
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Red-shift in fluorescence emission of D–A type asymmetrical Zn(II) complexes by extending the π–π stacking interaction†
Wen-Jie Wang,
Liang Hao,
Chao-Yuan Chen,
Qi-Ming Qiu,
Ke Wang,
Jian-Biao Song and
Hui Li *
*
Key Laboratory of Clusters Science of Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, 100081, P. R. China. E-mail: lihui@bit.edu.cn; Fax: +86-10-68914780-804; Tel: +86-10-81381366
First published on 10th April 2017
Abstract
Two D–A type asymmetrical Zn(II) coordination complexes, [ZnL1(C2H5OH)] (1) (H2L1 = 2,4-di-tert-butyl-6-((E)-((2-((E)-(2-hydroxybenzylidene)amino)-4-nitrophenyl)imino)methyl)phenol) and [ZnL2(DMF)]·DMF (2) (H2L2 = 1-((E)-((2-((E)-(3,5-di-tert-butyl-2-hydroxybenzylidene)amino)-5-nitrophenyl)imino)methyl)naphthalen-2-ol), were designed, synthesized, and studied. Their fluorescence properties were comprehensively analysed based on their single-crystal structures. The results showed that the red-shift of fluorescence emission from complex 1 to complex 2 was successfully pushed via the strategy of extending the π–π stacking interaction.
Introduction
In recent years, among the metallosalphen complexes, Zn(II) complexes have exhibited excellent electroluminescence and photoluminescence properties and have been particularly employed in the fabrication of fluorescent probes, organic solar cells, and OLEDs.1 Among these, asymmetrical Zn(II) salphen Schiff base complexes have gained extensive attention. To be specific, the design and study of Zn(II) salphen complexes bearing donor–acceptor groups have been the interesting field due to their tunable electronic properties that are mainly utilised for developing good luminescent materials, owing to ICT (intramolecular charge transfer).2 Besides modifying the push–pull electronic ability of the functional groups, other effective methods to tune the properties of D–A-type asymmetrical Zn(II) salphen Schiff base complexes have also been reported in the literature.3 One among these is extending the π-conjugation system in the ligands at the molecular structure level, which affects photoluminescence, conductivity, redox activity, and so on.4 In 2011, Araki et al. reported that the absorption band can red shift from 400 nm to 500–600 nm, owing to the conversion of benzene rings into naphthalene rings, for a series of salphen complexes.5 On the other hand, the macroscopic properties, especially the photophysical properties such as absorption, emission, or nonlinear optical properties, strongly depend on π–π stacking, H-bonding and/or other noncovalent interactions including those involved in the M–L coordination bond.6 Recently, studies on fluorescent dipeptide nanoparticles for targeted cancer cell imaging and real-time monitoring of drug release have been reported, which were inspired by the red-shift observed in the yellow fluorescent protein that results from π–π stacking and the enhanced fluorescence intensity observed in the green fluorescent protein mutant, which results from the structure rigidification by Zn(II).7 From molecular and crystal engineering point of view, ligand design must consider π-conjugation systems and electron donor and accepter motifs combined via noncovalent interactions to tailor molecular structure and control the pattern of molecular packing.In this study, two types of D–A type asymmetrical salphen ligands based on 4-nitrobenzene-1,2-diamine and 3,5-bis(1,1-dimethylethyl)-2-hydroxy-benzaldehyde with salicylaldehyde to form 2,4-di-tert-butyl-6-(((E)-((2-((E)-(2-hydroxybenzylidene)amino)-4-nitrophenyl)imino)methyl)phenol) (H2L1) or with naphthaldehyde to form 1-((E)-((2-((E)-(3,5-di-tert-butyl-2-hydroxybenzylidene)amino)-5-nitrophenyl)imino) methyl) naphthalene-2-ol) (H2L2) were designed (Scheme 1). The remarkable characteristic of the ligand is the presence of two different aldehydes that form asymmetrical salphen-type ligands. Most importantly, a synthesis strategy was developed to controllably construct the target compound with high yield, which is a great challenge in the synthesis of asymmetrical salphen-type ligands to avoid derivative mixtures. Moreover, their Zn(II) complexes were obtained. Based on single-crystal structure analysis, the fluorescence properties of the two coordination complexes [(ZnL1(C2H5OH)] (1) and [ZnL2(DMF)]·DMF (2) were comprehensively studied. The results showed that the fluorescence emission red-shift from complex 1 to complex 2 was successfully pushed via extending the π–π stacking interaction. In addition, aggregation-induced emission was observed in the crystallized solid state of complex 2.
 | ||
| Scheme 1 Schematic for the preparation of [ZnL1(C2H5OH)] (1) and [ZnL2(DMF)]·DMF (2). | ||
Experimental section
Materials
All chemicals used were of analytical grade, purchased from Alfa Aesar Chemical Company, and were used without further purification. Organic solvents of analytical grade were supplied via commercial sources and used as received.Synthesis of the precursor 1 and 2
Preparation of complex 1 and 2
[ZnL1(C2H5OH)] (1) was synthesized via a template method. A solution of precursor 1 (26 mg, 0.1 mmol) in ethanol (10 mL) and 3,5-bis(1,1-dimethylethyl)-2-hydroxy-benzaldehyde (26 mg, 0.1 mmol) in ethanol (2 mL) was mixed and stirred in a 25 mL-round-bottom flask. Then, Zn(OAc)2·2H2O (20 mg, 0.1 mmol) in ethanol (1 mL) was added to the reaction mixture. The resultant solution was stirred at 85 °C for 6 h. The orange precipitate was obtained via filtration. The pure product was obtained via recrystallization from ethanol in 43.6% yield. Orange prism single crystals suitable for X-ray diffraction analysis were obtained after 7 days via evaporation at room temperature (Fig. S8†). Anal. calcd for C28H29ZnN3O4: C, 62.63; H, 5.44; N, 7.83. Found: C, 62.47; H, 5.36; N, 7.77. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.57–1.21 (m, 18H), 6.58–6.50 (m, 1H), 6.75–6.63 (m, 1H), 7.40–7.22 (m, 3H), 7.48 (dd, J = 7.9, 1.7 Hz, 1H), 8.11 (t, J = 10.0 Hz, 1H), 8.27–8.17 (m, 1H), 8.76 (dd, J = 10.4, 2.3 Hz, 1H), 9.10 (d, J = 3.8 Hz, 1H), 9.19 (d, J = 9.7 Hz, 1H) (Fig. S5†).[ZnL2(DMF)]·DMF (2) has been prepared following the same procedure as described for complex 1. Red powder product was obtained with yield of 46.2%. Orange red block single crystals suitable for X-ray diffraction analysis were obtained by evaporating under room temperature after a week (Fig. S9†). Anal. calcd for C32H31ZnN3O4: C, 65.48; H, 5.32; N, 7.16. Found: C, 65.37; H, 5.36; N, 7.07. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.29 (s, 9H), 1.50 (s, 9H), 6.89 (d, J = 9.2 Hz, 1H), 7.29–7.20 (m, 2H), 7.41–7.35 (m, 1H), 7.51 (t, J = 7.1 Hz, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.80 (d, J = 9.3 Hz, 1H), 8.09 (d, J = 8.6 Hz, 1H), 9.10 (s, 1H), 9.79 (s, 1H) (Fig. S6†).
Physical measurements
Elemental analyses (C, H, and N) were performed via a EA3000 elemental analyzer. FT-IR spectrum was obtained by a Nicolet-360 FT-IR spectrometer using KBr pellets in 4000–400 cm−1 region. 1H NMR spectra were acquired via a 400 MHz Bruker FT-NMR spectrometer using DMSO-d6 solvent. UV-vis spectra were obtained via a TU-1950 spectrophotometer. The luminescent spectra for the liquid state were obtained at room temperature using a Hitachi F-7000 FL spectrophotometer with a xenon arc lamp ashen light source. In the measurements of emission spectra, the pass width was Ex = 5 nm and Em = 5 nm. In all the measurements, the sample concentration 5 × 10−5 M was maintained. Only freshly prepared solutions were used for the spectroscopic study, and all the experiments were carried out at room temperature (298 K). X-ray powder diffraction (XPRD) of the samples was carried out using a Japan Rigaku D/max γA X-ray diffractometer equipped with graphite-monochromatized Cu Kα radiation (λ = 0.154060 nm). Thermogravimetric analyses (TGA) were carried out using a DTG-60H thermal analyzer under nitrogen atmosphere from room temperature to 800 °C at the heating rate of 10 °C min−1. The SEM images were obtained via Hitachi JSM-7500F scanning electron microscopy and the confocal fluorescence images were obtained via FV 1000 laser scanning confocal microscopy.Single-crystal structure determination
Single-crystal X-ray diffraction data for [ZnL1(C2H5OH)] (1) and [ZnL2(DMF)]·DMF (2) were obtained using a Bruker-AXS CCD area detector with graphite monochromated molybdenum Kα (λ = 0.71073 Å) radiation at 298 K. Unit-cell parameters were determined from automatic centering of the reflections and refined by least-square method. The diffraction data were corrected for Lorentz and polarization effects and absorption (empirically from ψ scan data). The structure was solved using direct methods and refined using full-matrix least square techniques on F2 via program SHELXL-97. All non-hydrogen atomic positions were located on different Fourier maps and anisotropically refined. Some of the hydrogen atoms were placed at their geometrically generated positions and other hydrogen atoms were located from different Fourier maps and isotropically refined. Crystallographic data are given in Tables S1–S3.†Results and discussion
Synthetic strategy
In the synthesis methods for asymmetrical Schiff base, production of symmetrical by-products and asymmetrical mixtures are the main drawbacks, for which a controllable, specific, and effective synthesis strategy towards unique asymmetrical products needs to be developed. Several strategies have been reported to overcome the above mentioned drawbacks.8 The widely used method is applying an asymmetrical diamine: for example, 4-nitrobenzene-1,2-diamine or 2-(aminomethyl)anilin reacts with an aldehyde, wherein the aldehyde in the asymmetrical Schiff base is the same. Kleij and his coworkers have reported a practical approach to synthesize structurally diverse monoimine salts and asymmetrical metallosalphen complexes,9 which motivated us to develop monoimine as the precursor for the construction of asymmetrical Schiff base ligand and their coordination complexes. 4-Nitrobenzene-1,2-diamine has been widely used as an asymmetrical diamine for the construction of Schiff bases.10 The electron-withdrawing group –NO2 as a strong meta-orienting group makes –NH2 in the para-position inactive (Scheme 1). In the present study, monoimine Schiff bases were obtained as the only product with a high yield of about 70% (Fig. S3 and S4†), which were precursors for the synthesis of asymmetrical Schiff bases where in other active aldehydes reacted with –NH2 in the para-position of the precursors. Herein, the target Zn(II) asymmetrical Schiff bases coordination complexes were synthesized via a template method. Thus, a customized method for the synthesis of asymmetrical Schiff bases and their coordination complexes was developed taking the molecular merit of 4-nitrobenzene-1,2-diamine into account.Crystal structural description
The crystal structure of complex 1 was found to crystallize in the monoclinic C2/c space group. Zn atom was found to be five-coordinated with distorted square pyramidal coordination geometry (Fig. 1a), with the two phenoxy oxygen atoms (O1 and O2) and two imino nitrogen atoms (N2 and N3) occupying the basal coordination sites, while an C2H5OH molecule occupying the apical position. Note that the Zn atom deviated from the best coordination plane defined by the atoms N2, N3, O1, and O2 by 0.3647 Å in direction of the apical C2H5OH oxygen atom O3. The two strong H-bonds O3–H3A⋯O1 (1.806 Å and 2.618 Å, 170.9°) between two neighboring asymmetric units resulted in dimer formation that was a basic repeating unit (Fig. 1b). The linkage of two dimers was via π–π stacking (3.944 Å) between the benzene ring of salicylaldehyde and the benzene ring of the adjacent dimer that led to infinite 1D chain (Fig. 1c). These 1D chains in the crystal lattice were held together via the H-bond C29–H29B⋯O5 (2.898 Å, 3.851 Å, 171.5°) along 3D directions to form a 3D structure, where C29 and H29B originate from the methyl group of the coordinating C2H5OH molecule and O5 atom belongs to the electron-withdrawing –NO2 group (Fig. 1d). | ||
| Fig. 1 (a) ORTEP picture of complex 1. The hydrogen atoms are omitted for clarity. (b) The structure of the dimer of complex 1 linked by strong H-bonding (O3–H3A⋯O1: 1.806 Å, 2.618 Å, 170.9°, violet dotted line). (c) 1D chain in complex 1 generated from π–π stacking (3.944 Å, pink dotted line). (d) 3D supramolecular architecture of complex 1 formed by intra-chain H-bonding C29–H29B⋯O5 (2.898 Å, 3.851 Å, 171.5°, green dotted line) viewing from the c axis. | ||
The complex 2 has a similar structure as complex 1 and it crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The Zn atom in complex 2 is also five-coordinated. A DMF molecule was present in the axial position of the distorted square pyramidal geometry with two naphthyl oxygen atoms (O1 and O2) and two imino nitrogen atoms (N1 and N2) in the equatorial plane (Fig. 2a). The planarity of the Zn, 2N, and 2O (five) atoms in complex 2 was similar to that of those in complex 1 (0.3545 Å); however, they both were larger than those reported for the related five-coordinated Zn(II) salphen complexes.11 This indicated that the introduction of a benzene ring increased the rigidity of the conjugate system in the ligands; thus, large Zn atoms cannot perfectly occupy the N2O2 coordination pocket. This deviated planarity was not favourable for electron transfer and influenced optical properties further. π–π stacking (3.722 Å) between rings of naphthaldehyde of an adjacent molecule linked two mononuclear molecules into a supramolecular dimer (Fig. 2b). Moreover, π–π stacking interaction within the dimer (3.880 Å) led to the formation of a 1D chain (Fig. 2c). Compared to complex 1, these 1D chains in the crystal lattice of complex 2 were connected to each other via van der Waals interactions along the 2D and 3D directions (Fig. 2d).
space group. The Zn atom in complex 2 is also five-coordinated. A DMF molecule was present in the axial position of the distorted square pyramidal geometry with two naphthyl oxygen atoms (O1 and O2) and two imino nitrogen atoms (N1 and N2) in the equatorial plane (Fig. 2a). The planarity of the Zn, 2N, and 2O (five) atoms in complex 2 was similar to that of those in complex 1 (0.3545 Å); however, they both were larger than those reported for the related five-coordinated Zn(II) salphen complexes.11 This indicated that the introduction of a benzene ring increased the rigidity of the conjugate system in the ligands; thus, large Zn atoms cannot perfectly occupy the N2O2 coordination pocket. This deviated planarity was not favourable for electron transfer and influenced optical properties further. π–π stacking (3.722 Å) between rings of naphthaldehyde of an adjacent molecule linked two mononuclear molecules into a supramolecular dimer (Fig. 2b). Moreover, π–π stacking interaction within the dimer (3.880 Å) led to the formation of a 1D chain (Fig. 2c). Compared to complex 1, these 1D chains in the crystal lattice of complex 2 were connected to each other via van der Waals interactions along the 2D and 3D directions (Fig. 2d).
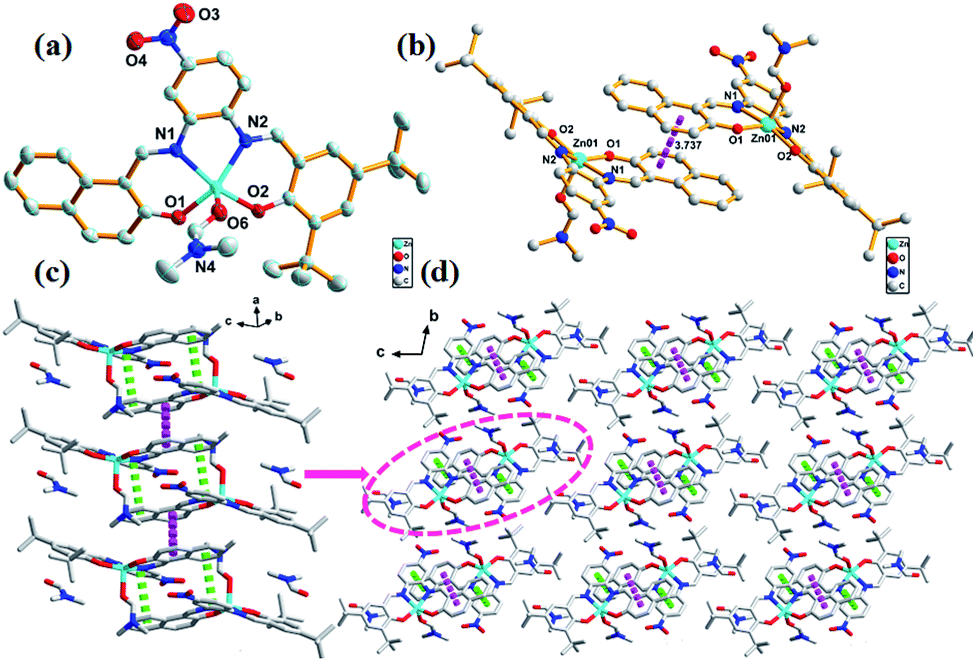 | ||
| Fig. 2 (a) ORTEP picture of complex 2. The hydrogen atoms are omitted for clarity. (b) The structure of the dimer of complex 2 linked by π–π stacking (3.722 Å, pink dotted line). (c) 1D chain in complex 2 generated from π–π stacking (3.880 Å, green dotted line). (d) 3D supramolecular architecture of complex 2 formed by van der Waals contacts, viewing from a axis. | ||
Clearly, the solvent molecule in the axial position in complex 1 is C2H5OH, which tends to form H-bonding, whereas DMF in the axial position in complex 2 does not. On the other hand, naphthalene rings in complex 2 have extended π-system, which has advantage in terms of π–π stacking. Accordingly, H-bonding is the dominant intermolecular interaction in complex 1, whereas π–π stacking is the main interaction in complex 2. From the viewpoint of molecular design and crystal engineering, subtle structural changes on ligand may modulate their molecular packing and their microcosmic structures (Fig. 3). Moreover, the extended π-system in 2 was confirmed by its red colors. The experimental and simulated XRD patterns of complex 1 and 2 have shown the phase purity (Fig. S7†).
 | ||
| Fig. 3 Images of complex 1 (a and b) and 2 (c and d) obtained via an optical microscope and a scanning electron microscope. | ||
Thermogravimetric analyses indicate two main thermal decomposition processes (Fig. S13†). For [ZnL1(C2H5OH)] (1), first weight loss of 8.13% in the range of 80–150 °C correspond to the removal of ethanol solvent molecule (calc. 8.20%). The second weight loss at 360–510 °C correspond to the complete decomposition of the framework and remaining weight loss correspond to that of ZnO (calc. 13.92%, obs. 13.81%). For [ZnL2(DMF)]·DMF (2), free DMF solvent molecule was removed in the temperature range of 103–210 °C (calc. 10.31%, obs. 10.11%). The complex collapsed at 370 °C.
Optical properties
UV-vis absorption spectra of both complex 1 and 2 show three main bands around 200–300 nm, 300–400 nm, and 400–600 nm, which can be ascribed to n → σ*, π → π*, and n → π* transitions, respectively (Fig. S12†). Moreover, the three absorption peaks at 246, 308, and 426 nm for complex 1 were relatively red-shifted to 251, 335, and 441 nm for complex 2 in THF solution. As π → π* transition is very sensitive to the environment, it can even red shift 27 nm for complex 1 (308 nm) and 2 (335 nm) because of stronger π–π stacking resulting from extended π–system.12 Relatively, the fluorescence properties of both complexes in THF solution and solid state were studied. The fluorescence emission at 426 nm of complex 2 red-shifted by 32 nm from that of complex 1 (394 nm), which was the mirror image of π → π* transition shown in the absorption spectra; as the energy of transition absorption is lower, transition energy from the excited state to the ground state is correspondingly lower (Fig. 4a). Note that complex 2 has an emission at 610 nm. This emission band can be attributed to the formation of excimers, according to the study reported by Yang and his colleagues.13 Because of the pairwise anthracene stacking, an excimer-like emission band in THF would arise in long-wavelength range. Herein, due to the existence of two types of π–π stacking, the pairwise stacking of complex 2 in THF solution can be confirmed by 610 nm emission band in the fluorescence spectra, which was not weak, indicating that the minimized non-radiative energy dissipation was due to excimer mechanism, thereby contributing to the enhanced emission as reported. The fluorescence lifetimes of complex 1 and 2 were measured to be 5.9 ns and 5.4 ns and their quantum yields were 0.16 and 0.23, respectively (Fig. S10 and S11†). Moreover, the fluorescence emission spectra of complex 1 and 2 in other solvents were also studied, but the fluorescence intensity was found to be too weak (Fig. S12†).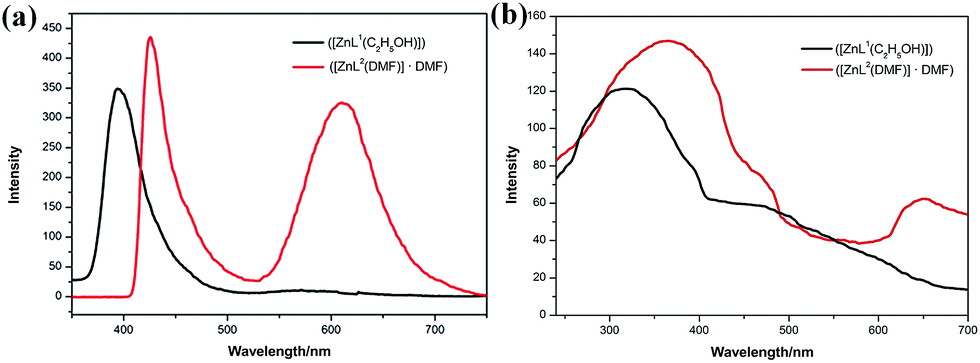 | ||
| Fig. 4 (a) The fluorescence spectra of complex 1 (λex = 308 nm, 5.0 × 10−5 mol L−1, THF) and complex 2 (λex = 335 nm, 5.0 × 10−5 mol L−1, THF); (b) solid state fluorescent spectra of 1 and 2. | ||
The solid state fluorescence spectra show that the maximum emissions of complex 1 and 2 are at 320 and 364 nm, indicating red shift as expected (Fig. 4b). Two π–π stacking interactions (3.722 Å and 3.880 Å) in the crystal lattice of complex 2 were found stronger than those of complex 1 (3.944 Å). Compared with the THF solution, the pairwise dimer in complex 2 was tied together and periodically extended to 3D structure in crystal lattice. Thus, the excimer-induced 610 nm fluorescence emission in solution red-shifted to 650 nm in the solid state, which has been represented in the confocal fluorescence images (Fig. 5).14 Moreover, visual evidence from laser scanning confocal microscope is shown in Fig. 5, in which complex 1 exhibited blue fluorescence and 2 exhibited red fluorescence with great red-shift. With respect to the abovementioned analysis and previous literature, the difference between fluorescence behavior could be attributed to the supramolecular state, which was often affected by the nature of ligands. There was no doubt that the supramolecular aggregation affected the functional properties of the compound, especially the molecular packing form affected the luminescent properties. Therefore, it was found to be an effective way to tune supramolecular aggregation via intermolecular interaction based on molecular structures and thereby influence the fluorescent properties.
 | ||
| Fig. 5 Confocal fluorescence images of complex 1 and 2 in fluorescent field (a and d), bright-field (b and e) and the superimposed images (c and f). | ||
Conclusion
Towards tailoring the molecular structure and controlling the pattern of molecular packing, two D–A type asymmetrical 4-NO2-salphen ligands with two different salicylaldehydes were designed and controllably synthesized with high yield. The fluorescent properties of their Zn(II) coordination complexes were studied based on the understanding of their crystal structures. Especially, complex 2 could be used in cell imaging. With respect to molecular engineering and crystal engineering, the extended π-system in ligands and enhanced π–π stacking in the crystal lattice were found to be effective ways to push the fluorescent emission to red-shift. The research results may help to design new luminescent materials.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21471017).References
- (a) H.-C. Lin, C.-C. Huang, C.-H. Shi, Y.-H. Liao, C.-C. Chen, Y.-C. Lin and Y.-H. Liu, Dalton Trans., 2007, 781–791 RSC; (b) K. G. Vladimirova, A. Y. Freidzon, O. V. Kotova, A. A. Vaschenko, L. S. Lepnev, A. A. Bagatur'yants, A. G. Vitukhnovskiy, N. F. Stepanov and M. V. Alfimov, Inorg. Chem., 2009, 48, 11123–11130 CrossRef CAS PubMed; (c) J. Cheng, K. Wei, X. Ma, X. Zhou and H. Xiang, J. Phys. Chem. C, 2013, 117, 16552–16563 CrossRef CAS; (d) G. Salassa, J. W. Ryan, E. C. Escudero-Adán and A. W. Kleij, Dalton Trans., 2014, 43, 210–221 RSC; (e) C. A. Barboza, J. C. Germino, A. M. Santana, F. J. Quites, P. A. N. M. Vazquez and T. D. Z. Atvars, J. Phys. Chem. C, 2015, 119, 6152–6163 CrossRef CAS; (f) J. Cheng, F. Gou, X. Zhang, G. Shen, X. Zhou and H. Xiang, Inorg. Chem., 2016, 55, 9221–9229 CrossRef CAS PubMed.
- (a) S. Di Bella, I. Fragalà, I. Ledoux, M. A. Diaz-Garcia and T. J. Marks, J. Am. Chem. Soc., 1997, 119, 9550–9557 CrossRef CAS; (b) K.-H. Chang, C.-C. Huang, Y.-H. Liu, Y.-H. Hu, P.-T. Chou and Y.-C. Lin, Dalton Trans., 2004, 1731–1738 RSC; (c) W.-L. Tong, S.-M. Yiu and M. C. Chan, Inorg. Chem., 2013, 52, 7114–7124 CrossRef CAS PubMed; (d) P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., 2016, 128, 5833–5838 CrossRef; (e) H. Xiao, P. Li, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 1588–1593 RSC.
- (a) S. Mizukami, H. Houjou, K. Sugaya, E. Koyama, H. Tokuhisa, T. Sasaki and M. Kanesato, Chem. Mater., 2005, 17, 50–56 CrossRef CAS; (b) G. Yin, Y. Ma, Y. Xiong, X. Cao, Y. Li and L. Chen, J. Mater. Chem. C, 2016, 4, 751–757 RSC; (c) Y.-C. Chen, C.-Y. Nien, K. Albert, C.-C. Wen, Y.-Z. Hsieh and H.-Y. Hsu, RSC Adv., 2016, 6, 44024–44028 RSC; (d) Y. Sheng, J. Ma, S. Liu, Y. Wang, C. Zhu and Y. Cheng, Chem.–Eur. J., 2016, 22, 9519–9522 CrossRef CAS PubMed; (e) I. P. Oliveri, S. Failla, A. Colombo, C. Dragonetti, S. Righetto and S. Di Bella, Dalton Trans., 2014, 43, 2168–2175 RSC.
- (a) T. Hirao, Coord. Chem. Rev., 2002, 226, 81–91 CrossRef CAS; (b) K. Yagi, M. Ito and H. Houjou, Macromol. Rapid Commun., 2012, 33, 540–544 CrossRef CAS PubMed; (c) E. Borré, J. F. Stumbé, S. Bellemin-Laponnaz and M. Mauro, Angew. Chem., Int. Ed., 2016, 55, 1313–1317 CrossRef PubMed.
- H. Houjou, M. Ito and K. Araki, Inorg. Chem., 2011, 50, 5298–5306 CrossRef CAS PubMed.
- (a) W. Xi, Y. Gong, B. Mei, X. Zhang, Y. Zhang, B. Chen, J. Wu, Y. Tian and H. Zhou, Sens. Actuators, B, 2014, 205, 158–167 CrossRef CAS; (b) F. Mandoj, A. D'Urso, S. Nardis, D. Monti, M. Stefanelli, C. M. Gangemi, R. Randazzo, F. R. Fronczek, K. M. Smith and R. Paolesse, New J. Chem., 2016, 40, 5662–5665 RSC.
- Z. Fan, L. Sun, Y. Huang, Y. Wang and M. Zhang, Nat. Nanotechnol., 2016, 11, 388–396 CrossRef CAS PubMed.
- (a) H.-L. Chen, S. Dutta, P.-Y. Huang and C.-C. Lin, Organometallics, 2012, 31, 2016–2025 CrossRef CAS; (b) J. E. Armstrong, P. M. Crossland, M. A. Frank, M. J. Van Dongen and W. R. McNamara, Dalton Trans., 2016, 45, 5430–5433 RSC; (c) A. H. Kianfar and M. Ebrahimi, Spectrochim. Acta, Part A, 2013, 115, 725–729 CrossRef CAS PubMed; (d) A. H. Kianfar, S. A. Khademi, R. H. Fath, M. Roushani and M. Shamsipur, J. Iran. Chem. Soc., 2013, 10, 347–355 CrossRef CAS.
- (a) E. C. Escudero-Adán, M. M. N. Belmonte, J. Benet-Buchholz and A. W. Kleij, Org. Lett., 2010, 12, 4592–4595 CrossRef PubMed; (b) D. Anselmo, E. C. Escudero-Adán, J. Benet-Buchholz and A. W. Kleij, Dalton Trans., 2010, 39, 8733–8740 RSC; (c) R. M. Haak, A. Decortes, E. C. Escudero-Adán, M. M. Belmonte, E. Martin, J. Benet-Buchholz and A. W. Kleij, Inorg. Chem., 2011, 50, 7934–7936 CrossRef CAS PubMed; (d) C. J. Whiteoak, G. Salassa and A. W. Kleij, Chem. Soc. Rev., 2012, 41, 622–631 RSC; (e) N. Kielland, E. C. Escudero-Adán, M. M. Belmonte and A. W. Kleij, Dalton Trans., 2013, 42, 1427–1436 RSC.
- (a) R. M. Clarke and T. Storr, Dalton Trans., 2014, 43, 9380–9391 RSC; (b) C. Wang, Y. Chen and W.-F. Fu, Dalton Trans., 2015, 44, 14483–14493 RSC; (c) B. Bugenhagen and M. Prosenc, Dalton Trans., 2016, 45, 7460–7468 RSC.
- (a) Q.-H. Meng, P. Zhou, F. Song, Y.-B. Wang, G.-L. Liu and H. Li, CrystEngComm, 2013, 15, 2786–2790 RSC; (b) L. Hao, T. Zhang, G. Chang and H. Li, Chin. J. Chem., 2015, 33, 425–430 CrossRef CAS.
- P. Weis, D. Wang and S. Wu, Macromolecules, 2016, 49, 6368–6373 CrossRef CAS.
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC.
- (a) Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (b) M. Yang, D. Xu, W. Xi, L. Wang, J. Zheng, J. Huang, J. Zhang, H. Zhou, J. Wu and Y. Tian, J. Org. Chem., 2013, 78, 10344–10359 CrossRef CAS PubMed; (c) J. Mei, Y. Hong, J. W. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed; (d) W. Guan, S. Wang, C. Lu and B. Z. Tang, Nat. Commun., 2016, 7 DOI:10.1038/ncomms11811; (e) M. Shimada, M. Tsuchiya, R. Sakamoto, Y. Yamanoi, E. Nishibori, K. Sugimoto and H. Nishihara, Angew. Chem., Int. Ed., 2016, 55, 3022–3026 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structure information, spectra (IR, fluorescence, TGA, 1H NMR), XRD, crystal photos and crystallographic data (cif). CCDC 1504319 and 1504320 for 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01135k |
| This journal is © The Royal Society of Chemistry 2017 |