and R. Rella, Sens. Actuators, B, 2004, 100, 88–93 CrossRef CAS.
- S. Jiang and M. Liu, Chem. Mater., 2004, 16, 3985–3987 CrossRef CAS.
- P. Muthukumar and S. A. John, Sens. Actuators, B, 2012, 174, 74–80 CrossRef CAS.
- K. Nakagawa, T. Aono, G. Ueda, C. Tsutsumi, N. Hayase, M. Mabuchi and Y. Sadaoka, Sens. Actuators, B, 2005, 108, 542–546 CrossRef CAS.
- F. W. Lin, X. L. Xu, L. S. Wan, J. Wu and Z. K. Xu, RSC Adv., 2015, 5, 30472–30477 RSC.
- Y.-Y. Lv, J. Wu and Z.-K. Xu, Sens. Actuators, B, 2010, 148, 233–239 CrossRef CAS.
- J. Lin, Y. Shang, B. Ding, J. Yang, J. Yu and S. S. Al-Deyab, Mar. Pollut. Bull., 2012, 64, 347–352 CrossRef CAS PubMed.
- M. Bognitzki, W. Czado, T. Frese, A. Schaper, M. Hellwig, M. Steinhart, A. Greiner and J. H. Wendorff, Adv. Mater., 2001, 13, 70–72 CrossRef CAS.
- R. Gentsch, B. Boysen, A. Lankenau and H. G. Borner, Macromol. Rapid Commun., 2010, 31, 59–64 CrossRef CAS PubMed.
- H. Fashandi and M. Karimi, Polymer, 2012, 53, 5832–5849 CrossRef CAS.
- C. L. Casper, J. S. Stephens, N. G. Tassi, D. B. Chase and J. F. Rabolt, Macromolecules, 2004, 37, 573–578 CrossRef CAS.
- S. Lu, W. Xu, Y. Chen, Y. Jiang, Q. Yao, F. Luo, Y. Wang and X. Chen, Sens. Actuators, B, 2016, 232, 585–594 CrossRef CAS.
- J. Y. Lin, B. Ding, J. M. Yang, J. Y. Yu and G. Sun, Nanoscale, 2012, 4, 176–182 RSC.
- J. Lin, F. Tian, Y. Shang, F. Wang, B. Ding, J. Yu and Z. Guo, Nanoscale, 2013, 5, 2745–2755 RSC.
- J. Huang, H. Deng, D. Song and H. Xu, Anal. Chim. Acta, 2015, 878, 102–108 CrossRef CAS PubMed.
- J. Lin, B. Ding, J. Yu and Y. Hsieh, ACS Appl. Mater. Interfaces, 2010, 2, 521–528 CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- M. Hu, W. Kang, B. Cheng, Z. Li, Y. Zhao and L. Li, Microchim. Acta, 2016, 183, 1713–1720 CrossRef CAS.
- P. Kalimuthu and S. Abraham John, Anal. Chim. Acta, 2008, 627, 247–253 CrossRef CAS PubMed.
- M. Evyapan and A. D. F. Dunbar, Sens. Actuators, B, 2015, 206, 74–83 CrossRef CAS.
- T. Balaji, M. Sasidharan and H. Matsunaga, Analyst, 2005, 130, 1162–1167 RSC.
- T. W. Sung and Y. L. Lo, Sens. Actuators, B, 2013, 188, 702–708 CrossRef CAS.
- T. H. Richardson, C. M. Dooling, L. T. Jones and R. A. Brook, Adv. Colloid Interface Sci., 2005, 116, 81–96 CrossRef CAS PubMed.
- K. Nakagawa, K. Tanaka, T. Kitagawa and Y. Sadaoka, J. Mater. Chem., 1998, 8, 1199–1204 RSC.
- G. R. Fonda, J. Opt. Soc. Am., 1936, 26, 316–322 CrossRef CAS.
- S. Wei, J. Liu, S. Zhang, X. Chen, Q. Liu, L. Zhu, L. Guo and X. Liu, Hydrometallurgy, 2016, 164, 219–227 CrossRef CAS.
- L. J. Rather, I. Shahid ul, M. A. Khan and F. Mohammad, J. Environ. Chem. Eng., 2016, 4, 1780–1796 CrossRef CAS.
- W. Kang, X. Ma, M. Hu, Z. Jia, H. Liu and B. Cheng, Anal. Methods, 2016, 8, 6489–6493 RSC.
- M. Cano, P. Castillero, J. Roales, J. M. Pedrosa, S. Brittle, T. Richardson, A. R. González-Elipe and A. Barranco, Sens. Actuators, B, 2010, 150, 764–769 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FESEM images of the TPPH2-PS NFMs with different TPPH2 content and the thermostability of the TPPH2-PS NFM. See DOI: 10.1039/c7ra02040f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Weimin Kang*ab,
Yixia Zhaob,
Jie Shia and
Bowen Cheng
a,
Weimin Kang*ab,
Yixia Zhaob,
Jie Shia and
Bowen Cheng