DOI:
10.1039/C7RA01212H
(Paper)
RSC Adv., 2017,
7, 17403-17416
Thermo-responsive fluorescence of AIE-active poly(N-isopropylacrylamides) labeled with highly twisted bis(N,N-dialkylamino)arenes†
Received
27th January 2017
, Accepted 13th March 2017
First published on 21st March 2017
Abstract
Highly twisted bis(N,N-dialkylamino)arenes, which represent a new class of viscosity-sensitive fluorophores with aggregation-induced emission (AIE) luminogens, were introduced as co-monomers and cross-linkers into poly(N-isopropylacrylamides) (PNIPAMs). Despite the excellent performance of these bis(N,N-dialkylamino)arenes as fluorophores that are sensitive to the steric environment, synthetic methods to endow them with other reactive groups have not yet been reported. This study presents short synthetic pathways to 9,10-bis(N,N-dialkylamino)anthracenes (BDAAs) and 1,4-bis(N,N-dialkylamino)-2,3-dimethylnaphthalenes (DMe-BDANs) with hydroxyl groups at their alkyl chains. These hydroxyl groups were acylated to afford methacrylate moieties, which were subsequently used for the co-polymerization with N-isopropylacrylamide. At T = 20 °C, the resulting BDAA-containing PNIPAMs exhibited faint fluorescence (Φfl ≈ 0.05) in THF, whereas DMe-BDAN-containing PNIPAMs showed substantially stronger fluorescence in THF (Φfl ≈ 0.32) and water (Φfl ≈ 0.55) relative to the corresponding monomers. Moreover, BDAA-containing PNIPAMs featured a sharp increase of fluorescence intensity and quantum yield at T = 27–35 °C, while DMe-BDAN-containing PNIPAMs exhibited a continuous decrease of fluorescence intensity and Φfl with increasing temperature.
Introduction
Due to their sensitivity toward local viscosity, aggregation, and the binding to macromolecules, as well as their encapsulation in cavities, two types of fluorophores, i.e., molecular rotors1–5 and luminogens with aggregation-induced emission (AIEgens),6–8 have received much attention regarding their fundamental and practical properties. Although various fluorescent molecular rotors and AIEgens have been developed, only few hold specific application monopolies. For example, in the area of molecular rotors, 9-(2-carboxy-2-cyano)vinyljulolidine (CCVJ) derivatives1 have been extensively applied in molecular biology,9,10 polymer science,1,12 contact mechanics,13 and fluid dynamics.14 In the area of AIEgens, tetraphenylethene (TPE)15 analogs are usually chosen in order to endow molecular systems with AIE functionality.6,16 Given their simple structures and facile functionalization, it is hardly surprising that CCVJs and TPEs were easily incorporated into larger, more elaborate molecular systems.1,10,12,13,16 From a practical perspective, it is thus very important that novel fluorophores with improved photophysical properties also exhibit the potential for facile subsequent functionalization.17
Some of our previous experimental and theoretical studies revealed that highly twisted N,N-dialkylamino groups at the para-positions of typical aromatic hydrocarbons induce AIE and viscosity-responsive fluorescence.18 This general design strategy enabled us to develop structurally simple AIEgens such as 9,10-bis(N,N-dialkylamino)anthracenes (BDAAs) and 1,4-bis(N,N-dialkylamino)-2,3-dimethylnaphthalenes (DMe-BDANs). In addition to their acute viscosity-responsiveness, BDAAs and DMe-BDANs feature brighter solid-state fluorescence compared to other simple AIEgens.18,19 Therefore, BDAAs and DMe-BDANs represent promising prospectives for microenvironmental probes. However, synthetic routes to highly twisted bis(N,N-dialkylamino)arenes with functional groups at their alkyl chains have not yet been reported, despite the fact that numerous studies have been reported regarding the synthesis of sterically congested N,N-diarylaminoarenes.20,21
Herein, we present a facile route to functionalize the alkyl chains of BDAAs and DMe-BDANs. These compounds were subsequently used as co-monomers and cross-linkers in the polymerization of N-isopropylacrylamides, which afforded the corresponding poly(N-isopropylacrylamides) (PNIPAMs). In aqueous solution, PNIPAMs undergo a coil-to-globule transition at T ≈ 31 °C.22 While the phase transition itself has been the subject of extensive research, the sharp phase-transition behavior close to the lower critical solution temperature (LCST) has been used to visualize intracellular temperature.23 For both fundamental and practical purposes, various fluorophores such as pyrene,24 3-hydroxyflavone,25 benzofurazan,26 rhodamine,27 and BODIPY28 have been introduced into PNIPAMs. In the same way, PNIPAMs containing BDAAs and DMe-BDANs should be expected to act as a microenvironmental probes. Furthermore, BDAAs and DMe-BDANs exhibit not only AIE but also viscosity-sensitive fluorescence. These fluorophores should therefore most likely be able to visualize weak restrictions of intramolecular motions (RIM), which result from specific interactions between PNIPAM chains, fluorophores, and H2O molecules, in addition to the severe RIM imposed by a coil-to-globule transition of the PNIPAM chains. Herein, we report the temperature-dependent fluorescence behavior of BDAA- and DMe-BDAN-labeled PNIPAMs and the corresponding gels.
Results and discussion
Synthesis of bis(N,N-dialkylamino)arenes functionalized at their alkyl chains
The syntheses of 9,10-bis((N-(3′-(N′′-(3′′-hydroxyprop-1′′-yl)-N′′-methylamino))prop-1′-yl)-N-methylamino)anthracene (6) and 1,4-bis(N-(hydroxypent-5-yl)-N-methylamino)-2,3-dimethylnaphthalene (14) with hydroxy groups at the terminal positions of their alkyl chains are outlined in Schemes 1 and 2, respectively. The hydroxyl groups in 6 and 14 were subsequently transformed into methacrylate moieties in order to prepare fluorescent monomers (ANTH-monomer and NAPH-monomer) and cross-linkers (ANTH-linker and NAPH-linker). Detailed synthetic procedures are described in the Experimental section. Alcoholamines, especially those with straight-chain structures, have been reported to deactivate palladium/phosphine catalyst systems by chelating palladium.29 For the N-arylation of alcoholamines, the copper iodide/2-isobutyrylcyclohexane system has been proposed as an alternative.30,31 However, a catalyst test on the coupling between 1-bromo-iodonaphthalene and 5-amino-1-pentanol revealed that this system yielded only trace amounts of the desired product. Therefore, the nucleophilicity of the hydroxyl group on the alcoholamine should be disabled prior to the C–N coupling. This may be accomplished by either functional group interconversion (FGI)32 or by protection–deprotection strategies.33,34
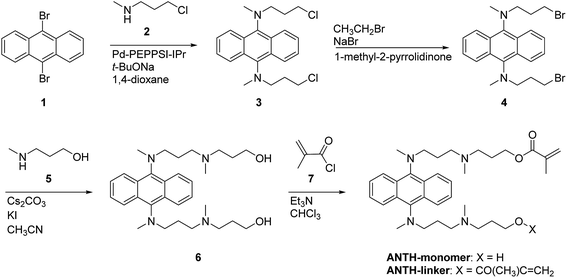 |
| | Scheme 1 Synthetic route to terminal-functionalized 9,10-bis(N,N-dialkylamino)anthracenes. | |
 |
| | Scheme 2 Synthetic route to terminal-functionalized 1,4-bis(N,N-dialkylamino)-2,3-dimethylnaphthalenes. | |
For the synthesis of terminal-functionalized 9,10-bis(N,N-dialkylamino)anthracene 6, the same catalyst system was employed as for the formation of the highly twisted N,N-dialkylaminoarenes,18,19 i.e., Pd-PEPPSI-IPr.20,35 As 9,10-diaminoanthracene and its N-monoalkylated derivatives are extremely sensitive to oxydation,18,36 a secondary amine (2) was chosen as the coupling partner for 9,10-dibromoanthracene (1). Subsequently, the chloride atoms in 3 were replaced with bromide atoms37 in order to enhance the reactivity toward a subsequent SN2 reaction with alcoholamine 5. The SN2 reaction proceeded selectively on the amine moiety to afford 6, which was confirmed by NOEs around the N-methyl group (Fig. S5†). In addition, functionalization with a methacrylate moiety endowed the resulting compounds with the characteristic absorption at 1717 cm−1, which clearly indicates the formation of an ester instead of an amide group. The overall yield of 6 with respect to starting material 1 was 53%.
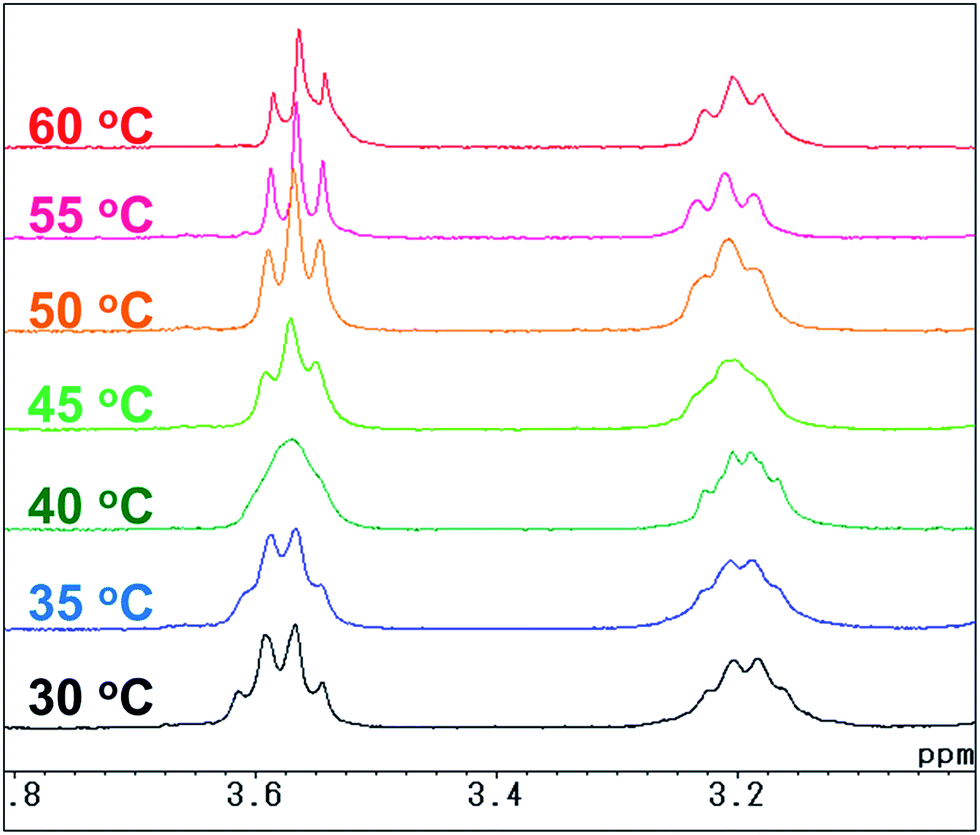
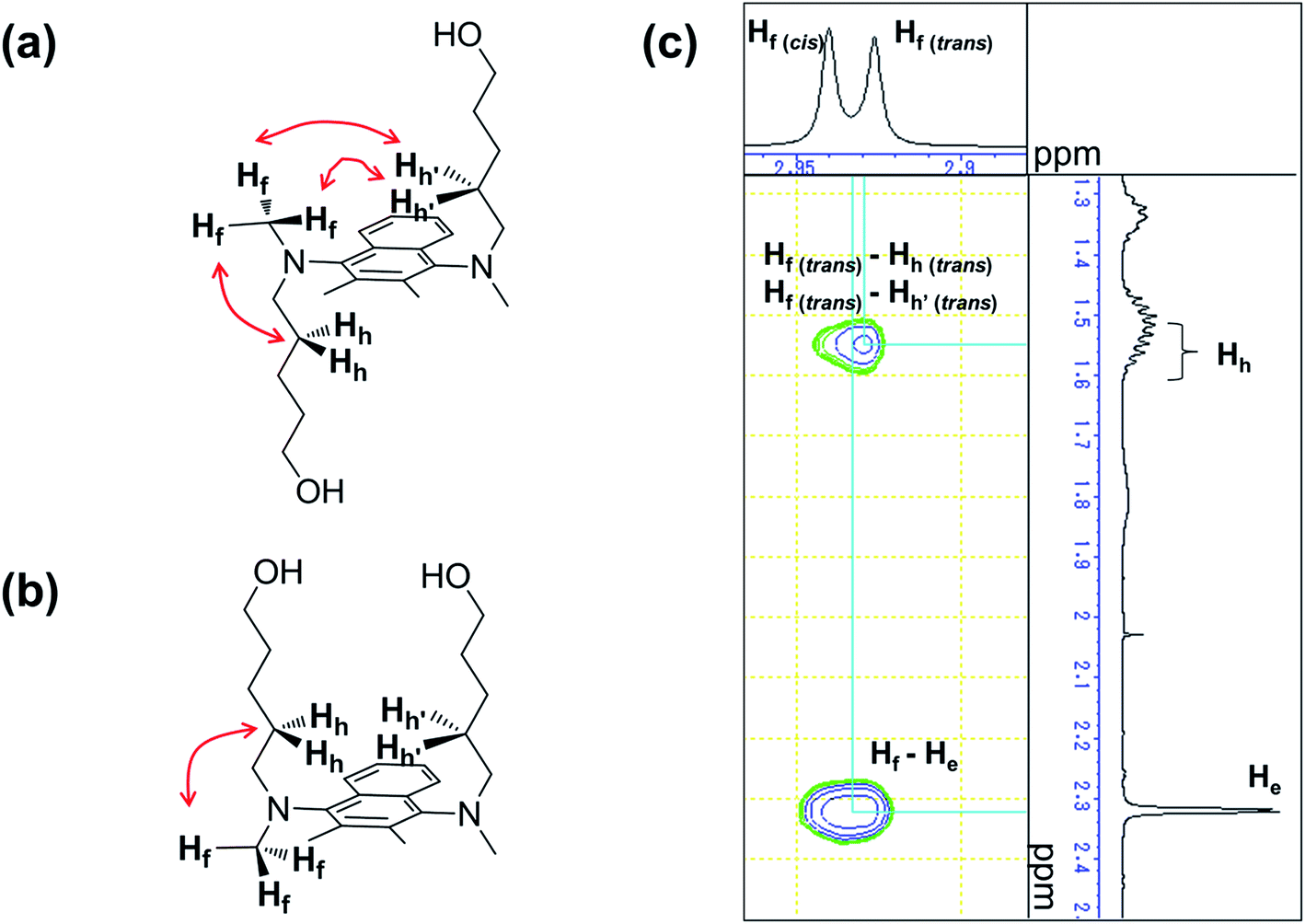
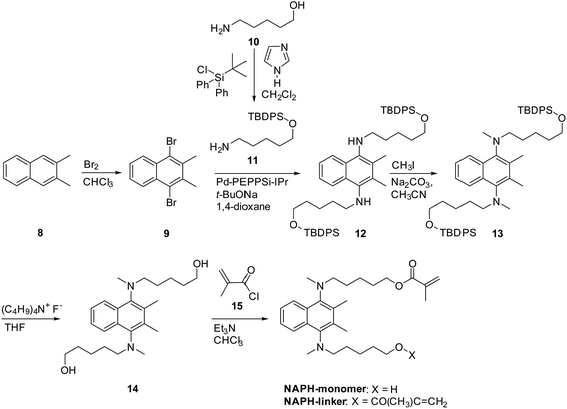
For the naphthalene-based system, a protection–deprotection strategy was employed in order to introduce hydroxyl groups at the terminal positions of the alkyl chains (Scheme 2). In this case, a route similar to Scheme 1 cannot be applied, as the secondary amine cannot be introduced at the 1- and 4-positions of 1,4-dibromo-2,3-dimethylnaphthalene by C–N coupling reactions.18 Furthermore, an FGI strategy as outlined in Scheme 1 would be more laborious than that described in Scheme 2. Firstly, 2,3-dimethylnaphthalene (8) was regioselectively brominated to give 9.38 Subsequently, 9 was combined with tert-butyldiphenylsilyl (TBDPS)-protected 5-amino-1-pentanol 11. It should be noted here that the C–N coupling between 9 and 11 proceeded successfully in the presence of trace amounts of moisture on the glass vessel, while the product yield was poor in the absence of moisture within the vessel after e.g. flame-drying in vacuo. The subsequent permethylation did not require adherence to strict stoichiometry between 13 and methyl iodide as the substantial steric hindrance around the nitrogen atoms prevents the further methylation of the tertiary amine moiety, even in the presence of an excess of methyl iodide. A final deprotection of the TBDPS group afforded 14 in 65% overall yield relative to 2,3-dimethylnaphthalene (8). The 1H and 13C NMR spectra of 14 showed pairs of peaks with comparable chemical shifts, identical peak multiplicities, and coupling constants (Fig. S14 and S15†). Further NMR studies based on DEPT-135, 1H–1H COSY, and 1H–13C HSQC techniques allowed a complete assignment of these peaks (Fig. S15–S17†). Variable-temperature (VT) NMR experiments revealed that these pairs of peaks originate from an equilibrium of two conformers (Fig. 1),39 while chemical-exchange peaks could not be discerned from the diagonal peaks in the NOESY spectra (Fig. S18†).40 These conformers are in fact two rotamers, which differ with respect to the rotation around the aryl-N bonds (Fig. 2a and b). As illustrated in Fig. 2c, the NOE signal of the Hh and Hh′ protons (1.59–1.49 ppm) intensified upon irradiating one of the Hf protons (2.93 ppm). In contrast to the cis-rotamer, the Hf protons in the trans-rotamer should be in close spatial proximity to both Hh and Hh′ protons (Fig. 2a and b). The observed stronger NOE for Hh and Hh′ thus indicates that the irradiated Hf proton peak should be attributed to the trans-rotamer. The slow equilibrium between the two rotamers on the NMR timescale reflects the severe steric restrictions imposed by the peri-hydrogen atoms and methyl groups at the 2- and 3-positions of the naphthalene core.
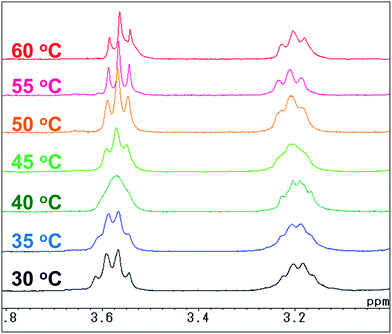 |
| | Fig. 1 Variable-temperature 1H NMR spectra (300 MHz, CDCl3) of 1,4-bis(N-(hydroxypent-5-yl)-N-methylamino)-2,3-dimethylnaphthalene (14). | |
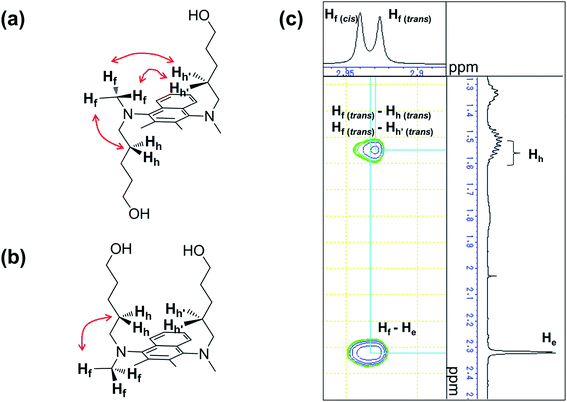 |
| | Fig. 2 Chemical structure of the (a) trans- and the (b) cis-rotamer of 1,4-bis(N-(hydroxypent-5-yl)-N-methylamino)-2,3-dimethylnaphthalene (14) (red arrows represent NOE signals). (c) NOESY spectrum (500 MHz, CDCl3) of 14 for the Hf peak area. | |
Co-polymerization of N-isopropylacrylamide with bis(N,N-dialkylamino)arene-based monomers and cross-linkers
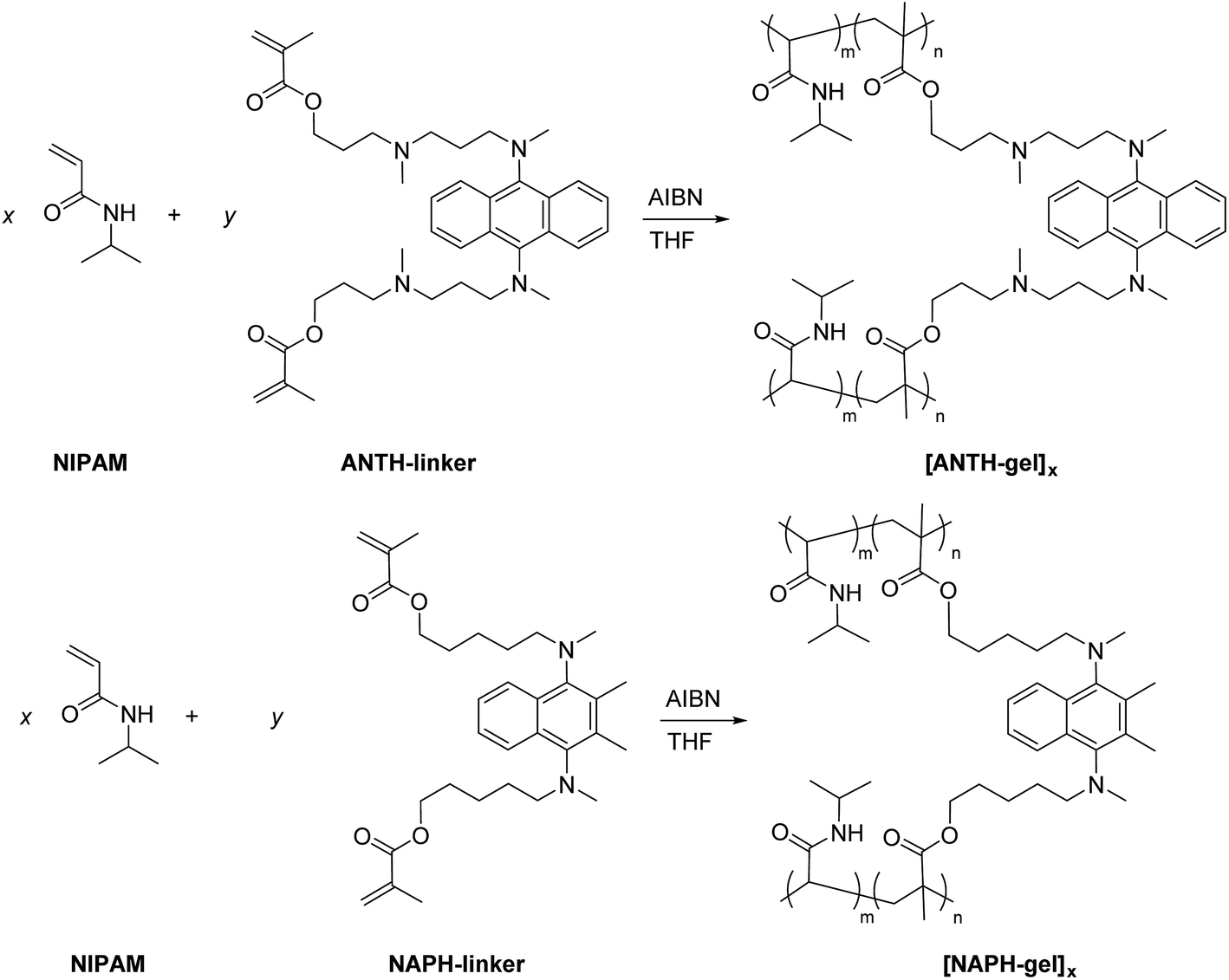
As outlined in Scheme 3, ANTH-monomer and NAPH-monomer were co-polymerized using various monomer feed ratios (x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y) to afford co-polymers [ANTH-polymer]x (yields: 61–84%) and [NAPH-polymer]x (yields: 91–96%). The thus obtained copolymers were purified by repeated reprecipitation into diethyl ether (for details, see the Experimental section). The 1H NMR spectra of [ANTH-polymer]x and [NAPH-polymer]x (Fig. S23 and S24†) are consistent with those previously reported for PNIPAMs,41 except for the small signals of the fluorophore moieties. Weight-average molecular weights (Mw) and polydispersity indexes (Mw/Mn; PDI) were determined by size exclusion chromatography (SEC; Fig. S25 and S26†). In general, [ANTH-polymer]x exhibited smaller Mw values and broader polydispersity values (Mw = 48000–66000 g mol−1, Mw/Mn = 2.51–2.61) than [NAPH-polymer]x (Mw = 65000–80000 g mol−1, Mw/Mn = 2.25–2.37). The copolymer composition m:n (Scheme 3) was calculated from the corresponding UV-Vis spectra. For that purpose, molar absorption coefficients of 6 (ε = 5330 M−1 cm−1 at λabs = 401 nm) and 14 (ε = 2700 M−1 cm−1 at λabs = 321 nm) were derived from calibration lines (Fig. S27†), and used to determine the concentration of the fluorophore moieties. While the m:n ratio of the [NAPH-polymers]x agreed well with the monomer feed ratio x:y, the concentration of the fluorophore moieties in [ANTH-polymer]x was significantly lower than expected (Table 1). Considering that the relative reactivity of ANTH-monomer and NAPH-monomer should be comparable, the physical properties of the ANTH-monomer, such as incompatibility with concentrated PNIPAMs, may be responsible for the lower molecular weight, the broader polydispersity, and the inefficient incorporation of the fluorophore moieties.
:y) to afford co-polymers [ANTH-polymer]x (yields: 61–84%) and [NAPH-polymer]x (yields: 91–96%). The thus obtained copolymers were purified by repeated reprecipitation into diethyl ether (for details, see the Experimental section). The 1H NMR spectra of [ANTH-polymer]x and [NAPH-polymer]x (Fig. S23 and S24†) are consistent with those previously reported for PNIPAMs,41 except for the small signals of the fluorophore moieties. Weight-average molecular weights (Mw) and polydispersity indexes (Mw/Mn; PDI) were determined by size exclusion chromatography (SEC; Fig. S25 and S26†). In general, [ANTH-polymer]x exhibited smaller Mw values and broader polydispersity values (Mw = 48000–66000 g mol−1, Mw/Mn = 2.51–2.61) than [NAPH-polymer]x (Mw = 65000–80000 g mol−1, Mw/Mn = 2.25–2.37). The copolymer composition m:n (Scheme 3) was calculated from the corresponding UV-Vis spectra. For that purpose, molar absorption coefficients of 6 (ε = 5330 M−1 cm−1 at λabs = 401 nm) and 14 (ε = 2700 M−1 cm−1 at λabs = 321 nm) were derived from calibration lines (Fig. S27†), and used to determine the concentration of the fluorophore moieties. While the m:n ratio of the [NAPH-polymers]x agreed well with the monomer feed ratio x:y, the concentration of the fluorophore moieties in [ANTH-polymer]x was significantly lower than expected (Table 1). Considering that the relative reactivity of ANTH-monomer and NAPH-monomer should be comparable, the physical properties of the ANTH-monomer, such as incompatibility with concentrated PNIPAMs, may be responsible for the lower molecular weight, the broader polydispersity, and the inefficient incorporation of the fluorophore moieties.
 |
| | Scheme 3 Co-polymerization of N-isopropylacrylamide with ANTH-monomer and NAPH-monomer. | |
Table 1 Monomer feed ratios (x:y), copolymer compositions (m:n), weight-average molecular weights (Mw), and polydispersity indexes (Mw/Mn) for [ANTH-polymer]x and [NAPH-polymers]x
| Entry |
x:y |
m:na |
Mwb [g mol−1] |
Mw/Mnb |
| Co-polymer composition based on the UV-Vis spectra (Fig. S27). Molecular weight and PDI based on SEC (DMF + 0.01 M LiBr) using calibrations with polystyrene standards. |
| [ANTH-polymer]167 |
167:1 |
180:1 |
48000 |
2.54 |
| [ANTH-polymer]83 |
83:1 |
90:1 |
66000 |
2.61 |
| [ANTH-polymer]56 |
56:1 |
84:1 |
53000 |
2.51 |
| [NAPH-polymer]167 |
167:1 |
169:1 |
65000 |
2.25 |
| [NAPH-polymer]83 |
83:1 |
83:1 |
76000 |
2.30 |
| [NAPH-polymer]56 |
56:1 |
56:1 |
80000 |
2.37 |
Under the same conditions used for the preparation of [ANTH-polymer]x and [NAPH-polymer]x, [ANTH-gel]x and [NAPH-gel]x were prepared by free-radical polymerizations (Scheme 4). In a preliminary experiment, the co-polymerization was examined for a monomer concentration of 1.0 M, using varying monomer feed ratios (x:y = 333:1, 167:1, and 83:1; Table 2). However, the preliminary experiment did not afford infusible and/or insoluble gels. Therefore, the monomer concentration (2.5 M) and the molar ratio of cross-linkers were increased (x:y = 167:1, 83:1 and 56:1). These changes successfully furnished PNIPAM gels ANTH-linker and NAPH-linker (see the Experimental section). The thus obtained PNIPAM gels were swollen in water and THF and their swelling ratio was consistent with the molar ratio of the cross-linkers (Table 2). Therefore, it can be concluded that ANTH-linker and NAPH-linker were successfully introduced as cross-linkers in the PNIPAM gels.
 |
| | Scheme 4 Co-polymerization of N-isopropylacrylamide with ANTH-linker and NAPH-linker. | |
Table 2 Experimental conditions for the preparation of the gels,a swelling ratios [(W′ − W)/W], as well as fluorescence quantum yields (Φfl) for the dried gels and those swollen in water and THF
| Entry |
Monomer [M] |
x:y |
Swelling ratio (W′ − W)/Wb |
Φfl (dried) |
Φfl (H2O)c |
Φfl (THF)d |
| 1.0 mol% AIBN, THF, T = 55 °C, t = 20 h. W: weight of the dry gel; W′: weight of the gel swollen in water. Immersed in water for 48 h. Immersed in THF for 48 h. |
| [ANTH-gel]333 |
1.0 |
333:1 |
No gelation (pale yellow powder) |
| [ANTH-gel]167 |
1.0 |
167:1 |
No gelation (pale yellow powder) |
| [ANTH-gel]83 |
1.0 |
83:1 |
No gelation (pale yellow powder) |
| [ANTH-gel]167 |
2.5 |
167:1 |
No gelation (yellow viscous liquid) |
| [ANTH-gel]83 |
2.5 |
83:1 |
14.2 |
0.24 |
0.22 |
0.07 |
| [ANTH-gel]56 |
2.5 |
56:1 |
3.57 |
0.21 |
0.20 |
0.05 |
| [NAPH-gel]167 |
2.5 |
167:1 |
17.1 |
0.52 |
0.53 |
0.33 |
| [NAPH-gel]83 |
2.5 |
83:1 |
4.43 |
0.46 |
0.47 |
0.28 |
| [NAPH-gel]56 |
2.5 |
56:1 |
2.87 |
0.49 |
0.53 |
0.28 |
Photophysical properties of [ANTH-polymer]x and [ANTH-gel]x
The absorption and fluorescence spectra, as well as the fluorescence quantum yields (Φfl) of ANTH-monomer and ANTH-linker were recorded in n-hexane, THF, acetonitrile, and methanol. A summary of the data is compiled in Fig. S28 and S29 and Table S1,† while data measured in THF are listed in Table 3 and Fig. 3. ANTH-monomer and ANTH-linker exhibited almost identical absorption and fluorescence spectra. These photophysical properties are comparable to those of BDAAs without functionalization at the alkyl chains.18 It can thus be concluded that the functionalization of alkyl chains of BDAAs does not affect their photophysical properties. Regardless of the monomer feed ratio x:y, [ANTH-polymer]x showed in both THF and aqueous solution identical absorption and fluorescence spectra as well as Φfl values (Table 3 and Fig. S30†). The spectra of [ANTH-polymer]x are similar to those of ANTH-monomer and ANTH-linker, except for the deviation of the absorption spectrum at λabs = ∼300 nm, which should be attributed to the absorption of PNIPAM.42 In aqueous solution, [ANTH-polymer]x exhibited a broader absorption band, a slight bathochromic shift of the fluorescence spectrum (Fig. 3), and enhanced Φfl (Table 2) than in THF solution. Although both THF and water represent good solvents for PNIPAM, the solvation mechanism for PNIPAM in water is substantially different from that in THF, and results in restricted motion of the side chains.43,44 Moreover, the hydrophobicity of the fluorophores45,46 and the relatively high viscosity47 of pure water most likely contribute to the enhanced Φfl. Similarly, the Φfl of [ANTH-gel]x increased upon swelling in water (0.20–0.24), while those of [ANTH-gel]x swollen in THF were comparable to those of ANTH-monomer and [ANTH-polymer]x in THF (Table 2). According to the swelling ratios, the concentration of PNIPAM and cross-linkers in swollen [ANTH-gel]x was much higher than in an aqueous solution of [ANTH-polymer]x (1 g L−1), which would explain the high Φfl observed for [ANTH-gel]x swollen in water.
Table 3 Photophysical parameters for ANTH-monomer, ANTH-linker, and [ANTH-polymer]x (x = 56, 83, 167)a
| Entry |
Solvent |
x:y |
λabs [nm] |
λflb [nm] |
Φflb |
| All measurements were conducted at T = 20 °C. Excitation wavelength corresponds to the maximum of the first absorption band λabs. |
| ANTH-monomer |
THF |
— |
400 |
527 |
0.05 |
| ANTH-linker |
THF |
— |
400 |
526 |
0.05 |
| [ANTH-polymer]x |
THF |
167:1 |
401 |
526 |
0.05 |
| THF |
83:1 |
401 |
525 |
0.05 |
| THF |
56:1 |
401 |
526 |
0.05 |
| H2O |
167:1 |
403 |
531 |
0.09 |
| H2O |
83:1 |
403 |
529 |
0.11 |
| H2O |
56:1 |
403 |
529 |
0.11 |
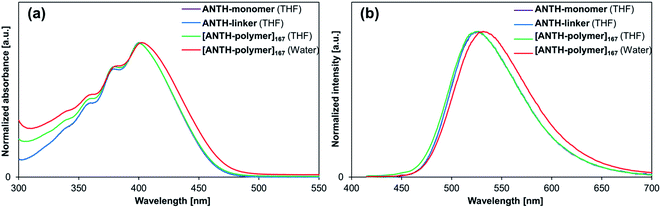 |
| | Fig. 3 (a) Absorption and (b) fluorescence spectra of ANTH-monomer (purple), ANTH-linker (blue), and [ANTH-polymer]167 in THF (green), as well as of [ANTH-polymer]167 in water (red). | |
[ANTH-polymer]167 exhibited the large enhancement of fluorescence intensity and Φfl at T = 27–35 °C (Fig. 4). Upon heating, the relative fluorescence intensity (I/I0) initially increased at T = 27–28 °C and peaked at T = 33–35 °C. Further heating led to a rapid decrease of I/I0 and at T = 50 °C, I/I0 fell below unity (T = 21 °C). A similar behavior of I/I0 was also observed for other monomer feed ratios x:y (Fig. S34†). This fluctuation of I/I0 coincided with the coil-to-globule transition of the PNIPAM chains;22 the transmittance of the [ANTH-polymer]x solutions began to decrease at T = 28 °C and finished at T = 40–45 °C (Fig. S32†). Peaking of I/I0, accompanied by a coil-to-globule transition of the polymer chains has been reported for some cases of fluorophore-labeled PNIPAM-based polymers27,48,49 and their microgels,50 although a decrease of fluorescence intensity at higher temperature is usually insignificant in diluted solution (≈0.01–0.02 g L−1).51,52 The Φfl values for [ANTH-polymer]x (Fig. 4b and S34†) showed a far less drastic decrease even at higher temperatures. Therefore, fast internal conversion or any other thermally induced deactivation processes should be insignificant for the decrease of I/I0. Given that the fluorescence intensity is a function of Φfl and the absorbance, the main reason for the decrease of I/I0 at higher temperatures should be associated with the less efficient absorption of the excitation light at T > 35 °C. This should be due to the aggregation of dehydrated [ANTH-polymer]x at higher temperature and a concomitantly high heterogeneity of the solution, through which the bulk of the excitation light could pass or be scattered from without absorption.
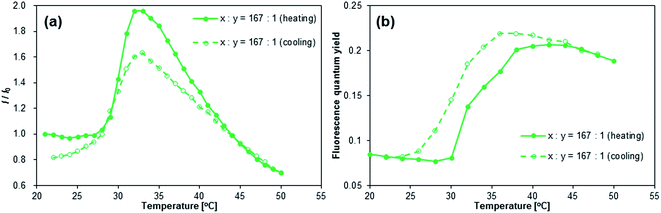 |
| | Fig. 4 Temperature dependence of (a) the fluorescence intensity (I) and (b) fluorescence quantum yields (Φfl) of [ANTH-polymer]167; I0 = fluorescence intensity at ∼530 nm; T = 21 °C, λex = 403 nm, and csolute = 1 g L−1. | |
Even though the peaking of I/I0 was also observed during the cooling, it was less pronounced than during the heating. In addition, Φfl exhibited a “hysteresis”-type behavior, i.e., the Φfl of [ANTH-polymer]x remained higher during the cooling than during the heating, but both values coincided eventually at T = 22 °C. In fact, the temperature-dependent transmittance (Fig. S32†) of [ANTH-polymer]167 during the cooling is almost identical to that during the heating. Therefore, the two hystereses observed during cooling should be attributed to microenvironments around the fluorophores rather than to abnormalities of the LCST behavior of [ANTH-polymer]x. As far as I/I0 is concerned, the heterogeneity of the optical density should be responsible for the observed hysteresis; immediately after hydration, the polymer chains should be in close spatial proximity with respect to each other, and consequently, absorption of the excitation lights should be inefficient. Higher Φfl values during the cooling should be ascribed to restrictions of the intramolecular motion of the fluorophores that persisted up to a late stage of hydration.
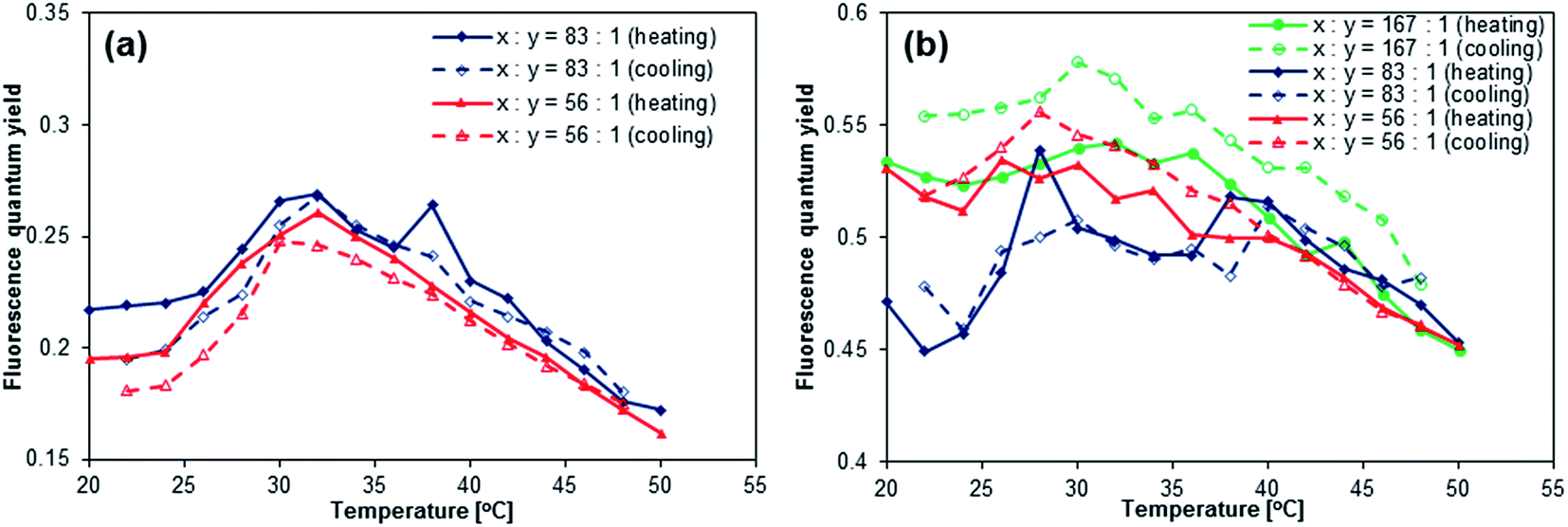
[ANTH-gel]x also exhibited an increase of Φfl at T = 25–32 °C (Fig. 5a). However, the increase of Φfl is less than half of that of [ANTH-polymer]x, which is due to the fact that [ANTH-gel]x is already fluorescent at T = 20 °C (Φfl = 0.20–0.22). Further heating (T > 32 °C) induced a significant decrease of Φfl, resulting in a peak of Φfl at T ≈ 32 °C. In the measurements of both [ANTH-gel]x and [NAPH-gel]x, experimental errors were inevitable due to the limited absorbance of these gel samples. At this stage, we are unable to unequivocally determine the cause of the decrease of Φfl at T > 32 °C, but the results nevertheless demonstrate that BDAA can be used to probe the coil-to-globule transition of the PNIPAM chains.
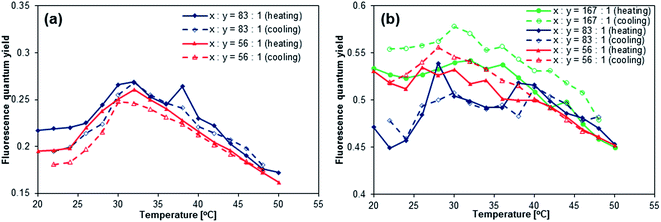 |
| | Fig. 5 Temperature dependence of the fluorescence quantum yields of (a) [ANTH-gel]83 (blue) and [ANTH-gel]56 (red) in H2O (λex = 403 nm) and (b) [NAPH-gel]167 (green), [NAPH-gel]83 (blue), and [NAPH-gel]56 (red) in H2O (λex = 323 nm). | |
Photophysical properties of [NAPH-polymer]x and [NAPH-gel]x
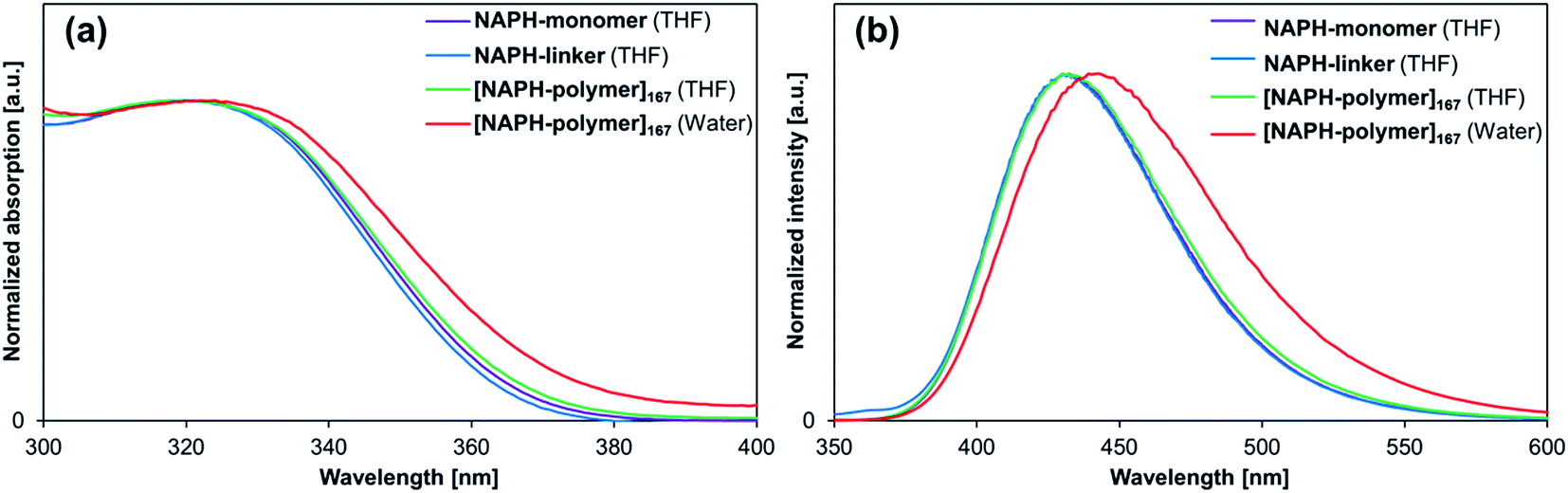
Absorption and fluorescence spectra, as well as Φfl values and fluorescence lifetimes were examined for NAPH-monomer and NAPH-linker in polar and nonpolar solvents (Table S1 and Fig. S31†). The photophysical parameters obtained from THF solutions are summarized in Table 4. Although the absorption and fluorescence spectra of NAPH-monomer and NAPH-linker were almost identical (Fig. 6), their Φfl values in THF were significantly higher (Φfl = 0.14–0.20) than that of DMe-BDAN (Φfl = 0.04).18 This difference may be attributed to the flexible alkyl chain of NAPH-monomer and NAPH-linker; our previous study suggested that flexible alkyl chains could induce planarity around the aryl-N bonds of N,N-dialkylaminoarenes, which may result in relatively large oscillator strengths for the S0 ← 1CT fluorescence transition.18 The co-polymerization or cross-linking with NIPAM resulted in a further increase of the Φfl for [NAPH-polymer]x in THF (Φfl = 0.32–0.34) and in H2O (Φfl = 0.54–0.59), as well as for [NAPH-gel]x in THF (Φfl = 0.28–0.33) and H2O (Φfl = 0.47–0.53). The relatively high Φfl value observed for [NAPH-polymer]x in THF coincides with the emergence of a long-lived excited-state species (τfl ≈ 6 ns), which indicates specific interactions between the PNIPAM chains and the fluorophore. However, considering the structural differences between NAPH-monomer and ANTH-monomer, i.e., shorter side chains, a smaller π-system, and the absence of aliphatic amino groups, it is difficult to determine precisely how the fluorophores in [NAPH-polymer]x interact with the PNIPAM chains in THF.
Table 4 Photophysical parameters of NAPH-monomer, NAPH-linker, and [NAPH-polymer]x (x = 56, 83, 167)a
| Entry |
Solvent |
x:y |
λabs [nm] |
λflb [nm] |
Φflb |
τflc [ns] |
| All measurements conducted at T = 20 °C. Excitation wavelength corresponds to the maximum of the first absorption band λabs. λex = 343 nm. |
| NAPH-monomer |
THF |
— |
320 |
432 |
0.20 |
2.94 |
| NAPH-linker |
THF |
— |
318 |
430 |
0.14 |
2.33 |
| [NAPH-polymer]x |
THF |
167:1 |
318 |
430 |
0.32 |
3.26 (0.25) |
| 5.80 (0.75) |
| THF |
83:1 |
320 |
430 |
0.32 |
3.83 (0.47) |
| 6.29 (0.53) |
| THF |
56:1 |
320 |
432 |
0.34 |
3.75 (0.42) |
| 6.21 (0.58) |
| H2O |
167:1 |
324 |
442 |
0.54 |
— |
| H2O |
83:1 |
322 |
439 |
0.58 |
— |
| H2O |
56:1 |
323 |
436 |
0.59 |
— |
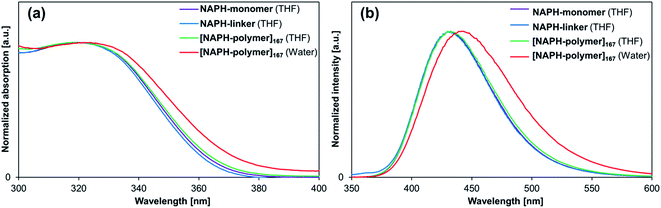 |
| | Fig. 6 (a) Absorption and (b) fluorescence spectra of NAPH-monomer (purple), NAPH-linker (blue), and [NAPH-polymer]167 (green) in THF, as well as of [NAPH-polymer]167 in H2O (red). | |
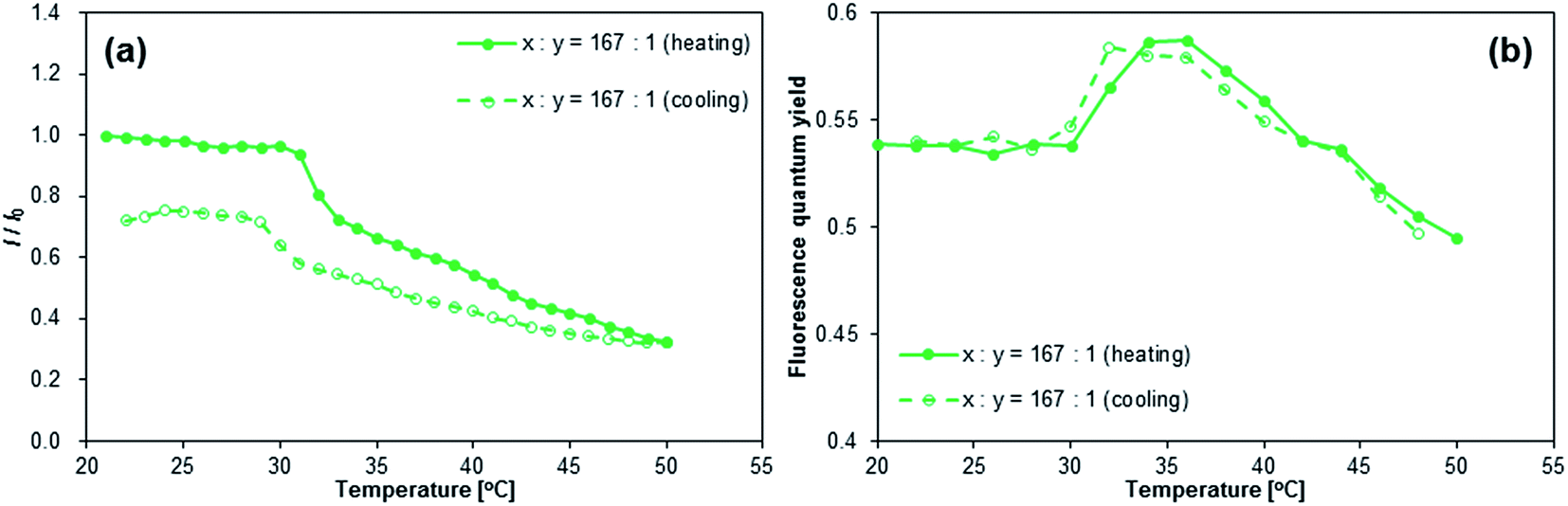
Given that the aqueous solutions of [NAPH-polymer]x are already fluorescent at T = 20 °C, the relative fluorescence intensity I/I0 declined during heating (Fig. 7a and S35†). Even though a small increase of Φfl at T = 30–35 °C was observed for [NAPH-polymer]167 (Fig. 7b), the Φfl of [NAPH-polymer]x (x = 56, 83) also decreased with increasing temperature (Fig. S35†). It should also be noted that the temperature-dependent transmittance of [NAPH-polymer]x, monitored at 650 nm, was largely affected by the incorporation of the fluorophore (Fig. S33†). For [NAPH-polymer]x, the decrease of transmittance occurred much slower and persisted up to higher temperatures than that of pristine PNIPAM.53 Especially [NAPH-polymer]x (x = 56 and 83) exhibited a steep drop of the transmittance, even at T > 40 °C. It has previously been reported that small amounts of hydrophobic moieties substantially affect the LCST behavior of the PNIPAM chains.45,48 Considering that the fluorophores in [NAPH-polymer]x is subject to specific interactions with the PNIPAM chains even in THF, it is reasonable to expect a severe perturbation of the LCST behavior of [NAPH-polymer]x. Similar to the Φfl of [NAPH-polymer]x, that of [NAPH-gel]x did not show any temperature dependence (Fig. 5b), i.e., although the Φfl of [NAPH-gel]x declined slightly with increasing temperature, the signal-to-noise ratio was too low to discern any reliable trends.
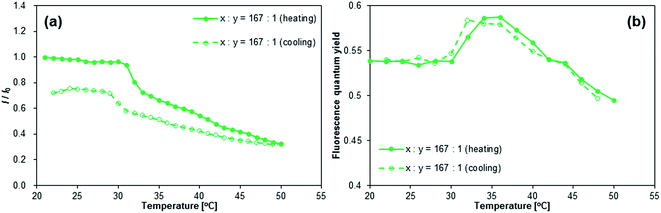 |
| | Fig. 7 Temperature dependence of (a) the fluorescence intensity (I) and (b) fluorescence quantum yields (Φfl) of [NAPH-polymer]167; I0 = fluorescence intensity at ∼440 nm; T = 21 °C, λex = 323 nm, and csolute = 1 g L−1. | |
Conclusions
In this study, we presented the synthesis of 9,10-bis((N-(3′-(N′′-(3′′-hydroxyprop-1′′-yl)-N′′-methylamino))prop-1′-yl)-N-methylamino)anthracene (6) and 1,4-bis(N-(hydroxypent-5-yl)-N-methylamino)-2,3-dimethylnaphthalene (14), which contain hydroxy groups at the terminal positions of their alkyl chains. While the former was obtained in 53% yield from a three-step protocol, the latter required four steps (yield: 65%). Due to the severe steric hindrance imposed by the peri-hydrogen and methyl groups at the 2- and 3-positions of 14, two rotamers with respect to the aryl-N bonds were observed. After endowing 6 and 14 with methacrylate moieties, ANTH-monomer, ANTH-linker, NAPH-monomer, and NAPH-linker were used for co-polymerizations with N-isopropylacrylamide to afford fluorophore-labeled PNIPAMs and the corresponding gels. While [ANTH-polymer]x exhibited low fluorescence, similar to ANTH-monomer and ANTH-linker, THF solutions of [NAPH-polymer]x exhibited stronger fluorescence (Φfl = 0.32–0.34) than those of NAPH-monomer and NAPH-linker (Φfl = 0.14–0.20); in aqueous solution, the fluorescence quantum yields of [NAPH-polymer]x increased even further (Φfl = 0.54–0.59). In a similar manner, [ANTH-gel]x was only weakly fluorescent in THF (Φfl = 0.05–0.07), while the fluorescence of [NAPH-gel]x was stronger in THF (Φfl = 0.28–0.33). The temperature-dependent fluorescence of [ANTH-polymer]x and [ANTH-gel]x revealed a steep increase of fluorescence intensity and Φfl at T = 27–35 °C, while [NAPH-polymer]x and [NAPH-gel]x did not exhibit an increase in fluorescence intensity upon heating. Thus, 9,10-bis(N,N-dialkylamino)anthracene (BDAA) is able to act as a useful probe for the microenvironment of PNIPAM chains In contrast, the sensitivity of 1,4-bis(N,N-dialkylamino)-2,3-dimethylnaphthalene (DMe-BDAN) toward the steric environment was hindered by interactions with the PNIPAM chains. Nevertheless, DMe-BDAN may be used to monitor other aspects of microenvironments, e.g. the local viscosity or molecular mobilities, upon introduction into suitable systems without such interactions.18 This study thus provides access to terminal-functionalized BDAA and DMe-BDAN, and these may be subjected to further modifications in order to facilitate their incorporation into larger and more complex molecular systems. Given their sensitivity and versatility, we expect these fluorophores to find a multitude of applications.
Experimental procedure
Variable-temperature photophysical studies
Unless otherwise noted, all measurements were conducted under ambient conditions and temperature (T = 293 K). For the photophysical measurements in THF, the solute concentration of [ANTH-polymer]x and [NAPH-polymer]x were adjusted so that the optical density (O.D.) at the maximum of the first absorption band was 0.1. For the variable-temperature photophysical measurements, aqueous solutions of [ANTH-polymer]x and [NAPH-polymer]x (csolute = 1 g L−1) were prepared and aliquots of 3.5 mL were transferred into quartz cells (1 cm). The samples of [ANTH-gel]x and [NAPH-gel]x used for the photophysical measurements were prepared by dipping ϕ 6 mm × 22 mm glass tubes in the co-polymerization reaction mixtures (for details, see the synthetic procedure), yielding cylindrical gels (ϕ 4 mm × 15 mm) after drying. These cylindrical gel samples were transferred into quartz cells (1 cm × 1 cm) and immersed in THF or water for 48 h. Immediately prior to the photophysical measurements, the swelling solvent was replaced with fresh solvent.
For variable-temperature fluorescence quantum yield measurements, the sample temperature was controlled with a water bath and an immersion heater. After the temperature of the water bath reached the target temperature, the quartz cells containing the samples were kept immersed in the water bath for 10 min. Then, their fluorescence quantum yields were measured (∼10 s) quickly right after retrieving the samples from the water bath. For the measurement of the fluorescence spectra, the temperature of the sample solutions was controlled by a UNISOKU CoolSpeK USP-203 unit, which was mounted within a JASCO FP-6500 spectrofluorometer. The stirred sample solutions were also incubated for 10 min at the respective target temperatures.
Synthesis and characterization
9,10-Bis(N-(3-chloropropyl)-N-methylamino)anthracene (3). A solution of potassium tert-butoxide (2.0 g, 21 mmol), PEPPSITM-IPr catalyst (142 mg, 0.21 mmol), N-methyl-3-chloropropylamine hydrochloride (2, 1.3 g, 9.2 mmol), and 9,10-dibromoanthracene (1, 1.4 g, 4.2 mmol) in dry and degassed 1,4-dioxane (70 mL) was stirred at 100 °C for 18 h. After cooling to room temperature, the reaction mixture was filtered and the filtrate was extracted with ethyl acetate/hexane (1:2, v/v). The combined organic layers were washed with water and brine, before being dried over magnesium sulfate. The solvent was removed in vacuo, and the residue was purified by column chromatography on silica gel using dichloromethane/hexane (1:4, v/v) to yield crude 3 (1.4 g) as a yellow solid. The crude product was used for the next step of the reaction without further purification. Yield: 87%; 1H NMR (300 MHz, CDCl3): δ 8.33 (dd, 4J = 3.3 Hz, 3J = 6.8 Hz, 4H), 7.46 (dd, 4J = 3.3 Hz, 3J = 6.8 Hz, 4H), 3.64 (t, 3J = 6.9 Hz, 4H), 3.60 (t, 3J = 6.5 Hz, 4H), 3.22 (s, 6H), 2.03 (quint, 3J = 6.8 Hz, 4H) ppm (Fig. S1†).
9,10-Bis(N-(3-bromopropyl)-N-methylamino)anthracene (4). The reaction apparatus was flame-dried in vacuo before being charged under an argon atmosphere with a mixture of 3 (3.5 g, 9.0 mmol) and sodium bromide (0.37 g, 3.6 mmol). Subsequently, 1-methyl-2-pyrrolidine (50 mL) and 1-bromoethane (13 mL) were deaerated by sparging with argon and added to this mixture, which was then stirred for 31 h at 65 °C, before more 1-bromoethane (6.7 mL) was added. The reaction mixture was stirred for further 16 h at the same temperature. After complete consumption of 3 (monitored by TLC), the reaction mixture was extracted with ethyl acetate and washed with water and brine. The combined organic layers were dried over magnesium sulfate before the solvent was removed in vacuo. The thus obtained residue was purified by column chromatography on silica gel using chloroform:hexane (1:3, v/v) to yield crude 4 (3.5 g) as a yellow solid. Yield: 81%; 1H NMR (300 MHz, CDCl3): δ 8.32 (dd, 4J = 3.3 Hz, 3J = 6.8 Hz, 4H), 7.46 (dd, 4J = 3.3 Hz, 3J = 6.8 Hz, 4H), 3.63 (t, 3J = 6.9 Hz, 4H), 3.45 (t, 3J = 6.7 Hz, 4H), 3.21 (s, 6H), 2.11 (quint, 3J = 6.8 Hz, 4H) ppm (Fig. S2†).
9,10-Bis((N-(3′-(N′′-(3′′-hydroxyprop-1′′-yl)-N′′-methylamino))prop-1′-yl)-N-methylamino)anthracene (6). A solution of 4 (0.50 g, 1.0 mmol), cesium carbonate (0.85 g, 2.6 mmol), and potassium iodide (0.17 g, 1.0 mmol) in deaerated acetonitrile was treated dropwise with 3-(N-methylamino)-1-propanol (5, 0.50 mL, 5.2 mmol). The reaction mixture was heated to reflux for 12 h under an atmosphere of argon. After cooling to room temperature, the reaction mixture was extracted with chloroform and the combined organic layers were washed with water and brine, before being dried over magnesium sulfate. Then, all volatiles were removed in vacuo. The residue was purified by column chromatography on silica gel using (i) hexane:chloroform (1:1, v/v), (ii) ethyl acetate, and (iii) ethyl acetate:methanol (5:1, v/v) with 5% triethylamine (v/v) to yield 6 (0.39 g) as a yellow solid. Yield: 76%; mp: 123.6–124.5 °C; 1H NMR (500 MHz, CDCl3): δ 8.34 (dd, 4J = 3.3 Hz, 3J = 6.8 Hz, 4H), 7.44 (dd, 4J = 3.3 Hz, 3J = 6.8 Hz, 4H), 3.71 (m, 4H), 3.49 (t, 4H, 3J = 7.1 Hz), 3.48 (s, 2H), 3.20 (s, 6H), 2.50 (t, 4H, 3J = 5.8 Hz), 2.40 (t, 4H, 3J = 7.4 Hz), 2.17 (s, 6H), 1.77 (quint, 4H, 3J = 7.1 Hz), 1.63–1.60 (m, 4H) ppm (Fig. S3†); 13C NMR (125 MHz, CDCl3): δ 143.3 (Ar), 131.4 (Ar), 125.6 (Ar), 124.6 (Ar), 64.6, 58.6, 56.2, 55.7, 50.8, 43.2, 41.9, 27.6 ppm (Fig. S4†); FT-IR (KBr): 3150 (OH), 1618 (Ar ring stretch), 1475 (Ar ring stretch), 1385 (Ar-N stretch), 776 (Ar-H) cm−1. The structure of 6 was also supported by a NOESY spectrum (Fig. S15–S18†).
Methacrylate-monofunctionalized 9,10-bis((N-(3′-(N′′-(3′′-hydroxyprop-1′′-yl)-N′′-methylamino))prop-1′-yl)-N-methylamino)anthracene (ANTH-monomer). The reaction apparatus was flame-dried in vacuo before being charged with a solution of 6 (1.1 g, 2.1 mmol) and dry triethylamine (3.0 mL, 21 mmol) in deaerated and dry chloroform (30 mL). Methacryloyl chloride (0.23 mL, 2.4 mmol) was added to this solution at 0 °C, and the mixture was stirred under an argon atmosphere for 10 min before being allowed to warm to room temperature, where stirring was continued for 5 h. The reaction mixture was consecutively washed with aqueous sodium hydroxide (1 M, 100 mL), water, and brine. The combined organic layers were dried over magnesium sulfate, before all volatiles were removed in vacuo. The residue was purified by column chromatography on silica gel using (i) ethyl acetate and (ii) ethyl acetate:methanol (5:1, v/v) with 5% triethylamine (v/v), which afforded crude ANTH-monomer as a yellow liquid. Further purification was achieved by high-performance liquid chromatography (HPLC), which furnished pure ANTH-monomer (0.32 g). Yield: 26%; 1H NMR (300 MHz, CDCl3): δ 8.35 (dd, 4J = 1.7 Hz, 3J = 4.2 Hz, 2H), 8.33 (dd, 4J = 1.7 Hz, 3J = 4.2 Hz, 2H), 7.46–7.40 (m, 4H), 6.06 (dq, 4J = 0.9 Hz, 3J = 1.7 Hz, 1H), 5.51 (dq, 4J = 1.5 Hz, 3J = 1.7 Hz, 1H), 4.98 (brs, 1H), 4.12 (t, 3J = 6.5 Hz, 2H), 3.72 (t, 3J = 5.3 Hz, 2H), 3.49 (t, 3J = 7.2 Hz, 4H), 3.20 (s, 6H), 2.49 (t, 3J = 5.7 Hz, 2H), 2.38 (t, 3J = 8.2 Hz, 2H), 2.36 (t, 3J = 7.9 Hz, 4H), 2.17 (s, 3H), 2.14 (s, 3H), 1.92 (dd, 4J = 0.9 Hz, 4J = 1.5 Hz, 3H), 1.79–1.70 (m, 6H), 1.66–1.60 (m, 2H) ppm (Fig. S6†); 13C NMR (75 MHz, CDCl3): δ 167.3 (CO), 143.4 (Ar), 143.2 (Ar), 136.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 131.4 (Ar), 131.4 (Ar), 125.6 (Ar), 125.5 (Ar), 125.1 (CCH2), 124.6 (Ar), 124.5 (Ar), 64.5, 63.1, 58.4, 56.2, 55.9, 55.8, 55.7, 54.3, 43.1, 43.1, 42.2, 41.9, 27.9, 27.7, 26.6, 18.2 ppm (Fig. S7†); FT-IR (neat): 2951, 1718 (CO ester stretch), 1637 (Ar ring stretch), 1392 (Ar-N stretch), 779 (Ar-H) cm−1; HRMS (FAB+) exact mass calculated for [M + H]+ (C34H51N4O3): m/z = 563.3961, found: m/z = 563.3965.
CH2), 131.4 (Ar), 131.4 (Ar), 125.6 (Ar), 125.5 (Ar), 125.1 (CCH2), 124.6 (Ar), 124.5 (Ar), 64.5, 63.1, 58.4, 56.2, 55.9, 55.8, 55.7, 54.3, 43.1, 43.1, 42.2, 41.9, 27.9, 27.7, 26.6, 18.2 ppm (Fig. S7†); FT-IR (neat): 2951, 1718 (CO ester stretch), 1637 (Ar ring stretch), 1392 (Ar-N stretch), 779 (Ar-H) cm−1; HRMS (FAB+) exact mass calculated for [M + H]+ (C34H51N4O3): m/z = 563.3961, found: m/z = 563.3965.
Methacrylate-difunctionalized 9,10-bis((N-(3′-(N′′-(3′′-hydroxyprop-1′′-yl)-N′′-methylamino))prop-1′-yl)-N-methylamino)anthracene (ANTH-linker). The reaction apparatus was flame-dried in vacuo before being charged with a solution of 6 (0.50 g, 1.0 mmol) and dry trimethylamine (1.4 mL, 10 mmol) in deaerated and dry chloroform (30 mL). Under an argon atmosphere, methacryloyl chloride (0.29 mL, 3.0 mmol) was added at 0 °C, and the reaction mixture was stirred at this temperature for 10 min before being allowed to warm to room temperature, where stirring was continued for 5 h. After the complete consumption of 6 (monitored by TLC), the reaction mixture was consecutively washed with aqueous potassium hydroxide (1 M, 100 mL), water, and brine. The combined organic layers were dried over magnesium sulfate, before the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel using ethyl acetate followed by ethyl acetate:methanol (5:1, v/v) to afford ANTH-linker (0.48 g) as a yellow liquid. Yield: 75%; 1H NMR (300 MHz, CDCl3): δ 8.34 (dd, 4J = 3.2 Hz, 3J = 6.8 Hz, 4H), 7.43 (dd, 4J = 3.2 Hz, 3J = 6.8 Hz, 4H), 6.07 (s, 2H), 5.53 (s, 2H), 4.12 (t, 3J = 6.0 Hz, 4H), 3.49 (t, 3J = 7.1 Hz, 4H), 3.19 (s, 6H), 2.42–2.37 (m, 8H), 2.18 (s, 6H), 1.92 (s, 6H), 1.79–1.75 (m, 8H) ppm (Fig. S8†); 13C NMR (75 MHz, CDCl3): δ 167.3 (CO), 143.3 (Ar), 136.3 (CCH2), 131.3 (Ar), 125.5 (Ar), 125.3 (CCH2), 124.6 (Ar), 62.9, 55.8, 55.5, 54.1, 43.0, 41.9, 27.5, 26.2, 18.3 ppm (Fig. S9†); FT-IR (neat): 2950, 1717 (CO ester stretch), 1638 (Ar ring stretch), 1392 (Ar-N stretch), 779 (Ar-H) cm−1; HRMS (FAB+) exact mass calculated for [M + H]+ (C38H55N4O4): m/z = 631.4223, found: m/z = 631.4210.
1,4-Dibromo-2,3-dimethylnaphthalene (9)38. A stirred solution of 2,3-dimethylnaphthalene (1.0 g, 6.4 mmol) in chloroform (10 mL) at 0 °C was gradually treated with a solution of bromine (0.69 mL, 13 mmol) in chloroform (6 mL). The reaction mixture was stirred for 10 min at 0 °C, before being allowed to warm to room temperature. The reaction mixture was set aside for 3 h before being quenched by addition of sodium thiosulfate monohydrate (5.5 g, 19 mmol). The resultant suspension was extracted with chloroform and the combined organic layers were washed with water and brine. Then, the organic fraction was dried over magnesium sulfate and the solvent was removed in vacuo to yield crude 9 (2.0 g) as a white solid. Yield: ≥99%; 1H NMR (300 MHz, CDCl3): δ 8.30 (dd, 4J = 3.4 Hz, 3J = 6.3 Hz, 2H), 7.54 (dd, 4J = 3.4 Hz, 3J = 6.3 Hz, 2H), 2.69 (s, 6H) ppm (Fig. S10†).
5-(tert-Butyldiphenylsiloxy)pentan-1-amine (11). At 0 °C, a stirred solution of 5-amino-1-pentanol (10; 2.9 g, 28 mmol) and imidazole (2.8 g, 41 mmol) in dichloromethane (50 mL) was treated dropwise with tert-butyldiphenylchlorosilane (8.5 mL, 33 mmol). After the exothermic stopped, the reaction temperature was allowed to raise to room temperature, where stirring was continued for 5 h, before the temperature was raised to 40 °C, were stirring was continued for 1 h. The reaction was quenched with water and washed with brine. The combined organic layers were dried over magnesium sulfate and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel consecutively using ethyl acetate:hexane (1:1, v/v) and ethyl acetate:methanol (5:1, v/v) with 5% triethylamine to afford crude 11 (6.8 g) as a colorless liquid. Yield: 72%; 1H NMR (300 MHz, CDCl3): δ 7.66 (dd, 4J = 1.9 Hz, 3J = 7.4 Hz, 4H), 7.40–7.33 (m, 6H), 4.29 (brs, 2H), 3.65 (t, 3J = 6.3 Hz, 2H), 2.75 (t, 3J = 7.2 Hz, 2H), 1.59–1.49 (m, 4H), 1.41–1.36 (m, 2H), 1.05 (s, 9H) ppm (Fig. S11†).
1,4-Bis(N-(5-(tert-butyldiphenylsiloxy)-pent-1-yl)amino)-2,3-dimethylnaphthalene (12). A solution of sodium tert-butoxide (1.5 g, 16 mmol), PEPPSITM-IPr catalyst (0.13 g, 0.19 mmol), 5-(tert-butyldiphenylsiloxy)pentan-1-amine (11; 2.7 g, 8.0 mmol), and 1,4-dibromo-2,3-dimethylnaphthalene (9; 1.0 g, 3.2 mmol) in deaerated and dry 1,4-dioxane (100 mL) was stirred for 15 min under argon at room temperature. The resulting mixture was heated to reflux for 72 h. After cooling to room temperature, the reaction mixture was extracted with ethyl acetate and the combined organic layers were washed with water and brine. Then, the organic layer was dried over magnesium sulfate and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel using hexane:ethyl acetate (10:1, v/v) to afford crude 12 (2.1 g) as a pale yellow liquid. Yield: 80%; 1H NMR (300 MHz, CDCl3): δ 8.03 (dd, 4J = 3.4 Hz, 3J = 6.4 Hz, 2H, ArH), 7.72–7.66 (m, 8H, ArH), 7.40–7.31 (m, 14H, ArH), 3.69 (t, 3J = 6.2 Hz, 4H, CH2), 3.10 (brs, 2H, NH), 2.98 (t, 3J = 7.0 Hz, 4H, CH2), 2.36 (s, 6H, ArCH3), 1.67–1.61 (m, 8H, CH2), 1.54–1.52 (m, 4H, CH2), 1.06 (s, 18H, C(CH3)3) ppm (Fig. S12†).
1,4-Bis(N-(5-(tert-butyldiphenylsiloxy)-pent-1-yl)-N-methylamino)-2,3-dimethylnaphthalene (13). A solution of 12 (0.96 g, 1.2 mmol) and sodium carbonate (0.31 g, 2.9 mmol) in deaerated acetonitrile (30 mL) was treated at room temperature with methyl iodide (0.22 mL, 3.5 mmol). The reaction mixture was warmed to 60 °C and stirred for 12 h under argon. Then, more methyl iodide (0.22 mL, 3.5 mmol) was added and the reaction mixture was stirred for 4 h at the same temperature. After the complete consumption of 12 (monitored by TLC), the reaction mixture was extracted with ethyl acetate and the combined organic layers were washed with water and brine. Then, the organic layer was dried over magnesium sulfate and the solvent was removed in vacuo. The thus obtained residue was purified by column chromatography on silica gel using hexane:ethyl acetate (20:1, v/v) to afford crude 13 (0.84 g) as a colorless liquid. Crude 13 was used in the next reaction step without further purification. Yield: 85%; 1H NMR (300 MHz, CDCl3): δ 8.13 (dd, 4J = 3.3 Hz, 3J = 6.1 Hz, 2H), 7.66 (dd, 4J = 2.0 Hz, 3J = 7.6 Hz, 8H), 7.37–7.31 (m, 14H), 3.64 (t, 3J = 6.4 Hz, 4H), 3.18–3.13 (m, 4H), 2.93 (s, 6H), 2.32 (s, 6H), 1.58–1.53 (m, 8H), 1.42–1.38 (m, 4H), 1.04 (s, 18H) ppm (Fig. S13†).
1,4-Bis(N-(hydroxypent-5-yl)-N-methylamino)-2,3-dimethylnaphthalene (14). A solution of TBDPS-protected alcohol 13 (1.4 g, 1.6 mmol) in dry THF (30 mL) was treated with a THF solution of tetrabutylammonium fluoride (6.4 mL, 1 M, 6.4 mmol). The reaction mixture was stirred for 21 h under argon at room temperature. After the complete consumption of 13 (monitored by TLC), the reaction mixture was extracted with ethyl acetate and the combined organic layers were washed with water and brine. Then, the organic layer was dried over magnesium sulfate and the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel using hexane:ethyl acetate (1:1, v/v) to afford crude 14 (0.59 g) as a colorless liquid. For further purification, column chromatography on silica gel was repeated. Yield: 95%; 1H NMR (500 MHz, CDCl3): δ 8.13 and 8.13 (dd, 4J = 3.3 Hz, 3J = 6.4 Hz, 2H, ArH of different rotamers), 7.37 (dd, 4J = 3.3 Hz, 3J = 6.4 Hz, 2H, ArH), 3.59 and 3.56 (t, 3J = 6.6 Hz, 4H, CH2 of different rotamers), 3.18 and 3.17 (t, 3J = 6.9 Hz, 4H, CH2 of different rotamers), 2.95 and 2.94 (s, 6H, NCH3 of different rotamers), 2.33 and 2.33 (s, 6H, ArCH3 of different rotamers), 1.66 (brs, 2H, OH), 1.59–1.49 (m, 8H, CH2), 1.39–1.33 (m, 4H, CH2) ppm (Fig. S14†); 13C NMR (125 MHz, CDCl3): δ 143.8 and 143.7 (Ar of different rotamers), 135.0 and 134.9 (Ar of different rotamers), 132.4 (Ar), 124.8 (Ar), 124.2 (Ar), 62.8 and 62.7 (CH2 of different rotamers), 56.7 and 56.6 (CH2 of different rotamers), 41.6 and 41.5 (NCH3 of different rotamers), 32.6 and 32.5 (CH2 of different rotamers), 29.4 and 29.4 (CH2 of different rotamers), 23.4 and 23.3 (CH2 of different rotamers), 16.2 and 16.2 (ArCH3 of different rotamers) ppm (Fig. S15†); HRMS (FAB+) exact mass calculated for [M]+ (C24H38N2O2): m/z = 386.2933, found: m/z = 386.2937. These 1H and 13C NMR signals were assigned on the basis of DEPT-135, 1H–1H COSY, 1H–13C HSQC, and NOESY spectra (Fig. S5†).
Methacrylate-monofunctionalized 1,4-bis(N-(hydroxypent-5-yl)-N-methylamino)-2,3-dimethylnaphthalene (NAPH-monomer). The reaction apparatus was flame-dried in vacuo before a stirred solution of 14 (0.50 g, 1.3 mmol) and dry triethylamine (1.8 mL, 13 mmol) in deaerated and dry chloroform (18 mL) was treated at 0 °C with methacryloyl chloride (0.14 mL, 1.4 mmol). Under an argon atmosphere, the reaction mixture was stirred at this temperature for 10 min, before being allowed to warm to room temperature, where stirring was continued for 3 h. After the complete consumption of 14 (monitored by TLC), the reaction mixture was washed consecutively with aqueous potassium hydroxide (1 M, 100 mL), water, and brine. The combined organic layers were dried over magnesium sulfate before the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel using ethyl acetate:hexane (1:1, v/v) to afford NAPH-monomer (0.25 g) as a colorless liquid. The NAPH-monomer was thoroughly purified by column chromatography on silica gel using chloroform as the eluent immediately prior to any photophysical measurements and polymerization reactions. Yield: 43%; 1H NMR (500 MHz, CDCl3): δ 8.13 and 8.12 (dd, 4J = 3.3 Hz, 3J = 6.5 Hz, 2H, ArH of different rotamers), 7.37 (dd, 4J = 3.3 Hz, 3J = 6.5 Hz, 2H, ArH), 6.06 (s, 1H, CCH2), 5.52 (s, 1H, CCH2), 4.11 and 4.10 (t, 3J = 6.5 Hz, 2H, CH2 of different rotamers), 3.57 (t, 3J = 6.5 Hz, 2H, CH2), 3.22–3.15 (m, 4H, CH2 of different rotamers), 2.94 and 2.93 (s, 6H, NCH3 of different rotamers), 2.33 and 2.33 (s, 6H, ArCH3 of different rotamers), 1.91 (s, 3H, COC(CH3)), 1.66–1.49 (m, 8H, CH2 of different rotamers), 1.41–1.34 (m, 4H, CH2 of different rotamers) ppm (Fig. S19†); 13C NMR (125 MHz, CDCl3): δ 167.5 (CO), 143.9 and 143.9 (Ar of different rotamers), 143.8 (Ar), 143.8 (Ar), 136.4 (CCH2), 135.0 and 135.0 (Ar of different isomers), 135.0 (Ar), 132.4 (Ar), 132.4 and 132.4 (Ar of different rotamers), 125.2 (CCH2), 124.8 (Ar), 124.7 (Ar), 124.2 and 124.2 (Ar of different rotamers), 64.8, 62.8, 56.7, 56.6, 41.5, 41.5, 32.6, 29.5, 29.4, 28.5, 23.8, 23.4, 18.3, 16.3, 16.2 ppm (Fig. S20†); FT-IR (neat): 2933, 1718 (CO ester stretch), 1637 (Ar ring stretch), 1577, 1387 (Ar-N stretch), 756 (Ar-H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C28H42N2O3): m/z = 454.3195, found: m/z = 454.3195.
Methacrylate-difunctionalized 1,4-bis(N-(hydroxypent-5-yl)-N-methylamino)-2,3-dimethylnaphthalene (NAPH-linker). The reaction apparatus was flame-dried in vacuo before a stirred solution of 14 (0.31 g, 0.81 mmol) and dry triethylamine (1.1 mL, 7.8 mmol) in deaerated and dry chloroform (11 mL) was treated at 0 °C with methacryloyl chloride (0.24 mL, 2.4 mmol). Under an argon atmosphere, the reaction mixture was stirred at this temperature for 10 min, before being allowed to warm to room temperature, where stirring was continued for 3 h. After complete consumption of 14 (monitored by TLC), the reaction mixture was consecutively washed with aqueous potassium hydroxide (1 M, 100 mL), water, and brine. The combined organic layers were dried over magnesium sulfate before the solvent was removed in vacuo. The thus obtained residue was purified by column chromatography on silica gel using ethyl acetate:hexane (1:4, v/v) to afford NAPH-linker (0.42 g) as a colorless liquid. Immediately prior to any photophysical measurements and polymerization reactions, the NAPH-linker was thoroughly purified by column chromatography on silica gel using chloroform as the eluent. Yield: 99%; 1H NMR (300 MHz, CDCl3): δ 8.12 (dd, 4J = 3.3 Hz, 3J = 6.5 Hz, 2H, ArH), 7.37 (dd, 4J = 3.3 Hz, 3J = 6.5 Hz, 2H, ArH), 6.06 (s, 2H, CCH2), 5.51 (s, 2H, CCH2), 4.11 (t, 3J = 6.6 Hz, 4H, CH2), 3.20 and 3.18 (two triplets from different isomers, 3J = 7.0 Hz, 4H, CH2), 2.94 (s, 6H, NCH3), 2.33 (s, 6H, ArCH3), 1.92 (s, 6H, COC(CH3)), 1.66 (quint, 3J = 6.9 Hz, 4H, CH2), 1.59 (quint, 3J = 7.3 Hz, 4H, CH2), 1.45–1.36 (m, 4H, CH2) ppm (Fig. S21†); 13C NMR (75 MHz, CDCl3): δ 167.3 (CO), 144.7 (Ar), 137.5 (CCH2), 135.7 and 135.7 (Ar from different isomers), 133.3 (Ar), 125.5 (CCH2), 125.3 (Ar), 125.1 (Ar), 65.1, 57.4, 41.8, 30.6, 30.2, 24.5, 18.4, 16.5 and 16.4 (from different isomers) ppm (Fig. S22†); FT-IR (neat): 2936, 1717 (CO ester stretch), 1638 (Ar ring stretch), 1387 (Ar-N stretch), 757 (Ar-H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C32H46N2O4): m/z = 522.3458, found: m/z = 522.3460.
Representative co-polymerization procedure
A 10 mL Schlenk tube with a stopcock in the sidearm was flame-dried in vacuo before being charged with N-isopropyl acrylamide (NIPAM) (1.13 g, 10.0 mmol), a solution of NAPH-monomer (5.0 × 10−2 M, 3.6 mL for a monomer feed ratio x:y = 56:1, 2.4 mL for x:y = 83:1, and 1.2 mL for x:y = 167:1), and 2,2′-azobisisobutyronitrile (AIBN, 16 mg, 0.10 mmol). Then, dry, deaerated THF was added under an argon atmosphere to reach a total volume of 5 mL for the reaction mixture. Subsequently, the reaction mixture was subjected to thorough argon sparging until the total volume of the reaction mixture decreased to 4 mL. The reaction mixture was kept for 20 h at 55 °C, before being transferred into diethyl ether at 0 °C. The thus obtained precipitate was collected by filtration and dissolved again in THF. This reprecipitation procedure was repeated three times to afford [ANTH-polymer]x and [NAPH-polymer]x as yellow and colorless solids, respectively. Yields for [ANTH-polymer]x: 75% (0.85 g) for x:y = 56:1, 61% (0.69 g) for x:y = 83:1, 84% (0.95 g) for x:y = 167:1; yields for [NAPH-polymer]x: 91% (1.0 g) for x:y = 56:1, 96% (1.1 g) for x:y = 83:1, 95% (1.1 g) for x:y = 167:1; 1H NMR (500 MHz, CDCl3): δ 6.80–6.20 (br, 1H, CONH), 4.00 (m, 1H, CH(CH3)2), 2.14 (m, 1H, CHCH2), 1.86–1.35 (m, 2H, CHCH2), 1.14 (m, 6H, CH(CH3)2) ppm.41 For a detailed assignment, see Fig. S23 and S24.†
Representative procedure for the preparation of gels
A 10 mL Schlenk tube with a stopcock in the sidearm was flame-dried in vacuo before N-isopropyl acrylamide (NIPAM, 1.13 g, 10.0 mmol), a THF solution of ANTH-linker (5.0 × 10−2 M, 3.6 mL for a monomer feed ratio x:y = 56:1, 2.4 mL for x:y = 83:1, 1.2 mL for x:y = 167:1), and 2,2′-azobisisobutyronitrile (AIBN, 16 mg, 0.10 mmol) were added. Dry, deaerated THF was added under an argon atmosphere to reach a total volume of 5 mL for the reaction mixture. Then, the reaction mixture was subjected to thorough argon sparging until the total volume of the reaction mixture decreased to 4 mL. The reaction mixture was kept at 55 °C for 20 h, before being transferred to THF (150 mL) in order to confirm the formation of a gel. The thus prepared PNIPAM gel was purified by Soxhlet extraction using THF as eluents to afford yellow ([ANTH-gel]x) or colorless ([NAPH-gel]x) gels.
References
- M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669–1678 CAS.
- N. Amdursky, Y. Erez and D. Huppert, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef CAS PubMed.
- L. Zhu and Y. Zhao, J. Mater. Chem. C, 2013, 1, 1059–1065 RSC.
- T. Liu, X. Liu, D. R. Spring, X. Qian, J. Cui and Z. Xu, Sci. Rep., 2014, 4, 5418 CAS.
-
(a) M. K. Kuimova, G. Yahioglu, J. A. Levit and K. Suhling, J. Am. Chem. Soc., 2008, 130, 6672–6673 CrossRef CAS PubMed;
(b) M. K. Kuimova, S. W. Botchway, A. W. Parker, M. Balaz, H. A. Collins, H. L. Anderson, K. Suhling and P. R. Ogilby, Nat. Chem., 2009, 1, 69–73 CrossRef CAS PubMed;
(c) V. I. Stsiapura, A. A. Maskevich, V. A. Kuzmitsky, V. N. Uversky, I. M. Kuznetsova and K. K. Turoverov, J. Phys. Chem. B, 2008, 112, 15893–15902 CrossRef CAS PubMed;
(d) M. Nishikawa, S. Kume and H. Nishihara, Phys. Chem. Chem. Phys., 2013, 15, 10549–10565 RSC;
(e) G. Vaccaro, A. Bianchi, M. Mauri, S. Bonetti, F. Meinardi, A. Sanguineti, R. Simonutti and L. Beverina, Chem. Commun., 2013, 49, 8474–8476 RSC;
(f) I. A. Karpenko, Y. Niko, V. P. Yakubovskyi, A. O. Gerasov, D. Bonnet, Y. P. Kovtun and A. S. Klymchenko, J. Mater. Chem. C, 2016, 4, 3002–3009 RSC;
(g) A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
-
(a) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed;
(b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed;
(c) R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC;
(d) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC;
(e) S. Sasaki, G. P. C. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC;
(f) Y. Niko and G. Konishi, J. Synth. Org. Chem., Jpn., 2012, 70, 918–927 CrossRef CAS.
-
(a) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed;
(b) C. Botta, S. Benedini, L. Carlucci, A. Forni, D. Marinotto, A. Nitti, D. Pasini, S. Righetto and E. Cariati, J. Mater. Chem. C, 2016, 4, 2979–2989 RSC;
(c) H. Zhou, Q. Ye, X. Y. Wu, J. Song, C. M. Cho, Y. Zong, B. Z. Tang, T. S. A. Hor, E. K. L. Yeow and J. W. Xu, J. Mater. Chem. C, 2015, 3, 11874–11880 RSC;
(d) R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Chem.–Eur. J., 2015, 21, 7231–7237 CrossRef CAS PubMed;
(e) H. Imoto, K. Nohmi, K. Kizaki, S. Watase, K. Matsukawa, S. Yamamoto, M. Mitsuishi and K. Naka, RSC Adv., 2015, 5, 94344–94350 RSC;
(f) H. Imoto, K. Kizaki, S. Watase, K. Matsukawa and K. Naka, Chem.–Eur. J., 2015, 21, 12105–12111 CrossRef CAS PubMed;
(g) J. Li, W. Yang, W. Q. Zhou, C. C. Li, Z. Q. Cheng, M. Y. Li, L. Q. Xie and Y. Y. Li, RSC Adv., 2016, 6, 35833–35841 RSC;
(h) Q. K. Sun, W. Liu, S. A. Ying, L. L. Wang, S. F. Xue and W. J. Yang, RSC Adv., 2015, 5, 73046–73050 RSC;
(i) T. Jiang, Y. Qu, B. Li, Y. T. Gao and J. L. Hua, RSC Adv., 2015, 5, 1500–1506 RSC;
(j) T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, RSC Adv., 2015, 5, 29878–29884 RSC.
-
(a) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC;
(b) J. Yang, N. Sun, J. Huang, Q. Q. Li, Q. Peng, X. Tang, Y. Q. Dong, D. G. Ma and Z. Li, J. Mater. Chem. C, 2015, 3, 2624–2631 RSC;
(c) Y. Okazawa, K. Kondo, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2015, 137, 98–101 CrossRef CAS PubMed;
(d) R. Y. Zhang, R. T. K. Kwok, B. Z. Tang and B. Liu, RSC Adv., 2015, 5, 28332–28337 RSC;
(e) D. Belei, C. Dumea, E. Bicu and L. Marin, RSC Adv., 2015, 5, 8849–8858 RSC;
(f) Z. F. Huang, X. Q. Zhang, X. Y. Zhang, C. K. Fu, K. Wang, J. Y. Yuan, L. Tao and Y. Wei, Polym. Chem., 2015, 6, 607–612 RSC;
(g) Z. Chen, X. Han, J. Zhang, D. Wu, G. A. Yu, J. Yin and S. H. Liu, RSC Adv., 2015, 5, 15341–15349 RSC;
(h) Q. Wan, K. Wang, H. L. Du, H. Y. Huang, M. Y. Liu, F. J. Deng, Y. F. Dai, X. Y. Zhang and Y. Wei, Polym. Chem., 2015, 6, 5288–5294 RSC;
(i) H. Yamane, S. Ito, K. Tanaka and Y. Chujo, Polym. Chem., 2016, 7, 2799–2807 RSC;
(j) A. Pandith, A. Kumar and H. S. Kim, RSC Adv., 2016, 6, 68627–68637 RSC;
(k) S. Sasaki, Y. Niko, K. Igawa and G. Konishi, RSC Adv., 2014, 4, 33474–33477 RSC.
-
(a) C. E. Kung and J. K. Reed, Biochemistry, 1989, 28, 6678–6686 CrossRef CAS PubMed;
(b) S. Sawada, T. Iio, Y. Hayashi and S. Takahashi, Anal. Biochem., 1992, 204, 110–117 CrossRef CAS PubMed.
-
(a) W. L. Goh, M. Y. Lee, T. L. Joseph, S. T. Quah, C. J. Brown, C. Verma, S. Brenner, F. J. Ghadessy and Y. N. Teo, J. Am. Chem. Soc., 2014, 136, 6159–6162 CrossRef CAS PubMed;
(b) M. A. Haidekker, T. Brady, K. Wen, C. Okada, H. Y. Stevens, J. M. Snell, J. A. Frangos and E. A. Theodorakis, Bioorg. Med. Chem., 2002, 10, 3627–3636 CrossRef CAS PubMed;
(c) T. Iwaki, C. Torigoe, M. Noji and M. Nakanishi, Biochemistry, 1993, 32, 7583–7592 CrossRef;
(d) K. Cao, M. Farahi, M. Dakanali, W. M. Chang, C. J. Sigurdson, E. A. Theodorakis and J. Yang, J. Am. Chem. Soc., 2012, 134, 17338–17341 CrossRef CAS PubMed.
-
(a) R. O. Loutfy, Pure Appl. Chem., 1986, 58, 1239–1248 CrossRef CAS;
(b) S. K. Dishari and M. A. Hickner, Macromolecules, 2013, 46, 413–421 CrossRef CAS;
(c) R. D. Priestley, P. Rittigstein, L. J. Broadbelt, K. Fukao and J. M. Torkelson, J. Phys.: Condens. Matter, 2007, 19, 205120 CrossRef.
-
(a) R. D. Priestley, C. J. Ellison, L. J. Broadbelt and J. M. Torkelson, Science, 2005, 309, 456–459 CrossRef CAS PubMed;
(b) M. K. Mundra, S. K. Donthu, V. P. Dravid and J. M. Torkelson, Nano Lett., 2007, 7, 713–718 CrossRef CAS PubMed.
- T. Suhina, B. Weber, C. E. Carpentier, K. Lorincz, P. Schall, D. Bonn and A. M. Brouwer, Angew. Chem., Int. Ed., 2015, 54, 3688–3691 CrossRef CAS PubMed.
- A. Mustafic, H.-M. Huang, E. A. Theodorakis and M. A. Haidekker, J. Fluoresc., 2010, 20, 1087–1098 CrossRef CAS PubMed.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Sun, J. Liu, Z. Li, S. Zhang, J. Sun, H. S. Kwok and B. Z. Tang, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- The applications of functionalized TPE are too widespread in order to be covered exhaustively herein. Therefore, only selected examples are given here; for further details, see: ref. 6.
(a) N. B. Shustova, B. D. McCarthy and M. Dinca, J. Am. Chem. Soc., 2011, 133, 20126–20129 CrossRef CAS PubMed;
(b) F. Ishiwari, H. Hasebe, S. Matsumura, F. Hajjaj, N. H. Hayashi, M. Nishi, T. Someya and T. Fukushima, Sci. Rep., 2016, 6, 24275 CrossRef CAS PubMed;
(c) X. Xue, Y. Zhao, L. Dai, X. Zhang, X. Hao, C. Zhang, S. Huo, J. Liu, C. Liu, A. Kumar, W.-Q. Chen, G. Zou and X.-J. Liang, Adv. Mater., 2014, 26, 712–717 CrossRef CAS PubMed;
(d) Y. Yuan, R. T. K. Kwok, R. Zhang, B. Z. Tang and B. Liu, Chem. Commun., 2014, 50, 11465–11468 RSC.
- B. Z. Tang, “Organic, Organometallic, and Metallic Luminogens” in Cutting-Edge Chemistry, ACS, 15 Aug 2016, https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/organic-organometallic-and-metallic-luminogens.html Search PubMed.
- S. Sasaki, S. Suzuki, W. M. C. Sameera, K. Igawa, K. Morokuma and G. Konishi, J. Am. Chem. Soc., 2016, 138, 8194–8206 CrossRef CAS PubMed.
- S. Sasaki, K. Igawa and G. Konishi, J. Mater. Chem. C, 2015, 3, 5940–5950 RSC.
- Y. Suzuki, N. Fukui, K. Murakami, H. Yorimitsu and A. Osuka, Asian J. Org. Chem., 2013, 2, 1066–1071 CrossRef CAS.
-
(a) W. Huang and S. L. Buchwald, Chem.–Eur. J., 2016, 22, 14186–14189 CrossRef CAS PubMed;
(b) S. Riedmüller, O. Kaufhold, H. Spreitzer and B. J. Nachtsheim, Eur. J. Org. Chem., 2014, 1391–1394 CrossRef;
(c) M. Prashad, X. Y. Mak, Y. Liu and O. Repic, J. Org. Chem., 2003, 68, 1163–1164 CrossRef CAS PubMed;
(d) S. M. Raders, J. N. Moore, J. K. Parks, A. D. Miller, T. M. Leißing, S. P. Kelley, R. D. Rogers and K. H. Shaughnessy, J. Org. Chem., 2013, 78, 4649–4664 CrossRef CAS PubMed;
(e) G. L. Duc, S. Meiries and S. P. Nolan, Organometallics, 2013, 32, 7547–7551 CrossRef.
-
(a) K. Kubota, S. Fujishige and I. Ando, Polym. J., 1990, 22, 15–20 CrossRef CAS;
(b) M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 CrossRef CAS.
-
(a) K. Okabe, N. Inada, C. Gota, Y. Harada, T. Funatsu and S. Uchiyama, Nat. Commun., 2012, 3, 705 CrossRef PubMed;
(b) C. Gota, K. Okabe, T. Funatsu, Y. Harada and S. Uchiyama, J. Am. Chem. Soc., 2009, 131, 2766–2767 CrossRef CAS PubMed;
(c) C. D. S. Brites, P. P. Lima, N. J. O. Silva, A. Millan, V. S. Amaral, F. Palacio and L. D. Carlos, Nanoscale, 2012, 4, 4799–4829 RSC.
- F. M. Winnik, Macromolecules, 1990, 23, 233–242 CrossRef CAS.
- C.-Y. Chen and C.-T. Chen, Chem. Commun., 2011, 47, 994–996 RSC.
- E. M. Graham, K. Iwai, S. Uchiyama, A. P. De Silva, S. W. Magennis and A. C. Jones, Lab Chip, 2010, 10, 1267–1273 RSC.
- Y. Shiraishi, R. Miyamoto, X. Zhang and T. Hirai, Org. Lett., 2007, 9, 3921–3924 CrossRef CAS PubMed.
- D. Wang, R. Miyamoto, Y. Shiraishi and T. Hirai, Langmuir, 2009, 25, 13176–13182 CrossRef CAS PubMed.
- M. C. Harris, X. Huang and S. L. Buchwald, Org. Lett., 2002, 4, 2885–2888 CrossRef CAS PubMed.
- A. Shafir, P. A. Lichtor and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3490–3491 CrossRef CAS PubMed.
- A. Shafir and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 8742–8743 CrossRef CAS PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, New York, 2nd edn, 2012, pp. 694–722 Search PubMed.
-
(a) R. S. Coleman and W. Chen, Org. Lett., 2001, 3, 1141–1144 CrossRef CAS PubMed;
(b) K. Lakshman, J. C. Keeler, J. H. Hilmer and J. Q. Martin, J. Am. Chem. Soc., 1999, 121, 6090–6091 CrossRef.
- O. J. Plante, S. L. Buchwald and P. H. Seeberger, J. Am. Chem. Soc., 2000, 122, 7148–7149 CrossRef CAS.
-
(a) M. G. Organ, S. Calimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383–2387 CrossRef CAS PubMed;
(b) C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- T. W. Campbell, V. E. Mccoy, J. C. Kauer and V. S. Foldi, J. Org. Chem., 1961, 26, 1422–1426 CrossRef.
- W. E. Willy, D. R. Mckean and B. A. Garcia, Bull. Chem. Soc. Jpn., 1976, 49, 1989–1995 CrossRef CAS.
- P. H. Gore, C. K. Thadani and S. Thorburn, J. Chem. Soc. C, 1968, 2502–2508 RSC.
- R. A. Al-Horani and U. R. Desai, Tetrahedron, 2012, 68, 2027–2040 CrossRef CAS PubMed.
- D. X. Hu, P. Grice and S. V. Ley, J. Org. Chem., 2012, 77, 5198–5202 CrossRef CAS PubMed.
-
(a) M. Ito and T. Ishizone, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4832–4845 CrossRef CAS;
(b) C. S. Biswas, V. K. Patel, N. K. Vishwakarma, V. K. Tiwari, B. Maiti, P. Maiti, M. Kamigaito, Y. Okamoto and B. Ray, Macromolecules, 2011, 44, 5822–5824 CrossRef CAS.
- Z. Wang, Z. Yang, T. Gao, J. He, L. Gong, Y. Lu, Y. Xiong and W. Xu, Anal. Methods, 2015, 7, 2738–2746 RSC.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossmann, W. Pan, M. Garcia-Garibay and N. J. Turro, Macromolecules, 1993, 26, 4577–4585 CrossRef CAS.
- J. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132, 154902 CrossRef PubMed.
- H. Ringsdorf, J. Venzmer and F. M. Winnik, Macromolecules, 1991, 24, 1678–1686 CrossRef CAS.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossman, W. Pan, M. Garcia-Garibay and N. J. Turro, J. Phys. Chem., 1993, 97, 12998–13005 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis, Boca Raton, 2006, p. 548 Search PubMed.
- L. Tang, J. K. Jin, A. Qin, W. Z. Yuan, Y. Mao, J. Mei, J. Z. Sun and B. Z. Tang, Chem. Commun., 2009, 4974–4976 RSC.
- M. Onoda, S. Uchiyama and T. Ohwada, Macromolecules, 2007, 40, 9651–9657 CrossRef CAS.
- K. Iwai, Y. Matsumura, S. Uchiyama and A. P. De Silva, J. Mater. Chem., 2005, 15, 2796–2800 RSC.
- T. Li, S. He, J. Qu, H. Wu, S. Wu, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 2964–2970 RSC.
- S. Uchiyama, Y. Matsumura, A. P. De Silva and K. Iwai, Anal. Chem., 2003, 75, 5926–5935 CrossRef CAS PubMed.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR charts, and data of photophysical studies. See DOI: 10.1039/c7ra01212h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Gen-ichi Konishi
and
Gen-ichi Konishi