
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct C(sp2)–H amination of aryl aldehyde-derived hydrazones via visible light promoted photoredox catalysis†
Xin Zhua,
Zhi Hea,
Qiu-Yan Lib and
Xiao-Jun Wang *bc
*bc
aCollege of Chemistry, Beijing Normal University, Beijing 100875, P. R. China
bJiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, School of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou 221116, P. R. China. E-mail: xjwang@jsnu.edu.cn
cKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 10th May 2017
Abstract
An unprecedented visible-light-induced direct C(sp2)–H amination of aryl aldehyde-derived hydrazones was developed by using N-acyloxyphthalimides as nitrogen-radical precursors. This reaction represents a new way to substituted hydrazones with amination of carbon–nitrogen π bonds. A radical C–H amination mechanism is proposed.
Aldehyde-derived hydrazones belong to an important class of chemicals because of their many applications in synthetic organic chemistry.1 Notably, they are attractive as umpolung carbonyl reagents because of the presence of the electron-releasing amino component that activates the azomethine (CH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) carbon atom position toward electrophilic substitution. Much research attention has been devoted to the preparation of functionalized aromatic aldehyde hydrazones.2 Recent attention has shifted toward the achievement of using the hydrazones' CN bond as various radical acceptors for their diverse functionalizations (Scheme 1).3–6 Along this line, Baudoin and co-workers have reported an efficient procedure for the CuCl catalyzed trifluoromethylation of N,N-dialkylhydrazones under Togni reagent.3 In 2016, the groups of Hashmi and Zhu presented a work of difluoroalkylation of aldehyde N,N-dialkylhydrazones by CF2 radical.4,5 Then Zhu's group reported a procedure for the C–H bond phosphonation of aldehyde N,N-dialkylhydrazones with the commercially available diphenylphosphine oxide.6 For the limited work of presented studies, substantial effort need to be devoted to the development of different radical (such as N, Si and B) to activate the hydrazones' C–H bond.‡
N) carbon atom position toward electrophilic substitution. Much research attention has been devoted to the preparation of functionalized aromatic aldehyde hydrazones.2 Recent attention has shifted toward the achievement of using the hydrazones' CN bond as various radical acceptors for their diverse functionalizations (Scheme 1).3–6 Along this line, Baudoin and co-workers have reported an efficient procedure for the CuCl catalyzed trifluoromethylation of N,N-dialkylhydrazones under Togni reagent.3 In 2016, the groups of Hashmi and Zhu presented a work of difluoroalkylation of aldehyde N,N-dialkylhydrazones by CF2 radical.4,5 Then Zhu's group reported a procedure for the C–H bond phosphonation of aldehyde N,N-dialkylhydrazones with the commercially available diphenylphosphine oxide.6 For the limited work of presented studies, substantial effort need to be devoted to the development of different radical (such as N, Si and B) to activate the hydrazones' C–H bond.‡
 | ||
| Scheme 1 Radical addition to carbon–nitrogen π bond. | ||
Due to the abundant, renewable, inexpensive and eco-friendly energy source, visible-light promoted photocatalysis has recently been employed as a powerful strategy in synthetic organic chemistry.7 Many novel visible-light mediated reactions were developed for the efficient and rapid preparation of fine chemicals over the past years.7,8 In the context, the addition of nitrogen-central radical generated by photo-catalysis to a π acceptor has been developed as a straightforward method to access structurally diverse N-containing compounds.9 Nevertheless, despite these important progress, existing methods for radical-type amination are largely limited to carbon–carbon π bonds (alkene, alkyne, or arene). For example, MacMillan et al. developed an elegant N-radical-mediated enantioselective direct α-amination of aldehydes by rationally merging visible-light photoredox catalysis with asymmetric aminocatalysis in photoredox-catalyzed C–H amination of arenes and heteroarenes with the use of N-acyloxyphthalimides as the N-radical precursor.10 Shortly thereafter, Yu et al. developed a similar strategy by using a readily accessible hydroxylamine derivative as an N-radical precursor, which proved to be an efficient and broadly applicable visible-light photoredox-catalyzed direct C–H bond amination of various heteroarenes, including indoles, pyrroles, and furans.11
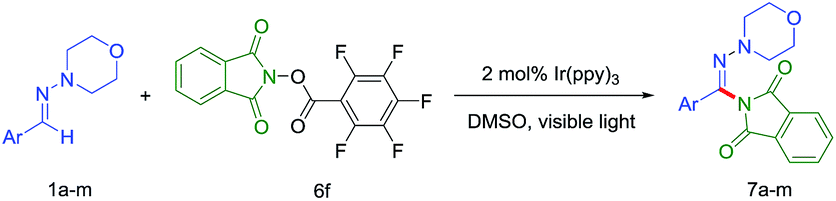
Inspired by the pioneering works, we wondered that a N-radical precursor with good leaving group could be easy to generate a N-mediated radical and here present a work of direct C–H amination of aldehyde N,N-dialkyl hydrazones by visible light irradiation. To the best of our knowledge, the direct C(sp2)–H amination of benzaldehyde or its synthetic equivalents (aromatic aldehyde hydrazones) through a radical way is unexplored, but could complement existing strategies. Herein we reported an efficient and mild procedure for the amination of aromatic aldehyde hydrazones based on N-(acyloxy)phthalimides as aminating reagents under visible-light irradiation.
The generation of a N-central radical by visible-light photoredox catalysis recently has emerged as a powerful tool in synthetic organic chemistry.7e On the basis of these previous reports, we initiated the reaction of N-morpholino-1-phenyl-methanimine (1a) and various nitrogen radical precursors by using fac-Ir(ppy)3 as photocatalyst owing to its superior reduction capacity upon excitation by visible light (Scheme 2). After irradiation by fluorescent light bulb for 24 h at room temperature, it was found only precursor 6b with strong electron-deficient group gave a slight yield (7%).
 | ||
| Scheme 2 Screening for an effective N-radical precursors. Reaction condition: 1a (0.1 mmol), nitrogen source (3 equiv.), Ir(ppy)3 (0.002 mmol, 2 mol%), MeCN (1.5 mL), at room temperature, fluorescent light bulb, 24h.aYields determined by GC-MS. | ||
Encouraged by this preliminary result, we further optimized the reaction condition. The desired product (7a) was obtained by employing aldehyde hydrazone (1a) and the N-(acyloxy)phthalimides as the model substrates and by using the photoredox catalyst Ir(ppy)3 with visible light in MeCN (Table 1). According to the Sanford group's report that the use of more electron-withdrawing R-substituents would enhance the leaving group ability of the carboxylate anion, thereby favoring fragmentation to release RCO2− with generation of the N-centered phthalimidyl radical (PhthN˙). The PhthN˙ then could participate in C–H amination of diverse substrates. Our initial efforts toward achieving this goal focused on the changing of leaving group of N-(acyloxy)phthalimides. To achieve this goal, we examined the C–H amination of aldehyde hydrazone (1a) with a series of electronically different N-(acyloxy)phthalimides in the presence of 2 mol% of Ir(ppy)3 and visible light in MeCN. As shown in Table 1, N-(acyloxy)phthalimide 6a (R = CH3, entry 1) did not react to afford C–H amination product 7a but with the starting materials consumed. However, the increase of electron-withdrawing ability of R group in 6 derivatives resulted in the improved yield. In particular, 6f gave the best yield (18%).
|
||
|---|---|---|
| Entry | R | Yieldb (%) |
| a Reaction condition: 1a (0.1 mmol), 2 (0.3 mmol, 3 equiv.), Ir(ppy)3 (0.002 mmol, 2 mol%), MeCN (1.0 mL), at room temperature, fluorescent light bulb, 24 h.b Yields determined by GC-mass, decahydronaphthalene used as internal standard.c Starting materials were consumed and no products were detected by GC-mass. nd = not detected. Bs = benzenesulfonyl, Ns = 4-nitrobenzensulfonyl. | ||
| 1 | CH3CO (6a) | ndc |
| 2 | CF3CO (6b) | 7 |
| 3 | PhCO (6c) | <5 |
| 4 | p-MeOPhCO (6d) | <5 |
| 5 | p-CF3PhCO (6e) | 11 |
| 6 | C6F5CO (6f) | 18 |
| 7 | Bs (6g) | 8 |
| 8 | Ns (6h) | <5 |

To optimize the reaction condition, the solvents were varied together with using different additions. After solvent screening, dimethyl sulfoxide (DMSO) was found to be the most suitable for this reaction (Table 2, entry 3). Contrary to expectations, the bases were bad for the reaction (Table 2, entries 5–8). We then speculated that the nitrogen precursor was easily decomposed under the inorganic bases and could not generate the N-radical. Control experiments showed that the reaction could not proceed in the absence of either visible-light irradiation or the photoredox catalyst (Table 2, entries 9 and 10). And we also found that the reaction got a good yield under visible light irradiation from blue LEDs instead of fluorescent light bulb after 48 h (Table 2, entries 12).
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yieldb (%) |
| a Reaction condition: 1a (0.1 mmol), 2a (0.3 mmol, 3 equiv.), base (0.15 mmol, 1.5 equiv.), Ir(ppy)3 (0.002 mmol, 2 mol%), solvent (1.0 mL), at room temperature, fluorescent light bulb, 24 h.b Yields determined by GC-mass, decahydro-naphthalene used as internal standard.c In dark.d Without Ir(ppy)3.e With blue LEDs (20 W).f With blue LEDs (20 W) and 48 h. | |||
| 1 | — | MeCN | 8 |
| 2 | — | DMF | <5 |
| 3 | — | DMSO | 52 |
| 4 | — | DCM | <5 |
| 5 | K2HPO4 | DMSO | <5 |
| 6 | KOAc | DMSO | <5 |
| 7 | NaHCO3 | DMSO | <5 |
| 8 | CsF | DMSO | <5 |
| 9c | — | DMSO | 0 |
| 10d | — | DMSO | 0 |
| 11e | — | DMSO | 56 |
| 12f | — | DMSO | 87 |

With the optimized condition established, we next investigated the effect of the N-substituent (Scheme 3). The results showed that this reaction did not tolerate secondary amino groups, as a complex mixture of product was obtained in the case of 1ac–1ab, even though the starting materials were consumed. These experimental results indicated that the N,N-dialkyl structural motif was crucial for this transformation.
 | ||
| Scheme 3 Investigation of the effect of the N-substituent. | ||
Furthermore, a plausible photo–catalytic cycle is proposed in Scheme 4. First, upon visible-light irradiation, the photocatalyst Ir(ppy)3 undergoes a metal to ligand charge-transfer (MLCT) process to produce the strongly reducing excited state  (step a). Single electron transfer from
(step a). Single electron transfer from  to 6f results in fragmentation of the N-(acyloxy)phthalimide with the generation of an N-centered phthalimidyl radical (PhthN˙) and Ir(ppy)3+ (step b). Subsequent addition of PhthN˙ to the CN π bond leads to an aminyl radical intermediate 9 (step c), which then undergoes oxidation and deprotonation of the aminyl cation, giving aminated product 7a. When 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) was added into the reaction mixture under the standard condition, only trace amount of product 7a can be detected (Scheme 5). It demonstrated that this reaction generated radical which could be trapped by TEMPO, which suggested the single electron transfer process during the reaction.
to 6f results in fragmentation of the N-(acyloxy)phthalimide with the generation of an N-centered phthalimidyl radical (PhthN˙) and Ir(ppy)3+ (step b). Subsequent addition of PhthN˙ to the CN π bond leads to an aminyl radical intermediate 9 (step c), which then undergoes oxidation and deprotonation of the aminyl cation, giving aminated product 7a. When 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) was added into the reaction mixture under the standard condition, only trace amount of product 7a can be detected (Scheme 5). It demonstrated that this reaction generated radical which could be trapped by TEMPO, which suggested the single electron transfer process during the reaction.
 | ||
| Scheme 4 Proposed the photocatalyzed reaction mechanism. | ||
 | ||
| Scheme 5 Investigation into the reaction mechanism. | ||
The amination of various aromatic N,N-dialkyl hydrazones under the optimized reaction conditions was next examined to gauge the scope of the reaction (Table 3). Aryl aldehyde-derivied hydrazones bearing either electron-donating or electron-withdrawing substituents furnished the corresponding products. And electron-withdrawing (7b,7c) gave a slightly better yield. The C–H amination reaction of heterocyclic hydrazones or aliphatic aldehyde-derived hydrazones was discouraged in the present condition (7l and 7m).

In conclusion, we have developed an efficient and mild visible-light photoredox-catalyzed C–H amination of aryl aldehyde-derived hydrazones for the first time, where N-acyloxyphthalimides were employed as nitrogen-radical precursors. And it is anticipated that the present strategy could be used for more useful transformation of C–N π bond in the future.
Acknowledgements
This work was financially supported from Beijing Normal University, National Natural Science Foundation of China (21302072) and Key Laboratory of Photochemical Conversion and Optoelectronic Materials, TIPC, CAS (PCOM201711).Notes and references
- (a) R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386–1434 CrossRef CAS PubMed; (b) R. Brehme, D. Enders, R. Fernandez and J. M. Lassaletta, Eur. J. Org. Chem., 2007, 5629–5660 CrossRef CAS.
- (a) G. K. Friestad, Tetrahedron, 2001, 57, 5461–5496 CrossRef CAS; (b) G. K. Friestad, Y. Shen and E. L. Ruggles, Angew. Chem., Int. Ed., 2003, 42, 5061–5063 CrossRef CAS PubMed; (c) K. Yamada and K. Tomioka, Chem. Rev., 2008, 108, 2874–2886 CrossRef CAS PubMed; (d) H. Miyabe, Synlett, 2012, 23, 1709–1724 CrossRef CAS.
- E. Pair, N. Monteiro, D. Bouyssi and O. Baudoin, Angew. Chem., Int. Ed., 2013, 52, 5346–5349 CrossRef CAS PubMed.
- J. Xie, T. Zhang, F. Chen, N. Mehrkens, F. Rominger, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 2934–2938 CrossRef CAS PubMed.
- P. Xu, G. Wang, Y. Zhu, W. Li, Y. Cheng, S. Li and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2939–2943 CrossRef CAS PubMed.
- P. Xu, Z. Wu, N. Zhou and C. Zhu, Org. Lett., 2016, 18, 1143–1145 CrossRef CAS PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (e) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- (a) Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-Y. Meng, L.-Z. Wu and C.-H. Tung, J. Am. Chem. Soc., 2016, 138, 10080–10083 CrossRef CAS PubMed; (b) L.-M. Zhao, Q.-Y. Meng, X.-B. Fan, C. Ye, X.-B. Li, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 3020–3024 CrossRef CAS PubMed; (c) G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed; (d) Q.-Y. Li, Z. Ma, W.-Q. Zhang, J.-L. Xu, W. Wei, H. Lu, X. Zhao and X.-J. Wang, Chem. Commun., 2016, 52, 11284–11287 RSC; (e) W.-Q. Zhang, Q.-Y. Li, Q. Zhang, Y. Lu, H. Lu, W. Wang, X. Zhao and X.-J. Wang, Inorg. Chem., 2016, 55, 1005–1007 CrossRef CAS PubMed; (f) Q.-Y. Meng, J.-J. Zhong, Q. Liu, X.-W. Gao, H.-H. Zhang, T. Lei, Z.-J. Li, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2013, 135, 19052–19055 CrossRef CAS PubMed.
- (a) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC; (b) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC; (c) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef PubMed; (d) S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed; (e) M. Louillat-Habermeyer, R. Jin and F. W. Patureau, Angew. Chem., Int. Ed., 2015, 54, 4102–4104 CrossRef CAS PubMed; (f) L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607–5610 CrossRef CAS PubMed; (g) H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2015, 54, 4055–4059 CrossRef CAS PubMed; (h) J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 127, 14223–14227 CrossRef; (i) T. W. Greulich, C. G. Daniliuc and A. Studer, Org. Lett., 2015, 17, 254–257 CrossRef CAS PubMed; (j) K. Miyazawa, T. Koike and M. Akita, Chem.–Eur. J., 2015, 21, 11677–11680 CrossRef CAS PubMed.
- G. Cecere, C. M. Konig, J. L. Alleva and D. W. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS PubMed.
- Q. Qin and S. Yu, Org. Lett., 2014, 16, 3504–3507 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experiment details and compounds characterization. CCDC 1543507. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01229b |
| ‡ Crystal data for 7b: CCDC 1543507, C20H16N3O3F3, Mr = 403.36, orthorhombic, space group Pbcn, T = 153 K, a = 19.520(4) Å, b = 13.946(3) Å, c = 13.312(3) Å, α = β = γ = 90°, V = 3623.8(13) Å3, λ = 0.71073 Å, Z = 8, independent reflections 4113, Rint = 0.0501, R1 [I > 2σ(I)] = 0.0669, wR2 (all data) = 0.1341, GOF = 1.251. |
| This journal is © The Royal Society of Chemistry 2017 |