
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bicyclic lactones and racemic mixtures of dimeric styrylpyrones from the leaves of Miliusa velutina†
N. Wongsaab,
K. Kanokmedhakul *a,
J. Boonmakc,
S. Youngmec and
S. Kanokmedhakula
*a,
J. Boonmakc,
S. Youngmec and
S. Kanokmedhakula
aNatural Products Research Unit, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: kwanjai@kku.ac.th
bDivision of Chemistry, Faculty of Science, Udon Thani Rajabhat University, Udon Thani 41000, Thailand
cMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand
First published on 11th May 2017
Abstract
A unique class of eight bicyclic lactones with a C18 carbon architecture, named velutinones A–H (1–8), three new dimeric styrylpyrones, velutinindimers A–C (9–11), five known compounds, the kawapyrone, yangonin (12), three flavonoids (13–15), and an acetogenin, cananginone H (16) were isolated from the leaves of Miliusa velutina. The absolute configurations of 2 and 5 were assigned by Mosher's method, whereas ECD, optical rotations, and X-ray crystallographic analysis indicated the racemic nature of compounds 10 and 11. Compounds 2–4 and 7–11 showed antimalarial activity with IC50 values in the range of 5.4–10.0 μM. Moreover, 1–4 and 6–8 displayed cytotoxicity against the KB, MCF7, and NCI-H187 cancer cell lines and Vero cell lines with IC50 values in the range of 4.0–24.1 μM.
1. Introduction
Miliusa velutina (Dunal) Hook. f. & Thomson belongs to the family Annonaceae. This plant is found widely in Thailand with local names ‘‘Khang hua mu” or “Kong kang”. A water decoction of the wood is used traditionally as a tonic and an aphrodisiac.1 The genus Miliusa comprises ca. 50 species distributed from India, South East Asia, to Australia. At least 19 species of Miliusa, have been found in Thailand.2,3 Eight of the Miliusa genera growing worldwide have been investigated for their phytochemistry and biological activities.4–19 Among these species, a Thai medicinal plant, M. velutina, has been shown to contain the acetogenin, goniothalamusin,17 an aporphine alkaloid, (+)-isocorydine α-N-oxide,18 and four alkaloids, reticuline, liriodenine, norcorydine, and isocorydine.19 Recently, the isolation and characterization of the linear acetogenins, cananginones A–I from the stem bark of M. velutina were reported.20,21 In a continued investigation of this plant, the crude n-hexane and EtOAc extracts from the leaves of this plant were found to exhibit activity towards Mycobacterium tuberculosis with 99.6 and 98.9% inhibition at a concentration of 50 μg mL−1, respectively. Herein the isolation, structural identification, and bioactivities of eight new bicyclic lactones (1–8), and three new cyclobutane dimers (9–11), as well as five known compounds (12–16) from the leaves of M. velutina are discussed (Fig. 1).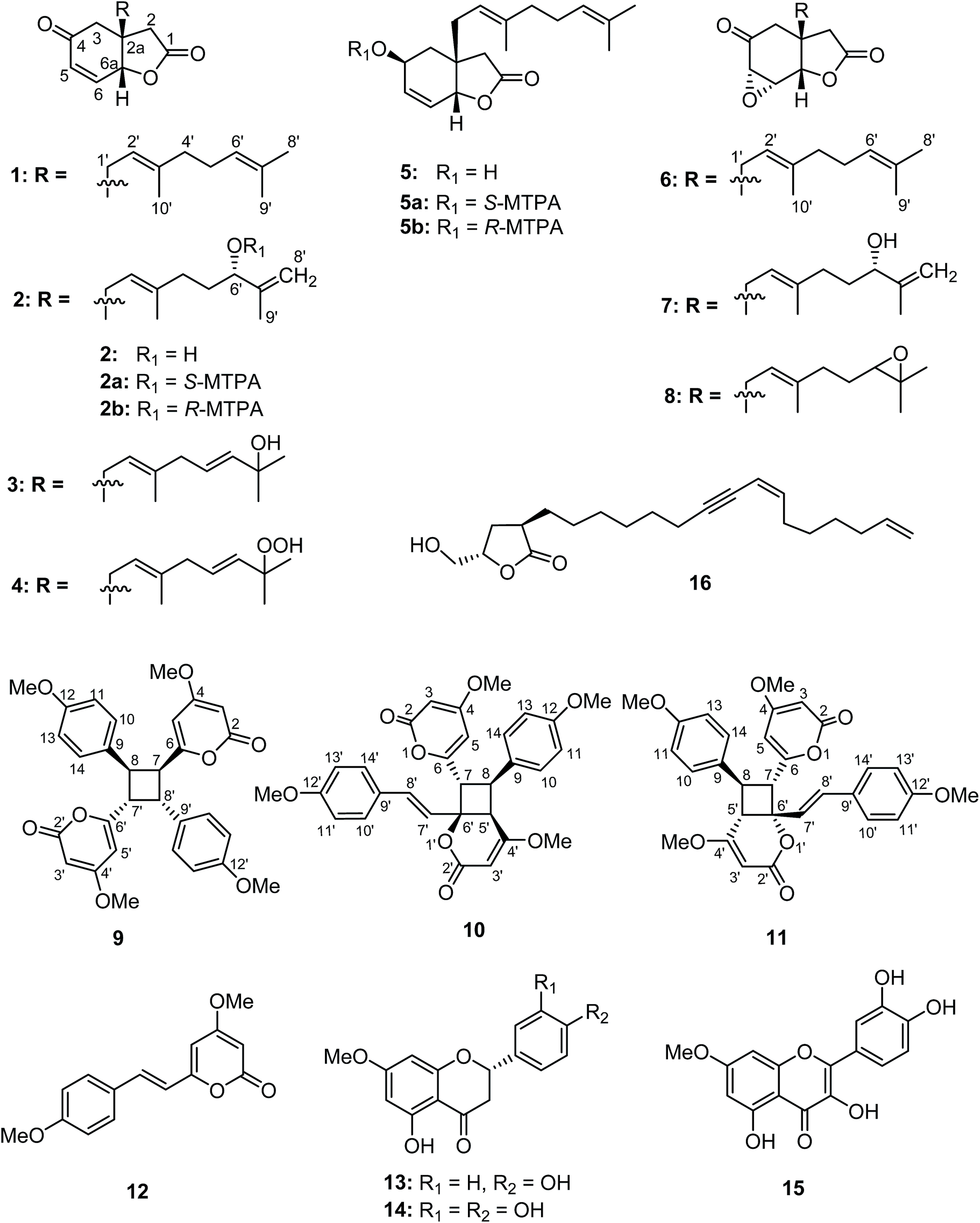 | ||
| Fig. 1 Structures of isolated compounds 1–16. | ||
2. Experimental section
2.1. General procedures
A Gallenkamp melting point apparatus (0–300 °C, 4 °C min−1, uncorrected) was used to measure melting points. Optical rotations were recorded on a JASCO P-1020 polarimeter and ECD spectra were recorded on a JASCO J-810 apparatus. UV spectra were recorded using an Agilent 8453 UV-visible spectrophotometer. FTIR spectra were recorded on a Bruker Tensor 27 spectrophotometer. The NMR spectra were acquired on a Varian Mercury Plus 400 spectrometer. HRESITOFMS spectra were recorded on a Micromass Q-TOF-2 mass spectrometer. Flash column chromatography (FCC) was performed on MERCK silica gel 60 (230–400 mesh) and LiChroprep® RP-18 (40–63 μm). Thin layer chromatography (TLC) was performed using precoated MERCK silica gel 60 PF254 and RP-18 F254S.2.2. Plant material
The leaves of M. velutina were collected in Nam Som district, Udon Thani province, Thailand in November 2010. The identification of the plant was performed by Prof. Pranom Chantaranothai. A voucher specimen (S. Kanokmedhakul-17) was deposited at the herbarium of the Department of Biology, Faculty of Science, Khon Kaen University, Thailand. It should be noted that, in our previous report20 on this plant, it was collected in different locations and was misidentified as Cananga latifolia because of incomplete material for species identification. It was identified based on the vegetative part (leaf and stem). Later in 2015, plants with fruit from both locations were collected and the samples identified and confirmed as Miliusa velutina, and this has been corrected as an erratum.21 Since the work has been published for some time, to avoid any future confusion, the names of the new compounds have not been changed.2.3. Extraction and isolation
The dried, milled leaves of M. velutina (2.5 kg) were extracted with n-hexane (3 × 10 L) and EtOAc (3 × 10 L) to give 127 g (5.1%) and 93 g (3.7%) of n-hexane and EtOAc extracts, respectively. The n-hexane extract was separated using silica gel flash column chromatography (FCC), eluted with a gradient system of n-hexane–EtOAc (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 90:10, 85:15, 70:30, 50:50, 40:60, 20:80, 0:100) and EtOAc–MeOH, EtOAc–MeOH (80:20, 50:50, 30:70, 15:85, 0:100) to afford 5 fractions, LH1–LH5. Fraction LH3 (48.2 g) was chromatographed on silica gel FCC, eluting with n-hexane–acetone (4:1) to yield 3 subtractions, LH3.1–LH3.3. Subfraction LH3.2 (32.8 g) was purified by LiChroprep RP-18 column chromatography, eluted with MeOH–H2O (4:1) to give 6 fractions, LH3.2.1–LH3.2.6. Further purification of subfraction LH3.2.1 (0.48 g) by silica gel FCC, eluting with n-hexane–EtOAc (4:1) gave compounds 4 (53.8 mg) and 2 (23 mg) as colorless viscous liquids. Subfraction LE3.2.2 (23.6 g) was separated on silica gel FCC, eluting with n-hexane–EtOAc (85:15) to give 1 (20.3 g) as a colorless viscous liquid. Subfraction LH3.2.3 (1.7 g) was chromatographed on silica gel FCC, eluting with n-hexane–EtOAc (85:15) to give 1 (1.59 g) and 6 (57.3 mg) as colorless viscous liquids. Subfraction LH3.3 (9.6 g) was purified by silica gel FCC, eluting with n-hexane–acetone (7:1) to afford 5 subfractions, LH3.3.1–LH3.3.5. Further purification of subfraction LH3.3.1 (0.28 g) by silica gel FCC, eluted with n-hexane–EtOAc (3:1) gave 8 (49.8 mg) as a colorless viscous liquid. Subfraction LE3.3.2 (0.49 g) was chromatographed on silica gel FCC, eluted with n-hexane–EtOAc (1:1) to afford 7 (408.5 mg) as a colorless viscous liquid. Subfraction LH3.4.4 (0.68 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (75:35) to give an extra amount of 2 (551.7 mg). Subfraction LH3.4.5 (0.56 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (1:1) to give 3 (507.4 mg) as a colorless viscous liquid. Fraction LH4 (6.7 g) was purified by silica gel FCC, eluting with acetone-CH2Cl2 (1:4) to yield 3 subfractions, LH4.1–LH4.3. Subfraction LH4.3 (3.0 g) was chromatographed on silica gel FCC, eluted with n-hexane–acetone (7:3) to give 3 subfractions, LH4.3.1–LH4.3.3. Solid in subfraction LH4.3.3 (1.6 g) was crystallized from CH2Cl2-n-hexane to give 12 (332 mg) as a yellow solid. Fraction LH5 (1.9 g) was separated on silica gel FCC, eluted with n-hexane–acetone (7:3) to give 2 subfractions, LH5.1–LH5.2. Solid in subfraction LH5.2 (0.62 g) was crystallized from MeOH-n-hexane to give 10 (30 mg) as colorless needles. The EtOAc extract was separated over silica gel FCC, using gradient elution with n-hexane–EtOAc (80:20 to 0:100) and EtOAc–MeOH (80:20 to 0:100) to afford 6 fractions, LE1–LE6. Fraction LE2 (6.3 g) was then purified by silica gel FCC, eluting with n-hexane–acetone (4:1) to give 2 subfractions, LE2.1–LE2.2. Further purification of subfraction LE2.2 (0.74 g) by silica gel FCC, eluting with n-hexane–EtOAc (4:1) gave 13 (552 mg) as a white solid. Fraction LE3 (3.6 g) was purified by silica gel FCC, eluting with n-hexane–acetone (3:1) to give 2 subfractions, LE3.1–LE3.2. Subfraction LE3.2 (0.31 g) was further separated by LiChroprep RP-18 column chromatography, eluting with MeOH–H2O (6:1) to give 16 (17.6 mg) as a colorless viscous liquid. Fraction LE4 (12.0 g) was separated by silica gel FCC, eluting with n-hexane–acetone (7:3) to give 4 subfractions, LE4.1–LE4.4. Subfraction LE4.1 (2.6 g) was purified by LiChroprep RP-18 column chromatography, eluting with MeOH–H2O (4:1) to give 2 subfractions, LE4.1.1–LE4.1.2. Subfraction LE4.1.1 (1.8 g) was separated on silica gel FCC, eluting with MeOH–CH2Cl2 (1:19) to give 3 subfractions, LE4.1.1.1–LE4.1.1.3. Solid in subfraction LH4.1.1.1 was crystallized from MeOH to give 12 (21 mg) as a yellow solid. Subfraction LE4.1.1.2 (1.4 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (1:1) to give an additional amount of 2 (214 mg). Subfraction LE4.1.2 (0.67 g) was purified by silica gel FCC, eluting with n-hexane–acetone (7:3) to give 5 (25.8 mg) as a colorless viscous liquid. Subfraction LE4.2 (3.0 g) was purified by silica gel FCC, eluting with n-hexane–acetone (7:3) to give 2 subfractions, LE4.2.1–LE4.2.2. Solid in subfraction LH4.2.1 was crystallized from MeOH to give an additional amount of 12 (105.3 mg). Solid in subfraction LH4.2.2 (0.54 g) was crystallized from MeOH to give 14 (30 mg) as a white solid. Subfraction LE4.3 (2.9 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (1:1), to give an additional amount of 3 (174 mg). Solid in subfraction LE4.4 (0.12 g) was crystallized from DMF–CH2Cl2 to give 15 (26 mg) as a yellow solid. Fraction LE5 (8.4 g) was purified by silica gel FCC, eluting with MeOH–CH2Cl2 (3:97) to give 2 subfractions, LE5.1–LE5.2. Solid in subfraction LE5.1 (1.6 g) was crystallized from MeOH–CH2Cl2 to give 10 (1.08 g) as colorless needles and the filtrate was further purified by silica gel FCC, eluting with EtOAc–CH2Cl2 (15:85) to yield 9 (22 mg) as a white solid and 11 (75 mg) as colorless needles. Solids in subfractions LE5.2 (6.2 g) and LE6 (10.9 g) were crystallized from DMF-CH2Cl2 to give 15 (931 mg) as a yellow solid.
:3); [α]23D −68.0 (c 0.20, CHCl3); ECD (80 μM, MeOH) λmax (Δε) 215 (−19.02) nm; IR (ATR) νmax 2966, 2916, 2854, 1781, 1685, 1440, 1418, 1383, 1161, and 997 cm−1; for 1H and 13C NMR spectroscopic data, see Table 1; HRESITOFMS m/z 311.1611 [M + Na]+ (calcd for C18H24O3 + Na, 311.1618).
:0, 90:10, 85:15, 70:30, 50:50, 40:60, 20:80, 0:100) and EtOAc–MeOH, EtOAc–MeOH (80:20, 50:50, 30:70, 15:85, 0:100) to afford 5 fractions, LH1–LH5. Fraction LH3 (48.2 g) was chromatographed on silica gel FCC, eluting with n-hexane–acetone (4:1) to yield 3 subtractions, LH3.1–LH3.3. Subfraction LH3.2 (32.8 g) was purified by LiChroprep RP-18 column chromatography, eluted with MeOH–H2O (4:1) to give 6 fractions, LH3.2.1–LH3.2.6. Further purification of subfraction LH3.2.1 (0.48 g) by silica gel FCC, eluting with n-hexane–EtOAc (4:1) gave compounds 4 (53.8 mg) and 2 (23 mg) as colorless viscous liquids. Subfraction LE3.2.2 (23.6 g) was separated on silica gel FCC, eluting with n-hexane–EtOAc (85:15) to give 1 (20.3 g) as a colorless viscous liquid. Subfraction LH3.2.3 (1.7 g) was chromatographed on silica gel FCC, eluting with n-hexane–EtOAc (85:15) to give 1 (1.59 g) and 6 (57.3 mg) as colorless viscous liquids. Subfraction LH3.3 (9.6 g) was purified by silica gel FCC, eluting with n-hexane–acetone (7:1) to afford 5 subfractions, LH3.3.1–LH3.3.5. Further purification of subfraction LH3.3.1 (0.28 g) by silica gel FCC, eluted with n-hexane–EtOAc (3:1) gave 8 (49.8 mg) as a colorless viscous liquid. Subfraction LE3.3.2 (0.49 g) was chromatographed on silica gel FCC, eluted with n-hexane–EtOAc (1:1) to afford 7 (408.5 mg) as a colorless viscous liquid. Subfraction LH3.4.4 (0.68 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (75:35) to give an extra amount of 2 (551.7 mg). Subfraction LH3.4.5 (0.56 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (1:1) to give 3 (507.4 mg) as a colorless viscous liquid. Fraction LH4 (6.7 g) was purified by silica gel FCC, eluting with acetone-CH2Cl2 (1:4) to yield 3 subfractions, LH4.1–LH4.3. Subfraction LH4.3 (3.0 g) was chromatographed on silica gel FCC, eluted with n-hexane–acetone (7:3) to give 3 subfractions, LH4.3.1–LH4.3.3. Solid in subfraction LH4.3.3 (1.6 g) was crystallized from CH2Cl2-n-hexane to give 12 (332 mg) as a yellow solid. Fraction LH5 (1.9 g) was separated on silica gel FCC, eluted with n-hexane–acetone (7:3) to give 2 subfractions, LH5.1–LH5.2. Solid in subfraction LH5.2 (0.62 g) was crystallized from MeOH-n-hexane to give 10 (30 mg) as colorless needles. The EtOAc extract was separated over silica gel FCC, using gradient elution with n-hexane–EtOAc (80:20 to 0:100) and EtOAc–MeOH (80:20 to 0:100) to afford 6 fractions, LE1–LE6. Fraction LE2 (6.3 g) was then purified by silica gel FCC, eluting with n-hexane–acetone (4:1) to give 2 subfractions, LE2.1–LE2.2. Further purification of subfraction LE2.2 (0.74 g) by silica gel FCC, eluting with n-hexane–EtOAc (4:1) gave 13 (552 mg) as a white solid. Fraction LE3 (3.6 g) was purified by silica gel FCC, eluting with n-hexane–acetone (3:1) to give 2 subfractions, LE3.1–LE3.2. Subfraction LE3.2 (0.31 g) was further separated by LiChroprep RP-18 column chromatography, eluting with MeOH–H2O (6:1) to give 16 (17.6 mg) as a colorless viscous liquid. Fraction LE4 (12.0 g) was separated by silica gel FCC, eluting with n-hexane–acetone (7:3) to give 4 subfractions, LE4.1–LE4.4. Subfraction LE4.1 (2.6 g) was purified by LiChroprep RP-18 column chromatography, eluting with MeOH–H2O (4:1) to give 2 subfractions, LE4.1.1–LE4.1.2. Subfraction LE4.1.1 (1.8 g) was separated on silica gel FCC, eluting with MeOH–CH2Cl2 (1:19) to give 3 subfractions, LE4.1.1.1–LE4.1.1.3. Solid in subfraction LH4.1.1.1 was crystallized from MeOH to give 12 (21 mg) as a yellow solid. Subfraction LE4.1.1.2 (1.4 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (1:1) to give an additional amount of 2 (214 mg). Subfraction LE4.1.2 (0.67 g) was purified by silica gel FCC, eluting with n-hexane–acetone (7:3) to give 5 (25.8 mg) as a colorless viscous liquid. Subfraction LE4.2 (3.0 g) was purified by silica gel FCC, eluting with n-hexane–acetone (7:3) to give 2 subfractions, LE4.2.1–LE4.2.2. Solid in subfraction LH4.2.1 was crystallized from MeOH to give an additional amount of 12 (105.3 mg). Solid in subfraction LH4.2.2 (0.54 g) was crystallized from MeOH to give 14 (30 mg) as a white solid. Subfraction LE4.3 (2.9 g) was purified by silica gel FCC, eluting with n-hexane–EtOAc (1:1), to give an additional amount of 3 (174 mg). Solid in subfraction LE4.4 (0.12 g) was crystallized from DMF–CH2Cl2 to give 15 (26 mg) as a yellow solid. Fraction LE5 (8.4 g) was purified by silica gel FCC, eluting with MeOH–CH2Cl2 (3:97) to give 2 subfractions, LE5.1–LE5.2. Solid in subfraction LE5.1 (1.6 g) was crystallized from MeOH–CH2Cl2 to give 10 (1.08 g) as colorless needles and the filtrate was further purified by silica gel FCC, eluting with EtOAc–CH2Cl2 (15:85) to yield 9 (22 mg) as a white solid and 11 (75 mg) as colorless needles. Solids in subfractions LE5.2 (6.2 g) and LE6 (10.9 g) were crystallized from DMF-CH2Cl2 to give 15 (931 mg) as a yellow solid.
:3); [α]23D −68.0 (c 0.20, CHCl3); ECD (80 μM, MeOH) λmax (Δε) 215 (−19.02) nm; IR (ATR) νmax 2966, 2916, 2854, 1781, 1685, 1440, 1418, 1383, 1161, and 997 cm−1; for 1H and 13C NMR spectroscopic data, see Table 1; HRESITOFMS m/z 311.1611 [M + Na]+ (calcd for C18H24O3 + Na, 311.1618).
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a Overlap of the signals. | ||||||||
| 1 | 174.2 | 174.3 | 174.2 | 174.4 | ||||
| 2 | 38.9 | 2.48, d (17.4), Hα/2.39, d (17.4), Hβ | 38.9 | 2.43, d (17.8), Hα/2.39, d (17.8), Hβ | 39.0 | 2.40, s | 39.0 | 2.42, s |
| 2a | 44.7 | 44.6 | 44.5 | 44.5 | ||||
| 3 | 42.6 | 2.52, s | 42.7 | 2.55, d (16.7), Hα/2.50, d (16.7), Hβ | 42.9 | 2.53, d (16.7), Hα/2.48, d (16.7), Hβ | 43.0 | 2.56, d (16.7), Hα/2.50, d (16.7), Hβ |
| 4 | 196.2 | 196.2 | 196.1 | 196.3 | ||||
| 5 | 131.2 | 6.17, dd (10.3, 1.2) | 131.2 | 6.13, dd (10.3, 1.2) | 131.2 | 6.12, dd (10.3, 1.2) | 131.2 | 6.18, dd (10.3, 1.2) |
| 6 | 141.4 | 6.75, dd (10.3, 3.3) | 141.4a | 6.74, dd (10.3, 3.3) | 141.3 | 6.74, dd (10.3, 3.3) | 141.4 | 6.76, dd (10.3, 3.3) |
| 6a | 77.8 | 4.87, dd (3.3, 1.2) | 77.9 | 4.87, d (3.3) | 77.9 | 4.86, dd, (3.3, 1.2) | 78.1 | 4.88, dd, (3.3, 1.2) |
| 1′ | 36.0 | 2.31, dd (14.4, 7.7), Ha/2.21, dd (14.4, 7.7), Hb | 36.2 | 2.31, dd (14.5, 7.7), Ha/2.22, dd (14.5, 7.7), Hb | 36.6 | 2.30, dd (14.4, 7.7), Ha/2.23, dd (14.4, 7.7), Hb | 36.8 | 2.32, dd (14.4, 7.7), Ha/2.26, dd (14.4, 7.7), Hb |
| 2′ | 116.9 | 5.10, td (7.7, 1.4) | 116.9 | 5.15, td (7.7, 1.4) | 117.5 | 5.11, td (7.7, 1.4) | 117.8 | 5.15, td (7.7, 1.4) |
| 3′ | 141.5 | 141.4a | 140.3 | 140.0 | ||||
| 4′ | 39.9 | 2.10–2.02, ma | 35.9 | 2.14–1.98, m | 42.4 | 2.68, d (6.5) | 42.5 | 2.73, d (5.5) |
| 5′ | 26.2 | 2.10–2.02, ma | 33.0 | 1.65–1.58, m | 123.8 | 5.51, dt (15.6, 6.6) | 128.0 | 5.62, dt (15.8, 6.0) |
| 6′ | 123.7 | 5.01, t (5.4) | 75.3 | 4.00, t (6.3) | 140.2 | 5.60, d, (15.6) | 135.9 | 5.56, d, (15.8) |
| 7′ | 131.9 | 147.3 | 70.4 | 81.8 | ||||
| 8′ | 25.7 | 1.65, s | 111.1 | 4.91, brs, Ha/4.82, t (1.4), Hb | 29.7 | 1.28, s | 24.3 | 1.31, s |
| 9′ | 17.7 | 1.58, s | 17.5 | 1.70, s | 29.7 | 1.28, s | 24.2 | 1.31, s |
| 10′ | 16.3 | 1.60, s | 16.4 | 1.61, s | 16.5 | 1.58, s | 16.6 | 1.60, s |
:1); [α]24D −61.2 (c 0.20, CHCl3); ECD (130 μM, MeOH) λmax (Δε) 215 (−28.90) nm; IR (ATR) νmax 3468, 2920, 2857, 1777, 1683, 1447, 1385, 1165, 1065, 995, and 990 cm−1; for 1H and 13C NMR spectroscopic data, see Table 1; HRESITOFMS m/z 305.1740 [M + H]+ (calcd for C18H24O4 + H+, 305.1747).:1); [α]23D −55.4 (c 0.20, CHCl3); ECD (50 μM, MeOH) λmax (Δε) 213 (−7.63) nm; IR (ATR) νmax 3447, 2972, 2929, 1778, 1682, 1481, 1385, 1155, and 975 cm−1; for 1H and 13C NMR spectroscopic data, see Table 1; HRESITOFMS m/z 327.1543 [M + Na]+ (calcd for C18H24O4 + Na, 327.1572).:1); [α]24D −58.4 (c 0.20, CHCl3); ECD (56 μM, MeOH) λmax (Δε) 213 (−9.74) nm; IR (ATR) νmax 3393, 2978, 2931, 1778, 1682, 1417, 1384, 1165, and 996 cm−1; for 1H and 13C NMR spectroscopic data, see Table 1; HRESITOFMS m/z 343.1472 [M + Na]+ (calcd for C18H24O5 + Na, 343.1521).:1); [α]23D −32.5 (c 0.20, CHCl3); ECD (62 μM, MeOH) λmax (Δε) 202 (−13.84) nm; IR (ATR) νmax 3442, 3030, 2967, 2921, 2856, 1771, 1668, 1446, 1419, 1377, 1328, 1167, 1069, 1030, and 990 cm−1; for 1H and 13C NMR spectroscopic data, see Table 2; HRESITOFMS m/z 291.1935 [M + H]+ (calcd for C18H26O3 + H+, 291.1955).
| No. | 5 | 6 | 7 | 8 | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a overlapping of the signals. | ||||||||
| 1 | 176.2 | 173.8 | 173.9 | 173.7 | ||||
| 2 | 37.1a | 2.38, d (17.4), Hα/2.19, d (17.4), Hβ | 38.3 | 2.32, d (17.4), Hα/2.28, d (17.4), Hβ | 38.4 | 2.30–2.24, ma | 38.4 | 2.30–2.24, ma |
| 2a | 42.3 | 47.5 | 47.4 | 47.4 | ||||
| 3 | 37.2a | 2.25–2.17, ma, Hα/1.58–1.51, m, Hβ | 39.9a | 2.86, d (13.9), Hα/2.14, d (13.9), Hβ | 39.9 | 2.82, d (13.9), Hα/2.11, d (13.9), Hβ | 39.9 | 2.84, d (13.9), Hα/2.12, d (13.9), Hβ |
| 4 | 63.7 | 4.31, m | 204.6 | 204.5 | 204.5 | |||
| 5 | 135.2 | 5.97, brd (10.2) | 58.6 | 3.65, d (3.6) | 58.5 | 3.62, d (3.6) | 58.5 | 3.62, d (3.7) |
| 6 | 125.2 | 6.75–5.65, m | 54.9 | 3.37, d (3.6) | 54.8 | 3.34, d (3.6) | 54.8 | 3.34, d (3.7) |
| 6a | 80.1 | 4.62, m | 76.6 | 4.71, d (3.6) | 76.6 | 4.68, s | 76.5 | 4.69, s |
| 1′ | 36.5 | 2.25–2.09, m | 36.9 | 2.29, dd (14.4, 7.7), Ha/2.20, dd (14.4, 7.7), Hb | 36.8 | 2.25–2.09, ma | 36.9a | 2.32–2.09, ma |
| 2′ | 117.5 | 5.12, td (7.7, 1.4) | 116.2 | 5.08, td (7.7, 1.4) | 116.3 | 5.10, td (7.8, 1.4) | 116.7 | 5.14, td (7.7, 1.4) |
| 3′ | 140.2 | 141.8 | 141.6 | 141.1 | ||||
| 4′ | 39.8 | 2.08–1.98, ma | 39.9a | 2.10–2.02, ma | 35.8 | 2.01–1.96, ma | 36.7a | 2.32–2.09, ma |
| 5′ | 26.3 | 2.08–1.98, ma | 26.2 | 2.10–2.02, ma | 32.8 | |||
| 6′ | 123.8 | 5.01, m | 123.6 | 5.04, t (6.6) | 75.1 | 1.64–1.55, m | 27.2 | 1.69–1.53, m |
| 7′ | 131.6 | 132.0 | 147.3 | 3.98, t (6.0) | 63.7 | 3.65, t (6.7) | ||
| 8′ | 25.6 | 1.63, s | 25.7 | 1.68, s | 111.0 | 4.79, brs, Ha/4.89, t (0.8), Hb | 58.2 | |
| 9′ | 17.6 | 1.56, s | 17.7 | 1.59, s | 17.5 | 1.68, s | 24.7 | 1.27, s |
| 10′ | 16.2 | 1.58, s | 16.4 | 1.61, s | 16.4 | 1.60, s | 18.7 | 1.23, s |
:3); [α]23D −29.5 (c 0.20, CHCl3); ECD (72 μM, MeOH) λmax (Δε) 207 (+4.83) nm; IR (ATR) νmax 2967, 2917, 2855, 1789, 1719, 1423, 1377, 1347, 1161, 1035, 857, and 802 cm−1; for 1H and 13C NMR spectroscopic data, see Table 2; HRESITOFMS m/z 327.1562 [M + Na]+ (calcd for C18H24O4 + Na, 327.1567).:3); [α]23D −25.6 (c 0.20, CHCl3); ECD (94 μM, MeOH) λmax (Δε) 205 (+11.89) nm; IR (ATR) νmax 3481, 2939, 2858, 1783, 1718, 1651, 1422, 1347, 1164, 1033, and 902 cm−1 for 1H and 13C NMR spectroscopic data, see Table 2; HRESITOFMS m/z 321.1690 [M + H]+ (calcd for C18H24O5 + H+, 321.1697).:3); [α]24D −23.5 (c 0.20, CHCl3); ECD (75 μM, MeOH) λmax (Δε) 215 (+11.84) nm; IR (ATR) νmax 2962, 2924, 2855, 1785, 1719, 1423, 1378, 1163, and 1034 cm−1; for 1H and 13C NMR spectroscopic data, see Table 2; HRESITOFMS m/z 321.1690 [M + H]+ (calcd for C18H24O5 + H+, 321.1697).:1); [α]28D + 0.08 (c 0.63, MeOH–CHCl3, 3:1); ECD (26 μM, MeOH) λmax (Δε) 285 (0.00) nm; UV (MeOH) λmax (logε) 226 (4.53), 285 (4.20); IR (ATR) νmax 3088, 2944, 2837, 1715, 1643, 1611, 1564, 1513, 1455, 1409, 1249, 1180, 1034, and 818 cm−1; for 1H and 13C NMR spectroscopic data, see Table 3; HRESITOFMS m/z 539.1651 [M + Na]+ (calcd for C30H28O8 + Na, 539.1682).
| No. position | 9 | 10 | 11 | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 2 | 164.0 | 163.9 | 164.1 | |||
| 3 | 87.7 | 5.21, d (2.2) | 88.6 | 5.33, d (2.2) | 88.9 | 5.45, d (2.2) |
| 4 | 170.1 | 170.5 | 170.6 | |||
| 5 | 101.3 | 5.71, d (2.2) | 102.5 | 5.89, d (2.2) | 102.0 | 5.99, d (2.2) |
| 6 | 162.9 | 158.9 | 158.7 | |||
| 7 | 43.0 | 4.35, dd (10.0, 7.6) | 55.0 | 4.09, d (10.8) | 54.5 | 3.64, d (10.3) |
| 8 | 45.5 | 4.16, dd (10.0, 7.6) | 38.7 | 4.26, dd (10.8, 9.9) | 45.6 | 4.00, dd (10.3, 9.7) |
| 9 | 129.4 | 127.7 | 131.4 | |||
| 10 | 128.5 | 7.19, d (8.7) | 128.6 | 7.16, d (8.7) | 127.4 | 7.22, d (8.6) |
| 11 | 113.9 | 6.82, d (8.7) | 113.8 | 6.85, d (8.7) | 114.2 | 6.89, d (8.6) |
| 12 | 158.6 | 159.2 | 159.0 | |||
| 13 | 113.9 | 6.82, d (8.7) | 113.8 | 6.85, d (8.7) | 114.2 | 6.89, d (8.6) |
| 14 | 128.5 | 7.19, d (8.7) | 128.6 | 7.16, d (8.7) | 127.4 | 7.22, d (8.6) |
| 2′ | 164.0 | 164.7 | 165.4 | |||
| 3′ | 87.7 | 5.21, d (2.2) | 91.7 | 5.29, s | 89.2 | 5.19, s |
| 4′ | 170.1 | 170.1 | 171.4 | |||
| 5′ | 101.3 | 5.71, d (2.2) | 45.8 | 3.55, d (9.9) | 44.2 | 3.20, d (9.7) |
| 6′ | 162.9 | 79.4 | 82.6 | |||
| 7′ | 43.0 | 4.35, dd (10.0, 7.6) | 122.2 | 6.42, d (15.8) | 125.2 | 6.18, d (15.9) |
| 8′ | 45.5 | 4.16, dd (10.0, 7.6) | 130.8 | 6.86, d (15.8) | 131.3 | 6.63, d (15.9) |
| 9′ | 129.4 | 127.7 | 127.9 | |||
| 10′ | 128.5 | 7.19, d (8.7) | 128.1 | 7.34, d (8.7) | 128.8 | 7.33, d (8.7) |
| 11′ | 113.9 | 6.82, d (8.7) | 114.1 | 6.85, d (8.7) | 114.0 | 6.84, d (8.7) |
| 12′ | 158.6 | 159.7 | 159.9 | |||
| 13′ | 113.9 | 6.82, d (8.7) | 114.1 | 6.85, d (8.7) | 114.0 | 6.84, d (8.7) |
| 14′ | 128.5 | 7.19, d (8.7) | 128.1 | 7.34, d (8.7) | 128.8 | 7.33, d (8.7) |
| 4-OMe | 55.7 | 3.75, s | 55.8 | 3.69, s | 55.8 | 3.76, s |
| 12-OMe | 55.2 | 3.67, s | 55.3 | 3.79, s | 55.27 | 3.79, s |
| 4′-OMe | 55.7 | 3.75, s | 55.5 | 3.32, s | 56.1 | 3.77, s |
| 12′-OMe | 55.2 | 3.67, s | 55.3 | 3.78, s | 55.30 | 3.80, s |
:1); [α]28D +0.08 (c 0.63, MeOH–CHCl3, 5:3); ECD (33 μM, MeOH) λmax (Δε) 265 (0.00) nm; UV (MeOH) λmax (logε) 265 (4.45); IR (ATR) νmax 2940, 2838, 1699, 1647, 1608, 1566, 1512, 1455, 1410, 1391, 1246, 1176, 1031 and 814 cm−1; for 1H and 13C NMR spectroscopic data, see Table 3; HRESITOFMS m/z 539.1666 [M + Na]+ (calcd for C30H28O8 + Na, 539.1682).:1); [α]27D +0.03 (c 0.23, MeOH–CHCl3, 9:1); ECD (22 μM, MeOH) λmax (Δε) 268 (0.00) UV (MeOH) λmax (logε) 268 (4.59); IR (ATR) νmax 2924, 2837, 1708, 1649, 1625, 1608, 1567, 1513, 1455, 1406, 1248, 1176, 1031, and 821 cm−1; for 1H and 13C NMR spectroscopic data, see Table 3; HRESITOFMS m/z 539.1664 [M + Na]+ (calcd for C30H28O8 + Na, 539.1682).2.4. Preparation of the (R)-and (S)-α-methoxy-α-(trifluoromethyl) phenyl acetate of 2
The determination of configuration for the stereogenic carbinol carbons was carried out following the method reported by Ohtani et al.22 A solution of (S)-MPTA-Cl (10 μL, 53.4 μmol) was added to a solution mixture of 2 (8 mg, 27.5 μmol) and DMAP (5 mg) in dry CH2Cl2 (1 mL) and stirred under N2 at room temperature for 6 h. Then the solvent was removed in vacuo. The residue was separated on preparative TLC (CH2Cl2) to give the (R)-ester (2a, 7 mg, 50%). The (S)-ester of 2 was prepared using the procedure described above [alcohol 2 (10 mg, 34.4 μmol), CH2Cl2 (1 mL), dimethylaminopyridine (5 mg), and (R)-MPTA-Cl (10 μL, 53.4 μmol)] to yield (S)-ester (2b, 12 mg, 71%).2.5. Preparation of the (R)-and (S)-α-methoxy-α-(trifluoromethyl) phenyl acetate of (5)
The esterification of 5 was carried out using the procedure described for the preparation of 2a and 2b to yield (S)-ester (5a, 7 mg, (50%)) and (R)-ester (5b, 12 mg, (71%)).2.6. X-ray crystallographic analyses of 10 and 11
The reflection data were collected on a Bruker D8 Quest PHOTON100 CMOS detector with graphite-monochromated MoKα radiation using the APEX2 program.23 Raw data frame integration was performed with SAINT,23 which also applied correction for Lorentz and polarization effects. An empirical absorption correction using the SADABS program23 was applied. The structure was solved by direct methods and refined by full-matrix least-squares method on F2 with anisotropic thermal parameters for all non-hydrogen atoms using the SHELXTL software package.24 All hydrogen atoms were placed in calculated positions and refined isotropically. Crystallographic data of 10 and 11 were deposited with the following Cambridge Crystallographic Data Centre codes: CCDC 1415288 and CCDC 1062125, respectively.470 reflections measured (6.204° ≤ 2Θ ≤ 52.798°), 10518 unique (Rint = 0.0929, Rsigma = 0.0445) which were used in all calculations. The final R1 was 0.0508 (I > 2σ(I)) and wR2 was 0.1225 (all data).![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.6555(5) Å, b = 12.1165(5) Å, c = 12.1203(5) Å, α = 113.9720(10), β = 102.4530(10), γ = 92.9570(10), V = 1508.54(11) Å3, Z = 2, T = 293(2) K, μ(MoKα) = 0.354 mm−1, Dcalc = 1.400 g cm−3, 61914 reflections measured, 7472 unique (Rint = 0.0262) which were used in all calculations. The final R1 was 0.0642 (I > 2σ(I)) and wR2 was 0.2211 (all data).
, a = 11.6555(5) Å, b = 12.1165(5) Å, c = 12.1203(5) Å, α = 113.9720(10), β = 102.4530(10), γ = 92.9570(10), V = 1508.54(11) Å3, Z = 2, T = 293(2) K, μ(MoKα) = 0.354 mm−1, Dcalc = 1.400 g cm−3, 61914 reflections measured, 7472 unique (Rint = 0.0262) which were used in all calculations. The final R1 was 0.0642 (I > 2σ(I)) and wR2 was 0.2211 (all data).2.7. Antimalarial assay
Antimalarial activity was performed against P. falciparum (K1, multidrug resistant strain, see ESI†), using the method of Trager and Jensen.25 Quantitative assessment of malarial activity in vitro was determined by means of the microculture radioisotope technique based upon the method described by Desjardins et al.26 The inhibitory concentration (IC50) represents the concentration that causes 50% reduction in parasite growth as indicated by the in vitro uptake of [3H]-hypoxanthine by P. falciparum. The standard compound was dihydroartemisinin.2.8. Antimycobacterial assay
Antimycobacterial activity was performed against M. tuberculosis H37Ra (purchased from ATCC) using the MicroplateAlamar Blue Assay (MABA).27 The standard drug streptomycin was used as reference substance.2.9. Cytotoxicity assay
Cytotoxicity assays against human epidermoid carcinoma (KB), human breast cancer (MCF7), and human small cell lung cancer (NCI-H187) cell lines human breast adenocarcinoma Resazurin microplate assay described by O'Brien and co-workers.28 The reference substances were ellipticine and doxorubicin. Cytotoxicity test against primate cell line (Vero) was performed using the green fluorescent protein detection method described by Hunt and co-workers.29 The reference substances used were ellipticine and doxorubicin. All cells were purchased from ATCC.3. Results and discussion
Chromatographic fractionation of n-hexane and EtOAc extracts yielded eight new bicyclic lactones, velutinones A–H (1–8), three cyclobutane dimers, velutinindimers A–C (9–11), and four known compounds (12–15), kawapyrone, yangonin (12),30 three flavonoids, sakuranetin (13),31 7-O-methyleriodictyol (14)32 and rhamnetin (15),33 and an acetogenin, cananginone H (16)20,21 (Fig. 1).The IR spectra of 1–4 showed absorption bands of a γ-lactone moiety at (1789–1771 cm−1) and a conjugated carbonyl functionality (1685–1862 cm−1) similar to the absorption bands of a synthetic bicyclic cyclohexenone.34,35
Compound 1 possessed the molecular formula C18H24O3 based on the 13C NMR and HRESITOFMS (m/z 311.1611 [M + Na]+) data, indicating seven indices of hydrogen deficiency. The 1H NMR data (Table 1) had resonances at δ 2.48 (d, J = 17.4 Hz, H-2α), 2.39 (d, 17.4 Hz, H-2β), 2.52 (s, 2H, H-3), 6.17 (dd, J = 10.3, 1.2 Hz, H-5), 6.75 (dd, J = 10.3, 3.3 Hz, H-6), and 4.87 (dd, J = 3.3, 1.2 Hz, H-6a). The 13C NMR data (Table 1), DEPT, and HMQC experiments indicated seven resonances which were associated with an α,β-unsaturated carbonyl (δ 196.2/C-4), a lactone carbonyl (δ 174.2/C-1), two olefinic (δ 131.2/C-5 and 141.4/C-6), two methylene (δ 38.9/C-2 and 42.6/C-3), one methine (δ 77.8/C-6a), and one quaternary (δ 44.7/C-2a) carbons. Interpretation of the COSY and HMBC correlations (Fig. 2) indicated that 1 has a core structure of a five-membered lactone ring fused to an α,β-unsaturated cyclohexanone ring. This arrangement is similar to that of a compound isolated from the fruit kernels of Otoba parvifolia35,36 and from a total synthesis of its core structure,34 except for the side chain at C-2a which was replaced by a geranyl moiety in 1. This geranyl side chain (C10H17) was evident from the 1H and 13C NMR spectroscopic data (Table 1). The COSY spectrum showed the connectivity of the geranyl side chain by correlations between H-1′ and H-2′, and amongst H-4′, H-5′ and H-6′. The HMBC spectrum exhibited correlations of H-1′ to C-2, C-3, C-2a, C-6a, C-2′and C-3′; H-2′ to C-2a, C-1′, C-3′, C-4′, and C-10′; H-4′ to C-2′, C-3′, C-5′, C-6′ and C-10′; H-6′ to C-4′, C-5′, C-8′, and C-9′ indicating that the geranyl group was linked to the stereogenic quaternary carbon C-2a (Fig. 2). The relative configuration at C-2a and C-6a was established as syn from the NOESY correlation of H-6a and H-1′. Based on the above evidence, the structure of 1, velutinone A, was defined as shown in Fig. 1.
 | ||
| Fig. 2 Selected HMBC correlations of 1. | ||
Compound 2 had the molecular formula C18H24O4 derived from the 13C NMR and HRESITOFMS (m/z 305.1740 [M + H]+) data, demonstrating the same index of hydrogen deficiency, but having one additional oxygen atom compared to 1. The 1H and 13C NMR spectroscopic data of 2 (Table 1) were similar to those of 1, except for the geranyl side chain being oxidized at C-6′ and having a 7′, 8′ terminal double bond. The NMR spectroscopic data displayed resonances for an olefinic methylene protons at δH 4.82 (t, J = 1.4 Hz, 4.91 (brs), H-8′)/δC 111.1 and an oxymethine at δH 4.00 (t, J = 6.3 Hz, H-6′)/δC 75.3. The HMBC correlations of H-5′ to C-3′, C-4′, C-6′, and C-7′; H-6′ to C-4′, C-5′, C-7′, C-8′, and C-9′; and H-8′ to C-6′, C-7′, and C-9′ confirmed the position of the terminal olefinic moiety and the hydroxy group in the side chain. The assignment of the (6′S) absolute configuration was done via the modified Mosher's ester method22(Fig. 3). Therefore, the structure of compound 2, velutinone B, was defined as shown in Fig. 1.
 | ||
| Fig. 3 Δδ values (Δδ = δS − δR in ppm) obtained for MTPA esters 2a and 2b. | ||
Compound 3 had the molecular formula C18H24O4, deduced from 13C NMR and HRESITOFMS (m/z 327.1543 [M + Na]+) data, implying the same index of hydrogen deficiency, but having one additional oxygen atom compared to 1. The 1H and 13C NMR spectroscopic data of 3 (Table 1) were similar to those of 1, except for the appearance of the olefinic protons at δH 5.51 (dt, J = 15.6, 6.6 Hz, H-5′) and δH 5.60 (d, J = 15.6 Hz, H-6′), and one additional oxygenated carbon signal at δC 70.4 (C-7′). The position of the hydroxy group at C-7′ on the side chain was confirmed by the HMBC correlations of H-5′ to C-3′, C-4′, C-6′, and C-7′; and H-6′ to C-4′, C-5′, C-7′ and C-8′/C-9′. Therefore, the structure of compound 3, velutinone C, was defined as shown (Fig. 1).
Compound 4 had the molecular formula C18H24O5 derived from the 13C NMR and HRESITOFMS (m/z 343.1472 [M + Na]+) data, demonstrating the same index of hydrogen deficiency, but having one additional oxygen atom compared to 3. The NMR spectroscopic data of 4 (Table 1) was similar to that of 3, except for the resonance of a C-7′ hydroxy group. The 13C NMR spectrum revealed the unusual downfield oxygenated carbon signal at δC 81.8 for C-7′, suggesting the presence of a hydroperoxy group.37 Thus the structure of compound 4, velutinone D, was determined as shown (Fig. 1).
Compound 5 had the molecular formula C18H26O3 derived from the 13C NMR and HRESITOFMS (m/z 291.1935 [M + Na]+) data, implying six indices of hydrogen deficiency. The IR spectrum showed hydroxy (3442 cm−1) and γ-lactone carbonyl (1771 cm−1) functionalities. The NMR data of 5 (Table 2) corresponded to those of 1, except that the ketone carbonyl resonance for C-4 was replaced by a resonance for an oxymethine group at δH 4.31 (m)/δC 63.7. The assignment of (4S) absolute configuration was done via the modified Mosher's ester method22 (Fig. 4). The configurations of (2aR and 6aR) were assigned by NOESY and a molecular modeling study. NOESY correlations were observed between H-4 and H-2α, H-6a and H-1′a, H-6a and H-3β, H-1′b and H-2β, and H-3α and H-2′ (Fig. 5). Therefore, the structure of compound 5, velutinone E, was established as shown (Fig. 1).
 | ||
| Fig. 4 Δδ values (Δδ = δS − δR in ppm) obtained for MTPA esters 5a and 5b. | ||
 | ||
| Fig. 5 Key NOESY correlations of 5, energy minimized using MM2. | ||
Compound 6 had the molecular formula C18H24O4 derived from the 13C NMR and HRESITOFMS (m/z 327.1562 [M + Na]+) data, implying the same index of hydrogen deficiency, but having one more oxygen atom than 1. The IR spectrum showed bands for γ-lactone (1789 cm−1 and a cyclohexanone (1719 cm−1) groups. The NMR data of 6 (Table 2) was similar to that of 1, except for the resonances of the C-5/6 double bond which were replaced by those of an epoxide moiety [δH 3.65 (d, J = 3.6 Hz, H-5)/δC 58.6 and δH 3.37 (d, J = 3.6 Hz, H-6)/δC 54.9)]. The relative configuration of the epoxide was assigned by the NOESY correlations between H-6 and H-6a. Hence, the structure of compound 6, velutinone F, was defined as shown (Fig. 1).
Compound 7 had the molecular formula C18H24O5, established from the 13C NMR and HRESITOFMS (m/z 321.1690 [M + Na]+) data, having the same index of hydrogen deficiency as in 2. The IR spectrum displayed an extra hydroxyl band at (3481 cm−1) which differed from that of 6. The NMR data of 7 (Table 2) corresponded to that of 2, except for the resonances of a C-5/6 double bond, which was replaced by resonances for an epoxide moiety [δH 3.62 (d, J = 3.6 Hz, H-5)/δC 58.5 and δH 3.34 (d, J = 3.6 Hz, H-6)/δC 54.8] as in that of 6. The 6′S configuration was assigned by comparison of the NMR data to that of 2. Thus the structure of 7, velutinone G, was defined as shown (Fig. 1).
Compound 8 possessed the molecular formula C18H24O5 from the 13C NMR and HRESITOFMS (m/z 321.1690 [M + H]+) data, having seven indices of hydrogen deficiency as in 6, but having one additional oxygen. The IR spectrum was also similar to that of 6. The NMR data of 8 (Table 2) corresponded to that of 6, except for the resonances for a C-6′/7′ double bond which were replaced by resonances for an epoxide [δH 3.65 (t, J = 6.7 Hz, H-6′/δC 63.7 and δC 58.2, C-7′)] at this position. The configuration at C-6′ of 8 was proposed to be S, based on its ring opening to give 7. Hence, structure of 8, velutinone H, was designated as shown in Fig. 1.
There are only two closely related bicyclic lactones, panamonons A and B,38 which have NMR data similar to those of compounds 1–8. However, the relative configuration at the ring junction (C-2a and C-6a) of 1–8 were assigned as 2aR and 6aR which different from panamonons A and B (2aR and 6aS).38 The ECD spectra of compounds 1–4, containing an α,β-unsaturated ketone, and compound 5, showed negative Cotton effects in the range of 202–215 nm. While, compounds 6–8 contained a saturated ketone, exhibited positive Cotton effects in the range of 205–215 nm (Fig. 6).
 | ||
| Fig. 6 ECD (a) and UV (b) spectra (in MeOH) of compounds 1–8. | ||
To confirm the natural occurrence of 1–8, the isolates velutinone A (1) with a geranyl side chain and velutinone H (8) with two epoxide rings were stirred with or without silica gel in EtOAc and MeOH for 4 days following the conditions of the separation process. No change on TLC was observed. We conclude that isolates 1–8 are natural occurring products not artefacts.
The putative biosynthetic pathway towards compounds 1–8 is shown in Fig. 7. The precursor, homogentisic acid,35,36 could be prenylated by geranyl diphosphate to form intermediate A 38 which may be reduced and lactonized to form compound 1. Reduction of 1 would produce 5, while epoxidation of 1 would afford 6 or intermediate B. Oxidation of 5 would give 4 which could be reduced to give 3 or B, the latter via intermediate C. Protonation and deprotonation of B may give 2 and 3. The hydroxy group of 3 could be oxidized to give 4. Compound 8 could be derived from 6 or B via an oxidation reaction. Further protonation and deprotonation of 8 would give 7.
 | ||
| Fig. 7 Plausible biogenetic pathway of 1–8. | ||
Compound 9 showed an [M + Na]+ ion peak at m/z 539.1651 in its HRESITOFMS, which in conjunction with the 13C NMR data indicated the molecular formula C30H28O8, requiring seventeen indices of hydrogen deficiency. The IR spectrum showed bands for unsaturated lactone (1715 cm−1) and aromatic (1643 cm−1) groups. The UV spectrum also indicated an aromatic moiety (286 nm). Since the NMR spectroscopic data of 9 (Table 3) displayed half the number of resonance signals expected for 28 protons and 30 carbons, the structure should be a symmetrical dimer. These NMR resonances corresponded to those of the cyclobutane dimer, achyrodimer A,39 except for hydroxy groups at C-12 and C-12′ of the aromatic rings which were replaced by methoxy groups. The 1H NMR spectroscopic data (Table 3) showed resonances for the para-substituted benzene rings at δ 6.82 and 7.19 (each 4H, d, J = 8.7 Hz), four methines of the cyclobutyl ring at δ 4.16 and 4.35 (each 2H, dd, J = 7.6, 10.0 Hz), four olefinic protons for the two α-pyrone moieities at δ 5.21 and 5.71 (each 2H, d, J = 2.2 Hz), and four methoxy groups at δ 3.75 and 3.67 (each 6H, s). The 13C NMR data (Table 3) showed resonances for the para-disubstituted benzene rings at δ 129.4 (C-9, 9′), 128.5 (C-10, 10′ and C-14, 14′), and 113.9 (C-11, 11′), the cyclobutyl methines at δ 43.0 (C-7, 7′), and 45.5(C-8, 8′) the α-pyrone methines at δ 87.7 (C-3, 3′), 101.3 (C-5, 5′) and methoxy groups at δ 55.7 (4, 4′-OMe) and 55.2 (12, 12′-OMe), and a carbonyl group at 162.9 (C-6, 6′). The correlations of H-7/7′ to C-5/5′, C-6/6′, C-8/8′, and C-9/9′, and of H-8/8′ to C-6/6′, C-7/7′, C-10/10′, and C-14/14′ from the HMBC spectrum revealed the connection of a cyclobutane ring to an α-pyrone ring, and benzene rings at C-7/7′ and C-8/8′, respectively. Resonances for two sets of methoxy protons at δH 3.75 and 3.67 showed correlations with C-4/4′ and C-12/12′, respectively, confirming the location of methoxy groups at C-4/4′ and C-12/12′. The correlations between H-7 (7′) and H-8 (8′) in the NOESY spectrum indicated the relative configuration on the cyclobutane ring as reported for achyrodimer A.39 The specific rotation value of 9 was almost zero [+0.08 (c 0.63, MeOH–CHCl3; 3:1)] which was also the same as that reported for a symmetric achyrodimer A.39 Moreover, the ECD spectrum of 9 showed no signal for a Cotton effect (Fig. 8).40 Based on this evidence the structure of compound 9 could contain a plane of symmetry. Hence, 9 was concluded to be a new symmetrical cyclobutane dimer of the isolated styrylpyrone, yangonin (12), and it was named velutinindimer A.
 | ||
| Fig. 8 ECD (a) and UV (b) spectra (in MeOH) of compounds 9, 10 and 11. | ||
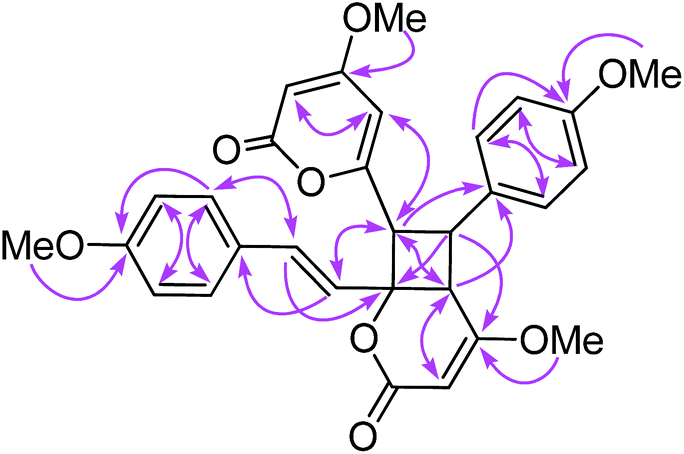
Compound 10 possessed the molecular formula C30H28O8 from the 13C NMR and HRESITOFMS (m/z 539.1666 [M + Na]+) data, having the same index of hydrogen deficiency as 9. The IR spectrum displayed bands for lactone (1699 cm−1) and aromatic (1647 cm−1) groups. The UV spectrum also supported an aromatic moiety (268 nm). The 1H NMR data of 10 (Table 3) showed resonances for two para-disubstituted benzene rings at δ 6.85, 7.16, and 7.34 and 6.85 (each 2H, d, J = 8.7 Hz), an E-double bond at δ 6.42 and 6.86 (each 1H, d, J = 15.8 Hz), α-pyrone ring at δ 5.33 and 5.89 (each 1H, d, J = 2.2 Hz, H-3 and H-5, respectively) and 5.29 (s, H-3′), three methine protons at δ 3.55 (d, J = 9.9 Hz, H-5′), 4.26 (dd, J = 10.8, 9.9 Hz, H-8), 4.09 (d, J = 10.8 Hz, H-7), and four methoxy groups at δ 3.32, 3.69, 3.78, and 3.79. The 13C NMR spectrum, DEPT and HMQC experiments of 10 showed 30 resonances, including two sets of p-disubstituted benzene rings, one α-pyrone ring, one olefinic, one cyclobutane ring, and four methoxy carbons. The HMBC spectrum displayed 3J correlations of H-3 to C-5; H-5 to C-3, and C-7; H-7 to C-5, C-9, C-5′, and C-7′; H-8 to C-6, C-10, C-14, C-4′, and C-6′; H-10, 14 to C-8, and C-12; H-11, 13 to C-9; H-3′ to C-5′; H-5′ to C-7, C-9, C-3′ and C-7′; H-7′ to C-7, C-5′, and C-9′; H-8′ to C-6′, C-10′, and C-14′; H-10′, 14′ to C-8′ and C-12′; H-11′, 13′ to C-9′ and C-12′; 4-OMe to C-4; 12-OMe to C-12; 4′-OMe to C-4′; and 12′-OMe to C-12′ confirming the structure of 10 (Fig. 9). The NMR data of 10 was comparable to the cyclobutane dimer achyrodimer D, reported from the aerial parts of Achyrocline bogotensis.39 It was found that 10 was the methoxy derivative of achyrodimer D. The relative configuration of 10 was determined from the relatively large coupling constants (9.9–10.8 Hz) between H-7 and H-8, and H-8 and H-5′, and the NOESY correlations between H-8 and H-5′, H-7 and H-14, H-8 and H-10, H-5 and H-7, H-7′ and H-10′, and H-8′ and H-14′. The magnitude of the coupling constant between H-7 and H-8 (Jtrans = 10.8 Hz), and H-8 and H-5′ (Jcis = 9.9 Hz) could be correlated to the dihedral angle between those protons, which corresponded with the values from the Karplus equation for a four membered ring.41 The ECD measurement of 10 in MeOH showed no signal of the Cotton effect40 (Fig. 8), and also the specific rotation value of 10 was almost zero (+0.08). These could suggest that compound 10 was a racemic mixture. Finally, the X-ray crystallographic analysis supported the structure of an isolated 10 containing asymmetric units of a racemic mixture (Fig. 10), and the one with the relative configuration (5′S, 6′R, 7S, and 8S) is shown in Fig. 1. Thus, the structure of 10 was an unsymmetrical cyclobutane dimer of the isolated yangonin (12), and it was named velutinindimer B.
 | ||
| Fig. 9 Selected HMBC correlations of 10. | ||
 | ||
| Fig. 10 ORTEP plot of the asymmetric units in 10. The thermal ellipsoids are shown at 40% probability. | ||
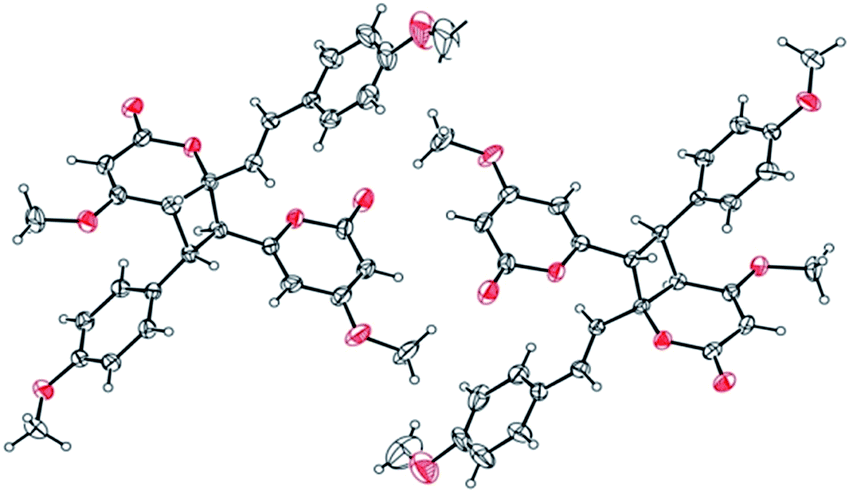
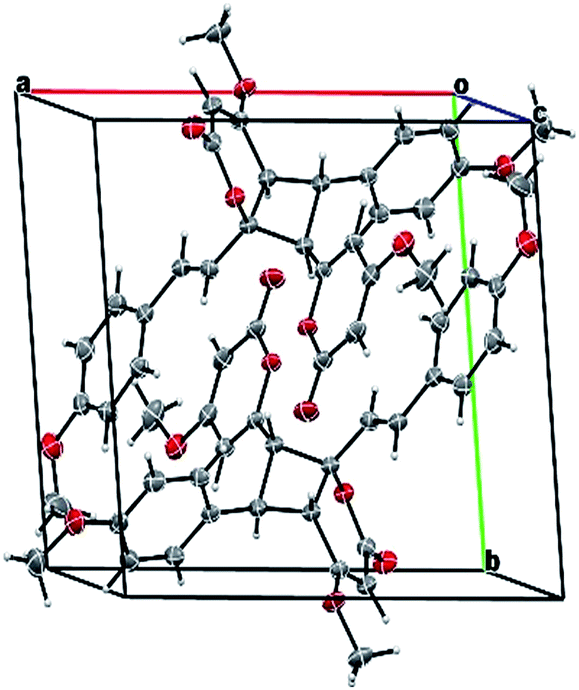
Compound 11 exhibited an [M + Na]+ peak at m/z 539.1664 in the positive HRESITOFMS corresponding to the molecular formula C30H28O8Na, the same as that of 10. The IR spectrum showed bands for a lactone moiety (1708 cm−1) and an aromatic ring (1649 cm−1). The UV spectrum indicated an aromatic moiety (273 nm). The NMR data (Table 3) and 2D NMR of 11 demonstrated a similar structure to a dimeric 10. Nevertheless, slight differences in chemical shifts around compounds 11 and 10 in the 1H and 13C NMR data at positions 7, 8, 9, 5′, 7′ and 8′ (Table 3) suggested different configurations at the cyclobutane ring between the two compounds. The large coupling constant of H-7 and H-8 (Jtrans = 10.3 Hz) and H-8 and H-5′ (Jtrans = 9.7 Hz) could be explained in the same way as for 10.41 The NOESY spectrum displayed correlations of H-5′ and H-7, H-7 and H-14, H-8 and H-10, H-7′ and H-10′, and H-8′ and H-14′, indicating the relative configuration of 11. Compound 11 also showed no signal of the Cotton effect (Fig. 7)40 and also its specific rotation value was almost zero (+0.3) suggesting that it should be a racemic mixture as compound 10. The X-ray crystallographic analysis confirmed that the isolated compound 11 was a racemic mixture as in 10, and the one with the relative configuration 5′R, 6′S, 7R, and 8S is shown in Fig. 1, 11 and 12. From the above evidence, the structure of 11 was determined to be another new dimeric styrylpyrone and it was named velutinindimer C.
 | ||
| Fig. 11 Packing structure of 11 showing the inverted racemic mixture. The chloroform solvate molecule is omitted for clarity. | ||
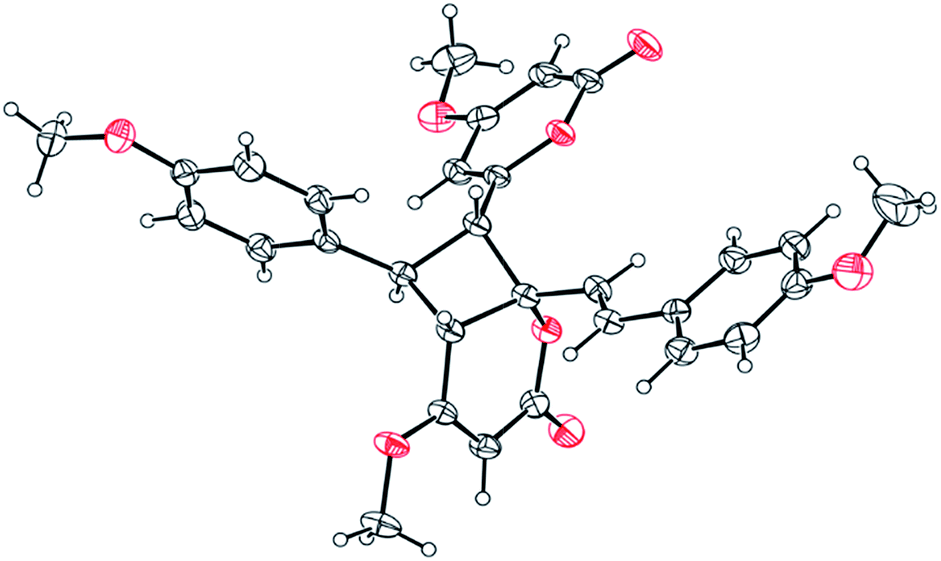 | ||
| Fig. 12 ORTEP plot of the asymmetric unit in 11. The thermal ellipsoids are shown at 30% probability level. The chloroform solvate molecule is omitted for clarity. | ||
To confirm the natural occurrence of styrylpyrone dimers, 9–11, the isolated yangonin (12), was stirred with silica gel in EtOAc and MeOH for a week, following the conditions for our separation process. The formation of 9, 10 and 11 was not observed. It is worth noting that isolated dimers 10 and 11 were racemic mixtures occurring from asymmetrical 2 + 2 cycloaddition of the isolated styrylpyrone, yangonin (12), while compound 9 has an axis of a symmetric dimer and so is not chiral.
The biological activities of the isolated compounds (purity > 95% from the NMR spectra) are shown in Table 4. Compounds 1 and 2, with MIC values of 43.4 and 82.1 μM respectively, should be responsible for the antimycobacterial activity against M. tuberculosis exhibited in the primary screening. From this result, the α,β-unsaturated carbonyl in the core structure plays an important role against TB, while the hydroxy or peroxide functionalities at C-6′ or C-7′ reduce this activity. Since our previous work reported the antimalarial activity and cytotoxicity of compounds from M. velutina,20,21 the compounds isolated herein have been further evaluated for their activities. Compounds 2–4 and 7–11 displayed antimalarial activity toward P. falciparum with IC50 values in the range of 5.4–10.0 μM. In addition, compounds 1–4 and 6–8 exhibited cytotoxicity against three cancer cell lines tested, with IC50 values in the range of 4.0–24.1 μM. Among these, 1, 2 and 8 exhibited moderate cytotoxicity against NCI-H187 cell lines with IC50 values of 4.2, 6.5 and 6.8 μM, respectively, which were close to the standard drug, ellipticine (5.6 μM). Compounds 1–4 and 6–8 exhibited cytotoxicity towards the Vero cell line with IC50 values in the range of 5.8–18.2 μM. However, dimeric styrylpyrones 9–11 showed no cytotoxicity in the test.
| Compound | Antimalarial | Anti-TB | Cytotoxicity (IC50, μM) | |||
|---|---|---|---|---|---|---|
| (IC50, μM) | (MIC, μM) | KBa | MCF7b | NCI-H187c | Vero celld | |
| a Human oral epidermoid carcinoma.b Human breast cancer cells.c Human lung cancer cells.d Normal African green monkey kidney cells. | ||||||
| 1 | Inactive | 43.4 | 4.0 | 4.8 | 4.2 | 5.8 |
| 2 | 9.6 | 82.1 | 9.6 | 12.9 | 6.5 | 8.8 |
| 3 | 10.0 | Inactive | 12.9 | 10.9 | 11.4 | 10.3 |
| 4 | 9.6 | Inactive | 10.5 | 15.2 | 8.7 | 11.7 |
| 6 | Inactive | Inactive | 14.5 | 20.7 | 11.5 | 11.2 |
| 7 | 9.8 | Inactive | 24.1 | 21.0 | 14.7 | 17.9 |
| 8 | 7.3 | Inactive | 10.5 | 11.9 | 6.8 | 18.2 |
| 9 | 6.4 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 10 | 5.4 | Inactive | Inactive | Inactive | Inactive | Inactive |
| 11 | 5.8 | Inactive | Inactive | Inactive | Inactive | Inactive |
| Dihydroartemisinin | 0.004 | |||||
| Streptomycin | 0.3–0.5 | |||||
| Ellipticine | 0.8 | 5.6 | 3.1 | |||
| Doxorubicin | 0.3 | 0.1 | 0.01 | |||
4. Conclusions
Isolation of the leaves extracts of M. velutina yielded an unique class of eight bicyclic lactones with a C18 carbons architecture, named velutinones A–H (1–8), three new dimeric styrylpyrones, velutinindimers A–C (9–11), five known compounds, the kawapyrone, yangonin (12), three flavonoids (13–15), and an acetogenin, cananginone H (16). Velutinindimers A–C (9–11) are dimmers occurring from symmetrical and asymmetrical 2 + 2 cycloaddition of the isolated styrylpyrone, yangonin (12). The structures of velutinindimers B and C (10 and 11) were identified as mixtures which were confirmed by X-ray crystallographic, ECD and specific rotation analyses. Biological activity of the isolated compounds had been evaluated. Compounds 2–4 and 7–11showed antimalarial activity with IC50 values in the range of 5.4–10.0 μM. Moreover, 1–4 and 6–8 displayed cytotoxicity against the KB, MCF7, and NCI-H187 cancer cell lines and Vero cell lines with IC50 values in the range of 4.0–24.1 μM.Acknowledgements
Financial support from the Thailand Research Fund and Khon Kaen University (Grant No. RTA 5980002) is gratefully acknowledged. We thanks the Center for Innovation in Chemistry (PERCH-CIC) for partial support. We are grateful to the Bioassay Research Facility of the BIOTEC, Thailand for biological activity assay.Notes and references
- W. Chuakul and N. Sornthornchareonon, Thai J. Phytopharm., 2003, 10, 25–32 Search PubMed.
- T. Smitinand, Thai Plant Names Revised Edition, Prachachon Co. Limited, Bangkok, 2001, p. 359 Search PubMed.
- T. Chaowasku and P. J. A. Keßler, Nord. J. Bot., 2013, 3, 680–699 CrossRef.
- G. G. Harrigan, A. A. L. Gunatilaka, D. G. I. Kingston, G. W. Chan and R. K. Johnson, J. Nat. Prod., 1994, 57, 68–73 CrossRef CAS.
- R. Wu, Q. Ye, N. Y. Chen and G. L. Zhang, Chin. Chem. Lett., 2001, 12, 247–248 CAS.
- C. Kamperdick, N. H. Van and T. V. Sung, Phytochemistry, 2002, 61, 991–994 CrossRef CAS PubMed.
- D. T. Huong, C. Kamperdick and T. V. Sung, J. Nat. Prod., 2004, 67, 445–447 CrossRef CAS PubMed.
- Y. Lei, L. Wu, H. Shi and P. Tu, Helv. Chim. Acta, 2008, 91, 495–500 CrossRef CAS.
- N. P. Thao, B. T. T. Luyen, B. H. Tai, N. M. Cuong, Y. C. Kim, C. V. Minh and Y. H. Kim, Bioorg. Med. Chem. Lett., 2015, 25, 3859–3863 CrossRef CAS PubMed.
- K. Sawasdee, T. Chaowasku and K. Likhitwitayawuid, Molecules, 2010, 15, 639–648 CrossRef CAS PubMed.
- K. Sawasdee, T. Chaowasku, V. Lipipun, T. H. Dufat, S. Michel and K. Likhitwitayawuid, Fitoterapia, 2013, 85, 49–56 CrossRef CAS PubMed.
- K. Sawasdee, T. Chaowasku, V. Lipipun, T. H. Dufat, S. Michel and K. Likhitwitayawuid, Tetrahedron Lett., 2013, 54, 4259–4263 CrossRef CAS.
- K. Sawasdee, T. Chaowasku, V. Lipipun, T. H. Dufat, S. Michel, V. Jongbunprasert and K. Likhitwitayawuid, Biochem. Syst. Ecol., 2014, 54, 179–181 CrossRef CAS.
- B. Chen, C. Feng, B. Li and G. Zhang, Nat. Prod. Res., 2003, 17, 397–402 CrossRef CAS PubMed.
- H. Zhang, C. Ma, N. V. Hung, N. M. Cuong, G. T. Tan, B. D. Santarsiero, A. D. Mesecar, D. D. Soejarto, J. M. Pezzuto and H. H. S. Fong, J. Med. Chem., 2006, 49, 693–708 CrossRef CAS PubMed.
- C. Naphong, W. Pompimon and P. Sombutsiri, Am. J. Appl. Sci., 2013, 10, 787–792 CrossRef CAS.
- S. Jumana, C. M. Hasan and M. A. Rashid, Fitoterapia, 2000, 71, 559–561 CrossRef CAS PubMed.
- C. M. Hasan, S. Jumana and M. A. Rashid, Nat. Prod. Lett., 2000, 14, 393–397 CrossRef CAS.
- S. Jumana, C. M. Hasan and M. A. Rashid, Biochem. Syst. Ecol., 2000, 28, 483–485 CrossRef CAS PubMed.
- N. Wongsa, S. Kanokmedhakul and K. Kanokmedhakul, Phytochemistry, 2011, 72, 1859–1864 CrossRef CAS PubMed.
- N. Wongsa, S. Kanokmedhakul and K. Kanokmedhakul, Phytochemistry, 2015, 109, 154 CrossRef CAS.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS.
- Bruker, APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, WI, USA, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef PubMed.
- W. Trager and J. B. Jensen, Science, 1967, 193, 673–675 Search PubMed.
- R. E. Desjardins, C. J. Canfield, J. D. Haynes and J. D. Chulay, Antimicrob. Agents Chemother., 1979, 16, 710–718 CrossRef CAS PubMed.
- D. A. Scudiere, R. H. Shoemaker, K. D. Paull, A. Monks, S. Tierney, T. H. Nofziger, M. J. Currens, D. Seniff and M. R. Boyd, Cancer Res., 1988, 48, 4827–4833 Search PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef.
- L. Hunt, M. Jordan, M. D. Jesus and F. M. Wurm, Biotechnol. Bioeng., 1999, 65, 201–205 CrossRef CAS PubMed.
- T. Hashimoto, M. Suganuma, H. Fujiki, M. Yamada, T. Kohno and Y. Asakawa, Phytomedicine, 2003, 10, 309–317 CrossRef CAS PubMed.
- Y. Ogawa, H. Oku, E. Iwaoka, M. Iinuma and K. Ishiguro, Chem. Pharm. Bull., 2007, 54, 675–678 CrossRef.
- J. M. J. Vasconcelos, A. M. S. Silva and J. A. S. Cavaleiro, Phytochemistry, 1998, 49, 1421–1424 CrossRef CAS.
- E. Lee, B. H. Moon, Y. Park, S. Hong, S. Lee, Y. Lee and Y. Lim, Bull. Korean Chem. Soc., 2008, 29, 507–510 CrossRef CAS.
- J. T. B. Ferreira, R. C. Boscaini and F. A. Marques, Nat. Prod. Lett., 1993, 2, 313–316 CrossRef CAS.
- A. G. Ferreira, J. B. Fernandes, P. C. Vieira, O. R. Gottlieb and H. E. Gottlieb, Phytochemistry, 1995, 40, 1723–1728 CrossRef CAS.
- A. G. Ferreira, M. Motidome, O. R. Gottlieb, J. B. Fernandes, P. C. Vieira, M. Cojocaru and H. E. Gottlieb, Phytochemistry, 1989, 28, 579–583 CrossRef CAS.
- A. G. Rücker, E. Schenkel, D. Manns, M. Falkenberg and A. Marek, Phytochemistry, 1996, 41, 297–300 CrossRef.
- Y.-S. Wang, R. Li, Y. Huang, W.-B. Shang, F. Chen, H.-B. Zhang and J.-H. Yang, Phytochem. Lett., 2013, 6, 26–30 CrossRef CAS.
- T. Sagawa, Y. Takaishi, Y. Fujimoto, C. Duque, C. Osorio, F. Ramos, C. Garzon, M. Sata, M. Okamoto, T. Oshikawa and S. U. Ahmed, J. Nat. Prod., 2005, 68, 502–505 CrossRef CAS PubMed.
- M. Fujiki, Symmetry, 2014, 6, 677–703 CrossRef CAS.
- http://www.nmr.ch.tum.de/home/dames/J_reich_uwisc.pdf, July 2015.
Footnote |
| † Electronic supplementary information (ESI) available: 1D NMR spectra of new compounds 1–11. CCDC 1415288 and 1062125. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01609c |
| This journal is © The Royal Society of Chemistry 2017 |