
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper(II)-mediated formation of oxazole-4-carbonitrile from acetophenone and coordinated cyanide anion via a radical coupling†
Congjun Xu‡
,
Mingze Qin‡,
Jun Yi,
Yanjing Wang,
Yanfeng Chen,
Bingfu Zhang,
Yanfang Zhao* and
Ping Gong *
*
Key Laboratory of Structure-based Drug Design and Discovery, Shenyang Pharmaceutical University, Ministry of Education, 103 Wenhua Road, Shenhe District, Shenyang 110016, People's Republic of China. E-mail: yanfangzhao@126.com; gongpinggp@126.com
First published on 5th May 2017
Abstract
A protocol for the direct synthesis of 5-aryloxazole-4-carbonitrile from acetophenone was first described with potassium ferricyanide as a cheap and low toxicity cyanide reagent, in which, multiple bond formation was implemented via an oxygen mediated radical mechanism. Potassium ferricyanide played a dual role as a “CN” source and also as a coupling partner for the cyclization of oxazole.
Oxazole derivatives, one of the most important heterocyclic compounds, are widely present in natural products,1 synthesized drugs2 and advanced materials.3 Meanwhile, a significant proportion of drugs used in clinical treatment contain a cyano group.4 In addition, the introduction of a cyano group to small-molecule lead compounds is one of the most critical strategies in drug development because the cyano group can be easily converted into amide, carboxylic acid, imido ester, amidine, and methyl ammonia groups.5,6 Thus, the introduction of a cyano group to an oxazole ring is an appealing idea.
Over the years, many strategies for the preparation of cyano-oxazoles have been developed, which includes the dehydration of carboxamides to nitriles,7–9 nucleophilic substitution on oxazoles10 and intramolecular condensation of β-acyloxyenamines (Scheme 1).11 These methods suffered from harsh reaction conditions, toxic reagents or unavailable starting materials. Herein, exploring the potential of non-toxic and efficient systems is imperative, which might provide a smooth approach to construct carbonitrile from ubiquitously present substrates.
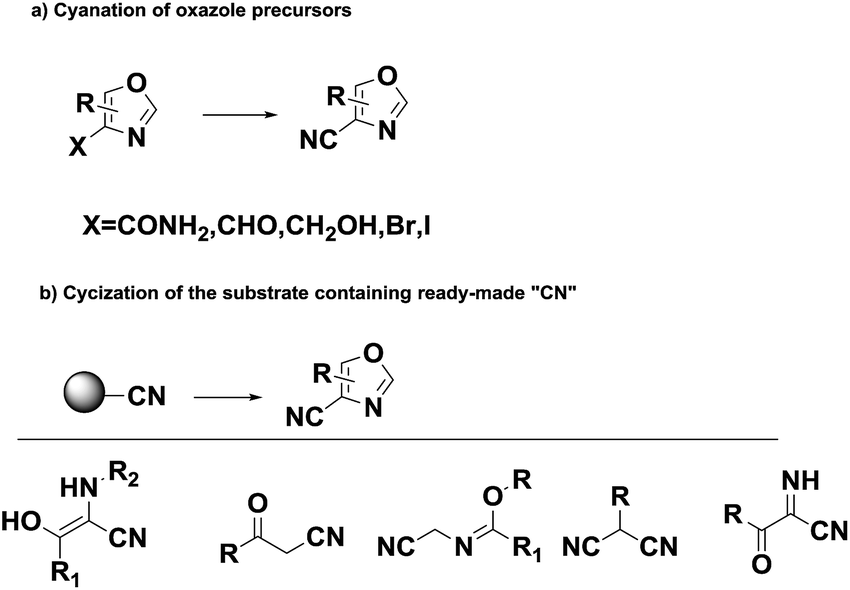 | ||
| Scheme 1 Synthesis of oxazole-4-carbonitrile. | ||
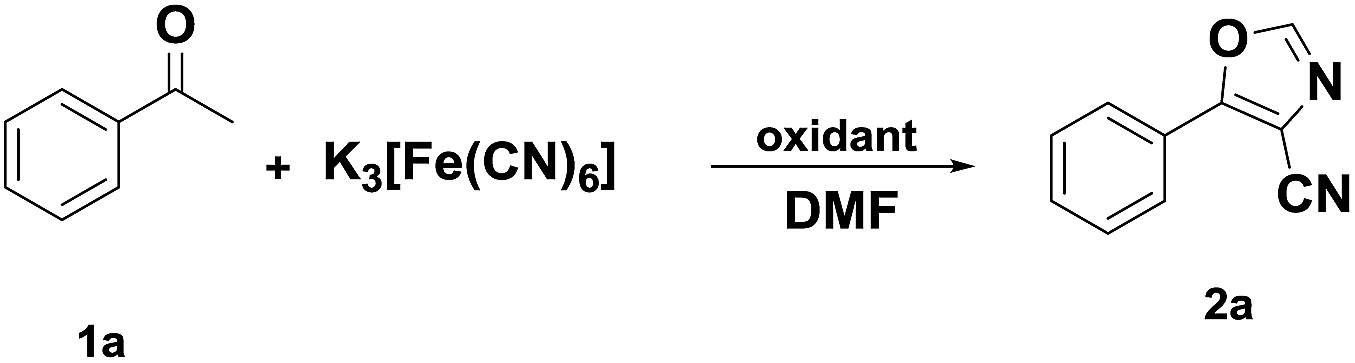
Initially, the authors tried to develop an efficient cyano substitution reaction via C–H bond activation12 with acetophenone as the substrate using nontoxic cyanide sources, such as potassium ferricyanide and potassium ferrocyanide (Scheme 2). Quite incidentally, 5-phenyloxazole-4-carbonitrile was obtained dramatically in the presence of CuI2 and Pd(OAc)2 with DMF as solvent (Scheme 2). The surprising results aroused our interesting and the reaction was investigated further. Subsequently, we found that 5-phenyloxazole-4-carbonitrile was obtained smoothly in the absence of Pd(AcO)2.
 | ||
| Scheme 2 A surprising result. | ||
To gain a better understanding of this attractive result, we systematically studied the function of oxidants and cyanide (Table 1). At the beginning of the investigation, the reaction of acetophenone (1a), DMF, and potassium ferricyanide was examined in the presence of CuI2 (1.0 equiv.) at 130 °C for 12 h. The expected 5-phenyloxazole-4-carbonitrile (2a) was obtained in 29% yield with the formation of side product N,N-dimethyl-2-oxo-2-phenylacetamide13 in 19% yield, and 40% of raw material was recovered (Table 1, entry 1). The reaction did not occur in the absence of CuI2 or K3[Fe(CN)6] (entries 2 and 3). The other oxidants such as CuI, FeBr3 and ZnBr2 were investigated (entries 4–10). The yield of 2a was up to 75% when CuBr2 was used as the oxidant (entry 10). It was found out that the effects of Cu(II) salts and Fe(III) salts was superior to Cu(I) salts in the system. Organic oxidants, such as NBS, were tested to enhance the conversion of this reaction. However, only inferior conversion was observed (entries 12). The use of KCN, CuCN or K4[Fe(CN)6] instead of K3[Fe(CN)6] also led to the formation (entry 15–17), but only using KCN showed good result (entry 16). From the safety and yield point of view, K3[Fe(CN)6] was chosen as the cyanide source. Interestingly, in an exclusive nitrogen atmosphere, the transformation was shut down (entry 18), which indicated that O2 play a crucial role in the reaction. In addition, when the reaction was performed under an O2 atmosphere, the result was the same as that under an air atmosphere (entry 19).
|
|||
|---|---|---|---|
| Entry | Oxidant | Cyanide | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), K3[Fe(CN)6] (0.4 mmol) or KCN (2.0 mmol) or CuCN (2.0 mmol), DMF (3 mL), oxidant (1.0 mmol), at 130 °C.b Isolated yield.c Nitrogen atmosphere. NR: no desired product was detected.d Oxygen balloon. | |||
| 1 | CuI2 | K3Fe(CN)6 | 29 |
| 2 | — | K3Fe(CN)6 | NR |
| 3 | CuI2 | — | NR |
| 4 | CuCl | K3Fe(CN)6 | NR |
| 5 | CuI | K3Fe(CN)6 | Trace |
| 6 | FeBr3 | K3Fe(CN)6 | 40 |
| 7 | ZnBr2 | K3Fe(CN)6 | NR |
| 8 | CuCl2 | K3Fe(CN)6 | 50 |
| 9 | CuBr | K3Fe(CN)6 | NR |
| 10 | CuBr2 | K3Fe(CN)6 | 75 |
| 11 | CuO/I2 | K3Fe(CN)6 | 26 |
| 12 | NBS | K3Fe(CN)6 | Trace |
| 15 | CuBr2 | CuCN | 39 |
| 16 | CuBr2 | KCN | 70 |
| 17 | CuBr2 | K4Fe(CN)6 | 43 |
| 18c | CuBr2 | K3Fe(CN)6 | NR |
| 19d | CuBr2 | K3Fe(CN)6 | 71 |
Next, we turned our attention to the effect of solvent on the reaction. It turned out that 5-phenyloxazole-4-carbonitrile was not detected when DMF was replaced with other solvents, such as acetonitrile, dimethyl sulfoxide, N-methyl-2-pyrrolidone or ethylene glycol, which indicated that the DMF is indispensable in the reaction. Considering that DMF has the capability to participate in the reaction,14 a control experiment with DMF-d7 as the solvent was carried out (Scheme 3). 5-Phenyloxazole-4-carbonitrile-2-d was not detected, which means that DMF participate in this reaction only as an excellent solvent involving coordinated cyanide anion.12a,15 Meanwhile, it turned out that the “CN” in K3[Fe(CN)6] also participated in the cyclization rather than DMF.
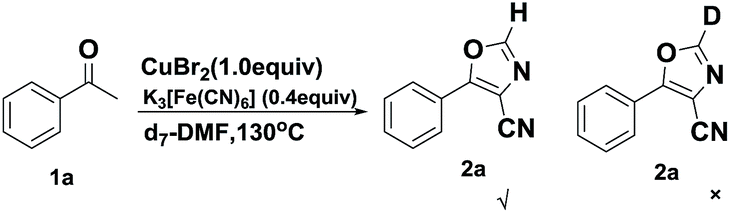 | ||
| Scheme 3 Control experiments for investigating the role of DMF. | ||
With the optimized conditions in hand (Table 1, entry 10), the scope of substrates for the reaction was explored. The structures and yields of the products are summarized in Scheme 4.
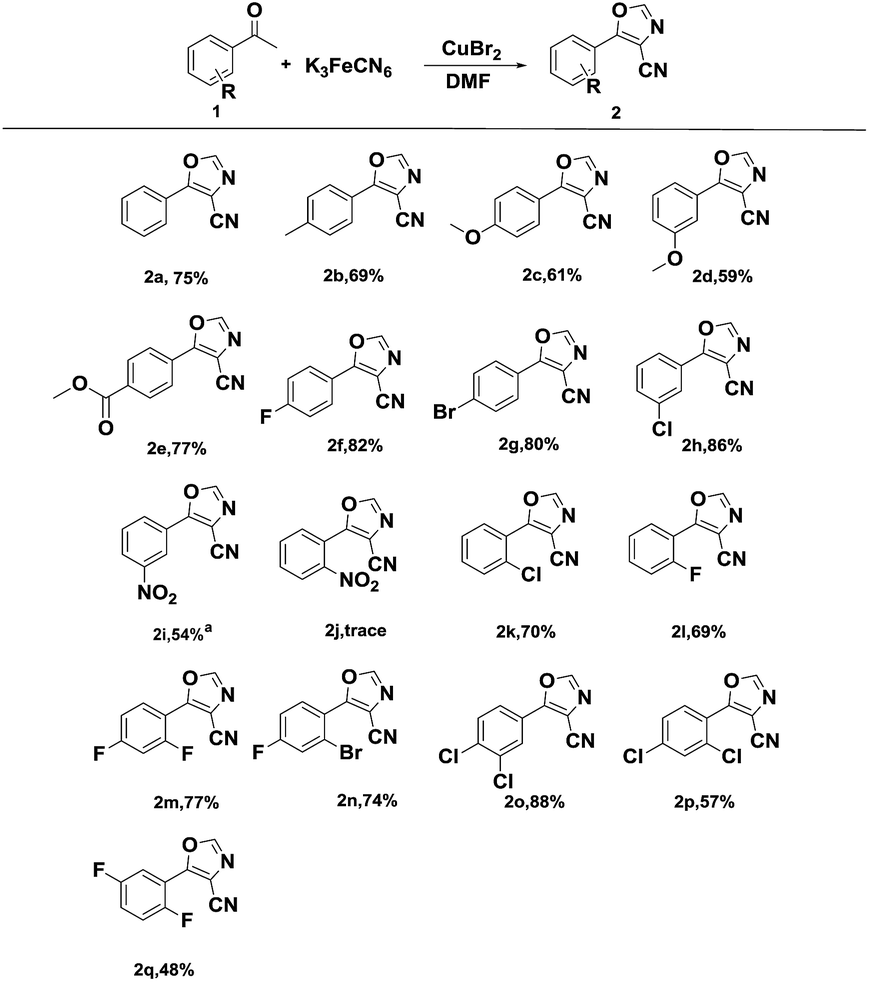 | ||
| Scheme 4 Scope of acetophenones. Reaction conditions: 1a (1.0 mmol), K3[Fe(CN)6] (0.4 mmol), DMF (3 mL), CuBr2 (1.0 mmol), at 130 °C. Isolated yield. aAt 140 °C. | ||
Almost all of the starting acetophenones bearing mono-substitution or poly-substitution were successfully transformed into the desired 4-carbonitrile products in moderate to good yields. In addition, the electronic properties of the substituents on the aromatic ring significantly influenced the efficiency of this transformation. The reactions of 4-fluoro, 4-bromo, and 3-chloro acetophenone proceeded smoothly to afford the corresponding products (2f–2h) in excellent yields. Furthermore, the optimized reaction conditions were also applicable to acetophenones with the electron-donating substituents (2b–2d). However, the strong electron-withdrawing groups on acetophenone, such as –NO2, hindered the conversion (2i and 2j) and the reaction required a high temperature. A low yield was observed in the case of 2-substituted acetophenones (2j–2l) due to the influence of steric hindrance. Besides, the reactions of methyl 4-acetylbenzoate proceeded smoothly to afford the corresponding products (2e), while, the starting acetophenones bearing aldehydes18a or acids were hardly transformed into the desired 4-carbonitrile products.
Subsequently, in order to gain insight into the reaction mechanism, several control experiments were performed as shown in Scheme 5. We proposed a plausible reaction pathway involving the formation of an intermediate, such as 2-bromo-1-phenylethan-1-one (3ab) and 3-oxo-3-phenylpropanenitrile (3ac) via halogenation16 and nucleophilic reaction.17 In subsequent experiments, both 3ab and 3ac transformed to the desired 2a in a comparative yields, which implied that the transfer undergo the halogenation and nucleophilic process. Furthermore, ethyl benzoylacetate could converse to the desired ethyl 5-phenyloxazole-4-carboxylate in this system (2ac).
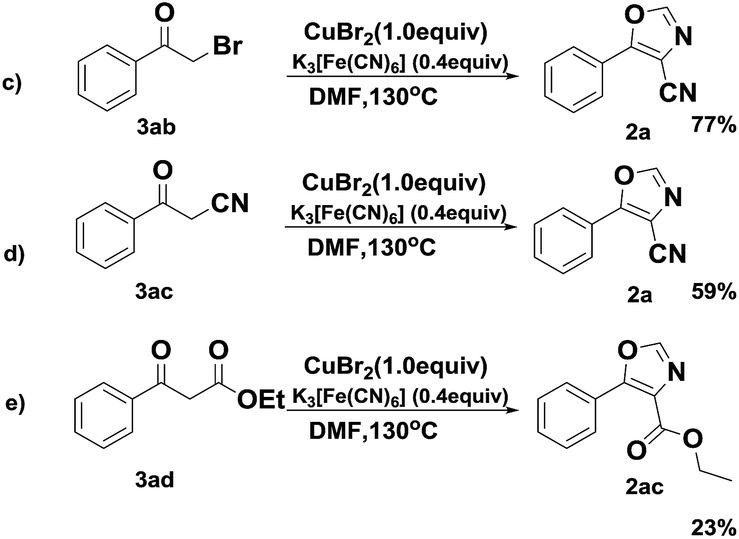 | ||
| Scheme 5 Control experiments for investigating the possible intermediates for the reaction. | ||
In addition, it is noteworthy that the reaction was inhibited in the presence of radical scavengers, suggesting the possibility of a radical pathway (Scheme 6).
 | ||
| Scheme 6 Control experiments for investigating the reaction type. | ||
On the base of experimental results and according to previous studies,16–20 a possible reaction mechanism is proposed and shown in Scheme 7. Firstly, acetophenone transferred to 3-oxo-3-phenylpropanenitrile (3ac) via halogenation14 and nucleophilic15 reaction in the presence of CuBr2 and K3[Fe(CN)6]. Next, CuII served as a single electron oxidant to convert the 3ac to the radical species 4.18 The CuICN18a species then combines with radical species 4 to generate the CuI-bonded radical intermediate 5. Immediately, intermediate 5 transformed to the radical intermediate 6 via cyclization,19 which was oxidized by CuII to form the cationic intermediate 7.20 Finally, the desired product 2a is obtained from 7 via elimination reaction.
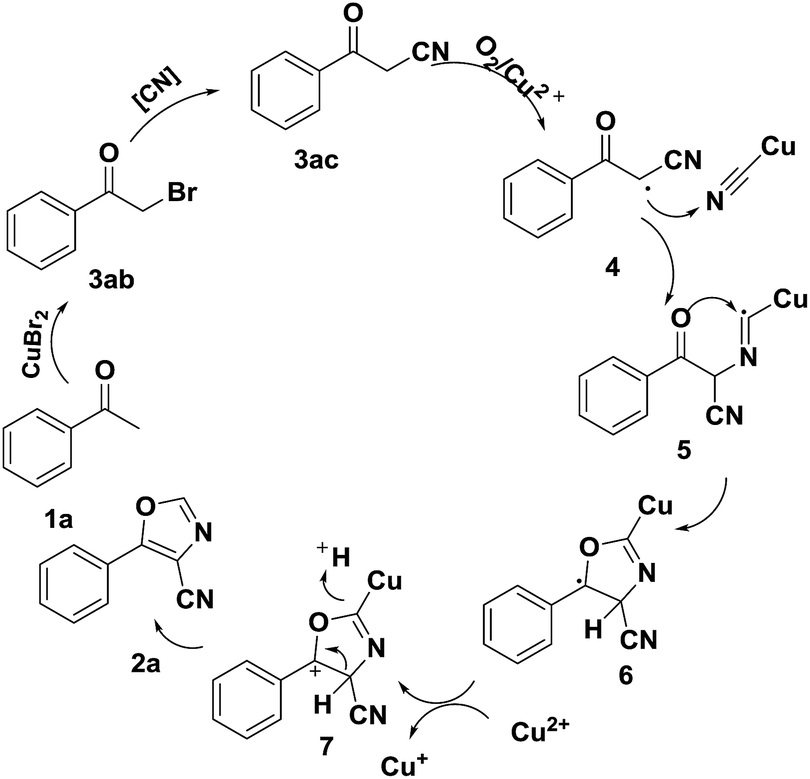 | ||
| Scheme 7 Proposed mechanism. | ||
In conclusion, an efficient, low-toxicity formation of 5-aryloxazole-4-carbonitrile from acetophenone was developed using potassium ferricyanide as a cyanide reagent with Cu(II) bromide as an oxidan, in which, potassium ferricyanide played a dual role as a “CN” source and also as a coupling partner for the cyclization of oxazole. The reaction conditions are mild and suitable for a wide range of substrates. Most of these substrates, such as 4-fluoro, 3-chloro, and 4-bromo acetophenone, provide the corresponding products in moderate to excellent yields. In addition, the dramatic multiple bond formation provides a practical one-pot access to various 5-aryloxazole-4-carbonitrile derivatives from accessible raw materials. In addition, a reasonable reaction mechanism was proposed.
Notes and references
- (a) W. Wang, D. Yao, M. Gu, M. Fan, J. Li, Y. Xing and F. Nan, Bioorg. Med. Chem. Lett., 2005, 15, 5284 CrossRef CAS PubMed; (b) N. Desroy, F. Moreau, S. Briet, G. Le Fralliec, S. Floquet, L. Durant, V. Vongsouthi, V. Gerusz, A. Denis and S. Escaich, Bioorg. Med. Chem., 2009, 17, 1276 CrossRef CAS PubMed; (c) S. Heng, K. R. Gryncel and E. R. Kantrowitz, Bioorg. Med. Chem., 2009, 17, 3916 CrossRef CAS PubMed; (d) D. S. Dalisay, E. W. Rogers, A. S. Edison and T. F. Molinski, Nat. Prod., 2009, 72, 732 CrossRef CAS PubMed; (e) Z. Jin, Nat. Prod. Rep., 2009, 26, 382 RSC; (f) Z. Jin, Nat. Prod. Rep., 2011, 28, 1143 RSC.
- (a) S. Heng, K. R. Gryncel and E. R. Kantrowitz, Bioorg. Med. Chem., 2009, 17, 3916 CrossRef CAS PubMed; (b) D. S. Dalisay, E. W. Rogers, A. S. Edison and T. F. Molinski, Nat. Prod., 2009, 72, 732 CrossRef CAS PubMed; (c) N. Desroy, F. Moreau, S. Briet, G. Le-Fralliec, S. Floquet, L. Durant, V. Vongsouthi, V. Gerusz, A. Denis and S. Escaich, Bioorg. Med. Chem., 2009, 17, 1276 CrossRef CAS PubMed; (d) S. Heng, K. R. Gryncel and E. R. Kantrowitz, Bioorg. Med. Chem., 2009, 17, 3916 CrossRef CAS PubMed.
- (a) A. Reiser, L. J. Leyshon, D. Saunders, M. V. Mijovic, A. Bright and J. Bogie, J. Am. Chem. Soc., 1972, 94, 2014 CrossRef; (b) T. Toyo'oka, H. P. Chokshi, R. S. Givens, R. G. Carlson, S. M. Lunte and T. Kuwana, Biomed. Chromatogr., 1993, 7, 208 CrossRef PubMed.
- (a) M. El-Kemary, J. A. Organero, L. Santos and A. J. Douhal, J. Phys. Chem. B, 2006, 110, 14128 CrossRef CAS PubMed; (b) C. A. Hughes, L. Robinson, A. Tseng and R. D. MacArthur, Expert Opin. Pharmacother., 2009, 10, 2445 CrossRef CAS PubMed; (c) Y. Fradet, Expert Rev. Anticancer Ther., 2004, 4, 37 CrossRef CAS PubMed; (d) E. Teodori, S. Dei, A. Garnier-Suillerot, F. Gualtieri, D. Manetti, C. Martelli, M. N. Romanelli, S. Scapecchi, P. Sudwan and M. Salerno, J. Med. Chem., 2005, 48, 7426 CrossRef CAS PubMed.
- (a) Y. Ju, F. Liu and C. Li, Org. Lett., 2009, 11, 3582 CrossRef CAS PubMed; (b) J. X. Qiao, X. Cheng, D. P. Modi, K. A. Rossi, J. M. Luettgen, R. M. Knabb, P. K. Jadhav and R. R. Wexler, Bioorg. Med. Chem. Lett., 2005, 15, 29 CrossRef CAS PubMed; (c) Z. Rappoport, The Chemistry of the Cyano Group, Interscience Publishers: London, 1970.
- F. F. Fleming, L. H. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed.
- (a) W. Bonrath, Ultrason. Sonochem., 2003, 10, 55 CrossRef CAS PubMed; (b) B. K. Srivastava, R. Soni, A. Joharapurkar, K. V. Sairam, J. Z. Patel, A. Goswami, S. A. Shedage, S. S. Kara, S. P. Salunke, S. B. Gugale, A. Dhawas, P. Kadam, B. Mishra, N. Sadhwanid, V. B. Unadkatd, P. Mitrad, M. R. Jainb and P. R. Patel, Bioorg. Med. Chem. Lett., 2008, 18, 963 CrossRef CAS PubMed.
- (a) F. A. Romero, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 14004 CrossRef CAS PubMed; (b) F. A. Romero, W. Du, I. Hwang, T. J. Rayl, F. S. Kimball, D. Leung, H. S. Hoover, R. L. Apodaca, J. G. Breitenbucher, B. F. Cravatt and D. L. Boger, J. Med. Chem., 2007, 50, 1058 CrossRef CAS PubMed; (c) F. Aquino, R. Karge, H. Pauling and W. Bonrath, Molecules, 1997, 2, 176 CrossRef CAS.
- (a) J. Linder, T. P. Garner, H. E. Williams, M. S. Searle and C. J. Moody, J. Am. Chem. Soc., 2011, 133, 1044 CrossRef CAS PubMed; (b) J. K. DeMartino, J. Garfunkle, D. G. Hochstatter, B. F. Cravatt and D. L. Boger, Bioorg. Med. Chem. Lett., 2008, 18, 5842 CrossRef CAS PubMed; (c) H. Wada, H. E. Williams and C. J. Moody, Angew. Chem., Int. Ed., 2015, 54, 15147 CrossRef CAS PubMed; (d) V. M. Prokopenko, S. G. Pil'o, A. N. Vasilenko and V. S. Brovarets, Russ. J. Gen. Chem., 2010, 80, 2358 CrossRef CAS.
- (a) D. Haas, M. Mosrin and P. Knochel, Org. Lett., 2013, 15, 6162 CrossRef CAS PubMed; (b) K. Burger, E. Hoss and K. Geith, Synthesis, 1990, 4, 360 CrossRef.
- (a) X. Liu, R. Cheng, F. Zhao, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2012, 14, 5282 CrossRef PubMed; (b) I. K. Khanna, Y. Yu, R. M. Huff, R. M. Weier, X. Xu, F. J. Koszyk and J. L. Masferrer, J. Med. Chem., 2000, 43, 3168 CrossRef CAS PubMed; (c) K. Matsumura, O. Miyashite, H. Shimadzu and N. Hashimoto, Chem. Pharm. Bull., 1976, 24, 948 CrossRef CAS; (d) T. P. Sycheva, T. K. Trupp and M. N. Shchukina, Zh. Obshch. Khim., 1962, 32, 1071 CAS; (e) J. W. Cornforth and H. T. Huang, J. Chem. Soc., 1948, 1969 RSC; (f) F. Freeman and D. S. Kim, Tetrahedron Lett., 1989, 30, 2631 CrossRef CAS.
- (a) X. F. Jia, D. P. Yang, W. H. Wang, F. Luo and J. Cheng, J. Org. Chem., 2009, 74, 9470 CrossRef CAS PubMed; (b) G. Yan, C. Kuang, Y. Zhang and J. Wang, Org. Lett., 2010, 12, 1052 CrossRef CAS PubMed; (c) Y. Ren, M. Yan, S. Zhao, J. Wang, J. Ma, X. Tian and W. Yin, Adv. Synth. Catal., 2012, 354, 2301 CrossRef CAS.
- (a) W. P. Mai, H. H. Wang, Z. C. Li, J. W. Yuan, Y. M. Xiao, L. R. Yang, P. Mao and L. B. Qu, Chem. Commun., 2012, 48, 10117 RSC; (b) Q. Zhao, T. Miao, X. Zhang, W. Zhou and L. Wang, Org. Biomol. Chem., 2013, 11, 1867 RSC.
- (a) J. Xiao, Q. Li, T. Chen and L. B. Han, Tetrahedron Lett., 2015, 43, 5937 CrossRef; (b) L. Zhang, P. Lu and Y. Wang, Org. Biomol. Chem., 2015, 30, 8322 RSC; (c) J. Kim, H. Kim and S. Chang, Org. Lett., 2012, 14, 3924 CrossRef CAS PubMed; (d) S. Ding and N. Jiao, J. Am. Chem. Soc., 2011, 133, 12374 CrossRef CAS PubMed.
- (a) O. Grossman and D. Gelman, Org. Lett., 2006, 8, 1189 CrossRef CAS PubMed; (b) Y. Z. Zhu and C. Cai, Synth. Commun., 2008, 38, 2753 CrossRef CAS; (c) Y. Ren, Z. Liu, S. He, S. Zhao, J. Wang, R. Niu and W. Yin, Org. Process Res. Dev., 2009, 13, 764 CrossRef CAS; (d) G. Yan, C. Kuang, Y. Zhang and J. Wang, Org. Lett., 2010, 12, 1052 CrossRef CAS PubMed.
- R. Aeluri, M. Alla, S. Polepalli and N. Jain, Eur. J. Med. Chem., 2015, 100, 18 CrossRef CAS PubMed.
- (a) S. Eagon, N. Ball-Jones, D. Haddenham, J. Saavedra, C. DeLieto, M. Buckman and B. Singaram, Tetrahedron Lett., 2010, 51, 6418 CrossRef CAS; (b) S. Kamila, D. Zhu, E. R. Biehl and L. Hua, Org. Lett., 2006, 8, 4429 CrossRef CAS PubMed.
- (a) Q. Wu, Y. Luo, A. Lei and J. You, J. Am. Chem. Soc., 2016, 138, 2885 CrossRef CAS PubMed; (b) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed; (c) G. G. Melikyan, Org. React., 1997, 49, 427 CAS.
- (a) Y. Yamamoto, M. Ohno and S. Eguchi, J. Org. Chem., 1996, 61, 9264 CrossRef CAS; (b) R. S. Liu, S. M. Ghorpade and P. D. Jadhav, Chem.–Eur. J., 2016, 22, 2915 CrossRef PubMed; (c) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 8, 1991 CrossRef; (d) H. Ishibashi, M. Higuchi, M. Ohba and M. Ikeda, Tetrahedron Lett., 1998, 39, 75 CrossRef CAS.
- (a) G. Qin, X. Chen, L. Yang and H. Huang, ACS Catal., 2015, 5, 2882 CrossRef CAS; (b) T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01983a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |