
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase diagram, stability and electronic properties of an Fe–P system under high pressure: a first principles study†
Ziyuan Zhaoa,
Lulu Liua,
Shoutao Zhanga,
Tong Yua,
Fei Lia and
Guochun Yang *ab
*ab
aCentre for Advanced Optoelectronic Functional Materials Research and Laboratory for UV Light-Emitting Materials and Technology of Ministry of Education, Northeast Normal University, Changchun 130024, China. E-mail: yanggc468@nenu.edu.cn
bState Key Laboratory of Superhard Materials, Jilin University, Changchun 130024, China
First published on 13th March 2017
Abstract
Fe–P binary compounds have attracted much attention, particularly under high pressure, since they are the constituents of the Earth's core. However, most studies focus on the single stoichiometry of Fe–P binary compounds at high pressure, and their whole phase diagram and relative stabilities have been unexplored thus far. Herein, first principles swarm structure predictions are performed to find stable structures of Fe–P compounds with various FexPy (x = 1–4 and y = 1, or x = 1 and y = 2) compositions. Then, their phase diagram and relative stabilities are reliably determined based on predicted structures. Specifically, the FeP, Fe2P and Fe4P compounds are found to be stable in the pressure range of 0–400 GPa. The Fe3P compound decomposes into Fe2P and Fe4P above 214 GPa. FeP2 becomes unstable above 82 GPa. Notably, two new phases (i.e. C2/c-structured Fe4P and Cmcm-structured Fe3P) are found to be more stable than the previously reported phases. In addition, the XRD pattern of the predicted Cmcm-structured Fe3P matches the experimental patterns, and we are awaiting future experimental confirmation. Electronic band calculations show that the Fe–P binary compounds are metallic, with a pronounced Fe 3d component crossing the Fermi level. Cmcm-structured Fe3P is ferromagnetic. Our study not only provides useful information for the further study of Fe–P binary compounds but also for the determination of the Earth's core components.
1. Introduction
Geochemical and geophysical studies show that light elements (i.e. hydrogen, oxygen, carbon, sulfur, phosphorus and silicon) play important roles in the Earth's core.1,2 Early models of the Earth's core consisted of a considerable amount of schreibersite, cohenite, and troilite.3 The Fe content of the Earth's core is rather rich (90.67%).3 Several phases of iron compounds, such as iron–sulfur, iron–silicon, and iron–oxygen, with light elements under high pressure have been determined by a large number of experimental and theoretical studies.4–7 Notably, the phosphorus content of the Earth's core was estimated to be around 0.20 wt%, and up to 4 wt% of phosphorus was found to be soluble in crystalline iron at 23 GPa in the Fe–S–P system.8,9 Since the structures of Fe–P compounds are similar to that of Fe–S, iron phosphides should have a stable composition in the cores. As a consequence, investigation of the structures and properties of iron-phosphorus (Fe–P) compounds is worthy of attention.To date, extensive studies have been carried out on Fe–P binary compounds, and although most of these studies were based on single stoichiometry, the relative stabilities of the Fe–P binary compounds remain undetermined, particularly at high pressure. For example, several stoichiometric compounds (i.e. Fe4P, Fe3P, Fe2P, FeP and FeP2) were separately investigated. Fe4P is known as melliniite, which is a mineral from Northwest Africa 1054 acapulcoite. The cubic polymorph with the P213 space group symmetry (AlAu4-type structure) was determined using single-crystal X-ray diffraction data (XRD).10 Under high pressure, the Fe4P phase is relatively stable compared to Fe3P + Fe.11 The tetragonal structure Fe3P was discovered from schreibersite at ambient pressure.12 Its possible high-pressure polymorph is either a P4/mnc or Pnma structure, as predicted by theoretical calculations, and the pressure-induced phase transition from I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) to P4/mnc has been observed at 64 GPa.13 For the Fe2P compound, it is a terrestrial garnet peridotite with implications for the Earth's core. A hexagonal structure of Fe2P with P
to P4/mnc has been observed at 64 GPa.13 For the Fe2P compound, it is a terrestrial garnet peridotite with implications for the Earth's core. A hexagonal structure of Fe2P with P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2m symmetry exists at ambient conditions, and the Co2Si-type structure (space group Pnma) is found to be more stable above 8.0 GPa (ref. 14) or 16.8 GPa,15 as observed through high-pressure and high-temperature X-ray diffraction experiments. Based on first principles calculations, another P
2m symmetry exists at ambient conditions, and the Co2Si-type structure (space group Pnma) is found to be more stable above 8.0 GPa (ref. 14) or 16.8 GPa,15 as observed through high-pressure and high-temperature X-ray diffraction experiments. Based on first principles calculations, another P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m-structured Fe2P was reported at 153 GPa.16 Several studies on FeP have been carried out because FeP is also an important member in the planetary cores. FeP crystallizes in the regular MnP-type structure (space group Pnma) at ambient conditions and undergoes a structural phase transition to a simple cubic FeSi-type phase (space group P213) at 87.5 GPa.17,18 FeP2 crystallizes in the regular marcasite structure (space group Pnnm) at ambient conditions with no unpaired spins in the low-spin configuration.19 Under high-pressure and high-temperature conditions, a new phase of FeP2 with I4/mcm symmetry was observed at 70 GPa.20 Recent studies show that Fe–P binary compounds (i.e. Fe3P, Fe2P, FeP, FeP2 and FeP4) are stable at ambient pressure,21 in which all the structures came from reported compounds. By all appearance, it is necessary to establish the whole phase diagram of Fe–P binary compounds under high pressure.
m-structured Fe2P was reported at 153 GPa.16 Several studies on FeP have been carried out because FeP is also an important member in the planetary cores. FeP crystallizes in the regular MnP-type structure (space group Pnma) at ambient conditions and undergoes a structural phase transition to a simple cubic FeSi-type phase (space group P213) at 87.5 GPa.17,18 FeP2 crystallizes in the regular marcasite structure (space group Pnnm) at ambient conditions with no unpaired spins in the low-spin configuration.19 Under high-pressure and high-temperature conditions, a new phase of FeP2 with I4/mcm symmetry was observed at 70 GPa.20 Recent studies show that Fe–P binary compounds (i.e. Fe3P, Fe2P, FeP, FeP2 and FeP4) are stable at ambient pressure,21 in which all the structures came from reported compounds. By all appearance, it is necessary to establish the whole phase diagram of Fe–P binary compounds under high pressure.
Recently, the first principles structural prediction method has become an irreplaceable tool to discover new materials and determine their stable structures.22–24 For example, a recent breakthrough in the superconductivity field is the observation of remarkably high superconductivity (with Tc up to 203 K),25 in which H3S is the main stoichiometry of the superconducting phase.26 This observation was achieved by direct investigation on the theoretical prediction of high-Tc superconductivity in compressed solid H2S.27 A new clathrate sodalite-like BN structure was predicted with a high energy density and large hardness.28 The reaction of iron with xenon has been predicted in the Earth's inner core environment through theoretical calculations, which provides a solution to the missing Xe paradox.29 Numerous unusual stoichiometric compounds with exotic physical or chemical properties have been discovered under high pressure.30–37
Herein, the most stable structures of the Fe–P binary compounds (Fe4P, Fe3P, Fe2P, FeP and FeP2) are reliably determined using the first principles calculations in combination with the swarm structural search technology.23,38 The relative stabilities of these considered compounds are reliably established. Two new phases (C2/c-structured Fe4P, and Cmcm-structured Fe3P) are found to be more stable than the previously reported phases. Cmcm-structured Fe3P meets the Stoner criterion and is ferromagnetic. Our study provides some useful information for the investigation of the constituents of the Earth's core.
2. Computational details
To search the stable structures for the given chemical compositions, structural predictions of the Fe–P system were performed at 0, 50, 100, 200, 300, and 400 GPa using the Crystal structure AnaLYsis by the Particle Swarm Optimization (CALYPSO) code.23,39 The CALYPSO method can successfully find stable structures just depending on chemical compositions and has demonstrated success in predicting the high-pressure structures of various systems.40–44 The ab initio structural relaxations and electronic calculations are based on the density functional theory (DFT) within the Perdew–Burke–Ernzerhof (PBE) of the generalized gradient approximation (GGA),45 as implemented in the Vienna ab initio simulation package (VASP) code.46 The electron–ion interaction was described using the all-electronic projector-augmented wave (PAW) method47 with 3s23p63d74s1 and 3s23p3 valence electrons for the Fe and P atoms, respectively. Hubbard U is necessary to correct the effective on-site electron–electron interaction of the transition metal, with valence electrons on the d and f orbits.48,49 Hubbard U parameters with U = 2 eV and J = 0.95 eV were applied to the Fe atoms in the Fe2P compounds.16 It successfully reproduces the experimental magnetic moments of Fe atoms.50 As a consequence, we chose Hubbard U with 2 eV at a fixed value of 0.95 eV (i.e. Ueff = U − J) to calculate the properties and fine structure optimization. U = 2.2 eV was verified to be suitable to elemental iron by the linear response approach.51 The cutoff energy of 700 eV and Monkhorst–Pack52 k-meshes with a grid spacing of 2π × 0.03 Å−1 were used to ensure that the total energy calculations converged well. The phonon calculations were carried out using the finite displacement approach to determine the dynamical stability of the predicted structures, as implemented in the Phonon code.53 To study the interatomic interaction, the integrated crystal orbital Hamilton populations (ICOHPs) were calculated as implemented in the LOBSTER package.543. Results and discussion
3.1. Phase stabilities of Fe–P compounds
Herein, we mainly considered Fe-rich phosphorus compounds (i.e. Fe4P, Fe3P, Fe2P, FeP and FeP2) since the Fe content of the Earth's core is estimated to be around 90.67% and P content is about 0.17%.3 In this study, the pressure ranges up to 400 GPa are considered. The predicted structures of Fe–P binary compounds were used to evaluate the formation enthalpy (ΔH) relative to the elemental solids Fe and P according to eqn (1), in order to explore the thermodynamic stabilities of Fe–P compounds.| ΔH(FemPn) = [H(FemPn) − mH(Fe) − nH(P)]/(m + n), (m = 1–4 and n = 1, or m = 1 and n = 2) | (1) |
, 8 formula units per cell) are a = b = 9.13 Å and c = 4.40 Å, which are in excellent agreement with the experimental values at a = b = 9.10 Å and c = 4.46 Å.12 These lattice parameter calculations support the validity of the pseudo potentials adopted in this study.
 | ||
| Fig. 1 Relative thermodynamic stabilities of Fe–P compounds. (a) Convex hulls of the calculated formation enthalpy per atom of the Fe–P system relative to the elemental solids Fe and P are shown as solid lines. Compounds corresponding to data points located on the convex hulls are thermodynamically stable. The bcc structure,57 and hcp structure58 of solid Fe, and the phases I (Cmca), II (Pmm), III (P6/mmm), IV (Imm), and V (I3d) of solid P59,60 are used to calculate the enthalpy of formation. Detailed information about the element solids can be found in Fig. S1.† (b) Pressure–composition phase diagram of the Fe–P system. | ||
Under high pressure, the relative stabilities of our considered compositions are significantly different from that at ambient pressure. Specifically, the FeP2 compound becomes unstable above 82 GPa, whereas the iron-rich compounds are relatively stable under high pressure. These phases of Fe4P, Fe2P and FeP are stable in the entire considered pressure range (0–400 GPa). Notably, the Fe3P phase is unstable compared above 214 GPa (Fig. S2†) and decomposes into Fe2P plus Fe4P. The pressure–composition phase diagram of the Fe–P system is shown in Fig. 1b. For all the predicted structures, the calculated phonon dispersion curves confirm their dynamical stability with no imaginary frequencies observed in the entire Brillouin zone (Fig. S4†).
3.2. Geometry structure of the stable phases
Two new phases (i.e. C2/c-structured Fe4P and Cmcm-structured Fe3P) were discovered by comparing previous structures and our predicted structures, and their structural characters are shown in Fig. 2. The predicted C2/c-structured Fe4P (4 formula units) is more stable than the phase with P213 symmetry above 37 GPa (Fig. S3a†). Its lattice parameters are a = 7.342 Å, b = 3.772 Å and c = 5.627 Å at 200 GPa. The structure contains two inequivalent Fe atoms occupying 8f (0.297, 0.337, 0.850) and 8f (0.900, 0.671, 0.551) sites and one equivalent P atom sitting at the 4e (0.000, 0.170, 0.750) position (Fig. 2a). To distinguish the two Fe atoms at different sites, we labelled the inequivalent Fe atoms with two different colors (Fig. 2c). In this structure, each P atom is in 12-fold coordination with the Fe atoms (Fig. 2b), forming a tetrakaidecahedron. The adjacent polyhedrons are interconnected through common edges or co-vertices. The distance of the two nearest Fe–Fe is 2.11 Å, which is smaller than the Fe–Fe distance of 2.48 Å in bulk Fe with Imm symmetry under ambient conditions57 and 2.44 Å in Fe2SH3 at 100 GPa.43 However, this structure is nonmagnetic, as will be discussed later. To determine the interatomic interaction in the C2/c-structure Fe4P, the integrated crystal orbital Hamilton populations (ICOHPs) for the Fe–P and Fe–Fe.
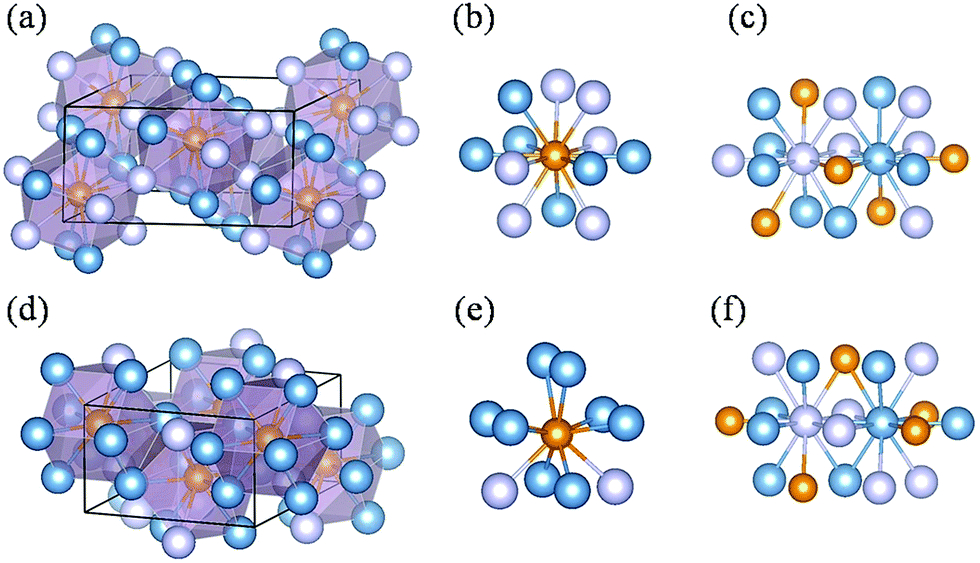 | ||
| Fig. 2 Crystal structures of the Fe4P and Fe3P compounds at 200 GPa. (a) Fe4P in the C2/c structure. (b) Coordination environment of the P atom in Fe4P. (c) Coordination environment of the Fe atoms in Fe4P. (d) Fe3P in the Cmcm structure. (e) Coordination environment of the P atom in Fe3P. (f) Coordination environment of the Fe atom in Fe3P. Here, the large shallow blue or dark blue and small orange spheres represent Fe and P atoms, respectively. | ||
Pairs at 200 GPa were calculated and are shown in Table S1.† The values of ICOHP between the Fe–P and Fe–Fe pairs are −1.80 and −4.97 eV, respectively. The ICOHP value of the Fe–Fe pair is very close to −4.90 eV in Fe2SH3.43 Apparently, the Fe–Fe interaction plays an important role in determining the structural stability of the compound. Further analysis indicates that the major contribution of the Fe–Fe interaction originates from 4s–3d, which corresponds to the decomposed ICOHP of −1.908 eV per pair.
The Fe3P compound with the I structure symmetry is stable at ambient conditions, and the first phase transition is observed at 64 GPa.13 This phase is assigned as the P4/mnc or Pnma structure. Notably, we discovered an orthorhombic structure of Fe3P (space group Cmcm, 4 formula units per cell) above 58 GPa. This structure is much more stable than the P4/mnc or Pnma structure in the pressure range of 0–400 GPa (Fig. S3b†). The lattice parameters of the Cmcm-structure Fe3P are a = 5.446 Å, b = 6.569 Å and c = 3.255 Å. This structure contains two inequivalent Fe atoms occupying the 4c (0.500, 0.900, 0.750), and 8g (0.784, 0.117, 0.750) positions, and one equivalent P occupying the 4c (0.000, 0.170, 0.250) site. For each P atom, there are 10 nearest neighbours of Fe atoms, forming a P–Fe hendecahedron (Fig. 2e). The adjacent polyhedrons have common edges. The two inequivalent Fe atoms are labelled with two different colors (Fig. 2f). The nearest Fe–Fe distance in the Cmcm phase is 2.10 Å, which is comparable to 2.14 Å in Fe4P. Unlike Fe4P with C2/c symmetry, the Cmcm-structured Fe3P structure is magnetic. An experimental study investigated the phase transitions of Fe3P, in which the P4/mnc or Pnma structure is stable above 64 GPa.13 However, our predicted Cmcm phase is more stable than the P4/mnc or Pnma phase at 64 GPa. The specific ΔH values of the three Fe3P phases can be found in Table S2.† To confirm the existence of the Cmcm phase, the simulated XRD pattern of the Cmcm phase is shown in Fig. 3 along with the experimental patterns. Several possible structures of Fe3P are also included in Fig. 3. Clearly, the main peak of Cmcm-structured Fe3P is close to the experimental peaks or that of the other phases. Thus, Cmcm-structured Fe3P might disappear under these experimental conditions.
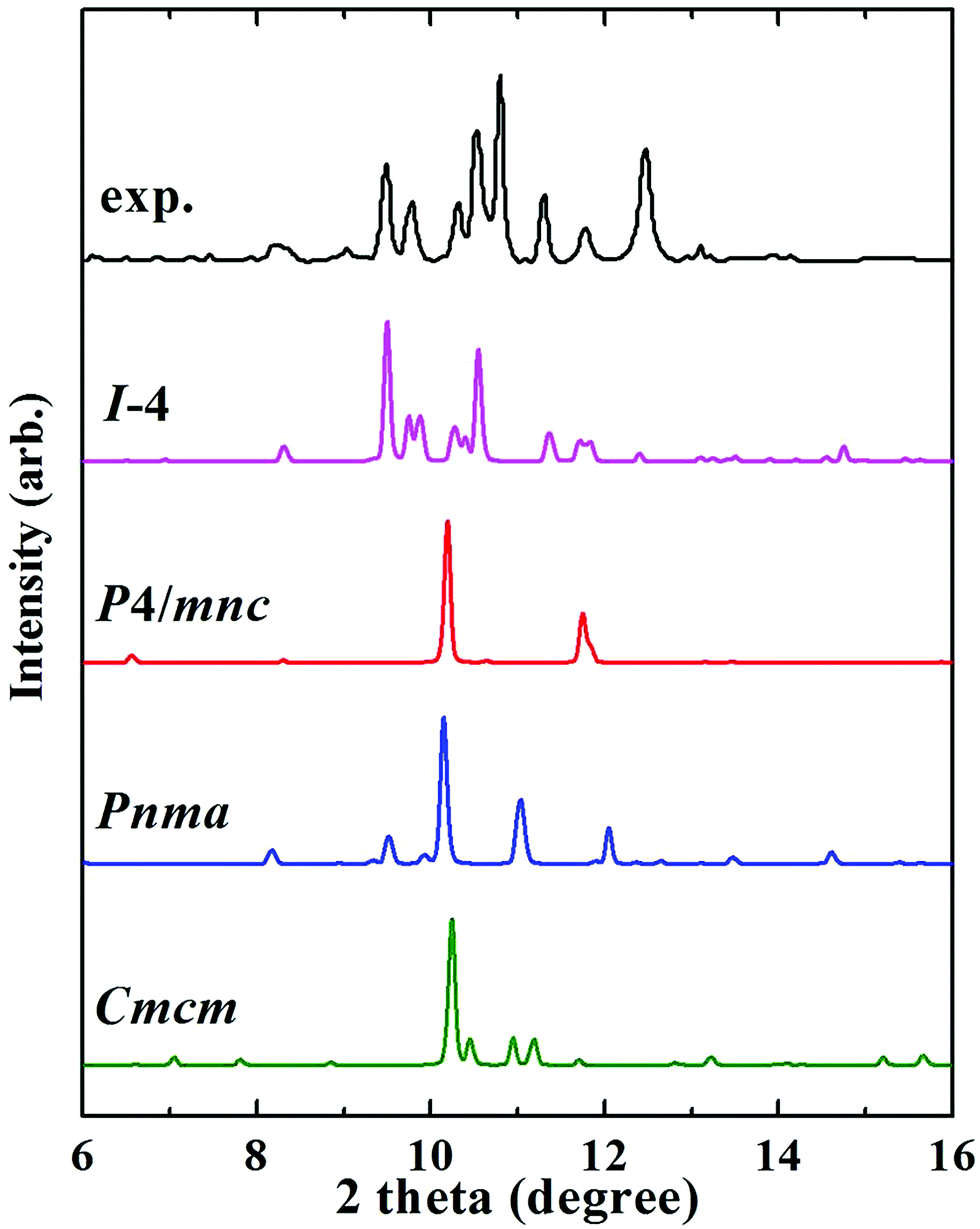 | ||
| Fig. 3 XRD patterns of Fe3P selected in synchrotron X-radiation with a fixed wavelength of 0.3344 Å at 64 GPa. The XRD pattern of Fe3P is described by a black line, which was collected in an experimental work.13 The XRD patterns of I, P4/mnc, Pnma, and Cmcm structures are labelled by pink, red, blue, and green lines, respectively. | ||
3.3. Electronic properties of stable phases
The electronic band structures and projected density of states (PDOS) of the Fe4P and Fe3P compounds are shown in Fig. 4. Similar to the electronic properties of the other Fe–P species,18 there are several bands crossing the Fermi level, which indicate that they are metallic. As can be seen in Fig. 4, there is a common character of a pronounced Fe 3d component near the Fermi level. Interestingly, the Cmcm-structured Fe3P is a ferromagnetic phase with the magnetic moment of 3.31μB per cell at 200 GPa by our calculations. Its electronic band structures and spin-polarized DOS based on spin up and spin down can be found in Fig. 4c and d. The calculated DOS per Fe atom at the Fermi level is about 1.0 eV−1 (Fig. 4d), which satisfies the Stoner criterion.61 | ||
| Fig. 4 Electronic band structures and projected density of states (PDOS) of the C2/c-structured Fe4P (a and b), and the Cmcm-structured Fe3P (c and d). The electronic band structures of spin up and spin down are described by the black and green lines, respectively. | ||
4. Conclusion
To find the stable phases of Fe–P binary compounds, we explored the crystal structures and stabilities of the Fe–P system with FexPy (x = 1–4 and y = 1, or x = 1 and y = 2) compositions under high pressure using first principles calculations combined with swarm structural searches. It is found that the FeP, Fe2P and Fe4P compounds are stable in the whole pressure range of 0–400 GPa, and Fe3P becomes unstable above 214 GPa. Several phases of iron-rich compounds can be synthesized at elevated conditions, which may be the possible phases in the Earth's cores. The Fe4P and Fe3P compounds are metallic, with a pronounced Fe 3d component across the Fermi level. Our study provides the theoretical basis for the further investigation of Fe–P binary compounds. It is also important to explore the constituents of the Earth's core.Acknowledgements
This study is supported by the Natural Science Foundation of China under No. 21573037; The Natural Science Foundation of Jilin Province (No. 20150101042JC); The Postdoctoral Science Foundation of China (under Grant No. 2013M541283). Parts of the calculations were performed in the High Performance Computing Centre of Jilin University.Notes and references
- J.-P. Poirier, Phys. Earth Planet. Inter., 1994, 85, 319–337 CrossRef CAS.
- A. S. Côté, L. Vačadlo and J. P. Brodholt, Geophys. Res. Lett., 2008, 35, L05306 CrossRef.
- H. S. Washington, Am. J. Sci., 1925, 53, 352–378 Search PubMed.
- S. Ono, A. R. Oganov, J. P. Brodholt, L. Vočadlo, I. G. Wood, A. Lyakhov, C. W. Glass, A. S. Côté and G. D. Price, Earth Planet. Sci. Lett., 2008, 272, 481–487 CrossRef CAS.
- D. Errandonea, D. Santamaría-Perez, A. Vegas, J. Nuss, M. Jansen, P. Rodríguez-Hernandez and A. Muñoz, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 94113 CrossRef.
- N. Sata, K. Hirose, G. Shen, Y. Nakajima, Y. Ohishi and N. Hirao, J. Geophys. Res.: Solid Earth, 2010, 115, B09204 Search PubMed.
- D. M. Sherman, Earth Planet. Sci. Lett., 1995, 132, 87–98 CrossRef CAS.
- W. F. Mcd, Treatise Geochem., 2003, 2, 547–568 Search PubMed.
- A. J. Stewart and M. W. Schmidt, Geophys. Res. Lett., 2007, 34, L13201 CrossRef.
- G. Pratesi, L. Bindi and V. Moggi-Cecchi, Am. Mineral., 2006, 91, 451–454 CrossRef CAS.
- X. Wu, M. Mookherjee, T. Gu and S. Qin, Geophys. Res. Lett., 2011, 38, L20301 Search PubMed.
- H. P. Scott, S. Huggins, M. R. Frank, S. J. Maglio, C. D. Martin, Y. Meng, J. Santillán and Q. Williams, Geophys. Res. Lett., 2007, 34, L06302 CrossRef.
- T. Gu, Y. Fei, X. Wu and S. Qin, Earth Planet. Sci. Lett., 2014, 390, 296–303 CrossRef CAS.
- P. Dera, B. Lavina, L. A. Borkowski, V. B. Prakapenka, S. R. Sutton, M. L. Rivers, R. T. Downs, N. Z. Boctor and C. T. Prewitt, Geophys. Res. Lett., 2008, 35, L10301 CrossRef.
- T. Gu, X. Wu, S. Qin, C. McCammon and L. Dubroksky, Eur. Phys. J. B, 2013, 83, 311 CrossRef.
- X. Wu and S. Qin, J. Phys.: Conf. Ser., 2010, 215, 12110 CrossRef.
- T. Gu, X. Wu, S. Qin and L. Dubrovinsky, Phys. Earth Planet. Inter., 2011, 184, 154–159 CrossRef CAS.
- H. Yan, Comput. Mater. Sci., 2015, 107, 204–209 CrossRef CAS.
- E. Dahl, Acta Chem. Scand., 1969, 23, 2677–2684 CrossRef CAS.
- T.-T. Gu, X. Wu, S. Qin, J. Liu, Y.-C. Li and Y.-F. Zhang, Chin. Phys. Lett., 2012, 29, 26102 CrossRef.
- J. Wu, X. Chong, R. Zhou, Y. Jiang and J. Feng, RSC Adv., 2015, 5, 81943–81956 RSC.
- P. F. McMillan, Chem. Soc. Rev., 2006, 35, 855–857 RSC.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 94116 CrossRef.
- Y. Wang, J. Lv, L. Zhu, S. Lu, K. Yin, Q. Li, H. Wang, L. Zhang and Y. Ma, J. Phys.: Condens. Matter, 2015, 27, 203203 CrossRef PubMed.
- A. P. Drozdov, M. I. Eremets, I. A. Troyan, V. Ksenofontov and S. I. Shylin, Nature, 2015, 525, 73–76 CrossRef CAS PubMed.
- D. Duan, Y. Liu, F. Tian, D. Li, X. Huang, Z. Zhao, H. Yu, B. Liu, W. Tian and T. Cui, Sci. Rep., 2014, 4, 6968 CrossRef CAS PubMed.
- Y. Li, J. Hao, H. Liu, Y. Li and Y. Ma, J. Chem. Phys., 2014, 140, 174712 CrossRef PubMed.
- Y. Li, J. Hao, H. Liu, S. Lu and J. S. Tse, Phys. Rev. Lett., 2015, 115, 105502 CrossRef PubMed.
- L. Zhu, H. Liu, C. J. Pickard, G. Zou and Y. Ma, Nat. Chem., 2014, 6, 644–648 CAS.
- W. Zhang, A. R. Oganov, A. F. Goncharov, Q. Zhu, S. E. Boulfelfel, A. O. Lyakhov, E. Stavrou, M. Somayazulu, V. B. Prakapenka and Z. Konopkova, Science, 2013, 342, 1502–1505 CrossRef CAS PubMed.
- M. S. Miao, X. L. Wang, J. Brgoch, F. Spera, M. G. Jackson, G. Kresse and H. Q. Lin, J. Am. Chem. Soc., 2015, 137, 14122–14128 CrossRef CAS PubMed.
- D. Pinkowicz, M. Rams, M. Misek, K. V. Kamenev, H. Tomkowiak, A. Katrusiak and B. Sieklucka, J. Am. Chem. Soc., 2015, 137, 8795–8802 CrossRef CAS PubMed.
- F. Peng, M. Miao, H. Wang, Q. Li and Y. Ma, J. Am. Chem. Soc., 2012, 134, 18599–18605 CrossRef CAS PubMed.
- H. Wang, J. S. Tse, K. Tanaka, T. Iitaka and Y. Ma, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6463–6466 CrossRef CAS PubMed.
- J. Botana, X. Wang, C. Hou, D. Yan, H. Lin, Y. Ma and M. S. Miao, Angew. Chem., Int. Ed., 2015, 127, 9412–9415 CrossRef.
- L. Zhu, H. Liu, C. J. Pickard, G. Zou and Y. Ma, Nat. Chem., 2014, 6, 644–648 CAS.
- X. Zhong, H. Wang, J. Zhang, H. Liu, S. Zhang, H. F. Song, G. Yang, L. Zhang and Y. Ma, Phys. Rev. Lett., 2016, 116, 57002 CrossRef PubMed.
- Y. Wang and Y. Ma, J. Chem. Phys., 2014, 140, 40901 CrossRef PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Phys. Rev. Lett., 2011, 106, 15503 CrossRef PubMed.
- L. Zhu, H. Wang, Y. Wang, J. Lv, Y. Ma, Q. Cui, Y. Ma and G. Zou, Phys. Rev. Lett., 2011, 106, 145501 CrossRef PubMed.
- M. Zhang, H. Liu, Q. Li, B. Gao, Y. Wang, H. Li, C. Chen and Y. Ma, Phys. Rev. Lett., 2015, 114, 15502 CrossRef PubMed.
- S. Zhang, L. Zhu, H. Liu and G. Yang, Inorg. Chem., 2016, 55, 11434–11439 CrossRef CAS PubMed.
- G. Yang, Y. Wang, F. Peng, A. Bergara and Y. Ma, J. Am. Chem. Soc., 2016, 138, 4046–4052 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- A. Jain, G. Hautier, C. J. Moore, S. Ping Ong, C. C. Fischer, T. Mueller, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2011, 50, 2295–2310 CrossRef CAS.
- C. Rdl, F. Fuchs, J. Furthmller and F. Bechstedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235114 CrossRef.
- A. Koumina, M. Bacmann, D. Fruchart, S. Niziol, P. Wolfers, M. Mesnaoui, J. Tobola, S. Kaprzyk, R. Zach and N. Techniques, Ann. Chimie Sci. Materiaux, 1998, 23, 177–180 CrossRef CAS.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B, 2005, 71, 35105 CrossRef.
- J. D. Pack and H. J. Monkhorst, Phys. Rev. B, 1977, 16, 1748–1749 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- R. Dronskowski and P. E. Blöchl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- R. Skála and M. Drábek, Mineral. Mag., 2003, 67, 783–792 CrossRef.
- S. Rundqvist and F. Jellinek, Acta Chem. Scand., 1959, 13, 425–432 CrossRef CAS.
- N. Kohlhaas, R. Dunner and P. Schmitz-Pranghe, Z. Angew. Phys., 1967, 23, 245 Search PubMed.
- T. Takahashi, W. a. Basett and H.-K. Mao, J. Geophys. Res., 1968, 73, 4717–4725 CrossRef CAS.
- T. Sugimoto, Y. Akahama, H. Fujihisa, Y. Ozawa, H. Fukui, N. Hirao and Y. Ohishi, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 24109 CrossRef.
- H. Katzke and P. Tolédano, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 24109 CrossRef.
- J. F. Janak, Phys. Rev. B, 1977, 16, 255–262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Pressure phase diagram of the elementary substances of Fe and P; the calculated enthalpy difference as a function of pressure, for the reaction equation: 2Fe3P ↔ Fe2P + Fe4P; enthalpy differences of the Fe3P and Fe4P compounds as a function of pressure; calculated phonon spectra for the Fe3P and Fe4P compounds; calculated ICOHPs of Fe–Fe, and Fe–P pairs in Fe4P. See DOI: 10.1039/c7ra01567d |
| This journal is © The Royal Society of Chemistry 2017 |