
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalyzed malonation of 2-arylquinazolines with 2-diazomalonates: double C–H functionalization†
Zhipeng Zhanga,
Xin Jianga,
Zhihong Denga,
Heng Wanga,
Jian Huanga and
Yiyuan Peng *ab
*ab
aKey Laboratory of Functional Small Organic Molecule, Ministry of Education, College of Chemical and Engineering, Jiangxi Normal University, Nanchang 330013, China. E-mail: yypeng@jxnu.edu.cn
bCAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, 330022, PR China
First published on 23rd May 2017
Abstract
Herein, rhodium-catalyzed malonation of 2-arylquinazolines with diazomalonates was described. A series of dimalonate-substituted 2-arylquinazolines were achieved through double C–H functionalization.
As privileged structural cores, quinazoline-based chemistry has been recognized as one of hot topics in the community of organic synthesis due to its ubiquity in many bioactive pharmaceuticals and agricultural chemistry molecules.1 For example, Erlotinib and Gefitinib are well-known lung cancer drugs.2 Trimetrexate has been used in the treatment of pneumocystis pneumonia3 and Prazosin is used for curing high blood pressure4 (Fig. 1).
 | ||
| Fig. 1 Quinazolines as core structures in pharmaceuticals. | ||
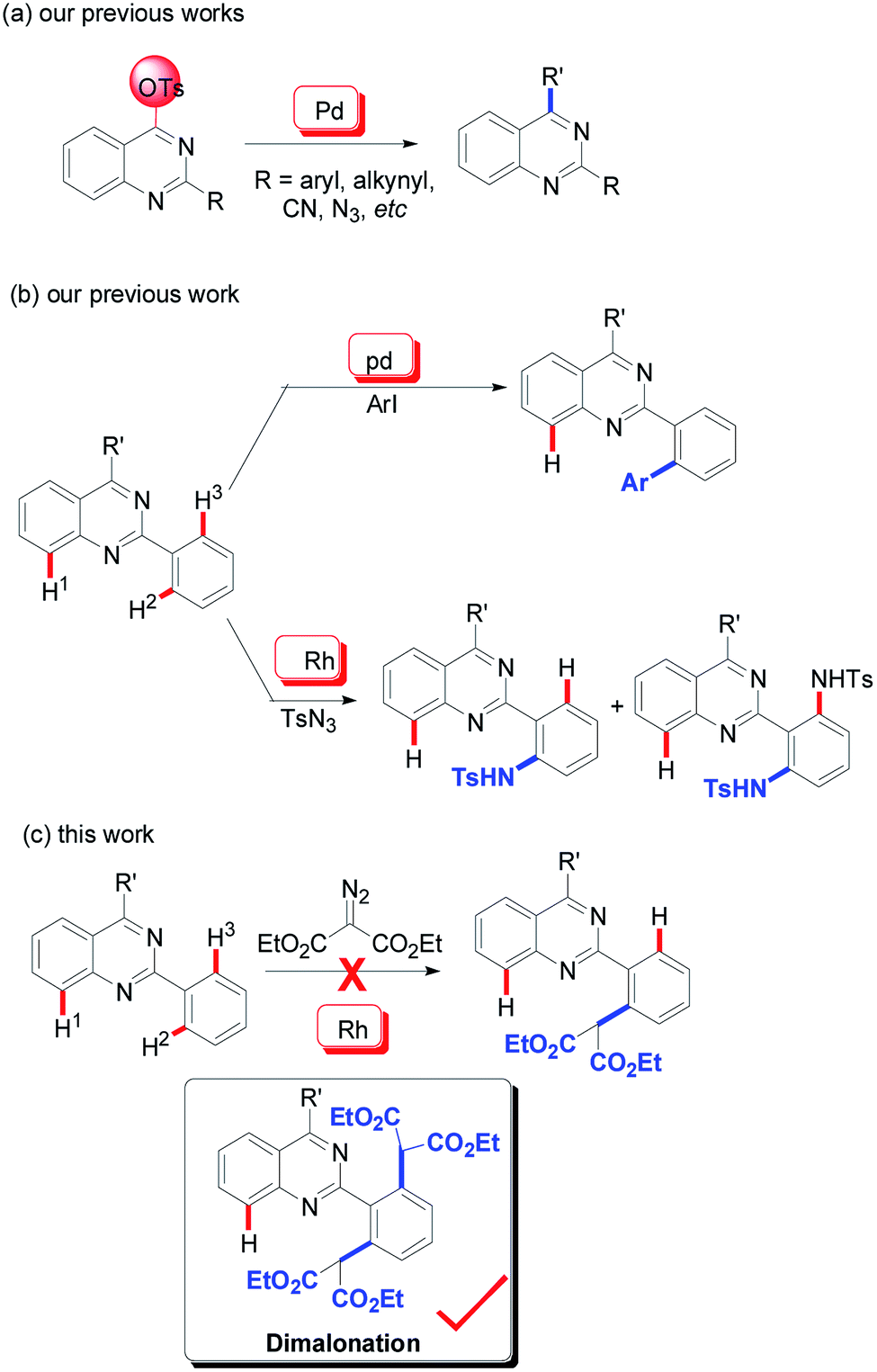
To date, it is reasonable that a particular and intensive emphasis has been place on the development of synthetic methodologies and the further bioactivity evaluation of the quinazoline architectures. Classical protocols prefer the use of condensation reactions between carbonyl-containing compounds and amines.5 With the development of modern organic synthesis, transition metal-catalyzed transformations (especially, copper and palladium) have been accepted as an efficient alternative method for the synthesis of quinazolines.6 Our group initiated studies on the development of this synthetic methodology since the year of 2009 and various 2,4-functionalized quinazolines were achieved via palladium-catalyzed C–O bond activation (Scheme 1a).7
 | ||
| Scheme 1 The proposed route for the rhodium-catalyzed malonation reaction. | ||
As an extension of our program, we were then pleased to discover that the quinazoline core is an efficient directing group in transition metal-catalyzed C–H functionalization (Scheme 1b). According to our findings, its directing properties are distinctive from that of pyridine and quinolines, and the regioselectivity observed in the palladium-catalyzed mono-iodination and mono-arylation of 2-phenylquinazolines specifically leads to the H2-functionalized product (Scheme 1b).8 Recently, amidation of 2-phenylquinazolines was also explored by our group using rhodium catalysis.9 In the reactions, sulfonyl azide was used as a nitrogen source and diamidation was mainly observed. Similar with our previous results, this rhodium-catalyzed mono-amidation was also regioselective, predominately giving the H2-functionalized products (Scheme 1b). The mono-amidation of the C–H2 bonds can be regulated using 2-phenylquinazolines with steric hinderance at the 2-phenyl group as the substrate.
Acceptably, rhodium catalysis is one of versatile and powerful tools used in the formation of carbon–carbon bonds and carbon–heteroatom bonds.10 In particular, rhodium catalysis plays a critical role in direct C–H functionalization, where the rhodium catalyst is readily inserted into C–H bonds due to its stronger electrophilicity.11 Encouraged by that mentioned above and our continuous interest in quinazoline-based chemistry, we would like to develop more rhodium-catalyzed quinazoline-based transformations and then further evaluate the bioactivities of the resulting quinazoline derivatives. We therefore focused our attention on rhodium-catalyzed malonation using diazomalonates as the reaction partners, where a rhodium carbene was designed as the key intermediate (Scheme 1c).12
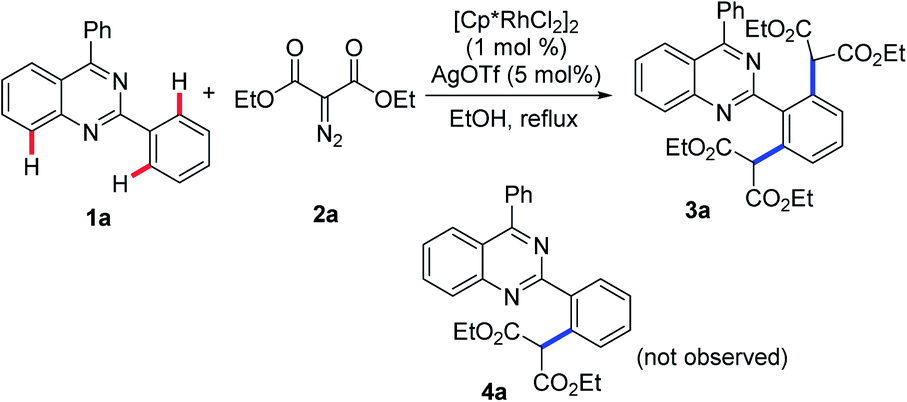
To verify this projected transformation, we selected the reaction of 2,4-diphenylquinazoline 1a and ethyl diazomalonate 2a as a model reaction. In our preliminary trials, a product was detected with high efficiency when the model reaction was carried out using 1 mol% of [Cp*RhCl2]2, 5 mol% of AgOTf, and 3 equiv. of diazomalonate in ethanol at reflux (Table 1, entry 1). Structural identification of the product by NMR, HRMS and X-ray diffraction revealed that the double malonated quinazoline 3a was observed and that dimalonation took place in the rhodium-catalyzed reaction of 2-phenylquinazoline 1a and ethyl diazomalonate 2a. The mono-malonation product 4a was not detected at all. We thought the observation was probably attributed to the use of excess of diazomalonate 2a. As a result, the reaction with 1.1 equiv. diazomalonate 2a was conducted under the standard conditions. However, we were surprised to find the unique occurrence of dimalonation of 2-phenylquinazoline 1a with high specificity, producing 3a in 32% yield (Table 1, entry 2). Based on the information in hand, it was confirmed that the double malonations mentioned here were stepwise and the second malonation was faster than that of the first one. This double C–H functionalization was not observed in our previous 2-phenylquinazoline-based transformations.8 This result prompted us to optimize the reaction conditions (see ESI†). Changing silver triflate to other silver salts greatly affected the yield of the reaction. When silver acetate was used as the additive, the reaction completely failed (Table 1 entry 3). Additionally, silver tetrafluoroborate drastically retarded the reaction and a 25% yield of product was obtained (Table 1, entry 4). Increasing of loading of silver triflate had a slight impact on the outcome of the reaction (Table 1, entry 5). The reaction was unfavorable when the loading of the rhodium catalyst was increased to 5 mol% and an inferior yield was afforded (Table 1 entry 6). To our surprise, the use of another rhodium catalyst exerted a great impact on the reaction. When the rhodium catalyst was switched to [Cp*Rh(BF4)2]2, the reaction did not occur (Table 1, entry 7). Decreasing the reaction temperature was also unfavorable, resulting in product 3a in 64% yield with no mono-malonation product 4a isolated (Table 1, entry 8). A detailed evaluation of the solvent, rhodium catalyst, additive, temperature and the equivalent ratio of substrate is presented in the ESI.†
|
||
|---|---|---|
| Entry | Conditions | Yield of 3ab (%) |
| a Standard conditions: quinazoline 1a (1.0 equiv.), diazomalonate 2a (3 equiv.), rhodium catalyst [Cp*RhCl2]2 (1 mol%), AgOTf (5 mol%), EtOH, reflux.b Isolated yield based on quinazoline 1a. | ||
| 1 | Standard conditions | 83 |
| 2 | 1.1 equiv. diazomalonate was used | 32 |
| 3 | AgOTf was replaced by AgOAc | 0 |
| 4 | AgOTf was replaced by AgBF4 | 25 |
| 5 | The loading of AgOTf was increased to 10 mol% | 82 |
| 6 | The loading of the rhodium catalyst was increased to 5 mol% | 72 |
| 7 | [Cp*Rh(BF4)2]2 instead of [Cp*RhCl2]2 | 0 |
| 8 | The reaction temperature was reduced to 65 °C | 64 |
With the optimized conditions in hand (1 mol% of [Cp*RhCl2]2, 5 mol% of AgOTf and 3 equiv. of diazomalonate in ethanol at reflux), we then examined the scope and generality of this rhodium-catalyzed dimalonation of 2-phenylquinazolines 1 and diazomalonates 2. The results are illustrated in Table 2.
|
|---|
| a Isolated yield based on quinazoline 1. |
 |

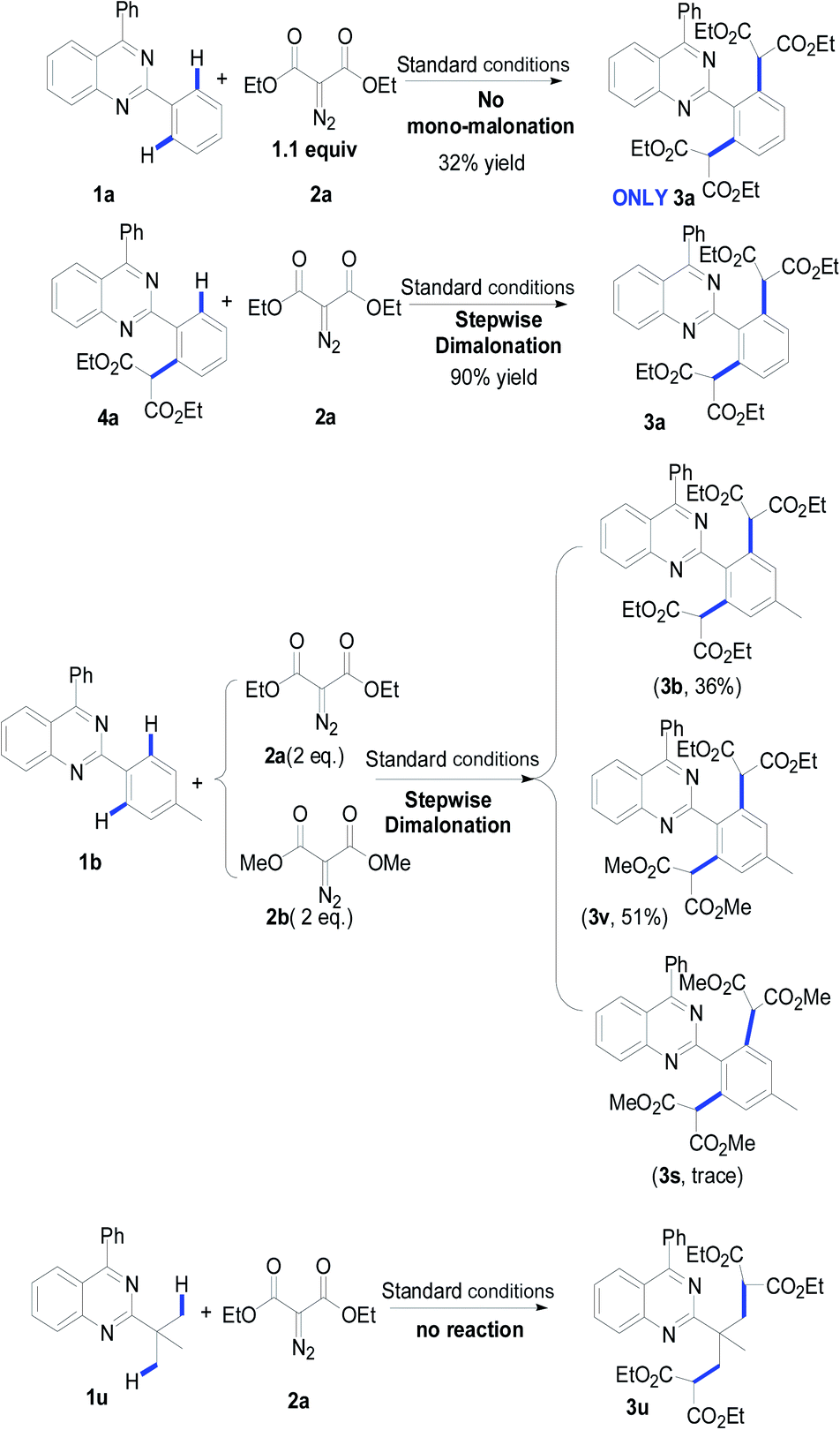
From the results in Table 2, a series of substituted dimalonated 2-phenylquinazoline 3 were achieved as expected. The electron-withdrawing/donating effects of the 2-phenyl group of the quinazolines drastically affected the yields of the reactions. In the reactions, the substrates attached a 2-phenyl group bearing electron-rich groups were more efficient than that of electron-deficient groups. For examples, the reaction of 2-(4-N,N-dimethylaminophenyl)quinazoline 1d with diazomalonate 2a produced the corresponding product 3d in 99% yield, while the reaction of 2-(4-trifluoromethylphenyl)quinazoline 1f gave the product 3f in 80% yield. The electron-withdrawing/donating effects of the R2 substituent slightly influenced the results. The corresponding products 3j–3n were delivered in excellent yields. To our delight, the R2 substituent could be expanded to the methyl group, yielding the desired product 3o in 83% yield. The steric hinderance of the substrate was also explored in the reaction. According to our previous findings, double C–H functionalization can be regulated by the use of sterically hindered 2-phenylquinazolines.9 Interestingly, the reaction of 2-(3-methyl)phenylquinazoline 1p still gave rise to the dimalonated product 3p in moderate yield, although the reaction of 2-(2-methyl)phenylquinazolines provided the mono-malonated product 3q. A heterocyclic unit at the 2-position of the quinazoline was also compatible in this reaction (entry 3r).
The effect of the 2-diazomalonates was then explored. The corresponding 2-diazomalonate product 3s was obtained in a yield of 78%, when 2-diazo-malonic acid dimethyl ester 2b was reacted with 1b. However the reactions of 2-diazo-malonic acid dipropyl 2c and 5-diazo-2,2-dimethyl-[1,3]dioxane-4,6-dione 2d gave only trace amounts of the desired products (not listed in Table 2). The reactions of 2-diazo-3-oxo-butyric acid ethyl ester 2e and diazo-phenyl-acetic acid ethyl ester 2f gave a complex mixture of products.
In order to achieve selective mono-malonation, we tried to install a TMS group at one of the ortho positions of 2-phenylquinazoline, which could then be removed using TBAF in a subsequent step. Thus, 2-(4-methyl-2-trimethylsilanyl)phenyl-4-phenyl-quinazoline (1t) was synthesized in our laboratory. When 1t was reacted with 1b, the mono-malonated product 3t was not obtained. However, the TMS group was removed and the reaction gave the double malonated product 3b in a yield of 63%.
To understand the possible mechanism, several control experiments were carried out (Table 3). As mentioned above, when reducing the equivalent of diazomalonate 2a, the dimalonated product 3a was uniquely detected in 32% yield. The reaction of the as-prepared product 4a with diazomalonate 2a under the standard conditions provided the desired product 3a in 90% yield. The reaction of 1b with a mixture of 2a and 2b (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gave a mixture of 3b (36%) and 3v (51%), and a trace amount of 3s. This information indicates that the double malonation was stepwise and the second malonation was faster than that of the first one. The reaction using 2-tert-butylquinazolines 1u did not produce the corresponding desired products. As illustrated in Scheme 2, the migration insertion of the rhodium carbene into the rhodium–carbon is the key step in the process.
:1) gave a mixture of 3b (36%) and 3v (51%), and a trace amount of 3s. This information indicates that the double malonation was stepwise and the second malonation was faster than that of the first one. The reaction using 2-tert-butylquinazolines 1u did not produce the corresponding desired products. As illustrated in Scheme 2, the migration insertion of the rhodium carbene into the rhodium–carbon is the key step in the process.
| a Isolated yield based on quinazoline 1. |
|---|
 |
 | ||
| Scheme 2 Possible mechanism. | ||
In conclusion, we have developed a novel rhodium-catalyzed dimalonation of 2-phenylquinazolines with diazomalonates with high efficiency and a good tolerance of functional groups. In this reaction, only 1 mol% of the rhodium catalyst was required. This protocol represents the first rhodium-catalyzed examples using the quinazolines as the directing group in the dimalonation reaction via double C–H functionalization. The application of the quinazoline as a directing group in other C–H functionalization is currently underway in our laboratory.
Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21162012 and 21362014), the Science Foundation of Education Department of Jiangxi province (grant no. KJLD13020), the open project program of CAS Key Laboratory of Molecular Recognition and Function, Chinese Academy of Sciences (No. 2012LMRF004) and Key Laboratory of Functional Small Organic Molecule, Ministry of Education, Jiangxi Normal University (No. KLFS-KF-201408) is gratefully acknowledged.Notes and references
- For selected examples, see: (a) B. Baruah, K. Dasu, B. Vaitilingam, P. Mamnoor, P. P. Venkata, S. Rajagopal and K. R. Yeleswarapu, Bioorg. Med. Chem., 2004, 12, 1991–1994 CrossRef CAS PubMed; (b) V. M. Sharma, P. Prasanna, K. V. A. Seshu, B. Renuka, C. V. L. Rao, G. S. Kumar, C. P. Narasimhulu and R. Rajagopalan, et al., Bioorg. Med. Chem. Lett., 2002, 12, 2303–2307 CrossRef CAS PubMed.
- R. Gundla, R. Kazemi, R. Sanam, R. Muttineni, J. A. R. P. Sarma, R. Dayam and N. Neamati, J. Med. Chem., 2008, 51, 3367–3377 CrossRef CAS PubMed.
- C. E. S. Short, Y. C. Gilleece, M. J. Fisher and D. R. Churchill, AIDS, 2009, 23, 1287–1290 CrossRef CAS PubMed.
- S. F. Campbell, M. J. Davey, J. D. Hardstone, B. N. Lewis and M. J. Palmer, J. Med. Chem., 1987, 30, 49–57 CrossRef CAS PubMed.
- Reviews see: (a) D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153–10202 CrossRef CAS; (b) A. Witt and J. Bergman, Curr. Org. Chem., 2003, 7, 659–677 CrossRef CAS; (c) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS; (d) A. Nefzi, J. M. Ostresh and R. A. Houghten, Chem. Rev., 1997, 97, 449–472 CrossRef CAS PubMed.
- X. Y. Wang and N. Jiao, Org. Lett., 2016, 18, 2150–2153 CrossRef CAS PubMed.
- (a) X. L. Ye, J. J. Yuan, Y. R. Zhou, Z. H. Deng, X. C. Mao and Y. Y. Peng, Synthesis, 2016, 48, 3976–3984 CrossRef CAS; (b) X. L. Ye, Z. H. Yuan, Y. R. Zhou, Q. Yang, Y. P. Xie, Z. H. Deng and Y. Y. Peng, J. Heterocycl. Chem., 2016, 53, 1956–1962 CrossRef CAS; (c) X. Chen, Q. Yang, Y. R. Zhou, Z. H. Deng, X. C. Mao and Y. Y. Peng, Synthesis, 2015, 47, 2055–2062 CrossRef CAS; (d) X. Zhao, Y. R. Zhou, Q. Yang, Y. P. Xie, Q. P. Ding and Y. Y. Peng, et al., Synthesis, 2013, 45, 3245–3250 CrossRef CAS; (e) Y. Y. Peng, P. Huang, Y. Wang, Y. R. Zhou, Q. Yang and X. Jiang, et al., Org. Biomol. Chem., 2014, 12, 5922–5927 RSC; (f) G. Y. S. Qiu, P. Huang, Q. Yang, H. Lu, J. S. Xu, Z. H. Deng, M. Zhang and Y. Y. Peng, Synthesis, 2013, 45, 3131–3136 CrossRef CAS; (g) S. Wacharasindhu, S. Bardhan, Z.-K. Wan, K. Tabei and T. S. Mansour, J. Am. Chem. Soc., 2009, 131, 4174–4175 CrossRef CAS PubMed.
- Y. Y. Peng, M. J. Zeng, H. M. Wang, J. Zhu, Q. Yang, Z. H. Deng and C. Y. Yu, Tetrahedron, 2015, 71, 9457–9462 CrossRef CAS.
- C. Y. Zhang, Y. R. Zhou, Z. H. Deng, X. Chen and Y. Y. Peng, Eur. J. Org. Chem., 2015, 1735–1744 CrossRef CAS.
- Reviews see: (a) G. Y. Song, F. Wang and X. W. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (b) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMedSelected recent examples, see: (c) S. J. Yu, S. Liu, Y. Lan, B. S. Wan and X. W. Li, J. Am. Chem. Soc., 2015, 137, 1623–1631 CrossRef CAS PubMed; (d) A. Geer, A. Serrano, B. Bruin, M. Ciriano and C. Tejel, Angew. Chem., Int. Ed., 2015, 54, 472–475 CAS; (e) K. Shibata and N. Chatani, Org. Lett., 2014, 16, 5148–5151 CrossRef CAS PubMed; (f) J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598–4601 CrossRef CAS PubMed; (g) R. Q. Ran, S. D. Xiu and C. Y. Li, Org. Lett., 2014, 16, 6394–6396 CrossRef CAS PubMed.
- (a) B. Parr and H. M. L. Davies, Org. Lett., 2015, 17, 794–797 CrossRef CAS PubMed; (b) R. Azpiroz, L. Rubio-Perez, A. D. Giuseppe, V. Passarelli, F. J. Lahoz, R. Castarlenas, J. J. Perez-Torrente and L. A. Oro, ACS Catal., 2014, 4, 4244–4253 CrossRef CAS; (c) X. C. Ma, J. Jiang, S. Y. Lv, W. F. Yao, Y. Yang, S. Y. Liu, F. Xia and W. H. Hu, Angew. Chem., Int. Ed., 2014, 53, 13136–13139 CrossRef CAS PubMed; (d) B. T. Parr and H. M. L. Davies, Nat. Commun., 2014, 5, 5455 CrossRef PubMed; (e) A. D. Giuseppe, R. Casrarlenas, J. J. Perez-Torrente, F. J. Lahoz and L. A. Oro, Chem.–Eur. J., 2014, 20, 8391–8403 CrossRef PubMed.
- (a) Y. Xia, S. Feng, Z. Liu, Y. Zhang and J. B. Wang, Angew. Chem., Int. Ed., 2015, 54, 7891–7894 CrossRef CAS PubMed; (b) X. Chen, X. W. Hu, S. Y. Bai, Y. F. Deng, H. F. Jiang and W. Zeng, Org. Lett., 2016, 18, 192–195 CrossRef CAS PubMed; (c) C. Song, C. Yang, F. F. Zhang, J. H. Wang and J. Zhu, Org. Lett., 2016, 18, 4510–4513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, 1H and 13C NMR spectra of compounds 3 and 3a. CCDC 1529457 and 1529463. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01131h |
| This journal is © The Royal Society of Chemistry 2017 |