
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An octanuclear Schiff-base complex with a Na2Ni6 core: structure, magnetism and DFT calculations†
Marek Machata,
Ivan Nemec ,
Radovan Herchel and
Zdeněk Trávníček*
,
Radovan Herchel and
Zdeněk Trávníček*
Department of Inorganic Chemistry, Regional Centre of Advanced Technologies and Materials, Faculty of Science, Palacký University, 17. listopadu 12, CZ-771 46 Olomouc, Czech Republic. E-mail: zdenek.travnicek@upol.cz
First published on 15th May 2017
Abstract
The octanuclear Na2Ni6 complex (Pr3NH)[Na2Ni6(L)4(Bza)5(HBza)(OH)2(ace)]·Et2O (1), where Pr3NH+ = the tripropylamonium cation, H2L = 2-[(E)-(2-hydroxybenzylidene)amino]phenol, HBza = benzoic acid and ace = acetone, was synthesized and characterized by the elemental analysis, FTIR spectroscopy, single crystal X-ray diffraction analysis, magnetic measurements and DFT calculations. All six NiII atoms are hexacoordinate with the {NiO6} or {NiNO5} chromophores forming two defective dicubane cores. The static magnetic data were fitted to the simplified spin Hamiltonian model which resulted in averaged ferromagnetic and antiferromagnetic exchange parameters J = +5.3 cm−1, and J = −9.2 cm−1, respectively, confirming the predominant role of the antiferromagnetic coupling in 1. The broken symmetry DFT method with various functionals (B3LYP, PBE0, TPPSh and CAM-B3LYP) was used to dissect information about magnetic coupling. As a result, the isotropic exchange parameters (Jab) derived by the PBE0 or B3LYP functionals seem to be the best to match the experimental magnetic data.
Introduction
Since the discovery of the first single-molecule magnets (SMMs),1 paramagnetic transition and inner transition metal complexes have been the subject of research interest in the field of molecular magnetism.2 In order to obtain SMMs, a non-zero spin ground state and non-negligible easy-axis magnetic anisotropy are required. Thus, two basic strategies, that can lead to the preparation of such compounds, can be recognized. The first approach is aimed at modulation of the magnetic anisotropy to favourable value and type (large, and axial, respectively) by careful choice of the ligands and symmetry of mononuclear complexes.3,4 In the recent works a very large single ion magnetic anisotropy of the NiII atoms was observed, nevertheless, the slow relaxation of magnetization in mononuclear NiII complexes is still observed only very rarely.5 The second strategy is aimed at preparation of the polynuclear complexes with well-separated and large ground spin state.6 Potential drawbacks of the latter strategy arise from predominant antiferromagnetic coupling7 and/or decrease of magnetic anisotropy due to different orientations of the local atomic D-tensors.2d,8,9 Thus, up to now, only a few homometalic and polynuclear NiII SMMs have been reported.7,10,11 Nevertheless, the research in the field of high-nuclear NiII complexes has brought the compounds with various structures such as wheels,12 grids,13 cages11b,14 or cubanes.7,11c–g,15 Interestingly, the majority of the NiII SMMs were reported for the cubanes.7,11c–g This might be attributed to the orthogonality of magnetic orbitals in such compounds as the bonding angles between the bridged metal centers are close to 90° favouring thus ferromagnetic coupling.16In our previous work9 we have reported on the crystal structure and magnetic properties of two new tetranuclear NiII compounds: [Ni4(L)4(CH3OH)3(H2O)] and (Pr3NH)2[Ni4(L)4(ac)2], where Pr3NH+ = the tripropylamonium cation, H2L = 2-[(E)-(2-hydroxybenzylidene)amino]phenol and Hac = acetic acid. We have shown that the used reaction solvent (i.e. CH3OH and/or CH2Cl2 in this case) strongly influence composition and topology of the polynuclear clusters despite using the same reactants and their ratios. Also, the magnetic properties of the above mentioned compounds differ significantly and (Pr3NH)2[Ni4(L)4(ac)2] exhibits a field-induced slow relaxation of magnetization. Furthermore, it must be noted that also other previously reported 3d,17 3d-4f18 or 4f19 polynuclear compounds with the H2L (and related) ligands exhibit interesting magnetic properties. With respect to the above mentioned facts and as a continuation of our ongoing study of polynuclear NiII compounds, we report here on the synthesis, crystal structure and magnetic properties of the octanuclear cubane-based complex of the formula (Pr3NH)[Na2Ni6(L)4(Bza)5(HBza)(OH)2(ace)]·Et2O (1), where HBza = benzoic acid, ace = acetone. The evaluation of magnetic properties was supported by thorough theoretical investigations utilizing a broken-symmetry DFT method with the B3LYP, PBE0, TPPSh and CAM-B3LYP functionals.
Results and discussion
Synthesis and crystal structure
Compound 1 (Fig. 1) was prepared by one-pot synthesis using NiCl2·6H2O, the Schiff base ligand H2L in the presence of sodium benzoate and Pr3N as bases (molar ratios: 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3:2), and acetone as the reaction solvent, see the eqn (1). Note: the molar ratios of the reactants mentioned above differ from those given in the eqn (1). This may be associated with the fact that the organic reactants behave as weak acids or bases which influences the chemical equilibria in solution during the reaction.
:1:3:2), and acetone as the reaction solvent, see the eqn (1). Note: the molar ratios of the reactants mentioned above differ from those given in the eqn (1). This may be associated with the fact that the organic reactants behave as weak acids or bases which influences the chemical equilibria in solution during the reaction.| 6NiCl2 + 4H2L + 8NaBza + 7Pr3N + 2H2O + ace ⇄ (Pr3NH)[Ni6Na2(L)4(Bza)5(HBza)(OH)2(ace)] + 6(Pr3NH)Cl + 6NaCl + 2HBza | (1) |
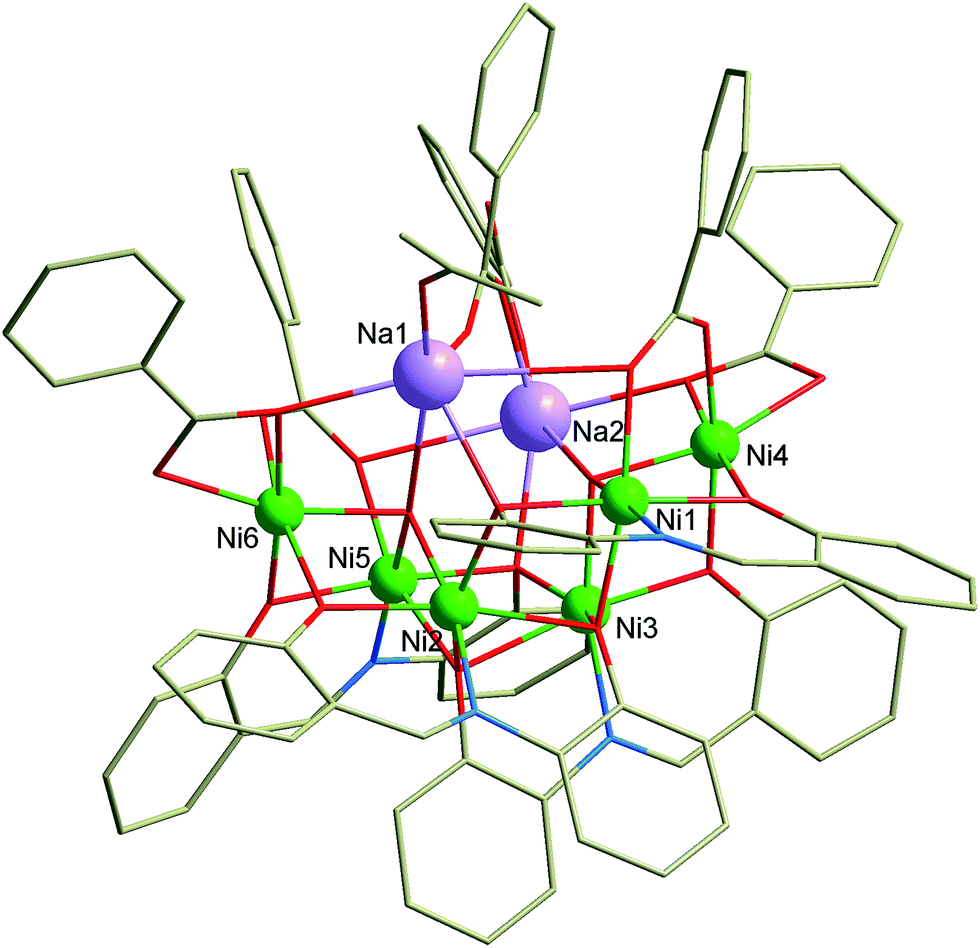 | ||
| Fig. 1 Molecular structure of the complex anion [Na2Ni6(L)4(Bza)5(HBza)(OH)2(ace)]−. Color code: bright green (Ni), violet (Na), red (O), blue (N), grey (C). Hydrogen atoms are omitted for clarity. | ||
The complex 1 crystallizes in the monoclinic space group P21/c and comprises the Pr3NH+ cation, octanuclear complex anion [Na2Ni6(L)4(Bza)5(HBza)(OH)2(ace)]− and co-crystalized Et2O molecule. Basic crystal data, structure refinement parameters, and selected bond lengths and angles are listed in Tables S1, S2, and Fig. S1 (in ESI†), respectively. The complex anion (Fig. 1) is composed of six NiII atoms, two Na+ atoms, four deprotonated L2− ligands, two μ3-OH− and five Bza− anionic ligands, one HBza and acetone (ace) coordinated molecules. All the NiII atoms are incorporated in two defective dicubane cores mutually connected by one face with the bridging (vertex) phenolate oxygen atoms (OPh) and the hydroxo ligands (OHy). The remaining coordination sites are occupied by the oxygen atoms originating from the carboxylate ligands (OCa), which adopt various bridging modes (2 × η2:η1:μ2, 1 × η1:η1:μ2, 2 × η1:η2:μ3 and 1 × η1, Fig. 1 and 2), and one acetone molecule. All the NiII atoms are hexacoordinate, but they differ in the composition of their coordination polyhedra: {NiO6} for the Ni4 and Ni6 atoms, {NiNO5} for the remaining ones (Fig. 1).
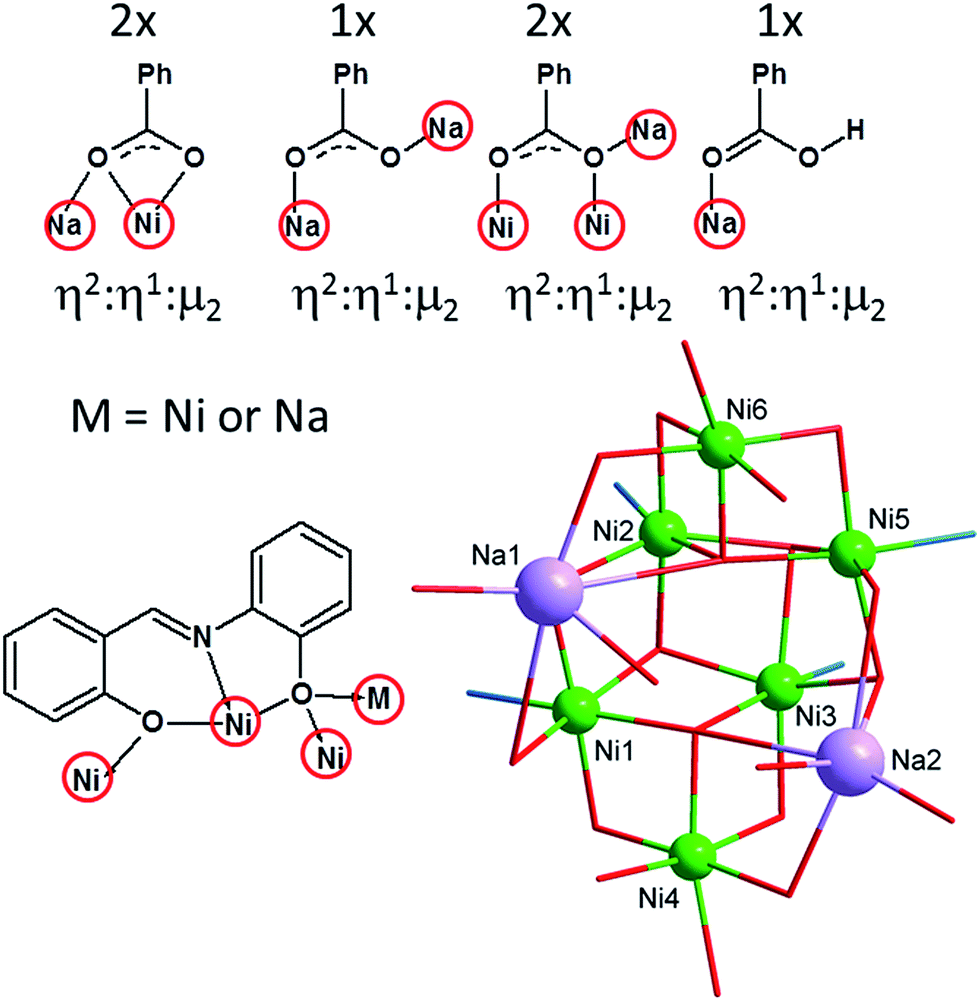 | ||
| Fig. 2 View of the bridging abilities of the benzoato (up) and H2L (down left) ligands, and simplified picture showing all the coordination polyhedrons and labelling of the metal atoms in 1 (down right). | ||
Furthermore, another difference between the Ni central atoms is observed. The coordination polyhedra of the Ni1 and Ni5 atoms are less angularly distorted (as defined by the parameter Σ,20 63.47° and 77.80°) than the remaining ones (96.8–119.4°). The bonding distances (in Å) within the coordination polyhedra of the NiII atoms vary in the range of 1.98–2.19 for the Ni–O and 1.99–2.03 for Ni–N bonds.
The Na+ atoms are hexacoordinate, coordinated solely by the oxygen atoms with the bond distances (in Å) ranging from 2.26 to 3.04. The OPh atoms are not equivalent from the view of their bridging functions and the asymmetry of L2−, which implies the aforementioned non-equivalency, as was discussed previously.18 In 1, the OPh atoms originating from the aldehydic part of the ligand L2− adopt μ bridging mode while the aminophenolic OPh atoms bridges three NiII atoms (μ3, Fig. 1 and 2). These atoms connect defective dicubane cores with Na+ atoms as well. Bond angles between the double oxo-bridged NiII atoms (in °) are ranging from 90.7 to 94.9 (Ni1–O–Ni2, Ni1–O–Ni4, Ni2–O–Ni3, Ni3–O–Ni5, Ni5–O–Ni6) and from 99.0 to 105.0 (Ni1–O–Ni3, Ni2–O–Ni5, Ni2–O–Ni6, Ni3–O–Ni4). Furthermore, the Ni atoms are not bridged solely by the double oxo-bridging oxygen atoms. Between the Ni1 and Ni4, and Ni5 and Ni6 atoms the additional η1:η2:μ3 bridging by the Bza− ligands (involving also bond with adjacent sodium atom, Fig. 2) takes place (Fig. 1).
The molecular structure of the complex anion is stabilized by two rather strong intramolecular hydrogen bonds between two μ3-OH groups and one OCa atom (from syn–anti – benzoate ligand bridging two Na atoms) with d(O⋯O) = 2.863(6) and 2.927(5) Å. The complexity of the molecular structure which was described above is not surprising in context of the bridging abilities of the H2L-type ligands (Fig. 2). The reactions of the NiII salts with the H2L-type ligands led previously to the preparation of cubane and defective dicubane Ni4 compounds,9,21 tetra and hexanuclear NaNi3 and NaNi5 compounds,17b or even Ni20 compound with bowl topology.22
There are no significant intermolecular contacts between the adjacent complex anions in the crystal structure and the shortest distance between the NiII atoms (in Å) of the adjacent complex anions is 9.6342(11). The only significant intermolecular contact is the N–H⋯O hydrogen bond between the tripropylamonium cation (donor) and the carboxylate group of the complex anion (acceptor) with the distance of 3.007(12) Å (Fig. S2 in ESI†).
Magnetic studies and DFT calculations
Variable-temperature dc magnetic data (Fig. 3, left) measured from 1.9 to 300 K showed room temperature value of μeff/μB equal to 7.55, which is consistent with six uncoupled NiII atoms with g = 2.16. The effective magnetic moment remains constant down to approximately 75 K and on further cooling it drops to 2.53 μB at T = 1.9 K. Such behaviour suggests presence of predominant antiferromagnetic coupling in 1.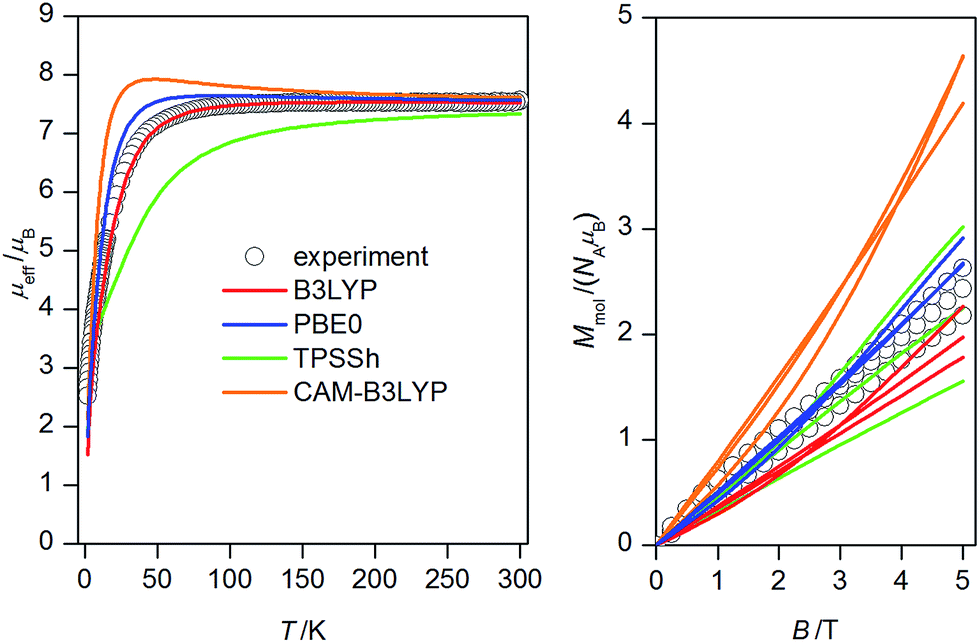 | ||
| Fig. 3 Magnetic data for 1: temperature dependence of the effective magnetic moment recorded under 0.1 T (left) and the isothermal magnetizations measured at 2, 5 and 10 K (right). Empty circles – experimental data, full lines – calculated data with Jab parameters resulted from DFT calculations listed in Table 1 and g = 2.16. | ||
Variable-field dc data (Fig. 3, right) measured up to 5 T do not display saturation most probably because there are close-lying spin states with various molecular spins affected also by magnetic anisotropy. In order to interpret magnetic data for 1, we postulated the following spin Hamiltonian
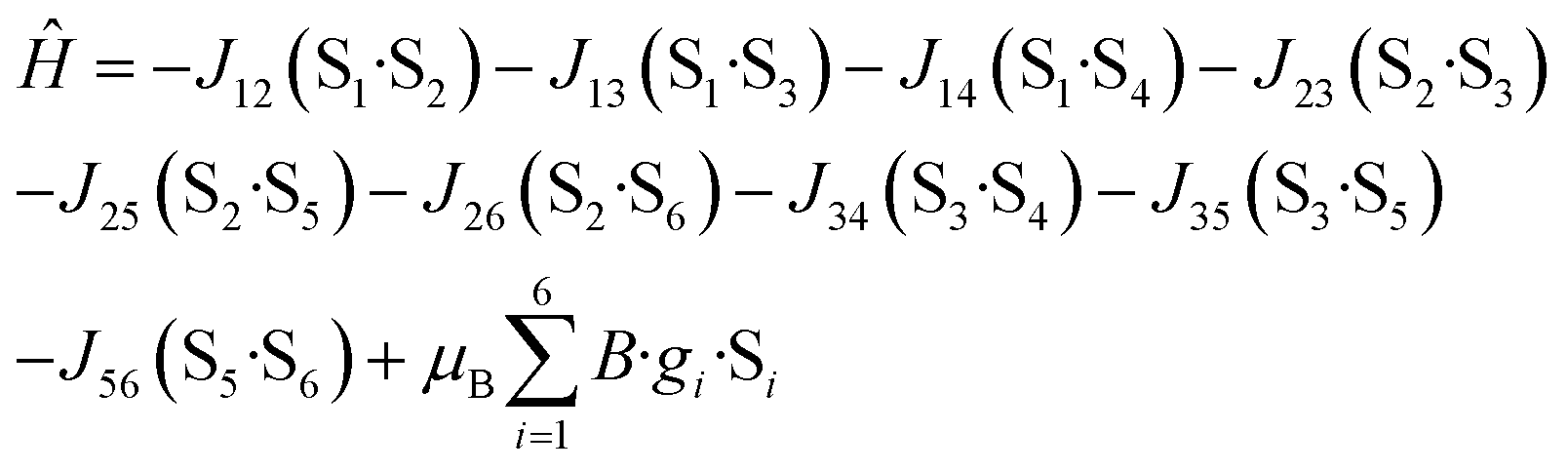 | (2) |
 | ||
| Fig. 4 Scheme of the magnetic coupling in 1. | ||
Therefore, we utilized Density Functional Theory (DFT) with the broken-symmetry approach to at least estimate values and nature of Jab in 1 and eventually facilitate the analysis of the magnetic properties following our previous works on polynuclear compounds.23 Herein, the ORCA computation package 3.0.3 was employed for all the calculations and polarized triple-zeta basis set def2-TZVP(-f) was used for all the atoms except for carbon and hydrogen atoms, for which def2-SVP basis set was used. The calculations were done for the molecular fragment (Pr3NH)[Na2Ni6(L)4(Bza)5(HBza)(OH)2(ace)] of 1 (Fig. 5). The broken-symmetry spin states (BS) need to be calculated and their energies compared to the energy of the high spin state (HS) in order to compute J-parameters. Herein, the Ruiz's approach was employed to derive the following relationships
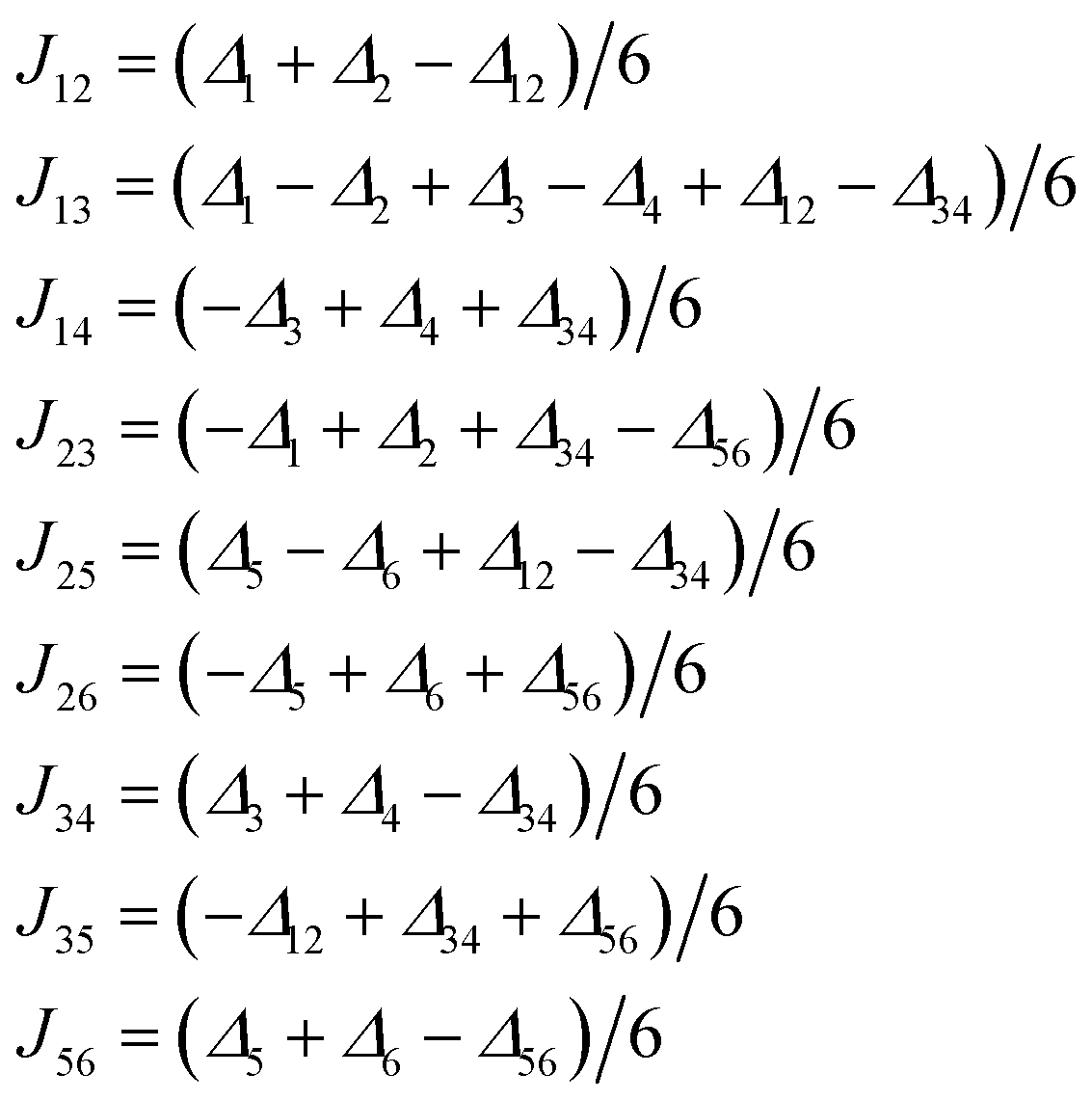 | (3) |
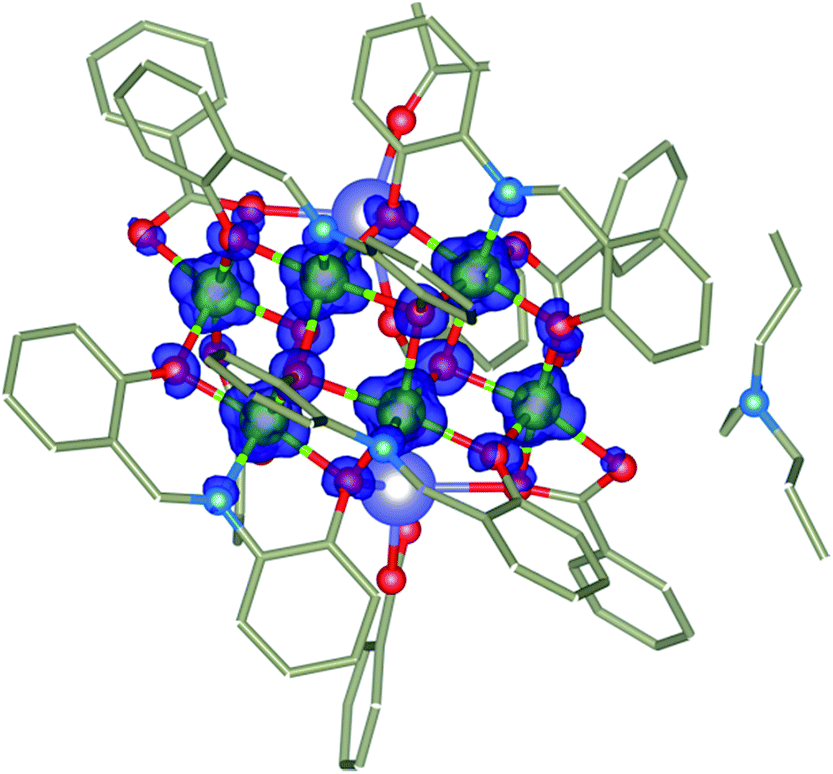 | ||
| Fig. 5 The molecular fragment (Pr3NH)[Na2Ni6(L)4(Bza)5(HBza)(OH)2(ace)] of 1 and B3LYP calculated spin isodensity surface for high spin state with the cutoff values of 0.01 e a0−3. Hydrogen atoms are omitted for clarity. | ||
The resulting Jab parameters are listed in Table 1 for all the above mentioned functionals and the details for the B3LYP functional are listed in Table 2. It follows from Table 1 that all the functionals (B3LYP, PBE0, TPPSh and CAM-B3LYP) provided consistent sets of J-parameters concerning their nature, concretely, J12, J14, J23, J35 and J56 are all ferromagnetic, and the J13, J25, J26 and J34 are all antiferromagnetic.
| B3LYP | PBE0 | TPSSh | CAM-B3LYP | |
|---|---|---|---|---|
| J12 | +10.5 | +8.86 | +9.63 | +9.39 |
| J13 | −6.72 | −4.69 | −13.2 | −2.50 |
| J14 | +16.9 | +15.1 | +14.7 | +15.6 |
| J23 | +13.8 | +11.9 | +13.3 | +12.0 |
| J25 | −12.4 | −8.87 | −20.7 | −7.11 |
| J26 | −14.2 | −9.26 | −25.9 | −6.49 |
| J34 | −14.1 | −9.47 | −25.0 | −7.02 |
| J35 | +7.48 | +6.77 | +4.64 | +7.52 |
| J56 | +16.0 | +13.8 | +14.8 | +14.2 |
| Spin state | 〈S2〉 | Δi (CM−1) |
|---|---|---|
| a Δi = εBS,i − εHS. | ||
| HS, |αααααα〉 | 42.02 | 0 |
| BS1, |βααααα〉 | 22.02 | 62.030 |
| BS2, |αβαααα〉 | 22.01 | −6.932 |
| BS3, |ααβααα〉 | 22.01 | 1.305 |
| BS4, |αααβαα〉 | 22.02 | 8.281 |
| BS5, |ααααβα〉 | 22.02 | 33.140 |
| BS6, |αααααβ〉 | 22.02 | 5.228 |
| BS12, |ββαααα〉 | 10.01 | −7.901 |
| BS34, |ααββαα〉 | 10.02 | 94.419 |
| BS56, |ααααββ〉 | 10.01 | −57.427 |
Next, the obtained four sets of J's values from each DFT functional were afterwards used to calculate magnetic properties of 1 with the spin Hamiltonian in the eqn (2). The calculated magnetic data are depicted in Fig. 3, from which we may conclude that PBE0 and B3LYP derived J-parameters delivered the best agreement with the experiment.
The CAM-B3LYP functional seems to slightly overestimate effect of the ferromagnetic exchanges, whereas TPPSh overestimates the effect of the antiferromagnetic exchanges. Furthermore, the attempt to find the magneto-structural correlations was done for PBE0-derived parameters as outlined in Fig. 6, where a linear equation of the form
| J (cm−1) = 196(30) − 1.99(31)·α | (4) |
 | ||
| Fig. 6 A magneto-structural correlation, J vs. α, α = < (Ni–O–Ni)av, for the PBE0-calculated J-parameters. The confidence interval with 95% is shown (dotted lines). The grey dashed lined represents predicted borderline between the ferromagnetic and the antiferromagnetic exchange parameters. | ||
Taking into the account that both PBE0 and B3LYP derived antiferromagnetic and ferromagnetic J-parameters are in more less narrow interval, the experimental magnetic data were analysed also with very simplified model using only two J-parameters, thus averaging the ferromagnetic exchanges (JF = J12 = J14 = J23 = J35 = J56) and the antiferromagnetic exchanges (JAF = J13 = J25 = J26 = J34). Under this assumption, good agreement with the experimental data was achieved for JF = +5.3 cm−1 and JAF = −9.2 cm−1 and g = 2.20 (Fig. 7). Both temperature and field dependent magnetic data are well described by this model, only calculated data for the isothermal magnetization at T = 2 K are a bit higher than the experimental one, which can be due the simplicity of the model and also due to expected significant single-ion zero-field splitting for NiII ions in deformed octahedral coordination environment, which usually leads to lower saturation values of the isothermal magnetization.25
 | ||
| Fig. 7 Magnetic data for 1: temperature dependence of the effective magnetic moment recorded under 0.1 T (left) and the isothermal magnetizations measured at 2, 5 and 10 K (right). Empty circles – experimental data, full lines – calculated data with JF = +5.3 cm−1 and JAF = −9.2 cm−1 and g = 2.20. | ||
Conclusions
In conclusion, we have prepared a heterobimetallic complex (Pr3NH)[Na2Ni6(L)4(Bza)5(HBza)(OH)2(ace)]·Et2O, (1). Its crystal structure revealed that the paramagnetic NiII atoms are arranged in two defective dicubane cores. Due to the structural complexity, implying the magnetic exchange interactions in 1, a few DFT functionals (B3LYP, PBE0, TPSSh, CAM-B3LYP) in combination with the def2-TZVP(-f) and def2-SVP basis sets were utilized for calculations of magnetic coupling parameters. It has been shown that the PBE0 and B3LYP methods provide the best estimates of Jab. The nature of the magnetic exchanges is governed by the average Nia-O-Nib angle with the estimated crossing point of the ferromagnetic and antiferromagnetic values at α = 98.5°, and this value is in good agreement with that obtained using the magneto-structural correlations (α ≈ 99°) reported for the Ni4-cubane compounds previously. Utilizing the theoretical calculations, the simplification of the spin Hamiltonian parameters was suggested leading to the fitted average isotropic exchange parameters, JF = +5.3 cm−1 and JAF = −9.2 cm−1 confirming the predominant role of the antiferromagnetic coupling in 1.Experimental section
Synthesis
All used chemicals and solvents were purchased from commercial sources and were used without any further purification. The ligand H2L was prepared as reported previously.26![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1614 (vs.), ν(CN), ν(CC) 1597, 1537 (vs.), ν(C–O) 1223 (s).
O) 1614 (vs.), ν(CN), ν(CC) 1597, 1537 (vs.), ν(C–O) 1223 (s).General methods
Elemental analysis (C, H, N) was performed on a ThermoScientific Flash 2000 CHNS–O Analyser. The infrared spectrum of the complex was recorded on a ThermoNicolet NEXUS 670 FT-IR spectrometer using the ATR technique on a diamond plate in the range of 400–4000 cm−1. The magnetic data were measured on powdered samples using a PPMS Dynacool system (Quantum Design) with VSM option. Experimental data were corrected for the diamagnetism of the constituent atoms by using Pascals' constants and for the diamagnetism of the sample holder.Acknowledgements
We acknowledge the financial support from the National Programme of Sustainability I (LO1305) of the Ministry of Education, Youth and Sports of the Czech Republic, and from Palacký University in Olomouc (PrF_2016_007).Notes and references
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- (a) R. E. P. Winpenny, Chem. Soc. Rev., 1998, 27, 447–452 RSC; (b) Y. Wei, H. Hou, Y. Fan and Y. Zhu, Eur. J. Inorg. Chem., 2004, 19, 3946–3957 CrossRef; (c) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS; (d) M. Nakano and H. Oshio, Chem. Soc. Rev., 2011, 40, 3239–3248 RSC; (e) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (f) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (g) A. K. Bar, C. Pichon and J.-P. Sutter, Coord. Chem. Rev., 2016, 308, 346–380 CrossRef CAS.
- (a) P.-H. Lin, N. C. Smythe, S. I. Gorelsky, S. Maguire, N. J. Henson, I. Korobkov, B. L. Scott, J. C. Gordon, R. T. Baker and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 15806–15809 CrossRef CAS PubMed; (b) S. Gomez-Coca, E. Cremades, N. Aliga-Alcalde and E. Ruiz, J. Am. Chem. Soc., 2013, 135, 7010–7018 CrossRef CAS PubMed; (c) L. Chen, J. Wang, J.-M. Wei, W. Wernsdorfer, X.-T. Chen, Y.-Q. Zhang, Y. Song and Z.-L. Xue, J. Am. Chem. Soc., 2014, 136, 12213–12216 CrossRef CAS PubMed; (d) S. Goméz-Coca, D. Aravena, R. Morales and E. Ruiz, Coord. Chem. Rev., 2015, 289–290, 379–392 CrossRef; (e) G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- I. Nemec, R. Herchel, I. Svoboda, R. Boča and Z. Trávníček, Dalton Trans., 2015, 44, 9551–9960 RSC.
- J. Miklovič, D. Valigura, R. Boča and J. Titiš, Dalton Trans., 2015, 44, 12484–12487 RSC.
- (a) A. M. Ako, V. Mereacre, Y. Lan, W. Wernsdorfer, R. Clérac, C. A. Anson and A. K. Powell, Inorg. Chem., 2010, 49, 1–3 CrossRef CAS PubMed; (b) T. Taguchi, W. Wernsdorgfer, K. A. Abboud and G. Christou, Inorg. Chem., 2010, 49, 10579–10589 CrossRef CAS PubMed; (c) M. Ibrahim, Y. Lan, B. S. Bassil, Y. Xiang, A. Suchopar, A. K. Powell and U. Kortz, Angew. Chem., Int. Ed., 2011, 50, 4708–4711 CrossRef CAS PubMed; (d) G. P. Guedes, S. Soriano, N. M. Comerlato, N. L. Spezialli, M. A. Novak and M. G. F. Vaz, Inorg. Chem. Commun., 2013, 37, 101–105 CrossRef CAS; (e) A. E. Thuijs, P. King, K. A. Abboud and G. Christou, Inorg. Chem., 2015, 54, 9127–9137 CrossRef CAS PubMed.
- S. T. Ochsenbein, M. Murrie, E. Rusanov, H. Stoeckli-Evans, C. Sekine and H. U. Gude, Inorg. Chem., 2002, 41, 5133–5140 CrossRef CAS PubMed.
- J. Ribas-Arino, T. Baruah and M. R. Pederson, J. Am. Chem. Soc., 2006, 128, 9497–9505 CrossRef CAS PubMed.
- R. Herchel, I. Nemec, M. Machata and Z. Trávníček, Dalton Trans., 2016, 45, 18622–18634 RSC.
- (a) G. Rogez, J.-N. Rebilly, A.-L. Barra, L. Sorace, G. Blondin, N. Kirchner, M. Duran, J. van Slageren, S. Parsons, L. Ricard, A. Marvilliers and T. Mallah, Angew. Chem., Int. Ed., 2005, 44, 1876–1879 CrossRef CAS PubMed; (b) S. Gómez-Coca, E. Cremades, N. Aliaga-Alcalde and E. Ruiz, Inorg. Chem., 2014, 53, 676–678 CrossRef PubMed.
- (a) C. Cadiou, M. Murrie, C. Paulsen, V. Villar, W. Wernsdorfer and R. E. P. Winpenny, Chem. Commun., 2001, 24, 2666–2667 RSC; (b) A. Bell, G. Aromí, S. T. Teat, W. Wernsdorfer and R. E. P. Winpenny, Chem. Commun., 2005, 22, 2808–2810 RSC; (c) G. Aromí, S. Parsons, W. Wernsdorfer, E. K. Brechin and J. L. McInnes, Chem. Commun., 2005, 22, 5038 RSC; (d) G. Aromí and E. K. Brechin, Struct. Bonding, 2006, 122, 1–67 CrossRef; (e) R. T. W. Scott, L. F. Jones and I. S. Tidmarsh, et al., Chem.–Eur. J., 2009, 15, 12389 CrossRef CAS PubMed; (f) S. Petit, P. Neugebauer, G. Pilet, G. Chastanet, A.-L. Barra, A. B. Antunes, W. Wernsdorfer and D. Luneau, Inorg. Chem., 2012, 51, 6645–6654 CrossRef CAS PubMed; (g) S. Hameury, L. Kayser, R. Pattacini, P. Rosa, A.-L. Barra and P. Braunstein, ChemPlusChem, 2015, 80, 1312–1320 CrossRef CAS.
- (a) G. E. Lewis and C. S. Kraihanzel, Inorg. Chem., 1983, 22, 2895–2899 CrossRef CAS; (b) A. J. Blake, C. M. Grant, S. Parsons, J. M. Rawson and R. E. P. Winpenny, J. Chem. Soc., Chem. Commun., 1994, 20, 2363–2364 RSC; (c) M. L. Tong, M. Monfort, J. M. Clemente-Juan, X.-M. Chen, X.-H. Bu, M. Ohba and S. Kitagawa, Chem. Commun., 2005, 2, 233–235 RSC; (d) D. Fouguet-Albiol, K. A. Abboud and G. Christou, Chem. Commun., 2005, 34, 4282–4284 RSC.
- (a) V. Niel, V. A. Milway and L. N. Dawe, et al., Inorg. Chem., 2008, 47, 176–189 CrossRef CAS PubMed; (b) K. V. Shuvaev, S. S. Tandon, L. N. Dawe and L. K. Thompson, Chem. Commun., 2010, 46, 4755–4757 RSC; (c) L. K. Thompson and L. N. Dawe, Coord. Chem. Rev., 2015, 289–290, 13–31 CrossRef CAS.
- B. A. Breeze, M. Shanmugam, F. Tuna and R. E. P. Winpenny, Chem. Commun., 2007, 48, 5185–5187 RSC.
- (a) M. Murrie, D. Biner, H. Stoeckli-Evans and H. U. Gudel, Chem. Commun., 2003, 8, 230–231 RSC; (b) J. Zhang, P. Teo, R. Pattacini, A. Kermagoret, R. Welter, G. Rogez, T. S. Andy Hor and P. Braustein, Angew. Chem., Int. Ed., 2010, 49, 4443–4446 CAS; (c) S. T. Meally, C. McDonnald, G. Karotsis, G. S. Papaefstathiou, E. K. Brechin, P. W. Dunne, P. McArdle, N. P. Power and L. F. Jones, Dalton Trans., 2010, 39, 4809–4816 RSC; (d) S.-H. Zhang, N. Li, C.-M. Ge, C. Feng and L.-F. Ma, Dalton Trans., 2011, 40, 3000–3007 RSC.
- M. A. Halcrow, J.-S. Sun, J. C. Huffman and G. Christou, Inorg. Chem., 1995, 34, 4167–4177 CrossRef CAS.
- (a) E. Loukopoulos, B. Berkoff, K. Griffiths, V. Keeble, V. N. Dokorou, A. C. Tsipis, A. Escuer and G. E. Kostakis, CrystEngComm, 2015, 17, 6753–6764 RSC; (b) K. Griffiths, A. Escuer and G. E. Kostakis, Struct. Chem., 2016, 27, 1703–1714 CrossRef CAS.
- (a) I. Nemec, M. Machata, R. Herchel, R. Boča and Z. Trávníček, Dalton Trans., 2012, 41, 14603–14610 RSC; (b) P. Bag, J. Goura, V. Mereacre, G. Novitchi, A. K. Powell and V. Chandrasekhar, Dalton Trans., 2014, 43, 16366–16376 RSC; (c) K. Griffiths, V. N. Dokorou, J. Spencer, A. Abdul-Sada, A. Vargas and G. E. Kostakis, CrystEngComm, 2016, 18, 704–713 RSC; (d) K. Griffiths, G. Novitchi and G. E. Kostakis, Eur. J. Inorg. Chem., 2016, 2016, 2750–2756 CrossRef CAS; (e) H. Ke, W. Zhu, S. Zhang, G. Xie and S. Chen, Polyhedron, 2015, 87, 109–116 CrossRef CAS; (f) V. Chandrasekhar, P. Bag, W. Kroener, K. Gieb and P. Müller, Inorg. Chem., 2013, 52, 13078–13086 CrossRef CAS PubMed; (g) W.-W. Kuang, C.-Y. Shao and P.-P. Yang, J. Coord. Chem., 2015, 68, 1412–1422 CrossRef CAS; (h) E. Loukopoulos, B. Berkoff, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, A. Escuer and G. E. Kostakis, Eur. J. Inorg. Chem., 2015, 2015, 2646–2649 CrossRef CAS.
- (a) R.-X. Zhang, Y.-X. Chang, H.-Y. Shen, W.-M. Wang, X.-P. Zhou, N.-N. Wang, J.-Z. Cui and H.-L. Gao, Dalton Trans., 2016, 45, 19117–19126 RSC; (b) W.-M. Wang, S.-Y. Wang, H.-X. Zhang, H.-Y. Shen, J.-Y. Zou, H.-L. Gao, J.-Z. Cui and B. Zhao, Inorg. Chem. Front., 2016, 3, 133–141 RSC; (c) H. Sun, L. Wu, W. Yuan, J. Zhao and Y. Liu, Inorg. Chem. Commun., 2016, 70, 164–167 CrossRef CAS; (d) H. Zhang, J. Zhang, Y. Li, Y. Qin, Y. Chen, W. Liu, D. Gao and W. Li, J. Coord. Chem., 2015, 68, 2798–2809 CrossRef CAS; (e) W.-W. Kuang, L.-L. Zhu, Y. Xu and P.-P. Yang, Inorg. Chem. Commun., 2015, 61, 169–172 CrossRef CAS; (f) W.-M. Wang, S.-Y. Wang, H.-X. Zhang, B. Zhao, J.-Y. Zou, H.-L. Gao and J.-Z. Cui, Inorg. Chim. Acta, 2016, 439, 106–110 CrossRef CAS; (g) W.-M. Wang, H.-X. Zhang, S.-Y. Wang, H.-Y. Shen, H.-L. Gao, J.-Z. Cui and B. Zhao, Inorg. Chem., 2015, 54, 10610–10622 CrossRef CAS PubMed; (h) K. C. Mondal, G. E. Kostakis, Y. Lan, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2011, 50, 11604–11611 CrossRef CAS PubMed.
- P. J. van Konigsbruggen, Y. Maeda and H. Oshio, Top. Curr. Chem., 2004, 233, 259–324 CrossRef.
- (a) S. Saha, S. Pal, C. J. Gómez-García, J. M. Clemente-Juan, K. Harms and H. P. Nayek, Polyhedron, 2014, 74, 1–5 CrossRef CAS; (b) P. S. Perlepe, A. A. Athanasopoulou, K. I. Alexopoulou, C. P. Raptopoulou, V. Psycharis, A. Escuer, S. P. Perlepes and T. C. Stamatatos, Dalton Trans., 2014, 43, 16605–16609 RSC.
- K. I. Alexopoulou, A. Terzis, C. P. Raptopoulou, V. Psycharis, A. Escuer and S. P. Perlepes, Inorg. Chem., 2015, 54, 5615–5617 CrossRef CAS PubMed.
- (a) R. Herchel, I. Nemec, M. Machata and Z. Trávníček, Inorg. Chem., 2015, 54, 8625–8638 CrossRef CAS PubMed; (b) J.-D. Leng, S.-K. Xing, R. Herchel, J.-L. Liu and T. L. Ming, Inorg. Chem., 2014, 53, 5458–5466 CrossRef CAS PubMed; (c) L. Mathivathanan, K. Al-Ameed, K. Lazarou, Z. Trávníček, Y. Sanakis, R. Herchel, J. E. McGrady and R. G. Raptis, Dalton Trans., 2015, 44, 20685–20691 RSC.
- A. Das, F. J. Klinke, S. Demeshko, S. Meyer, S. Dechert and F. Meyer, Inorg. Chem., 2012, 51, 8141–8149 CrossRef CAS PubMed.
- R. Boča, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef.
- T. Tunc, M. Sari, M. Sadikoglu and O. Büyükgüngör, J. Chem. Crystallogr., 2009, 39, 672–676 CrossRef CAS.
- CrysAlis CCD and CrysAlis RED, Version 1.171.33.52, Oxford Diffraction Ltd, England, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- (a) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391–1400 CrossRef CAS; (b) E. Ruiz, A. Rodríguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982–989 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS; (b) R. Izsak and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–127639 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1530435. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01374d |
| This journal is © The Royal Society of Chemistry 2017 |