
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of Heck coupling on 6-bromo[2,3-d]thienopyrimidines for construction of new EGFR inhibitor lead structures†
F. Bysting‡
a,
S. Bugge§
a,
E. Sundby b and
B. H. Hoff*a
b and
B. H. Hoff*a
aDepartment of Chemistry, Faculty of Natural Sciences and Technology, Norwegian University of Science and Technology (NTNU), NO-7491 Trondheim, Norway. E-mail: Bard.Helge.Hoff@chem.ntnu.no
bDepartment of Materials Science and Engineering, Norwegian University of Science and Technology (NTNU), NO-7491 Trondheim, Norway
First published on 28th March 2017
Abstract
With the aim of identifying new lead structures for EGFR inhibition, a study of palladium catalysed Heck coupling between (R)-6-bromo-N-(1-phenylethyl)thieno[2,3-d]pyrimidin-4-amine and various acrylates was performed. The Heck coupling was highly dependent on type of catalyst, solvent, base type and the use of tetrabutylammonium chloride as additive. The products were stable in the dark, but underwent trans–cis isomerization upon exposure to light. Kinase profiling indicate that acrylates grafted on the [2,3-d]thienopyrimidines is an attractive scaffold for identification of potent and highly selective EGFR inhibitors.
Introduction
The epidermal growth factor receptor tyrosine kinase (EGFR) is an attractive target in EGFR driven diseases such as non-small-cell lung cancer,1,2 EGFR positive breast cancer,3 and in pancreatic cancer.4 Inhibition of the intracellular ATP-binding site can be achieved using properly designed small molecular structures. Scaffolds employed includes mainly quinazolines,5,6 pyrrolopyrimidines,7–11 and furopyrimidines.12,13 Additionally, our laboratory and others14,15 have also identified potent thienopyrimidine based EGFR inhibitors (Fig. 1).
One drawback of some of the previously designed structures is the two carbon-based aromatic rings, which increases crystallinity, lowers solubility and in general could lead to a higher toxicity in vivo.17 In an attempt to substitute the carbon based aromatic ring at the thiophene moiety (C-6), we envisioned that alkenes could be employed. This moiety was planned inserted by a palladium catalysed Heck coupling. However, the Heck reaction is extremely sensitive to the choice of catalyst, co-catalyst, solvent, reaction temperature etc.18 Therefore, each new Heck reaction often needs careful optimisation of the reaction conditions. Herein, our efforts to identify new EGFR lead structures, and an investigation of the Heck coupling is described. The chemical part of the study has evaluated the effect of catalyst/ligand systems, solvent, base and additives, and also describes the challenges encountered during handling of these materials. Finally, the initial evaluation of the new structures as kinase inhibitors is presented.
Result and discussion
Investigation of the Heck reaction
As a starting point for the study of the Heck-reaction, (R)-6-bromo-N-(1-phenylethyl)thieno[2,3-d]pyrimidin-4-amine (1) and methyl acrylate were employed as coupling partners, see Scheme 1. Compound 1 was selected since it is a previously identified low molecular weight structure with nanomolar EGFR potency.19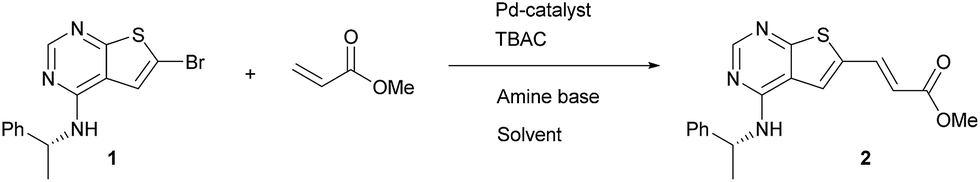 | ||
| Scheme 1 Model system for investigation of the Heck reaction. | ||
First, six catalysts systems were evaluated, namely palladium on carbon (Pd/C), palladium(II) diacetate (Pd(OAc)2), tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4), bis(tri-tert-butylphosphine)palladium(0) (Pd(t-Bu3P)2), [1,1′-bis(di-tert-butylphosphino)ferrocene]-dichloropalladium(II) (PdCl2(dtbpf)) and Xphos Pd 3rd generation catalyst, all in 2 mol%. Other conditions adapted from Murray et al.20 included tetrabutylammonium chloride (TBAC), N,N-dimethylacetamide (DMAc) as solvent and N,N-dicyclohexylmethylamine (Cy2NMe) as base. A ten-fold excess of methyl acrylate was employed. The reaction progress as followed by 1H NMR spectroscopy is shown in Fig. 2.
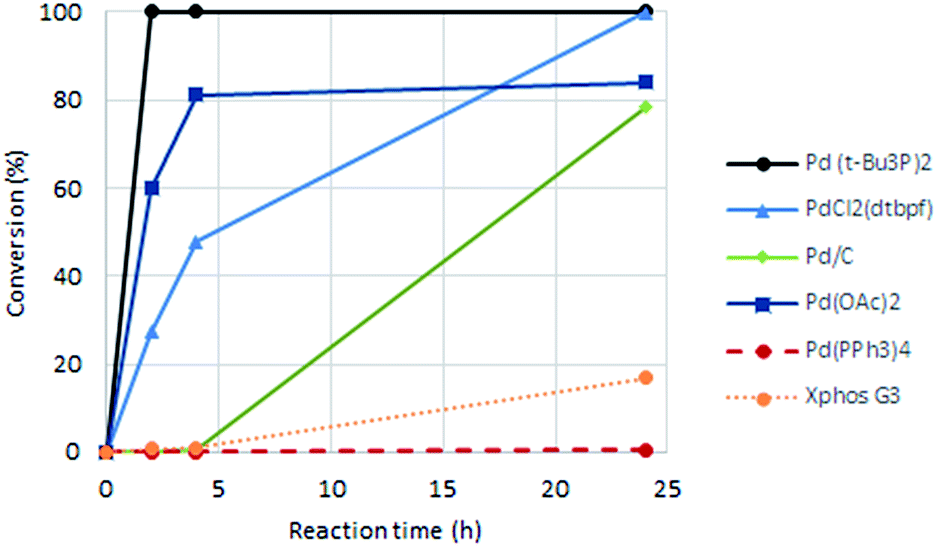 | ||
| Fig. 2 Effect of palladium catalyst on degree of conversion of 1 to the Heck product acrylate 2. | ||
Clear differences were seen in rate for the different catalysts, with Pd(t-Bu3P)2 being the most effective system. The reaction catalysed by palladium diacetate showed a high initial rate, however, conversion ceased around 80%, possibly due to catalyst precipitation. On the other hand, the ferrocene based catalyst PdCl2(dtbpf) showed lower initial rate, but managed to catalyse the reaction fully within 24 hours. The cross-coupling mediated by Pd/C had a slow onset, but attained 78% conversion within 24 hours, whereas the use of Xphos Pd G3 and Pd(PPh3)4 worked poorly under these conditions. Overall, Pd(t-Bu3P)2 appeared to be the catalyst of choice. One by-product was seen in these reaction, namely the cis-isomer 3. This was proved by the light induced interconversion of these compounds (see next Section), the 1H NMR coupling constants, MS analysis, and that reduction of the isomeric mixture yielded only compound 4, see Scheme 2.
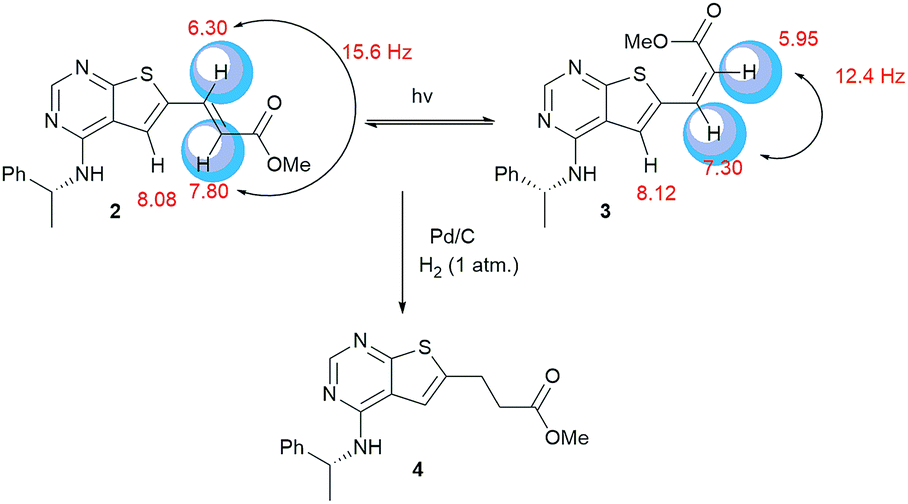 | ||
| Scheme 2 When exposed to light compound 2 converts to compound 3. Reduction of these compounds gave the saturated derivative 4. | ||
Following the initial catalyst selection, the effect of the amount of methyl acrylate on conversion was investigated using 2-, 4-, and a 6-fold excess, see Table 1.
The amount of methyl acrylate used in the reaction had a dramatic effect on degree of conversion. For the coupling with two equivalents 86% conversion was obtained in 24 hours, while with four equivalents full conversion was obtained in 24 hours. In contrast the Heck coupling employing 6 equivalents of methyl acrylate went to completion in only two hours. The trans/cis ratio of the product varied without any apparent trend. When the same reaction was run in the absence of inert atmosphere the reaction ceased at 8% conversion.
The choice of solvent can have a dramatic effect on rate in such coupling reactions. Therefore, the use of eight additional solvents was evaluated. The degree of conversion after 4 hours is compared in Table 2. Although couplings in all these solvents were slower than in DMAc, reactions in 1,2-dimethoxyethane (DME), 2-methyltetrahydrofuran (2-MeTHF), 1,4-dioxane, toluene and THF reached full conversion in 24 hours. The initial rate in both acetonitrile and neat methyl acrylate was comparable to that of the best solvent systems. However, in these medias the conversion ceased around 60%, indicating catalyst decomposition. The reactions in ethanol and tert-butyl methyl ether (TBME) also halted, but at even lower degree of conversion. Employing trimethylamine as both a base and a solvent lead to a slow reaction, which only reached 9% conversion in 4 hours. The isomeric ratio varied from 9/1 to 24/1 without any apparent trend with respect to solvent polarity.
| Entry | Solvent | Conv.a (%) 4 h | 2a | 3a | Ratio 2/3 |
|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy.b 40-fold excess of methyl acrylate.c N,N-Dicyclohexylmethylamine was not used.d Ratio could not be determined due to low conversion. | |||||
| 2 | 2-MeTHF | 97 | 93 | 5 | 20 |
| 3 | DME | 95 | 91 | 4 | 22 |
| 4 | Toluene | 80 | 72 | 8 | 9 |
| 5 | THF | 74 | 69 | 5 | 14 |
| 6 | ACN | 60 | 58 | 2 | 24 |
| 7 | Methyl acrylateb | 60 | 57 | 3 | 20 |
| 8 | EtOH | 32 | 29 | 3 | 10 |
| 9 | NEt3c | 9 | 9 | 0 | d |
| 10 | TBME | 5 | 5 | 0 | d |
Often, the Heck reaction is sensitive to the nature of co-catalysts and the base. Therefore, additional experiments were performed where TBAC was omitted and trimethylamine was used instead of Cy2NMe, see Table 3.
| Entry | Conditions | 4 hours | 24 hours | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Solvent | Base | TBAC | Conv.a (%) | 2a (%) | 3a (%) | Conv.a (%) | 2a (%) | 3a (%) | |
| a Determined by 1H NMR spectroscopy. | |||||||||
| 1 | DMAc | Cy2NMe | 0 | 100 | 93 | 7 | 100 | 96 | 4 |
| 2 | DMAc | NEt3 | 0.1 | 99 | 95 | 4 | 100 | 95 | 5 |
| 3 | DMAc | NEt3 | 0 | 91 | 88 | 3 | 91 | 86 | 5 |
| 4 | DME | NEt3 | 0.1 | 55 | 54 | 1 | 75 | 72 | 2 |
| 5 | DME | NEt3 | 0 | 11 | 10 | 1 | 23 | 21 | 2 |
| 6 | 2-MeTHF | Cy2NMe | 0.1 | 55 | 53 | 1 | 92 | 91 | 2 |
| 7 | 2-MeTHF | Cy2NMe | 0 | 30 | 28 | 2 | 35 | 33 | 2 |
| 8 | 2-MeTHF | NEt3 | 0.1 | 62 | 54 | 7 | 100 | 93 | 7 |
When performing the reaction in DMAc with Cy2NMe as base in the absence of the phase transfer catalyst (entry 1), no major change in conversion was noted. However, in experiments using trimethylamine as base in DMAc and DME, lower conversion was observed without TBAC. This was also the case using 2-MeTHF as solvent in combination with Cy2NMe as base. Obviously, the TBAC additive improves the reaction rate of the Heck reaction, and especially so in solvents of low polarity. The role of the phase transfer catalyst is somewhat unclear, but it has been suggested that the chloride anion play a role as a supporting ligand in generating the active Pd(0) complex.21–23 An alternative explanation proposed is that the phase-transfer catalyst brings about an encapsulation of Pd(0), preventing formation of palladium black.24,25 As the initial investigations indicated that the Heck coupling was fairly robust, the reaction was scaled starting with 1 gram of compound 1 using DMAc, triethylamine and TBAC and reacting for 24 hours. Full conversion was obtained and a trans/cis ratio of 15/1 was noticed. As a high trans/cis ratio was targeted, the purity of the material was improved by precipitation from dichloromethane followed by re-crystallization from the same solvent. This lead to a trans/cis ratio of 32 (97% by HPLC), however in only 27% yield (Table 4, entry 1). We were unable to purify similar prepared materials efficiently using silica-gel chromatography or preparative HPLC.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Conv.a (%) | Purityb | Ratio trans/cisc | Yield (%) | Product |
| a Conversion after 24 hours determined by 1H NMR spectroscopy.b Purity (area%) determined by RP-HPLC.c Isomer ratio by 1H NMR spectroscopy.d Purified by crystallization.e Purified by silica gel-column chromatography. | ||||||
| 1 | OMe | >99 | 97 | >30 | 27d | 2 |
| 2 | OCH2CH2OMe | >99 | 96 | 21 | 39e | 5 |
| 3 | OCH2CH2OH | 68 | 92 | >30 | 47e | 6 |
| 4 | NH2 | 0 | — | — | — | 7 |
| 5 | NMe2 | 92 | 99 | >30 | 27d | 8 |
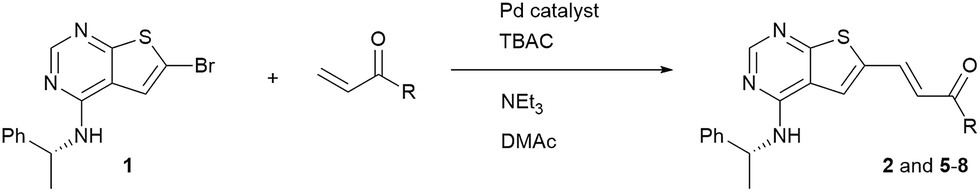
Four other Heck reactions were attempted in 0.2 g scale (entries 2–5). Of these, conversion to the 2-methoxyethyl ester 5 (entry 2) proceeded very efficiently, but anyhow the isolated yield was mediocre due to a difficult separation of the cis isomer, the presence of polymers and most likely trans–cis isomerization during the purification. The process to compound 6 (entry 3) was run for 24 hours, but was observed to stop already at 4 hours. Also disappointingly, the coupling with acrylamide (entry 4) failed completely. As Heck coupling with acrylamides have been performed with other Pd-catalyst systems,26,27 this result can most likely be ascribed to the delicate nature of this coupling method. In contrast, N,N-dimethylacrylamide (Table 4, entry 5) was well tolerated as substrate.
As compound 7 could not be made by the Heck reaction, it was formed by ammonolysis of 2 (Scheme 3). This process gave after crystallization 30 mg of compound 7, but the trans/cis ratio was 5/1.
 | ||
| Scheme 3 Ammonolysis of compound 2. | ||
Stability of the product
As judged from 1H NMR spectroscopy the reaction proceeded with a low level of cis-configurated product. However, upon final purification, HPLC analysis revealed this product to be difficult to remove completely. Apparently, the isomerization process occurred during handling. Fig. 3 shows 1H NMR of the same NMR sample prior and after four day storage in daylight.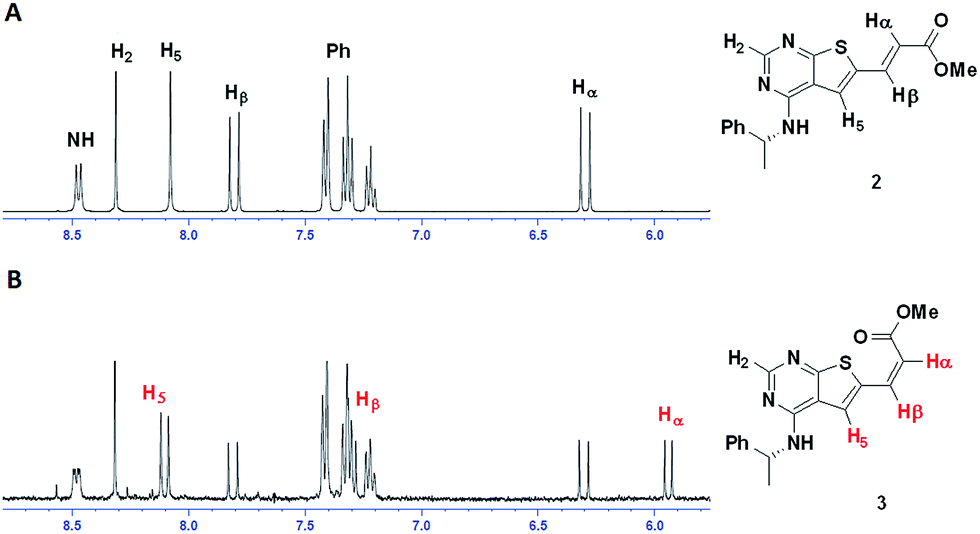 | ||
| Fig. 3 1H NMR before (A) and after (B) light exposure of compound 2. | ||
To shed light on the isomerization process, stability testing was performed in various solvents in the dark and at the lab bench exposed to light. The levels of the main impurity as analysed by HPLC (area%) is shown in Table 5.
When stored in the dark no major change of the level of the cis-isomer 3 was detected (entries 1–4). However, in benchtop daylight a steady rise in the level of cis-3 was seen in all solvents except in water. Compounds 5–8 were submitted to similar experiments in acetonitrile and exposed to daylight. After 48 hours the level of the cis impurity increased to a level of 23–37%. This clearly shows that such materials must be handled with extreme care and should be protected from light during production, purification and storage. In retrospect, these Heck-couplings could benefit from some of the solid supported catalyst systems developed.28–30 This would ease purification, probably lower the degree of trans–cis isomerisation and amount of palladium in the end-product.
EGFR kinase inhibitory properties
Compounds 2 and 4–8 were profiled in an epidermal growth factor receptor tyrosine kinase (EGFR) assay at 100 nM concentration using an ATP concentration equal to KM, and benchmarked with the corresponding phenyl substituted analogues 9 and 10, see Table 6.
|
|||||
|---|---|---|---|---|---|
| Entry | Comp. | R | Inhi.a (%) | IC50b (nM) | Mp (°C) |
| a Mean value of two measurements at 100 nM concentration.b Mean value of two titration curves (20 data points) and standard deviation.c See structure above.d Data is taken from Bugge et al.16e Data taken from Knesl et al.31 | |||||
| 1 | 2 | OMe | 78 | — | 98–100 |
| 2 | 4 | c | 49 | — | Oil |
| 3 | 5 | OCH2CH2OMe | 85 | — | Oil |
| 4 | 6 | OCH2CH2OH | 94 | 7.0 ± 0.3 | 144–145 |
| 5 | 7 | NH2 | 90 | 13 ± 0.2 | 246–251 |
| 6 | 8 | NMe2 | 73 | — | 154–155 |
| 7 | 9 | c | 58 | 58 ± 2d | 171–173d |
| 8 | 10 | c | 86 | 7 ± 1d | 215–217d |
| 9 | Erlotinib | 102 | 0.4 ± 0.1 | 159–160e | |
All compounds had promising inhibitory activity. Interestingly, the unsaturated compound 2 was more potent than the saturated analogue 4. The 2-methoxyethyl ester 5, and especially the 2-hydroxyethyl ester 6 showed a further improvement in potency. For the two amides assayed the unsubstituted derivative 7 was more potent that the dimethyl analogue 8. The two most active derivatives were also assayed by IC50 titration (Fig. 4).
 | ||
| Fig. 4 EGFR IC50 titration curves of compound 6 (squares) and 7 (triangles) compared with Erlotinib (circles). | ||
Compound 6 was found slightly more potent than the unsubstituted amide 7 and was equipotent with the 4-hydroxymethylphenyl substituted compound 9. Whereas the amide 7 had a rather high melting point, the ester derivatives and the dimethyl amide 8 appears less crystalline than the phenyl analogues 9 and 10.
Compound 5, 6 and 7 were subjected to docking studies using the EGFR crystal structure 2J6M (wild-type EGFR). 2D interaction plot for the best docking poses and corresponding docking score energies for all compounds are shown in S1–S3 (ESI†). These poses were subsequently exposed to 10 ns dynamic simulations, and diagrams showing the most important ligand–protein interactions for compound 5 and 6 are shown in Fig. 5. Binding of both compounds seem to depend heavily on a hydrogen-bond interaction from methionine 793 and threonine 854 (via a water molecule bridge) as well as a cation–π interaction between lysine 745 and the phenyl ring in the amine part of the ligand. However, no clear difference in terms of ligand–protein affinity were evident after these simulations. When evaluating the best docked poses of 5 and 6, an overlay of the two (ESI†) shows that the only notable difference was in the alcohol and ether side-chain, which might explain the difference in docking scores (5: −10.057 kcal mol−1 and 6: −9.408 kcal mol−1). A root mean square fluctuation analysis after 10 ns of dynamic simulations also showed that the flexibility of these side chains was quite similar when bound to the protein (ESI†).
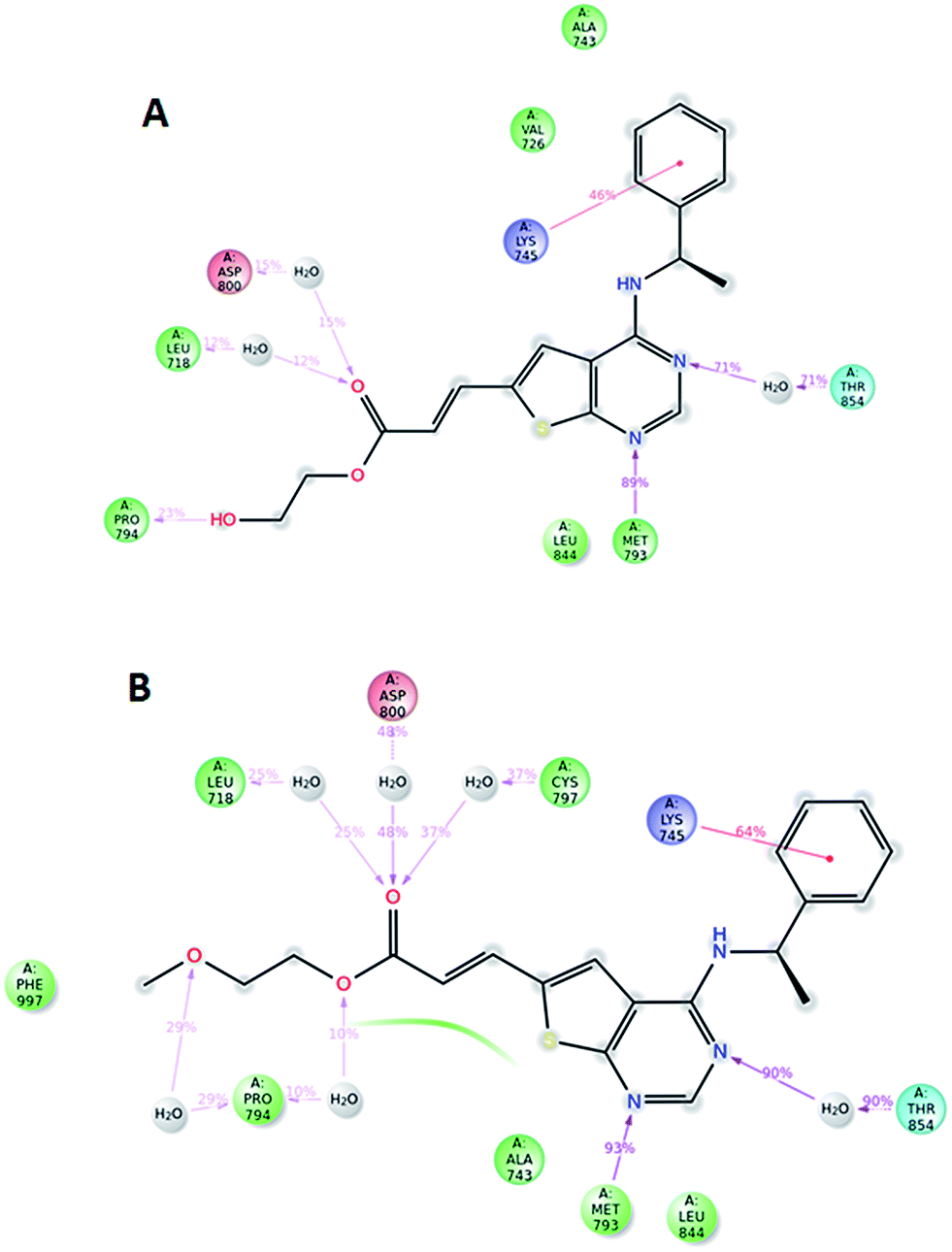 | ||
| Fig. 5 Ligand-EGFR contacts for compounds 6 (A) and 5 (B) after docking and 10 ns dynamic simulation. Only those interactions are shown that occur more than 10% of the 10 ns simulation time. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article). | ||
Potentially, these structures could be suitable as irreversible inhibitors by reaction between the β-carbon and the thiol moiety of Cysteine 797. However, the moderate increase in activity going from the saturated to the unsaturated derivatives, using one-hour incubation time indicate that this is not the case. Moreover, docking experiments show that the distance from the thiol to the β-carbon is ca. 7 Å, which is too distant to expect covalent binding (ESI†).
As an indication of the kinase selectivity displayed by this new class of inhibitors the inhibitor 5 was assayed towards a panel of 50 additional kinases at 500 nM test concentration. The analysis revealed a remarkable high selectivity for EGFR over all other kinases evaluated. The % inhibition towards the main off-targets are shown in Fig. 6.
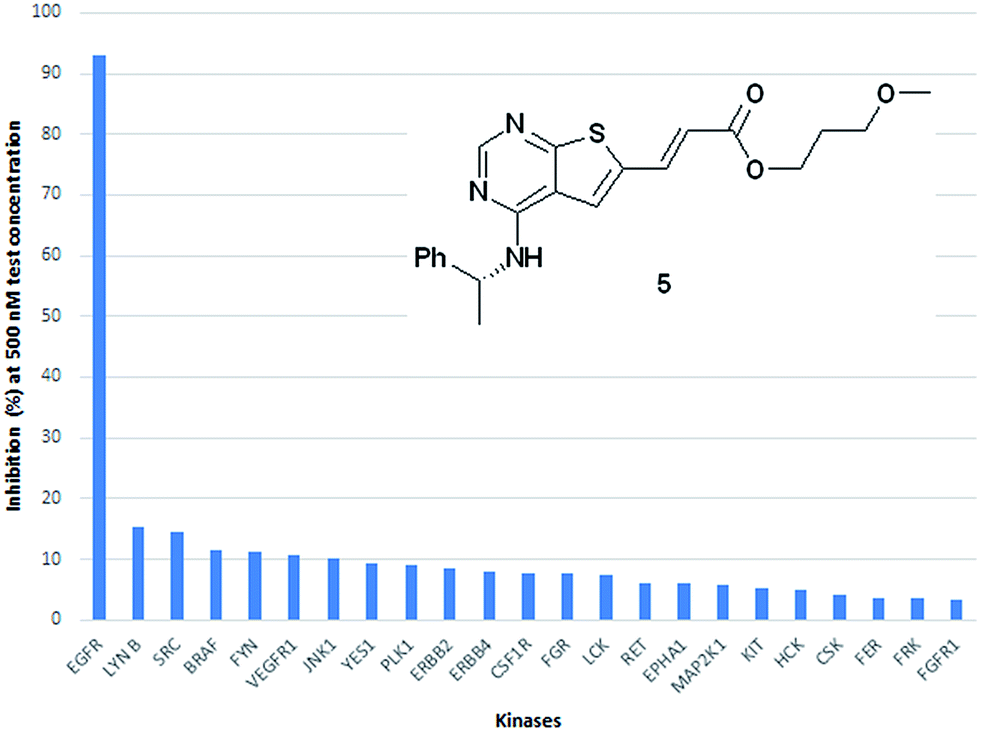 | ||
| Fig. 6 Kinase profiling towards a panel of 50 additional kinase showed a high selectivity for EGFR. The main off targets and their degree of inhibition is shown. | ||
Experimental
General
Compounds 1 was made in a previous study,16 while all other chemicals were from Sigma Aldrich. Silica-gel column chromatography was performed using silica gel 60A from Fluka, pore size 40–63 μm. Celite 545 from Fluka was also used. 1H and 13C NMR spectra were recorded with Bruker Avance 400 spectrometer operating at 400 MHz and 100 MHz, respectively. Coupling constants are in hertz. HPLC (Agilent 110-Series) with a G1379A degasser, G1311A Quatpump, G1313A ALS autosampler and a G1315D Agilent detector (230 nm) was used to determine the purity of the synthesised compounds. Conditions: Method A:Poroshell C18 (100 × 4.6 mm) column, flow rate 0.8 mL min−1, elution starting with water/CH3CN (90/10), 5 min isocratic elution, then linear gradient elution for 30 min ending at CH3CN/water (100/0). Accurate mass determination (ESI) was performed on an Agilent G1969 TOF MS instrument equipped with a dual electrospray ion source, or EI (70 eV) using a Finnigan MAT 95 XL. Accurate mass determination in positive and negative mode was performed on a “Synapt G2-S” Q-TOF instrument from Waters. Samples were ionized by the use of an ASAP probe, no chromatography separation was used before the mass analysis. FTIR spectra were recorded on a Thermo Nicolet Avatar 330 infrared spectrophotometer. All melting points are uncorrected and measured by a Stuart automatic melting point SMP40 apparatus.Synthesis of methyl (R,E)-3-(4-((1-phenylethyl)amino)thieno[2,3-d]pyrimidin-6-yl)acrylate (2)
(R)-6-Bromo-N-(1-phenylethyl)thieno[2,3-d]-pyrimidin-4-amine (1) (1.00 g, 2.99 mmol), bis(tri-tert-butylphospine)palladium(0) (31.9 mg, 0.06 mmol) and tetrabutylammonium chloride (90 mg, 0.32 mmol) were mixed under a nitrogen atmosphere. N,N-Dimethylacetamide (15 mL), triethylamine (0.625 mL, 4.49 mmol) and methyl acrylate (1.62 mL, 18.0 mmol) were added, and the mixture was stirred at 80 °C for 24 h. The reaction mixture was added ethyl acetate (50 mL) and washed with water (4 × 40 mL). The organic phase was dried with brine (50 mL), then over anhydrous Na2SO4, and filtered. The filtrate was concentrated under reduced pressure, resulting in a yellow oil. Repeated solvent displacement using dichloromethane resulted in 1.19 g of a yellow-brown solid. The crude product was washed with cold dichloromethane (15 mL), resulting in a less coloured solid (0.82 g). This was followed by crystallized from dichloromethane (10 mL) resulting in 270 mg (0.795 mmol, 27%) of a yellow solid; mp 98–100 °C; purity (HPLC): 97%, tR = 23.2 min; [α]20D = −351.2 (c 1.01, DMSO). 1H NMR (400 MHz, DMSO-d6): 8.48 (d, J = 7.8, 1H, NH), 8.32 (s, 1H, H-2), 8.08 (s, 1H, H-5), 7.81 (d, J = 15.6, 1H, Hβ), 7.42–7.40 (m, 2H, Ph), 7.34–7.30 (m, 2H, Ph), 7.24–7.20 (m, 1H, Ph), 6.30 (d, J = 15.6, 1H, Hα), 5.54–5.47 (m, 1H, CH), 3.74 (s, 3H, OCH3), 1.55 (d, J = 7.0, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): 166.1, 166.0, 156.0, 155.3, 144.3, 137.4, 133.5, 128.3 (2C), 126.7, 126.1 (2C), 125.3, 118.3, 116.6, 51.7, 49.1, 22.3; IR (cm−1): 3239 (w, br), 1715 (m), 1574 (s), 1502 (m), 1442 (m), 1299 (s), 1264 (s), 1163 (s), 962 (m), 694 (s); HRMS (ASAP+, m/z): found 340.1122, calcd for C18H18N3O2S [M + H]+ 340.1120.Synthesis of methyl (R)-3-(4-((1-phenylethyl)amino)thieno[2,3-d]pyrimidin-6-yl)propanoate (4)
Ethanol (20 mL) and Pd/C (10%, 20 mg) were mixed and then added methyl (R,E)-3-(4-((1-phenylethyl)amino)thieno[2,3-d]-pyrimidin-6-yl)-acrylate (2) (193 mg, 0.570 mmol). Hydrogen gas was bubbled through the solution and then applied as atmosphere (1 atm). The mixture was allowed to stir for 168 h at 20 °C, with additional addition of ethanol (10 mL) and Pd/C (10%, 20 mg) as well as refilling of H2. The reaction mixture was filtered through celite and concentrated under reduced pressure. The crude product (183 mg) was purified by silica-gel column chromatography (ethyl acetate/n-pentane 1/1). This gave 78 mg (0.229 mmol, 40%) as a light yellow oil; purity (HPLC) > 99%, tR = 21.5 min; [α]20D = −140.5 (c 1.03, DMSO). 1H NMR (400 MHz, DMSO-d6): 8.22 (s, 1H, H-2), 8.09 (d, J = 8.0, 1H, NH), 7.52 (s, 1H, H-5), 7.40–7.39 (m, 2H, Ph), 7.32–7.29 (m, 2H, Ph), 7.22–7.19 (m, 1H, Ph), 5.51–5.46 (m, 1H, CH), 3.62 (s, 3H, OCH3), 3.13 (t, J = 7.1, 2H, CH2), 2.76 (t, J = 7.1, 2H, CH2), 1.53 (d, J = 7.9, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): 172.1, 165.0, 155.3, 153.2, 144.8, 139.2, 128.2 (2C), 126.6, 126.0 (2C), 116.6, 116.2, 51.5, 48.9, 34.3, 25.5, 22.5; IR (cm−1): 3280 (w, br), 2975 (w, br), 1734 (s), 1578 (s), 1514 (s), 1436 (m), 1351 (m), 1203 (m), 1169 (m), 1024 (m), 866 (m), 698 (s); HRMS (ASAP+, m/z): found 342.1274, calcd for C18H20N3O2S [M + H]+ 342.1276.2-Methoxyethyl (R,E)-3-(4-((1-phenylethyl)amino)thieno[2,3-d]pyrimidin-6-yl)acrylate (5)
(R)-6-Bromo-N-(1-phenylethyl)thieno[2,3-d]-pyrimidin-4-amine (1) (208 mg, 0.622 mmol) and bis(tri-tert-butylphospine)palladium(0) (8.0 mg, 0.015 mmol) and tetrabutylammonium chloride (17 mg, 0.06 mmol) were mixed under a nitrogen atmosphere. N,N-Dimethylacetamide (3 mL), triethylamine (0.125 mL, 0.9 mmol) and 2-methoxyethyl acrylate (0.80 mL, 6.22 mmol) were added, and the mixture was stirred at 80 °C for 24 h. The reaction mixture was added ethyl acetate (20 mL) and washed with water (3 × 15 mL). The organic phase was dried with brine (20 mL), then over anhydrous Na2SO4, and filtered. The filtrate was concentrated under reduced pressure, resulting in 172 mg of the crude product. The material (151 mg, 0.40 mmol) was purified by silica-gel column chromatography (diethyl ether, Rf = 0.55) gave 93 mg (0.243 mmol, 39%) of a yellow oil; purity (HPLC): 96%, tR = 22.8 min; [α]20D = −360.0 (c 0.77, DMSO); 1H NMR (400 MHz, DMSO-d6): 8.46 (d, J = 7.9, 1H, NH), 8.32 (s, 1H, H-2), 8.10 (s, 1H, H-5), 7.81 (d, J = 15.6, 1H, Hβ), 7.42–7.40 (m, 2H, Ph), 7.34–7.30 (m, 2H, Ph), 7.24–7.20 (m, 1H, Ph), 6.31 (d, J = 15.6, 1H, Hβ), 5.54–5.47 (m, 1H, CH), 4.29–4.26 (m, 2H, CH2), 3.61–3.58 (m, 2H, CH2), 3.29 (s, 3H, CH3), 1.55 (d, J = 7.0, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): 166.1, 165.5, 156.0, 155.4, 144.3, 137.6, 133.4, 128.3 (2C), 126.7, 126.1 (2C), 125.3, 118.3, 116.6, 69.8, 63.4, 58.1, 49.1, 22.3; IR (cm−1): 3357 (w, br), 2976 (w, br), 1710 (m), 1574 (s), 1506 (m), 1265 (s), 1166 (s), 1034 (m), 964 (m), 732 (s), 698 (s); HRMS (ASAP+, m/z): found 384.1379, calcd for C20H22N3O3S [M + H]+ 384.1382.Synthesis of 2-hydroxyethyl (R,E)-3-(4-((1-phenylethyl)amino)-thieno[2,3-d]pyrimidin-6-yl)acrylate (6)
Compound 6 was prepared as described for 5, but starting with (R)-6-bromo-N-(1-phenylethyl)thieno[2,3-d]-pyrimidin-4-amine (1) 2-hydroxyethyl acrylate (0.69 mL, 6.01 mmol). The crude product (139 mg) was purified by a silica-gel column chromatography (ethyl acetate, Rf = 0.35) giving 103 mg (0.28 mmol, 47%) as a light yellow solid; mp 144–145 °C; purity (HPLC): 92%, tR = 19.4 min; [α]20D = −362.9 (c 1.01, DMSO). 1H NMR (400 MHz, DMSO-d6): 8.46 (d, J = 7.9, 1H, NH), 8.32 (s, 1H, H-2), 8.09 (s, 1H, H-5), 7.81 (d, J = 15.6, 1H, Hβ), 7.42–7.41 (m, 2H, Ph), 7.33–7.30 (m, 2H, Ph), 7.23–7.21 (m, 1H, Ph), 6.30 (d, J = 15.6, 1H, Hα), 5.53–5.48 (m, 1H, CH), 4.88–4.86 (m, 1H, OH), 4.17 (t, J = 4.8, 2H, CH2), 3.63 (q, J = 4.8, 2H, CH2), 1.56–1.55 (d, J = 7.0, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): 166.6, 165.6, 156.0, 155.3, 144.3, 137.3, 133.5, 128.3 (2C), 126.7, 126.1 (2C), 125.2, 118.6, 116.7, 66.1, 59.0, 49.1, 22.3; IR (cm−1): 3485 (w, br), 3422 (w, br), 3244 (w, br), 2357 (w), 1678 (m), 1619 (m), 1583 (s), 1540 (m), 1509 (m), 1454 (m), 1281 (m), 1178 (s), 1066 (m), 844 (s), 695 (m); HRMS (ASAP+, m/z): found 370.1226, calcd for C19H20N3O3S [M + H]+ 370.1225.Synthesis of (R,E)-3-(4-((1-phenylethyl)amino)thieno[2,3-d]pyrimidin-6-yl)acrylamide (7)
Methyl (R,E)-3-(4-((1-phenylethyl)amino)thieno[2,3-d]pyrimidin-6-yl)acrylate (2) (202 mg, 0.597 mmol) was added to a pressure tube and dissolved in 25% aq. ammonia (15 mL, 200 mmol) and THF (4 mL), and stirred at 30 °C for 96 h. The product was extracted with CH2Cl2 (3 × 15 mL) and dried with brine (10 mL) and then over anhydrous Na2SO4. After concentration, the crude product (102 mg) was crystallized from acetonitrile (7 mL) resulting in 30 mg (0.093 mmol, 16%) of white needle crystals; mp 246–251 °C (decomp.); purity (HPLC): 98%, tR = 17.3 min; [α]20D = −396.7 (c 0.97, DMSO). 1H NMR (400 MHz, DMSO-d6): 8.40 (d, J = 7.8, 1H, NH), 8.29 (s, 1H, H-2), 7.95 (s, 1H, H-5), 7.62 (s, 1H, NH), 7.54 (d, J = 15.4, 1H, Hβ), 7.42–7.40 (m, 2H, Ph), 7.34–7.30 (m, 2H, Ph), 7.24–7.20 (m, 1H, Ph), 7.20 (s, 1H, NH), 6.41 (d, J = 15.4, 1H, Hα), 5.54–5.47 (m, 1H, CH), 1.55 (d, J = 7.0, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): 167.4, 165.8, 155.8, 154.9, 144.5, 134.7, 132.2, 128.3 (2C), 126.7, 126.1 (2C), 123.5, 123.2, 116.7, 49.1, 22.4; IR (cm−1): 3447 (m), 3276 (m, br), 3071 (w, br), 1667 (s), 1587 (m), 1573 (m), 1499 (m), 1309 (s), 1106 (m), 950 (s), 777 (m), 697 (m); HRMS (ASAP+, m/z): found 325.1121, calcd for C17H17N4OS [M + H]+ 325.1123.Synthesis of (R,E)-N,N-dimethyl-3-(4-((1-phenylethyl)amino)thieno-[2,3-d]pyrimidin-6-yl)acrylamide (8)
Compound 8 was prepared as described for 5, but reacting (R)-6-bromo-N-(1-phenylethyl)thieno[2,3-d]-pyrimidin-4-amine (1) with N,N-dimethylacrylamide. The crude product (381 mg) was first purified by silica-gel column chromatography (tetrahydrofuran/n-pentane, 2/3) yielded 8 (234 mg) as a light yellow oil. The oil solidified upon addition of diethyl ether followed by evaporation. The material was dissolved in hot CH2Cl2 (8 mL) and crystallised by cooling giving 57 mg (0.16 mmol, 27%) of a white solid; mp 154–155 °C; purity (HPLC): 99%, tR = 19.1 min; [α]20D = −396.6 (c 1.01, DMSO). 1H NMR (400 MHz, DMSO-d6): 8.37 (d, J = 7.9, 1H, NH), 8.29 (s, 1H, H-2), 7.97 (s, 1H, H-5), 7.60 (d, J = 15.0, 1H, Hβ), 7.42–7.40 (m, 2H, Ph), 7.34–7.30 (m, 2H, Ph), 7.24–7.20 (m, 1H, Ph), 6.88 (d, J = 15.0, 1H, Hα), 5.54–5.47 (m, 1H, CH), 3.14 (s, 3H, CH3), 2.94 (s, 3H, CH3), 1.55 (d, J = 7.0, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): 166.2, 164.7, 155.9, 154.9, 144.5, 134.9, 133.9, 128.3 (2C), 126.7, 126.1 (2C), 123.3, 119.5, 116.6, 49.1, 36.9, 35.4, 22.4; IR (cm−1): 3262 (w, br), 1644 (m), 1574 (s), 1506 (m), 1305 (m), 1263 (m), 1137 (m), 1104 (m), 688 (m), 557 (m); HRMS (ASAP+, m/z): found 353.1431, calculated for C19H21N4OS [M + H]+ 353.1431.In vitro EGFR (ErbB1) inhibitory potency
The compounds were supplied in a 10 mM DMSO solution, and enzymatic EGFR (ErbB1) inhibition potency was determined by Invitrogen (LifeTechnology) using their Z′-LYTE® assay technology.32 All compounds were first tested for their inhibitory activity at 100 nM in duplicates. The potency observed at 100 nM was used to set starting point of the IC50 titration cure, in which three levels were used 100, 1000 or 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM. The IC50 values reported are based on the average of at least 2 titration curves (minimum 20 data points), and were calculated from activity data with a four parameter logistic model using SigmaPlot (Windows Version 12.0 from Systat Software, Inc.). Unless stated otherwise the ATP concentration used was equal to KM. The average standard deviation for single point measurements were <4%. The inhibitory potency towards EGFR mutants was determined in the same way.
000 nM. The IC50 values reported are based on the average of at least 2 titration curves (minimum 20 data points), and were calculated from activity data with a four parameter logistic model using SigmaPlot (Windows Version 12.0 from Systat Software, Inc.). Unless stated otherwise the ATP concentration used was equal to KM. The average standard deviation for single point measurements were <4%. The inhibitory potency towards EGFR mutants was determined in the same way.
Molecular modelling
The X-ray crystal structures of the protein 2J6M (Wild-type EGFR) were prepared using the protein preparation wizard, which is part of the Maestro software package (Schrödinger Release 2016-4: Glide, Schrödinger, LLC, New York, NY, 2016). Bond orders and formal charges were added for het-groups, and hydrogens were added to all atoms in the system. Water molecules beyond 5 Å from het-groups were removed. To alleviate steric clashes that may exist in the original PDB structures, an all-atom constrained minimization was carried out using the OPLS-3 force field. The minimization was terminated when the energy converged or the RMSD reached a maximum cutoff of 0.30 Å. The resulting protein structures were used in the following docking study. Ligands were drawn using the 2D sketcher within the Maestro package and were prepared using LigPrep (LigPrep, v2.2; Schrödinger, LLC). For the computational investigation of the receptor–inhibitor structures, the energy minimized structures of 2J6M and ligands were subsequently docked using Glide (Schrödinger Release 2016-4: Glide, Schrödinger, LLC, New York, NY, 2016). Selected low-energy protein–ligand poses were carried forward for 10 ns dynamic simulations. For dynamic simulation, the best poses from docking were used as starting points when building the model systems. Dynamic simulations were conducted for 10 ns simulation time using Maestros Desmond suite33 the OPLS-3 force field and a TIP4P solvent model. Briefly, this was performed by putting the docked protein–ligand complex inside a minimized solvent box and adding ions (Na+ or Cl−) in order to have an electrical neutral system. Finally, NaCl was added to a total concentration of 0.15 M, which is approximately the physiological concentration of monovalent ions. This gave normally a system of approximately 39000 atoms. Molecular dynamics were then calculated on these systems using the isothermal–isobaric (NPT) ensemble at 300 K and 1.01325 bar. Trajectory analysis were performed using Desmond's Simulation Interactions Diagram tool and all the graphical pictures were made using Maestro.
Kinase panel
The compounds were supplied in a 10 mM DMSO solution, and enzymatic kinase inhibition potency was determined by Invitrogen (LifeTechnology) using their Z′-LYTE® assay technology,32 at 500 nM in duplicates. ATP concentration used was equal to KM, except when this service was not provided and other concentrations had to be used.Conclusions
Undesirable physical–chemical properties can prohibit development of potent molecules to drugs. In the thienopyrimidine series of EGFR inhibitors, the 6-phenyl group does not contribute much to activity, but rather acts as a framework for grafting other activity inducing substituent. Thus, finding substitutes for the phenyl core is highly desirable. It was envisioned that substituted alkenes could be employed in this regard, which triggered a study of the Heck reaction. Palladium catalysed couplings between (R)-6-bromo-N-(1-phenylethyl)thieno[2,3-d]pyrimidin-4-amine and various acrylates were highly dependent on all parameters evaluated in line with past observations. Bis(tri-tert-butylphosphine)palladium(0) was found to be the most suited catalyst with N,N-dimethylacetamide as reaction medium. Other solvents could also be used, but conversion rate is lowered when substituting N,N-dicyclohexylmethylamine as base or excluding tetrabutylammonium chloride as additive. The conversion rate was also affected by acrylate structure and reaction with acrylamide was prohibited. The different reactions conditions gave a variable trans/cis ratio of product. However, the major challenge was that the products undergo a light induced trans–cis isomerization, which complicates purification and lower yield of the trans product. The products were however stable in the dark, allowing for bioevaluation of these compounds. Profiling towards EGFR and a panel of 50 kinases indicate that acrylate substituents grafted on thieno[2,3-d]pyrimidines is an attractive scaffold for identification of potent and highly selective EGFR inhibitors. These leads also possess promising physical chemical properties. Provided a way is found to make these structures configurational stable, highly efficient EGFR inhibitors could be developed.Acknowledgements
Susana Villa Gonzalez is thanked for the HRMS experiments, and Roger Aarvik are acknowledged for technical support. Anders Jahres Foundation is thanked for financial support.Notes and references
- W. Pao and J. Chmielecki, Nat. Rev. Cancer, 2010, 10, 760–774 CrossRef CAS PubMed
.
- M. R. Brewer, C. H. Yun, D. Lai, M. A. Lemmon, M. J. Eck and W. Pao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3595–E3604 CrossRef CAS PubMed
- H. J. Lee, A. N. Seo, E. J. Kim, M. H. Jang, Y. J. Kim, J. H. Kim, S. W. Kim, H. S. Ryu, I. A. Park, S. A. Im, G. Gong, K. H. Jung, H. J. Kim and S. Y. Park, Br. J. Cancer, 2015, 112, 103–111 CrossRef CAS PubMed
- N. Cook, K. K. Frese and M. Moore, Gastrointest. Cancer: Targets Ther., 2014, 4, 23–37 Search PubMed
- S. N. Li and H. Q. Li, Expert Opin. Ther. Pat., 2014, 24, 309–321 CrossRef CAS PubMed
- Y. J. Liu, C. M. Zhang and Z. P. Liu, Anti-Cancer Agents Med. Chem., 2012, 12, 391–406 CrossRef CAS
- P. Traxler, P. R. Allegrini, R. Brandt, J. Brueggen, R. Cozens, D. Fabbro, K. Grosios, H. A. Lane, P. McSheehy, J. Mestan, T. Meyer, C. Tang, M. Wartmann, J. Wood and G. Caravatti, Cancer Res., 2004, 64, 4931–4941 CrossRef CAS PubMed
- Y. W. Park, M. N. Younes, S. A. Jasser, O. G. Yigitbasi, G. Zhou, C. D. Bucana, B. N. Bekele and J. N. Myers, Clin. Cancer Res., 2005, 11, 1963–1973 CrossRef CAS PubMed
- P. LoRusso, K. Venkatakrishnan, E. G. Chiorean, D. Noe, J. T. Wu, S. Sankoh, M. Corvez and E. A. Sausville, Invest. New Drugs, 2014, 32, 160–170 CrossRef CAS PubMed
- T. Doi, H. Takiuchi, A. Ohtsu, N. Fuse, M. Goto, M. Yoshida, N. Dote, Y. Kuze, F. Jinno, M. Fujimoto, T. Takubo, N. Nakayama and R. Tsutsumi, Br. J. Cancer, 2012, 106, 666–672 CrossRef CAS PubMed
- J. Han, S. Henriksen, K. G. Nørsett, E. Sundby and B. H. Hoff, Eur. J. Med. Chem., 2016, 124, 583–607 CrossRef CAS PubMed
- Y. H. Peng, H. Y. Shiao, C. H. Tu, P. M. Liu, J. T.-A. Hsu, P. K. Amancha, J. S. Wu, M. S. Coumar, C. H. Chen, S. Y. Wang, W. H. Lin, H. Y. Sun, Y. S. Chao, P. C. Lyu, H. P. Hsieh and S. Y. Wu, J. Med. Chem., 2013, 56, 3889–3903 CrossRef CAS PubMed
- J. Han, S. J. Kaspersen, S. Nervik, K. G. Nørsett, E. Sundby and B. H. Hoff, Eur. J. Med. Chem., 2016, 119, 2778–2799 CrossRef PubMed
- S. Bugge, A. F. Buene, N. Jurisch-Yaksi, I. U. Moen, E. M. Skjoensfjell, E. Sundby and B. H. Hoff, Eur. J. Med. Chem., 2016, 107, 255–274 CrossRef CAS PubMed
- T. Beckers, A. Sellmer, E. Eichhorn, H. Pongratz, C. Schaechtele, F. Totzke, G. Kelter, R. Krumbach, H. H. Fiebig, F. D. Boehmer and S. Mahboobi, Bioorg. Med. Chem., 2012, 20, 125–136 CrossRef CAS PubMed
- S. Bugge, S. J. Kaspersen, S. Larsen, U. Nonstad, G. Bjørkøy, E. Sundby and B. H. Hoff, Eur. J. Med. Chem., 2014, 75, 354–374 CrossRef CAS PubMed
- N. A. Meanwell, Chem. Res. Toxicol., 2011, 24, 1420–1456 CrossRef CAS PubMed
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed
- S. Bugge, I. U. Moen, K. O. Kragseth Sylte, E. Sundby and B. H. Hoff, Eur. J. Med. Chem., 2015, 94, 175–194 CrossRef CAS PubMed
- P. M. Murray, J. F. Bower, D. K. Cox, E. K. Galbraith, J. S. Parker and J. B. Sweeney, Org. Process Res. Dev., 2013, 17, 397–405 CrossRef CAS
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed
- C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254–278 CrossRef CAS
- C. S. Consorti, F. R. Flores and J. Dupont, J. Am. Chem. Soc., 2005, 127, 12054–12065 CrossRef CAS PubMed
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC
- E. Amadio, A. Scrivanti, M. Bortoluzzi, M. Bertoldini, V. Beghetto, U. Matteoli and G. Chessa, Inorg. Chim. Acta, 2013, 405, 188–195 CrossRef CAS
- M. B. Ibrahim, R. Suleiman, M. Fettouhi and B. El Ali, Transition Met. Chem. (Dordrecht, Neth.), 2017, 42, 1–8 CrossRef CAS
- H. Wu, C. Jin, G. L. Huang, L. J. Wang, J. L. Jiang and L. Y. Wang, Sci. China: Chem., 2011, 54, 951–956 CrossRef CAS
- F. Dehghani Firuzabadi, Z. Asadi and F. Panahi, RSC Adv., 2016, 6, 101061–101070 RSC
- S. Jadhav, A. Jagdale, S. Kamble, A. Kumbhar and R. Salunkhe, RSC Adv., 2016, 6, 3406–3420 RSC
- N. Nishiwaki, S. Hamada, T. Watanabe, S. Hirao, J. Sawayama, H. Asahara, K. Saigo, T. Kamata and M. Funabashi, RSC Adv., 2015, 5, 4463–4467 RSC
- P. Knesl, D. Roeseling and U. Jordis, Molecules, 2006, 11, 286–297 CrossRef CAS PubMed
- B. A. Pollok, B. D. Hamman, S. M. Rodems and L. R. Makings, Optical probes and assays, WO 2000066766 A1, 2000
- v. Desmond Molecular Dynamics System, D. E. Shaw Research, Maestro-Desmond Interoperability Tools, version 3.9, Schrödinger, New York, NY, 2014 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Contains kinase screen data and molecular docking and dynamics figures. See DOI: 10.1039/c7ra01961k |
| ‡ Current address: Pronova BioPharma Norge AS, NO-3202 Sandeford, Norway. |
| § Current address: Dynea AS, Svelleveien 33, NO-2004 Lillestrøm, Norway. |
| This journal is © The Royal Society of Chemistry 2017 |