
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel one-pot process for the preparation of linear and hyperbranched polycarbonates of various diols and triols using dimethyl carbonate†
Jingjiang Suna,
Kamal Ibrahim Alyb and
Dirk Kuckling *a
*a
aUniversity of Paderborn, Chemistry Department, Warburger Str. 100, D-33098 Paderborn, Germany. E-mail: dirk.kuckling@uni-paderborn.de
bPolymer Lab. 122, Chemistry Department, Faculty of Science, Assiut University, Assiut 71516, Egypt
First published on 21st February 2017
Abstract
A new eco-friendly strategy for the preparation of linear and hyperbranched polycarbonates was developed. Our work referred to a one-pot condensation polymerization of various alcohols (diols and triols) with equivalent amounts of eco-friendly dimethyl carbonate (DMC) at 120 °C, atmospheric pressure and in 1,4-dioxane solution using 4-dimethylaminopyridine (DMAP) or lithium acetylacetonate (LiAcac) as a catalyst. Polymer chains were built by pure transesterification of hydroxyl and methyl carbonate chain ends, and the single byproduct (methanol) was removed using a pressure-equalized addition funnel filled with 4 Å molecular sieves as the crucial equipment in this work. Using this strategy, hyperbranched polycarbonates with high molar masses (Mn up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and Mw up to 64000 g mol−1) and high hydroxyl end group contents (up to 94%) were successfully prepared using dimethyl carbonate instead of toxic phosgene or phosgene-based monomers for the first time. In addition, linear aliphatic polycarbonates of various diols were also synthesized with Mn up to 16000 g mol−1 and low molar mass distributions (ĐM < 1.70). Another eco-friendly aspect of this work was the use of equimolar amounts of DMC to avoid waste and the disposal of excess DMC; in a classic 2-step polycondensation for polycarbonate synthesis excess DMC is a prerequisite in order to obtain high molar masses.
000 g mol−1 and Mw up to 64000 g mol−1) and high hydroxyl end group contents (up to 94%) were successfully prepared using dimethyl carbonate instead of toxic phosgene or phosgene-based monomers for the first time. In addition, linear aliphatic polycarbonates of various diols were also synthesized with Mn up to 16000 g mol−1 and low molar mass distributions (ĐM < 1.70). Another eco-friendly aspect of this work was the use of equimolar amounts of DMC to avoid waste and the disposal of excess DMC; in a classic 2-step polycondensation for polycarbonate synthesis excess DMC is a prerequisite in order to obtain high molar masses.
Introduction
Hyperbranched polymers (HBPs) have attracted significant attention in recent decades due to their unique three-dimensional highly branched topologies and their chemical and physical properties.1–8 The concept of hyperbranched polymerization was presented by Paul J. Flory in 1941.9 Compared to the preparation of dendrimers, hyperbranched polymers are usually synthesized in one-pot reactions.2,7 A hyperbranched polymer is composed of three types of structural units: dendritic units (D), linear units (L) and terminal units (T). The dendritic and linear units are randomly located in the polymer frameworks, while the terminal units are always placed at the terminals.2,10However, most intensively investigated hyperbranched polymers terminated with OH-groups are polyethers (e.g., polyglycerol)11–13 and polyesters (e.g., based on 2,2-bis(hydroxymethyl)propionic acid).1,14,15 Their medical applications are limited because they are non-degradable or have poor biocompatibilities.16,17
In past decades, aromatic polycarbonates have been widely used as engineering plastics18,19 because of their attractive mechanical properties, e.g., low moisture absorption, high impact strength, high elastic modulus, creep resistance and good thermal stability. Compared to traditional aromatic polycarbonates aliphatic polycarbonates received little interest because of their poor thermal stability and high susceptibility to hydrolysis.18,20–25 In recent years, aliphatic polycarbonates have attracted significantly increasing attention for biomedical applications, e.g., for the construction of biomedical implants and as drug delivery devices, due to their biodegradability, low toxicity and good biocompatibility.18,24,26–35 Although aliphatic polycarbonates are advantageous for use as biomaterials, there are still only a few papers concerning hyperbranched polycarbonates (HBPC).7,17,36 HBPCs made via the polycondensation of aromatic A2B or AB2 monomers have been reported by Bolton and Wooley.37,38 Recently, Nishikubo et al. reported similar aromatic HBPCs with Mn up to 7000 g mol−1 by an A2 + B3 polycondensation using di-tert-butyltricarbonate (DBTC) and 1,1,1-tris(4-hydroxyphenyl)ethane as the A2 and B3 monomers.36 Zhuo et al. developed a catalyst-free method to synthesize aliphatic HBPC based on 5-ethyl-5-hydroxymethyl-1,3-dioxan-2-one via ring-opening-polymerization (ROP).17,39 Parzuchowski et al. described the synthesis of HBPC from 5-(4-hydroxybutyl)-1,3-dioxan-2-one and its application.7
However, DBTC is not available commercially and has to be prepared from hazardous triphosgene. Cyclic carbonate monomers, which are very expensive due to their low synthetic yields, are the basic premise of the synthesis of HBPCs via ROP.40–42 The best strategy for large-scale preparation of aliphatic HBPCs is the condensation polymerization of dimethyl carbonate (DMC) and aliphatic triols. DMC is a non-petroleum-based and eco-friendly and has low toxicity (1000 times less than phosgene). Hence, it is an ideal candidate instead of phosgene or diphenyl carbonate (DPC) for the synthesis of polycarbonates. DMC is prepared industrially on a large scale using carbon dioxide and methanol.40,43,44
In our previous work,45 we successfully prepared linear aliphatic polycarbonates with high molar masses (up to 52000 g mol−1) via a two-step condensation polymerization using organo-catalysts (Scheme 1). In the first step, the oligomers were synthesized via transesterification from diol and DMC. In the second step, the polymer chains were grown through elimination of methanol or DMC by the transesterification or the reaction between the two methyl carbonate end groups under reduced pressure and at elevated temperature. Unfortunately, the two-step polycondensation for the synthesis of HBPCs did not work due to the very fast gelation in the second step under reduced pressure.
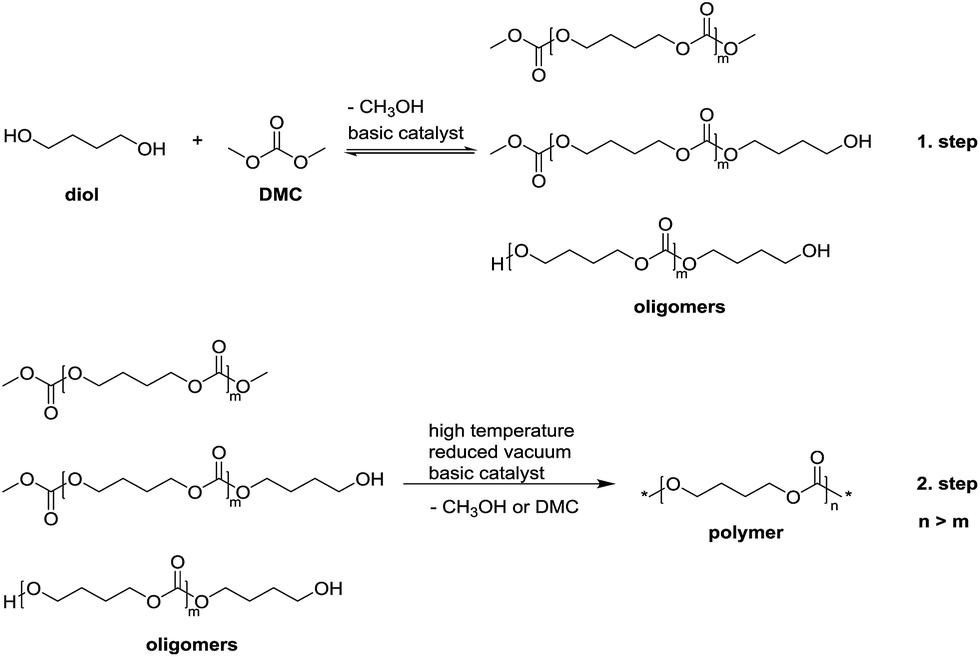 | ||
| Scheme 1 General method for the preparation of polycarbonates via a two-step condensation polymerization. | ||
In this work, we report a novel method for the preparation of linear or hyperbranched polycarbonates using a one-pot condensation polymerization. Compared with the traditional two-step method, the polycondensation in this work was carried out at a relatively low temperature and atmospheric pressure, and the methanol byproduct was removed via adsorption on molecular sieve instead of vacuum distillation at high temperature. Moreover, an equimolar amount of DMC was used to avoid waste and the disposal of excess DMC. LiAcac and DMAP were investigated as catalysts for polycarbonates synthesis. The results showed that LiAcac is a more effective catalyst for synthesis of poly(trimethylene carbonate) and hyperbranched polycarbonates, while DMAP is suitable to prepare other linear polycarbonates. The resulting linear and hyperbranched aliphatic polycarbonates were characterized by NMR spectroscopy, size exclusion chromatography (SEC) measurements, differential scanning calorimetry (DSC) and ESI-mass spectrometry. Moreover, the hydrolytic and enzymatic degradation investigations for linear and hyperbranched polycarbonates were evaluated under various conditions as well.
Experimental
Material
1,3-Propanediol (Alfa Aesar, 99%), 1,4-butanediol (Acros Organics, 99+%) and 1,5-pentanediol (Fluka, 96%) were vacuum distilled using a short path distillation apparatus and dried over 4 Å molecular sieves (from Merck) before use. 1,6-Hexanediol (Sigma-Aldrich, 99%) was dried under vacuum over night before use. 4-Dimethylaminopyridine (DMAP) (Fluka, >99%) was recrystallized from toluene and dried under vacuum. 1,4-Dioxane (Grüssing, 99.5%) was dried over 4 Å molecular sieves before use. 1,1,1-Tris(hydroxymethyl)ethane (Alfa Aesar, 97%) was dissolved in hot tetrahydrofuran, purified by precipitation in n-hexane, and dried under vacuum overnight before use. Dimethyl carbonate (DMC) (Acros Organics, 99+%), lithium acetylacetonate (LiAcac) (Alfa Aesar, 99.5%), cyclohexanedimethanol (Alfa Aesar, 99%), N-phenyldiethanolamine (TCI), dimethylformamide (DMF) (Acros Organics, 99.8%), dimethyl sulfoxide (DMSO) (Acros Organics, 99.7%) and lipase solution from Thermocyces languginosus (Sigma-Aldrich) were available commercially and used as received. The 4 Å molecular sieves were dried at 250 °C under vacuum overnight and stored in an argon atmosphere.Measurements
1H and 13C NMR spectra were recorded using Bruker AV 500 spectrometer at 500 MHz and 125 MHz, respectively. Chloroform-d (CDCl3, 99.8 D%) or dimethylsulfoxide-d6 (DMSO-d6, 99.5 D%) were used as solvent for NMR measurements. The molar masses and dispersities (ĐM) of the linear polymers were analyzed employing a size exclusion chromatography (SEC) system equipped with four consecutive columns (PSS-SDV columns filled with 5 μm gel particles with a defined porosity of 106 Å, 104 Å, 103 Å and 102 Å, respectively) and a Shodex RI-detector (RI-101) at 30 °C. The system was operated at a flow rate of 0.75 mL min−1 with chloroform as solvent. Polystyrene (PS) standards were used for calibration. The molar masses and ĐM of HBPCs were analysed on a SEC equipped with three consecutive columns (PSS-GRAM columns filled with 10 μm gel particles with a defined porosity of 104 Å, 103 Å and 102 Å, respectively), a Waters RI-detector (RI 2410) and a differential viscometer (PSS ETA2010) at 50 °C. As eluent, dimethylacetamide (DMAc) was used with a flow rate of 0.5 mL min−1. Molar masses were obtained by using universal calibration. Differential scanning calorimetry (DSC) was performed with Netzsch DSC 204 F1 Phönix® at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The glass transition temperature (Tg) and the melting point (Tm) values were recorded during the second run.ESI-ToF-mass spectra were measured on a SYNAPT G2 HDMS™ from Waters. The mass spectrometric parameters were the following: capillary voltage: 2.5 kV; sampling cone voltage: 50 V; extraction cone voltage: 1 V; cone gas flow: 30 L h−1; source temperature: 120 °C; desolvation gas flow: 650 L h−1; desolvation temperature: 350 °C; helium cell gas flow: 180 mL min−1; IMS gas flow: 90 mL min−1; IMS wave velocity: 460 m s−1; IMS wave height: 40 V. The PBC sample was dissolved in acetonitrile (2 g L−1) and then mixed with NaI 0.1 g L−1 in methanol and methanol in the ratio of 5:5:990. Data were obtained and processed using Polymerix Software.
General procedure for the synthesis of linear aliphatic homo- and copolycarbonates from different diols and DMC
All polymerizations were carried out in a two-necked flask equipped with a pressure-equalized addition funnel filled with 16 g of 4 Å molecular sieve, a cold reflux condenser and connected to a Schlenk line with vacuum and argon gas lines. The diol(s) (23.4 mmol), DMC, catalyst and 1,4-dioxane were added to the flask under argon atmosphere. The reaction mixture was stirred under reflux (approximate oil bath temperature of 120 °C) for 72 or 96 h. The mixture was then cooled to room temperature and diluted with dichloromethane. The polymer was isolated via precipitation in methanol and dried in vacuum.General procedure for the synthesis of HBPCs from different triols and DMC
In a two-necked flask equipped with the above-mentioned apparatus, triol (23.4 mmol), DMC, catalyst and solvent were added under argon atmosphere. The mixture was stirred under reflux (approximate oil bath temperature of 120 °C) for 2 to 18 h. The mixture was then cooled to room temperature and diluted with acetone or THF. The polymer was isolated by precipitation in water/methanol (v/v = 9:1) or in water and centrifugation and dried in vacuum at room temperature to give a colorless solid.
Polymer analysis for linear and hyperbranched polycarbonates from NMR spectra
The results of the linear polycarbonates analysis based on 1H and 13C NMR spectra can be found in the ESI.† The structures of the HBPCs are analysed in the section “Results of the synthesis of HBPC from aliphatic triols” (Fig. 3 and ESI-11†).Hydrolytic degradation investigation
In a vial (25 mL, 50 mm × 30 mm), 90 mg of the polycarbonate sample was dissolved in 5 mL dichloromethane. A polymer film was prepared via solvent evaporation, and 10 mL samples of buffer solutions with various pH values were added to the dry film in the vial. The hydrolytic degradation was performed at 37 °C or 55 °C with gentle stirring. The polymer samples were recovered at various time intervals by filtration, washed with 50 mL water and dried in vacuum at room temperature to a constant weight. The hydrolytic degradation was monitored by the decreases in molar mass and weight. All degradation experiments were carried out in triplicate.Enzymatic degradation investigation
In a vial (5 mL, 40 mm × 20 mm), 45 mg of the polycarbonate sample was dissolved in 2 mL dichloromethane. A polymer film was prepared via solvent evaporation, and 2 mL lipase solution was then added to the dry film in the vial. The lipase solutions were refreshed every four days to maintain the lipase activity. The enzymatic degradation was performed at 37 °C with gentle shaking. The polymer samples were recovered by filtration, washed with 50 mL water and dried under vacuum at room temperature to a constant weight. The enzymatic degradation process was monitored by the decreases in molar mass and weight. All degradation experiments were carried out in duplicate.Results and discussion
Strategy for a one-pot polycondensation at atmospheric pressure
In a classic two-step polycondensation for the synthesis of polycarbonates, low molar mass oligomers (Mn < 1000 g mol−1) are obtained in the first step. In the next step, the polymer chains are extended via transesterification reactions between hydroxyl and methyl carbonate (–OC(O)–OCH3) chain ends or mainly between two methyl carbonate chain ends due to their higher reactivities than the hydroxyl end groups.40,44 For this reason, excess DMC (e.g., diol:DMC = 1:3)24 is used in order to obtain oligomers mostly terminated with methyl carbonate groups. Condensation polymerization is then conducted at elevated temperature (170–200 °C) and under vacuum to remove the freshly generated major byproduct dimethyl carbonate and minor byproduct methanol to achieve high molar masses.
Here, we developed a new strategy for the preparation of linear and hyperbranched polycarbonates (HBPC) by a one-pot synthesis (Scheme 2). In contrast to a classic two-step polycondensation (Scheme 1), the polymerizations were carried out with equivalent amounts of diol and DMC in bulk or in solution at atmospheric pressure in the presence of basic catalysts. The polymer chains grow by pure transesterification between hydroxyl (–OH) and methyl carbonate (–OC(O)–OCH3) chain ends, and the methanol byproduct was removed using 4 Å molecular sieve in a pressure-equalized addition funnel.
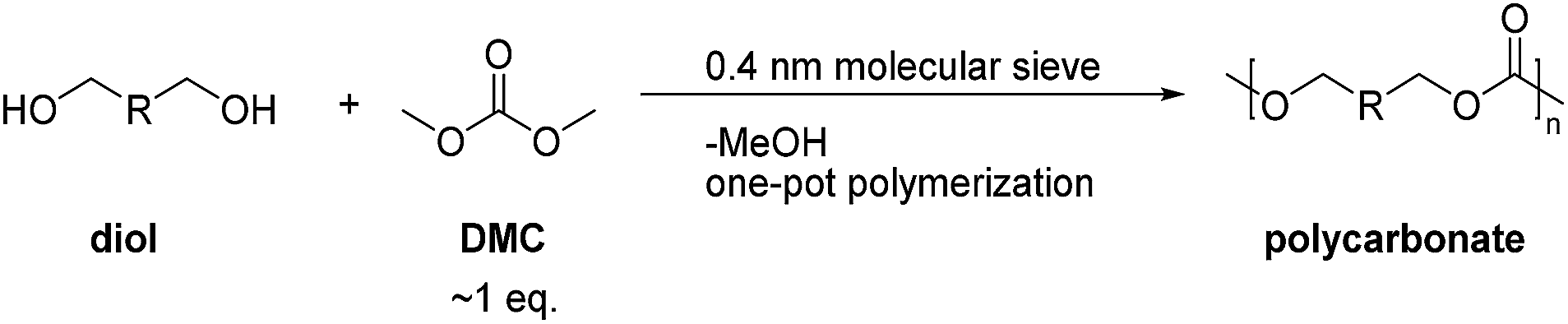 | ||
| Scheme 2 Strategy for the one-pot synthesis of polycarbonates. | ||
Results of polycarbonate synthesis from 1,4-butanediol
Recently, we surveyed various organo-catalysts for the synthesis of linear aliphatic polycarbonates.44 Among them, DMAP (1 mol% based on diol) showed the best catalytic activity. However, DMAP was not suitable for the synthesis of poly(trimethylene carbonate) (PTMC), leading to side reactions at high temperatures in the one-pot synthesis. Wang and Zheng reported that lithium acetylacetonate (LiAcac) was an effective catalyst for the synthesis of polycarbonate due to its strong coordination with carbonyl groups. According to their results, LiAcac at 0.1 percent by weight based on 1,4-butanediol (0.1 wt%) was also investigated.In this work, a variety of polymerizations were evaluated to optimize the reaction conditions. Table 1 summarizes the most significant results of the one-pot polycarbonate syntheses based on 1,4-butanediol (BD). The yields were calculated using eqn (1).
 | (1) |
| Catalysts | Solvents | [BD]:[DMC]:[cat.] |
T (°C) | Time (h) | Mna (g mol−1) | ĐMa | Yields (%) | |
|---|---|---|---|---|---|---|---|---|
| a Determined using SEC in chloroform solution with PS standards.b 0.1 wt% LiAcac based on BD was used as the catalyst in PBC 2–6. | ||||||||
| PBC 1 | DMAP | — | 1:1.2:0.01 |
125 | 72 | 2300 | 1.36 | 42 |
| PBC 2 | LiAcac | — | 1:1.2:0.1 wt%b |
125/180 | 48/24 | 6300 | 1.70 | 60 |
| PBC 3 | LiAcac | DMSO | 1:1.2:0.1 wt%b |
125/180 | 48/24 | — | — | — |
| PBC 4 | LiAcac | DMF | 1:1.2:0.1 wt%b |
125/160 | 48/24 | — | — | — |
| PBC 5 | LiAcac | Toluene | 1:1.2:0.1 wt%b |
130 | 72 | — | — | — |
| PBC 6 | LiAcac | 1,4-Dioxane | 1:1.2:0.1 wt%b |
120 | 72 | 1300 | 1.40 | 69 |
| PBC 7 | DMAP | 1,4-Dioxane | 1:1.2:0.01 |
130 | 72 | — | — | — |
| PBC 8 | DMAP | 1,4-Dioxane | 1:1.2:0.01 |
100 | 72 | 1500 | 1.33 | 33 |
| PBC 9 | DMAP | 1,4-Dioxane | 1:1.2:0.01 |
120 | 72 | 4300 | 1.53 | 75 |
| PBC 10 | DMAP | 1,4-Dioxane | 1:1.1:0.01 |
120 | 72 | 6300 | 1.67 | 77 |
| PBC 11 | DMAP | 1,4-Dioxane | 1:1.075:0.01 |
120 | 72 | 7100 | 1.70 | 77 |
| PBC 12 | DMAP | 1,4-Dioxane | 1:1.05:0.01 |
120 | 72 | 9800 | 1.66 | 77 |
| PBC 13 | DMAP | 1,4-Dioxane | 1:1.025:0.01 |
120 | 72 | 14000 |
1.66 | 87 |
| PBC 14 | DMAP | 1,4-Dioxane | 1:1:0.01 |
120 | 72 | 5300 | 1.67 | 74 |
Initially, to determine the influence of the temperature and solvent on the molar mass, a number of polymerizations were carried out. DMAP and LiAcac were active for the one-pot synthesis after a reaction time of 72 h. A number average molar mass (Mn) of 2300 g mol−1 was recorded using DMAP as the catalyst in the bulk (PBC 1), while little product was formed at T > 125 °C because of the dominant side reactions. In contrast, LiAcac was more stable than DMAP even at a high temperature of 180 °C. The polycondensation using LiAcac was carried out initially at 125 °C for 48 h to avoid loss of DMC due to its low bowling point and then stirred at 180 °C for another 24 h. The resulting poly(butylene carbonate) (PBC) had a higher molar mass of 6300 g mol−1 and a dispersity of 1.70 (PBC 2).
Solvents with high boiling points, such as dimethyl sulfoxide (DMSO, bp = 189 °C), dimethylformamide (DMF, bp = 153 °C), toluene (bp = 111 °C) and 1,4-dioxane (bp = 101 °C) were also investigated. The polycondensation could be carried out only in 1,4-dioxane solution, and samples with Mn of 1300 g mol−1 and 4300 g mol−1 were obtained using LiAcac and DMAP as catalysts, respectively, indicating that DMAP was more effective for synthesis of PBC under this polymerization condition. No polymer could be isolated after the reaction in the presence of DMSO, DMF and toluene. (PBC 3–8) Moreover, no product or polymer with low Mn formed in 1,4-dioxane solution at 130 °C and 100 °C, respectively.
To investigate the influence of the initial feed ratio on the polymer molar mass, the initial [DMC]:[BD] ratios were gradually varied from 1.2:1 to 1:1 (entries 8–13). The Mn increased slowly from 4300 g mol−1 to 7100 g mol−1 throughout the feed ratios from 1.2:1 to 1.075:1. Afterwards, the increase of Mn was more pronounced with Mn = 14000 g mol−1 for a polymerization with a feed ratio of 1.025:1. When the initial feed ratio of [DMC]:[BD] was adjusted to 1.0, Mn decreased to 5300 g mol−1. The last polymer possessed a high hydroxyl end group content of 94%, which could probably be attributed to a small loss of DMC due to the low boiling point. These results indicate that our strategy was effective for the synthesis of PBC, and with the initial feed ratios close to 1.0, the highest Mn of the resulting polymers could be achieved. All PBC samples generated at 120 °C had high yields (up to 87%) and dispersities below 1.70.
Detailed information about the polymerization process was obtained using 1H NMR spectroscopy (Fig. 1a). After 2 h, the reaction signals from 1,4-butanediol at 1.47, 3.44 and 4.35 ppm decreased due to the formation of poly(butylene carbonate) oligomers, while the methanol side product was detected at 3.21 ppm as doublet signal. After 16 and 41 h, the intensities of the signals for 1,4-butanediol and methanol decreased continuously, indicating that the chain growth proceeded and the produced methanol was successfully removed using 4 Å molecular sieves. The 1H NMR spectrum of purified PBC 10 is shown in Fig. 1b. Both the CH2-groups in the polymer backbone were detected at 1.77 and 4.15 ppm. The small signals at 3.68 and 3.77 ppm were attributed to terminal butanol groups and methyl carbonate groups, respectively. Moreover, a desorption process of used molecular sieve was conducted after the polymerization at 150 °C in vacuum. 1.52 g methanol were collected, which was close to the expected value (1.59 g at 100% conversion of DMC). Combining the result of the desorption investigation with the kinetic investigation, we confirm that, the generated methanol can be removed successfully from the reaction mixture using 4A molecular sieve.
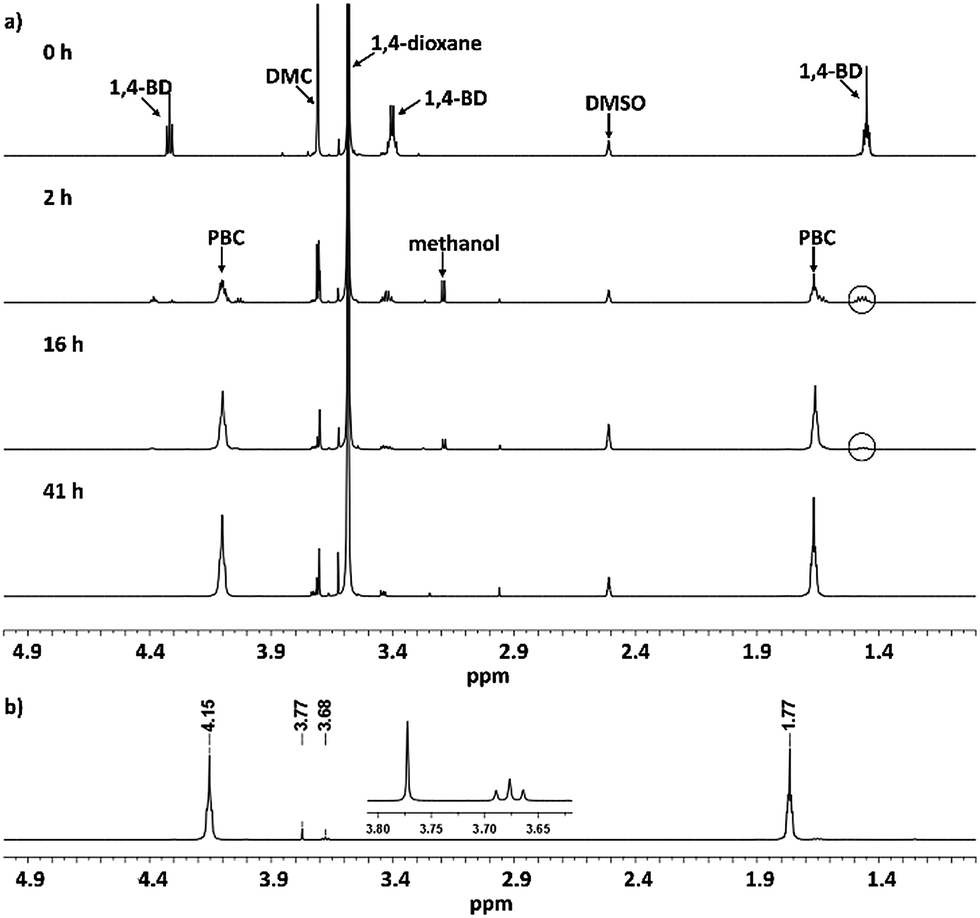 | ||
| Fig. 1 (a) 1H NMR spectra for the kinetic investigation of PBC 10 at different polymerization times (0, 2, 16 and 41 h) (b) 1H NMR spectrum of purified PBC 10. | ||
A typical ESI-ToF-MS spectrum of PBC 9, which had about 95% methyl carbonate end group according to the 1H NMR analysis, in the m/z region of 400 to 2000 Da is shown in Fig. 2. The data were processed using Polymerix Software and peaks were assigned to different series (S1–S6), which are shown in ESI-Fig. 13 and 14† in detail. ESI-ToF-MS analysis shows main populations corresponding to methyl carbonate terminated C2H3O3–(PBC)n–CH3·zNa+ (z = 1–3) with repeating PBC units of 116.047 g mol−1. The measured m/z of S1 and S2 are compared with calculated values. As shown in Fig. 2, the measured m/z correspond very closely to the calculated values. For example, the most intense signal was detected at m/z: 925.350 Da, denoting the C2H3O3–(PBC)n–CH3·Na+ series containing 7 repeating monomer units and two methyl carbonate end groups, corresponding to m/z = [7 × 116.047(MPBC) + 75.008(MC2H3O3) + 15.024(MCH3)] + 22.990(MNa). The second expected population (S5 and S6, ESI-Fig. 13 and 14†) were attributed to PBC with both hydroxyl and methyl carbonate end groups. The structure analysis based on ESI-MS is consistent with 1H NMR analysis. The obtained molar mass from ESI-MS was 3300 g mol−1 with ĐM of 1.2. In addition, cyclic polycarbonate with very low intensity was detected as well, indicating the presence of intramolecular transesterification side reaction.
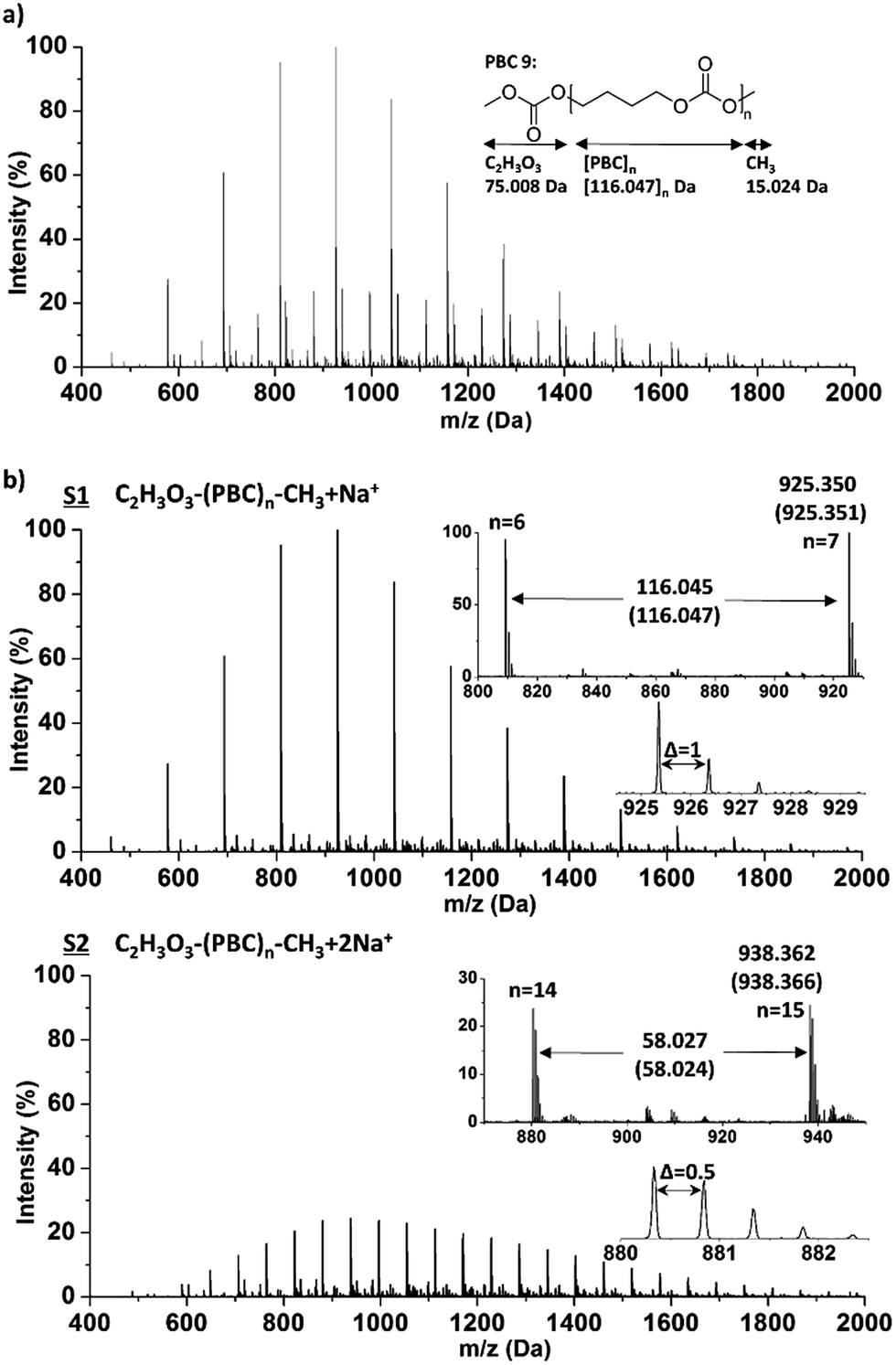 | ||
| Fig. 2 (a) ESI-ToF mass spectrum of PBC 9 in the m/z region of 400 to 2000; (b) separated spectra (S1 and S2) using Polymerix Software in the m/z region of 400 to 2000 with measured and calculated (in parenthesis) values. | ||
Results of polycarbonate synthesis from other aliphatic diols with DMC
Additionally, to study the versatility of this new strategy, the synthesis of poly(trimethylene carbonate) (PTMC), poly(pentamethylene carbonate) (PPC), poly(hexamethylene carbonate) (PHC), poly(cyclohexan-1,4-dimethylene carbonate) (PCDMC) and poly(diethylphenylamine carbonate) (PDEAC) from commercially available aliphatic diols (Scheme 3) with DMC were attempted. Because of the different purities of these diols, the initial [DMC]:[diol] ratio was adjusted to a slightly higher value of 1.05:1. The polymerization temperature was maintained at 120 °C and the polymerization time for 1,5-pentanediol and 1,6-hexanediol was extended to 4 days in order to obtain higher molar masses of the resulting polymers. LiAcac was used as the catalyst for the preparation from 1,3-propanediol because when DMAP was used as the catalyst, the polymerization solution changed to dark brown and no polymer was isolated after purification (Table 2, entry 1).
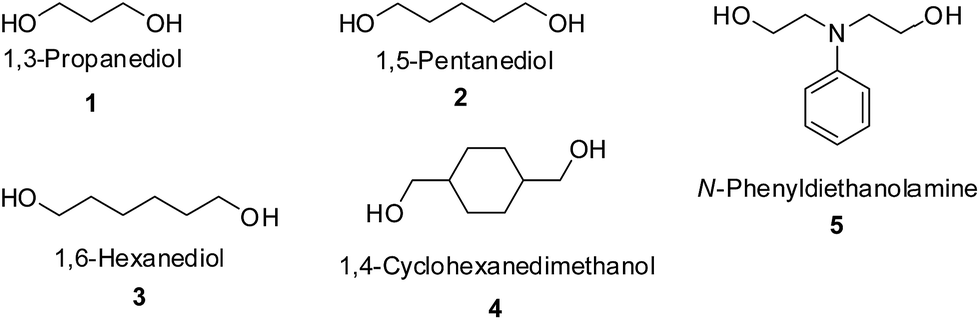 | ||
| Scheme 3 Various aliphatic diols used for the investigation of condensation polymerization. | ||
| Diol | [BD]:[DMC]:[cat.] |
Time | Mna (g mol−1) | ĐMa | pb (%) | Yields (%) | |
|---|---|---|---|---|---|---|---|
| a Determined using SEC in chloroform solution with PS standards.b Conversion, calculated from Carothers equation using Mn from SEC.c LiAcac was used as the catalyst. | |||||||
| PTMC 1 | 1 | 1:1.025:0.01 |
3 d | — | — | — | — |
| PTMC 2c | 1 | 1:1.025:0.1 wt% |
3 d | 5200 | 1.69 | 98.5 | 70 |
| PPC 3 | 2 | 1:1.05:0.01 |
4 d | 16000 |
1.55 | 99.2 | 77 |
| PHC 6 | 3 | 1:1.05:0.01 |
4 d | 13000 |
1.55 | 98.9 | 80 |
| PCDMC 1 | 4 | 1:1.05:0.01 |
3 d | 13000 |
1.57 | 98.9 | 82 |
| PDEAC 1 | 5 | 1:1.05:0.01 |
3 d | 4300 | 1.56 | 95.2 | 70 |
Using this method, PTMC with Mn of 5200 g mol−1 was generated. When 1,5-pentanediol and 1,6-hexanediol were used as monomers, after a polymerization time of 4 days, polycarbonates with higher molar masses (Mn = 16000 and 13000 g mol−1, respectively) were also obtained, while lower Mn of 4400 g mol−1 and 8500 g mol−1 were obtained for PPC and PHC, respectively, after a polymerization time of 3 days only. For 1,4-cyclohexanedimethanol, polymers with Mn of 13000 g mol−1 formed within 3 days. The polycarbonate based on N-phenyldiethanolamine had a relatively low Mn of 4300 g mol−1 due to its lower reactivity. Conversions were achieved more than 95%, which were calculated according to the Carothers equation from Mn. The yields (>70%) were lower than the conversions due to the loss in the purification step. All polycarbonate samples generated in 1,4-dioxane solution possessed relatively narrow molar mass distributions (ĐM < 1.70).
Results of copolymerization from BD mixed with various diols and DMC
Copolycarbonates of BD and different diols were also prepared by the one-pot polycondensation strategy in 1,4-dioxane solution. The feed ratio of [BD]:[diol]:[DMC] was maintained 0.8:0.2:1.05. The results are summarized in Table 3.
The molar masses were obtained in the range of 3500 g mol−1 and 7500 g mol−1 with dispersities below 1.70. The compositions of the resulting copolymers were determined by comparing the peak integrals of each repeating unit at 1.76 ppm for PBC and 1.46 ppm, 1.40 ppm, 1.01 ppm and 4.26 ppm for PPC, PHC, PCDMC and PDEAC, respectively. The calculated values correspond to the feed ratios fairly closely.
Results of the synthesis of HBPCs from aliphatic triols
Due to fast gelation during the two-step polycondensation, the one-step procedure was applied for the synthesis of HBPCs. High-molecular weight HBPCs were obtained by feeding triols and DMC into a 1,4-dioxane solution at 120 °C. The reaction is depicted in Scheme 4. Initially, various catalysts including organo-catalysts, such as DMAP, 4-pyrrolidinopyridine (PPY) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), and metal salts, such as LiAcac, Zn(OAc)2 and NaOMe were surveyed in the polycondensation of THE and DMC in the bulk at 130 °C (data not shown). Most of them showed slow polymerization rates. After a polymerization time of 18 h, only HBPCs with low molar masses were obtained. However, LiAcac showed very high activity among them for this polycondensation (Table 4, entry 1). Hence, 0.1 wt% LiAcac (based on triol) was used as the catalyst instead of DMAP in these preparations. The products were obtained by dilution with acetone or THF and further precipitation in H2O for THE and in H2O/MeOH (v/v = 9:1) for THP. The reaction conditions and results are summarized in Table 4.
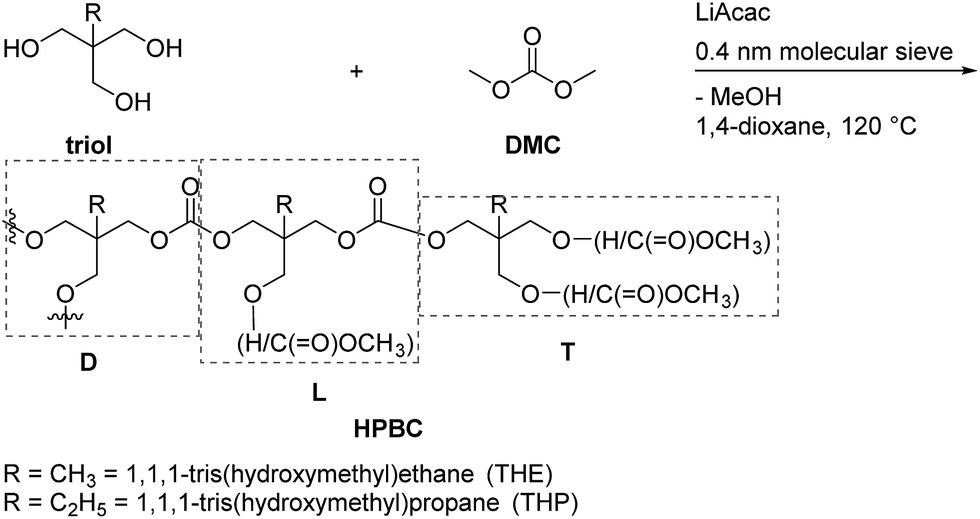 | ||
| Scheme 4 Strategy for the synthesis of HBPCs from triols and DMC. | ||
| [Triol]:[DMC] |
Time | Mna (g mol−1) | Mwa (g mol−1) | ĐMa | DBb | Yield (%) | End groupb (OH%) | |
|---|---|---|---|---|---|---|---|---|
| a Determined using SEC in DMAc solution with universal calibration.b Calculated from the 1H NMR spectrum.c The polymerization was carried out in bulk at 130 °C.d Gelation occurred after a polymerization time of 18 h, some solid precipitated and was not soluble in acetone. DB = degree of branching; PTHEC = hyperbranched poly(1,1,1-tris(hydroxymethyl)ethyl carbonate); PTHPC = hyperbranched poly(1,1,1-tris(hydroxymethyl)propyl carbonate). | ||||||||
| PTHEC 1c,d | 1:1.5 |
18 h | 4400 | 21000 |
4.87 | 0.50 | 42 | 81 |
| PTHEC 2 | 1:1.5 |
4 h | 2900 | 4000 | 1.39 | 0.31 | 41 | 68 |
| PTHEC 3 | 1:1.5 |
6 h | 2900 | 3800 | 1.33 | 0.31 | 36 | 74 |
| PTHEC 4 | 1:1.5 |
8 h | 3000 | 5600 | 1.81 | 0.36 | 40 | 72 |
| PTHEC 5 | 1:1.5 |
10 h | 8200 | 14000 |
1.67 | 0.43 | 42 | 73 |
| PTHEC 6 | 1:1.5 |
12 h | 7200 | 123000 |
17.2 | 0.46 | 59 | 70 |
| PTHEC 7 | 1:1.0 |
18 h | 10000 |
15000 |
1.47 | 0.46 | 25 | 94 |
| PTHPC 1 | 1:1.5 |
15 h | 9300 | 64000 |
6.89 | 0.50 | 75 | 68 |
Cross-linking phenomenon appeared in the bulk polymerization after a reaction time of 18 h. Some solid substances precipitated and were not soluble in acetone or THF. The resulting HBPC (acetone soluble part) had a Mn of 4400 g mol−1 and relatively broad dispersity (ĐM = 4.87). The cross-linking reaction was attributed to competitive intramolecular transesterification between the hydroxyl end groups and methyl carbonate end groups or polycarbonate backbone at high conversion. When 1,4-dioxane was used as the solvent for the same reaction, the molar masses increased significantly with narrower molar mass distributions (entries 2–7). Nevertheless, the prolonged reaction times lead to the generation of cross-linked gel products as well.
To determine the influence of the reaction time on the molar mass and cross-linking reaction, a kinetic study was performed (Table 4, entries 2–6). A series of polycondensations for THE and DMC were investigated with the same reaction conditions (120 °C, 1,4-dioxane) but with different reaction times from 4 to 12 h. The molar mass of the polymer was shown to increase very slowly throughout the initial 8 h. After a reaction time of 8 h, a Mn of 3000 g mol−1 with ĐM of 1.81 was obtained. When the polycondensation was further conducted for another 2 h, the increase in the molar mass became faster, with Mn = 8200 g mol−1. Afterwards, an explosive increase in the dispersity (ĐM = 17.2) was observed because the gel product from the cross-linking reaction started to form. Moreover, with the feed ratio of 1:1 a PTHEC sample with Mn = 10000 g mol−1 and ĐM = 1.47 was obtained.
The hydroxyl end group (–OH) contents could be calculated from the 1H NMR spectra by comparing the integration of the hydroxyl and methyl carbonate end groups. For the polycondensation, the OH end group contents were approximately 70% in 1,4-dioxane solution and 81% in bulk. When the feed ratio of THE:DMC was 1:1, the OH content increased to 94% but with a lower yield of 25%, because increasing the hydroxyl end group content enhanced the solubility of HBPC in water. Using the same reaction conditions, an HBPC from THP was generated. The polymer formed within 15 h with Mn of 9300 g mol−1, ĐM of 6.89 and OH end group content of 68%. In contrast, a higher yield of 75% was obtained due to the more hydrophobic property of the polymer backbone. The PTHPC spectrum is shown in Fig. SI-11.†
The fine structure of PTHEC was confirmed based on the analysis of the 1H NMR spectrum. As Scheme 5 shows, a hyperbranched polycarbonate structure consists of dendritic units (D), two linear structures (L) with hydroxyl (L1) and methyl carbonate (L2) side groups, respectively, and terminal units (T) including three possible structures. The terminal unit with two hydroxyl end groups (T1) and a mixture of methyl carbonate and hydroxyl end groups (T2) appeared in the 1H NMR spectrum, while the two methyl carbonate terminated structure (T3) could be neglected. This follows from the analysis of the 13C NMR spectrum by comparing the peak integrals of the different structures (Fig. SI-12†).
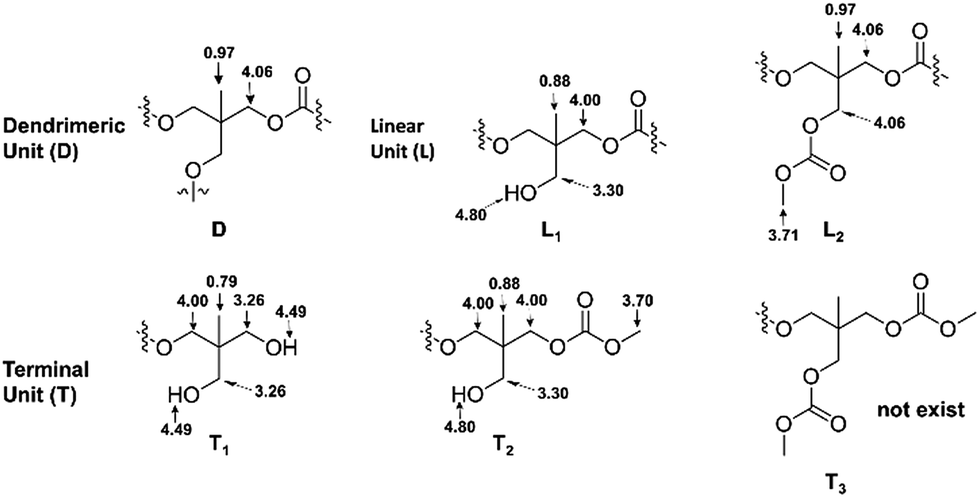 | ||
| Scheme 5 Possible chemical structures of PTHEC with chemical shifts (1H NMR) in DMSO-d6. | ||
The 1H NMR spectrum of PTHEC 1 is shown in Fig. 3. The three signals at 0.79 ppm, 0.88 ppm and 0.96 ppm were attributed to the methyl groups in the T1, L1 + T2 and D + L2 structures, respectively. The signals between 3.20 ppm and 3.30 ppm were assigned to the CH2OH groups. Two singlet peaks at 3.70 ppm and 3.71 ppm indicated the existence of terminal methyl carbonate groups (OCH3) in the T2 and L2 structures, respectively. The peaks between 3.90 ppm and 4.10 ppm corresponded to the CH2OC(O)O groups in the polymer backbone. Furthermore, the hydroxyl end groups from T1 with two protons and L1 + T2 with one proton were observed at 4.49 ppm and 4.80 ppm as two broad peaks. The calculations of the contents of dendritic (D), linear (L) and terminal (T) units was possible using the following 1H NMR analysis.
| D = CH3(D + L2) (0.97 ppm) − OCH3(L2) (3.71 ppm) = 1.52 |
| T = CH3(T1) (0.79 ppm) + OCH3(T2) (3.70 ppm) = 1.72 |
| L = CH3(T1) (0.79 ppm) + CH3(L1 + T2) (0.87 ppm) + CH3(D + L2) (0.96 ppm) − D − T = 2.99 |
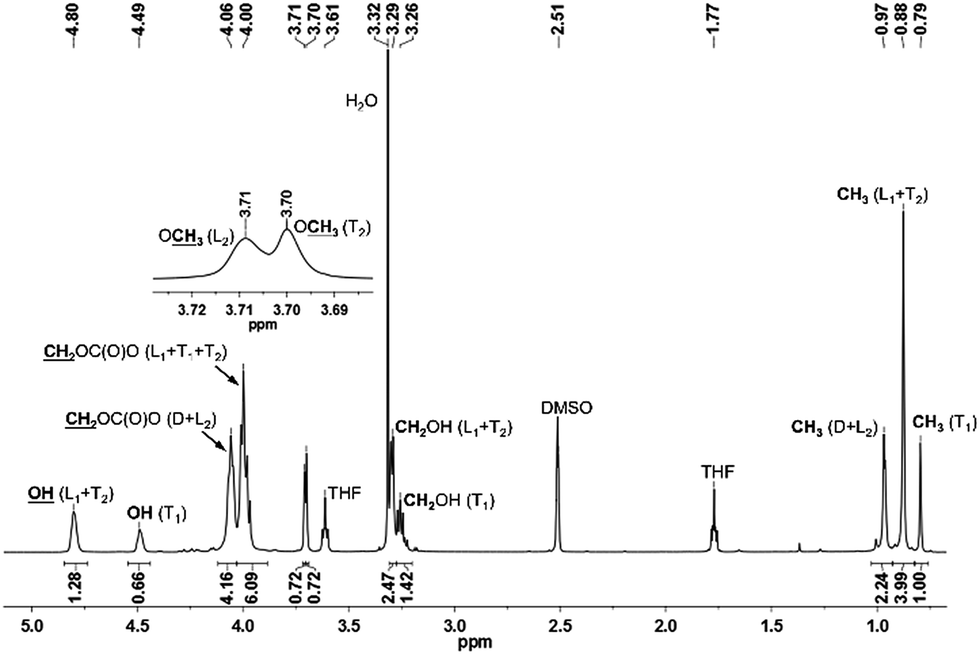 | ||
| Fig. 3 1H NMR spectrum of PTHEC. | ||
The degree of branching (DB) was calculated using 1H NMR from the following equation reported by Frey et al.46
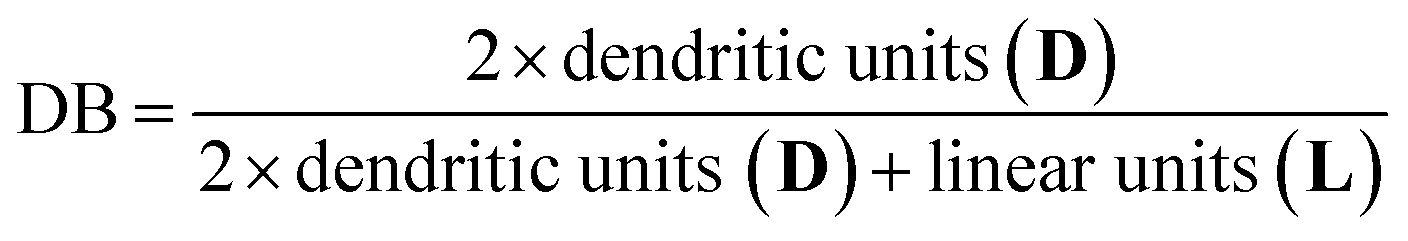
This equation is universally applicable for hyperbranched polymers with low and high molar masses. Using these calculated values, a DB for PTHEC 1 of 0.50 was obtained. According to the 1H NMR analysis, DBs of 0.31–0.51 were calculated for the HBPC of THE. The DB was clearly influenced by the reaction time. With increasing reaction time, a higher DB was obtained.
Results of thermal properties
As shown in Table 5, the thermal properties of the synthesized linear and hyperbranched samples were evaluated using DSC measurements. The PTMC, PBC and PPC samples displayed glass transition temperatures (Tg) of −21 to −40 °C. The Tg decreased as the number of carbon atoms increased in repeating units and can be explained by the increase in chain flexibility. PDEAC and PCDMC tended to higher Tg of 21 and 31 °C, respectively, due to their increasing rigidity from the phenyl side group in PDEAC and the rigid cyclic structure in the PCDMC backbone. By incorporating 20 mol% of a more flexible diol (1,5-propanediol and 1,6-hexanediol), with BD the Tg of the resulting polymers were decreased to −40 and −45 °C, respectively. In contrast, the polycarbonates based on PBC copolymerized with PDEAC and PCDMC have higher Tg detected at −23 and −32 °C, respectively. The PBC, PPC and PHC samples were semi-crystalline materials with melting points (Tm) of 63 °C, 49 °C and 55 °C, respectively, while the melting points of other homo- or copolycarbonates were not detected, indicating that the polymers were amorphous. For hyperbranched polycarbonates, PTHEC and PTHPC, higher Tg of 4 °C and −10 °C were observed compared to −38 °C for PBC sample. In comparison to linear polycarbonates, the Tg of HBPCs increased as a result of the large number of hydroxyl end groups in the hyperbranched structure, which lead to increases in polarity.47| Mna (g mol−1) | Tgb (°C) | Tmb (°C) | |
|---|---|---|---|
| a Determined using SEC in chloroform with PS standards.b Tg and Tm were measured by DSC. n.d.: not detected. | |||
| PTMC 2 | 5200 | −21 | n.d. |
| PBC 12 | 9800 | −38 | 63 |
| PPC 1 | 16000 |
−40 | 49 |
| PHC 1 | 13000 |
n.d. | 55 |
| PDEAC 1 | 4300 | 21 | n.d. |
| PCDMC 1 | 13000 |
31 | n.d. |
| PBC-co-PPC | 6300 | −40 | n.d. |
| PBC-co-PHC | 7500 | −45 | n.d. |
| PBC-co-PDEAC | 3500 | −23 | n.d. |
| PBC-co-PCDMC | 7100 | −32 | n.d. |
| PTHEC 7 | 10000 |
4 | n.d. |
| PTHPC 1 | 9300 | −10 | n.d. |
Firstly, the hydrolytic degradation of PBC specimen at 37 °C with pH values from 1.0 to 13.0 was investigated. The results in Fig. 4a show that the PBC specimens did not degrade for up to 30 days in acidic or weakly basic conditions, but in the buffer solution with pH 13.0, a mass loss of 15% was observed. To highlight the effect of pH on the degradation of polycarbonates, an accelerated experiment was performed at 55 °C in buffer solutions with pH values of 1.0, 7.4 and 13.0. As shown in Fig. 4a, the PBC specimens in the buffer solutions with pH values of 1.0 and 7.4 showed weight losses of 2.1 ± 0.9% and 0.3 ± 1.0%, respectively, after 30 days, while the specimen in basic condition with pH value of 13.0 showed a weight loss of 89.5 ± 6.3%. Moreover, the weight changes of PBC specimen during 30 d incubation at 37 °C, pH 7.4 and 55 °C pH 13.0 is compared in ESI-Fig. 15.† The results show that the sample weight remained constant at 37 °C and pH 7.4, while the PBC specimen was degraded at 55 °C pH 13.0 during 30 days according to zero order with an erosion rate of 2.9 wt% per day (correlation coefficient R2 = 0.9726).
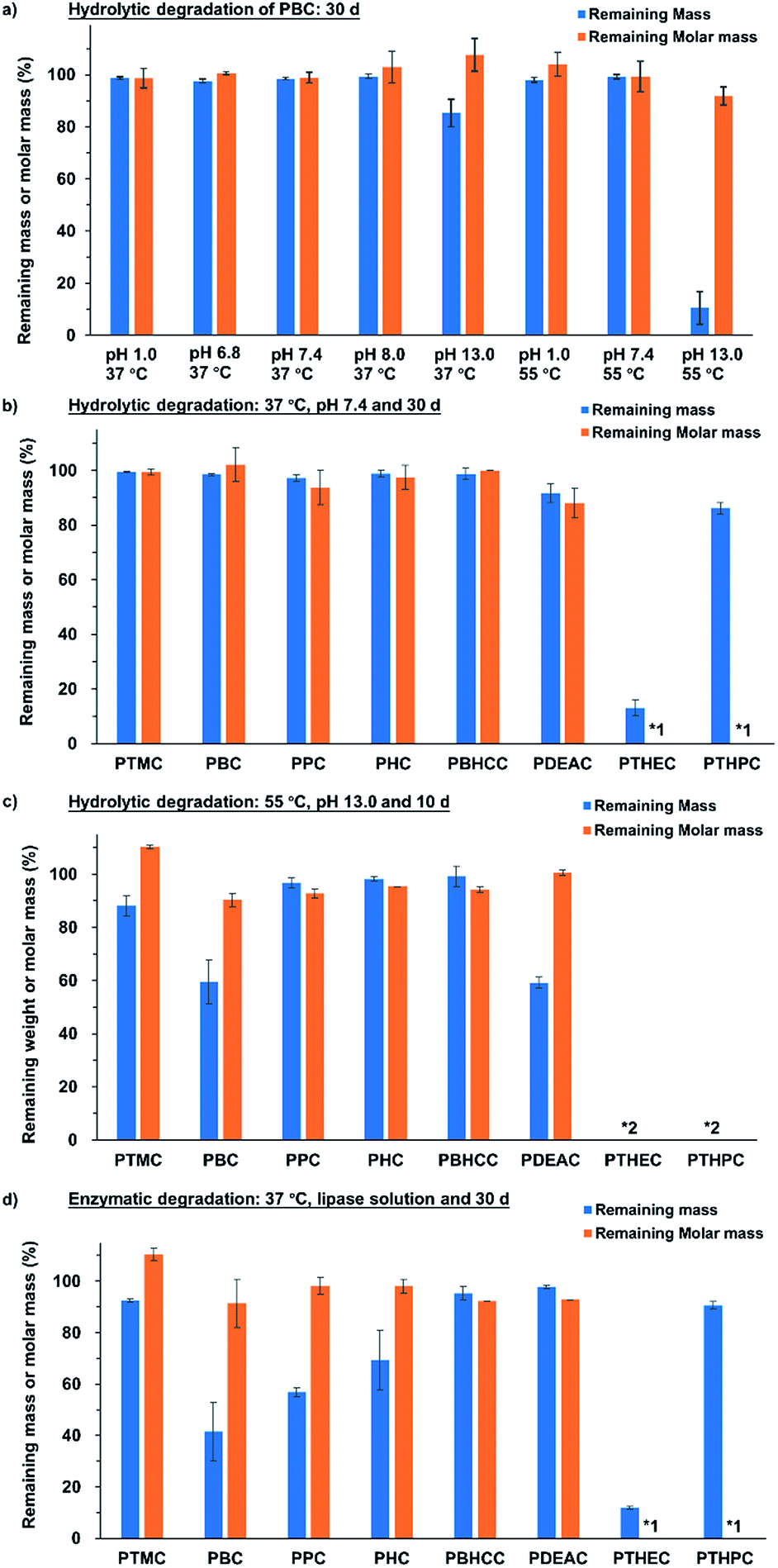 | ||
| Fig. 4 Results of the hydrolytic and enzymatic degradation for linear and hyperbranched polycarbonate specimens. (a) Mass and molar mass loss of PBC specimen under different conditions after 30 days. (b) Mass and molar mass loss of linear and hyperbranched polycarbonate specimens under the biological condition, 37 °C pH 7.4, after 30 days and (c) under the accelerated condition, 55 °C pH 13.0, after 10 days. (d) Mass and molar mass loss of linear and hyperbranched polycarbonate specimens in lipase solution after 30 days. *1, hyperbranched polymers were cross-linked after 30 days; *2, no polymer was recovered under this condition. | ||
All of the other linear and hyperbranched polycarbonates were also evaluated under the same conditions (at 37 °C pH 7.4 for 30 days and 55 °C pH 13.0 for 10 days) in order to determine the influence of polymer structures on the degradation rate. Fig. 4b shows that most of the linear polycarbonate specimens showed no decrease in either weight or molar mass. An exception is that PDEAC lost 8.2 ± 3.5% weight, which is probably attributed to the heteroatom in the polycarbonate backbone. The two hyperbranched specimens presented completely different phenomena. Only 13.0 ± 2.8% weight was recovered for PTHEC, while PTHPC was found to show 86.2 ± 2.1% mass remaining on day 30. The huge mass loss could be ascribed to the more hydrophilic property of PTHEC, leading to the dissolution in buffer solution because of the lack of a methylene group on the side chain in comparison to the PTHPC specimen. Both hyperbranched polymers were cross-linked after the investigation and did not dissolve in acetone or THF.
As shown in Fig. 4c, the degradation rate of the polycarbonates was also strongly influenced by the polymer structure. For linear polycarbonates under the accelerated condition, the mass losses of PTMC, PBC and PDEAC were 11.9 ± 3.8%, 40.5 ± 8.2% and 40.9 ± 2.1%, respectively, after 10 days. However, PPC, PHC and PCDMC showed higher stabilities under this condition. The hyperbranched polycarbonate specimens, PTHEC and PTHPC, were cooled to 0 °C for several hours after the degradation to reduce the error in the solubility. No hyperbranched polymer was recovered after 10 days, indicating that the hyperbranched structure could accelerate the hydrolytic degradation process probably due to its high density of functional groups in comparison to linear aliphatic polycarbonates.
Fig. 4d shows the mass and molar mass losses of linear and hyperbranched polycarbonates specimens after 30 days incubation time using lipase from Thermocyces languginosus (solution from Sigma-Aldrich L0777, ≥100000 U g−1, pH 6.2). All linear polycarbonates except PDEAC showed more or less degradability in the enzymatic degradation investigation. PBC had the fastest degradation rate. After 30 days, 58.5 ± 11.4% of the weight had been lost. Enzymatic degradation rates could be significantly affected by polymer structures. Amorphous PTMC showed only 7.4 ± 0.7% weight loss, which is consistent with previous literature.48 In contrast, linear polycarbonates with higher crystallinity were degraded more quickly. The degradation rates decreased with the increase of carbon atoms of used alcohols (from PBC to PHC) and with the increase of rigidity (PCDMC). The mass losses of PPC, PHC and PCDMC were found to be 43.2 ± 1.8%, 30.6 ± 11.6% and 4,7 ± 2.6%, respectively. Different from other linear polycarbonates, PDEAC displayed higher stability and lower degradation rate in lipase solution. The possible reason is that the presence of nitrogen atoms in the polymer backbone reduced the lipase activity. Both the hyperbranched polycarbonates showed similar results as hydrolytic degradation at 37 °C and pH 7.4. Since significant losses of mass were observed while molar mass remained constant after the enzymatic and basic hydrolytic degradations, both the erosion processes must be considered as surface erosion process.
Conclusions
In summary, we developed a universal new strategy for the synthesis of aliphatic linear and hyperbranched polycarbonates. In contrast to the classic two-step polycondensation in melt requiring a high polymerization temperature and high vacuum to remove unreacted monomers and byproducts, in our work, the one-pot polycondensation was carried out in 1,4-dioxane solution and under relatively mild polymerization conditions (T = 120 °C and at atmospheric pressure) using DMAP or LiAcac as catalysts. The only side product was methanol, which was removed by using 4 Å molecular sieve in a pressure-equalized addition funnel. This study is the first to report a one-pot polycondensation at atmospheric pressure for the preparation of linear and hyperbranched polycarbonates using dimethyl carbonate (DMC) instead of phosgene. As expected, the polycarbonates were prepared only by the transesterification reaction between hydroxyl and methyl carbonate chain ends. Consequently, a nearly equal molar ratio of diol or triol to DMC was used to reach higher molar masses and avoid wasting excess DMC during the polycondensation. Using this strategy, poly(trimethylene carbonate) (PTMC), poly(1,4-tetramethylene carbonate) (PBC), poly(1,5-pentamethylene carbonate) (PPC), poly(1,6-hexametylene carbonate) (PHC), poly(diethylphenylamine carbonate) (PDEAC) and poly(cyclohexan-1,4-dimethylene carbonate) (PCDMC) were successfully prepared with number averaged molar mass (Mn) up to 16000 g mol−1, dispersities below 1.70 and high yields above 70%. Additionally, the hyperbranched polycarbonates based on 1,1,1-tris(hydroxymethyl)ethane (THE) and 1,1,1-tris(hydroxymethyl)propane (THP) were also obtained with Mn up to 10000 g mol−1, Mw up to 64000 g mol−1 and high OH end group contents approximately 70%.
The hydrolytic degradations of the synthesized linear and hyperbranched polycarbonates were evaluated at 37 °C and 55 °C with various pH values, and the linear aliphatic polycarbonates were relatively stable under acidic to weakly basic conditions even at elevated temperature. In contrast, linear polycarbonates could only be hydrolytically degraded under strongly basic condition (pH 13.0). For the hyperbranched polycarbonates, no polymer was recovered after 10 days under the accelerated condition (55 °C, pH 13.0), indicating that the hyperbranched structure could accelerate the hydrolytic degradation process probably due to its high density of functional groups in comparison to linear aliphatic polycarbonates. Compared with hydrolytic degradation, linear polycarbonates were degraded much faster in lipase solution from Thermocyces languginosus at 37 °C. The degradation rates were strongly dependent on polymer structures. Both the enzymatic and basic hydrolytic degradations showed that linear polycarbonate specimens degraded by surface erosion.
In addition, this new strategy can theoretically be applied to synthesize other types of hyperbranched polymers, such as polyesters, via A2 + B3 condensation polymerization to obtain high molar masses and avoid cross-linking reactions with water or methanol as the byproduct.
Acknowledgements
The authors gratefully acknowledge financial support from the German Ministry of Education and Research (BMBF Grand No. IB-070) and the Science and Technology Development Fund (STDF, EGYPT) (GERF-Call-3) No. 5086.References
- E. Žagar and M. Žigon, Prog. Polym. Sci., 2011, 36, 53–88 CrossRef.
- A.-M. Caminade, D. Yan and D. K. Smith, Chem. Soc. Rev., 2015, 44, 3870–3873 RSC.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- M. G. McKee, S. Unal, G. L. Wilkes and T. E. Long, Prog. Polym. Sci., 2005, 30, 507–539 CrossRef CAS.
- M.-a. K. M. Jikei, Prog. Polym. Sci., 2001, 26, 1233–1285 CrossRef CAS.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2679–2699 CrossRef CAS.
- M. Tryznowski, K. Tomczyk, Z. Fraś, J. Gregorowicz, G. Rokicki, E. Wawrzyńska and P. G. Parzuchowski, Macromolecules, 2012, 45, 6819–6829 CrossRef CAS.
- H. E. Rogers, P. Chambon, S. E. R. Auty, F. Y. Hern, A. Owenb and S. P. Rannard, Soft Matter, 2015, 11, 7005–7015 RSC.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- H. Jia, H. Wang, C. Liu, C. Li, J. Yang, X. Xu, J. Feng, X. Zhang and R. Zhuo, Soft Matter, 2012, 8, 6906–6912 RSC.
- A. Sunder, R. Mülhaupt and H. Frey, Macromolecules, 2000, 33, 309–314 CrossRef CAS.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- R. Haag, J. Stumbé, A. Sunder, H. Frey and A. Hebel, Macromolecules, 2000, 33, 8158–8166 CrossRef CAS.
- H. Magnusson, E. Malmström and A. Hult, Macromolecules, 2000, 33, 3099–3104 CrossRef CAS.
- E. Malmstroem, M. Johansson and A. Hult, Macromolecules, 1995, 28, 1698–1703 CrossRef CAS.
- K. Fu, D. W. Pack, A. M. Klibanov and R. Langer, Pharm. Res., 2000, 17, 100–106 CrossRef CAS.
- W. Su, X. Luo, H. Wang, L. Li, J. Feng, X. Zhang and R. Zhuo, Macromol. Rapid Commun., 2011, 32, 390–396 CrossRef CAS PubMed.
- J. Feng, R. Zhuo and X. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef CAS.
- H. Schobert and C. Song, Fuel, 2002, 81, 15–32 CrossRef CAS.
- A. Davis and J. H. Golden, J. Macromol. Sci., Polym. Rev., 1969, 3, 49–68 CrossRef CAS.
- A. Welle, M. Kröger, M. Döring, K. Niederer, E. Pindel and I. S. Chronakis, Biomaterials, 2007, 28, 2211–2219 CrossRef CAS PubMed.
- A. P. Pêgo, D. W. Grijpma and J. Feijen, Polymer, 2003, 44, 6495–6504 CrossRef.
- W. Kuran, M. Sobezak, T. Listos, C. Debek and Z. Florjanczyk, Polymer, 2000, 41, 8531–8541 CrossRef CAS.
- P. U. Naik, K. Refes, F. Sadaka, C.-H. Brachais, G. Boni, J.-P. Couvercelle, M. Picquet and L. Plasseraud, Polym. Chem., 2012, 3, 1475–1480 RSC.
- F. Suriano, O. Coulembier, J. L. Hedrickb and P. Duboisa, Polym. Chem., 2011, 2, 528–533 RSC.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- I. Engelberg and J. Kohn, Biomaterials, 1991, 12, 292–304 CrossRef CAS PubMed.
- W. H. Carothers and F. J. v. Natta, J. Am. Chem. Soc., 1930, 52, 314–326 CrossRef CAS.
- K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 24, 1736–1740 CrossRef CAS.
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC.
- L. Yang, J. Li, W. Zhang, Y. Jin, J. Zhang, Y. Liu, D. Yi, M. Li, J. Guo and Z. Gu, Polym. Degrad. Stab., 2015, 122, 77–87 CrossRef CAS.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Duboisb and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- J. Mindemark, B. Sun and D. Brandella, Polym. Chem., 2015, 6, 4766–4774 RSC.
- T. S. Kristufek, S. L. Kristufek, L. A. Link, A. C. Weems, S. Khan, S. Lim, A. T. Lonnecker, J. E. Raymond, D. J. Maitland and K. L. Wooley, Polym. Chem., 2016, 7, 2639–2644 RSC.
- A. K. Reitz, Q. Sun, R. Wilhelm and D. Kuckling, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 820–829 CrossRef CAS.
- M. Miyasaka, T. Takazoe, H. Kudo and T. Nishikubo, Polym. J., 2010, 42, 852–859 CrossRef CAS.
- D. H. Bolton and K. L. Wooley, Macromolecules, 1997, 30, 1890–1896 CrossRef CAS.
- D. H. Bolton and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 823–835 CrossRef CAS.
- Y. Zheng, S. Li, Z. Wenga and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- J. H. Park, J. Y. Jeon, J. J. Lee, Y. Jang, J. K. Varghese and B. Y. Lee, Macromolecules, 2013, 46, 3301–3308 CrossRef CAS.
- H. R. Kricheldorf and A. Mahler, Polymer, 1996, 37, 4383–4388 CrossRef CAS.
- H. R. Kricheldorf and A. Mahler, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 2399–2406 CrossRef CAS.
- S. Bigot, N. Kébir, L. Plasseraud and F. Burel, Polymer, 2015, 66, 127–134 CrossRef CAS.
- Q. Li, W. Zhu, C. Li, G. Guan, D. Zhang, Y. Xiao and L. Zheng, JJ. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1387–1397 CrossRef CAS.
- J. Sun and D. Kuckling, Polym. Chem., 2016, 7, 1642–1649 RSC.
- D. Holtel, A. Burgath and H. Frey, Acta Polym., 1997, 48, 30–35 CrossRef.
- Y. H. Kim and R. Beckerbauer, Macromolecules, 1994, 27, 1968–1971 CrossRef CAS.
- Z. Zhang, R. Kuijerb, S. K. Bulstrab, D. W. Grijpmaa and J. Feijen, Biomaterials, 2006, 27, 1741–1748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of polycarbonates, ESI-ToF-MS of PBC 9 and result of hydrolytic degradation. See DOI: 10.1039/c7ra01317e |
| This journal is © The Royal Society of Chemistry 2017 |