
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophilic–hydrophobic phase transition of photoresponsive linear and macrocyclic poly(2-isopropyl-2-oxazoline)s†
Yongseok Jung,
Jaehyuk Nam,
Joo-Ho Kim and
Woo-Dong Jang *
*
Department of Chemistry, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea. E-mail: wdjang@yonsei.ac.kr
First published on 3rd February 2017
Abstract
A series of macrocyclic and linear poly(2-isopropyl oxazoline)s containing a photoresponsive azobenzene moiety (Az-PiPOxs) were synthesized by Cu(I)-catalyzed cycloaddition reaction between azide-functionalized azobenzene and alkyne-bearing poly(2-isopropyl oxazoline)s. Upon temperature elevation, both macrocyclic and linear Az-PiPOx exhibited a sharp hydrophilic-to-hydrophobic phase transition: the initially clear solution became turbid at specific temperatures. The photoisomerization of the azobenzene moiety resulted in the large change of the thermal transition temperature of the polymers. cis isomers exhibited a higher thermal transition temperature than trans isomers. Thus, the phase transition can be controlled by 365 and 254 nm UV irradiation.
Introduction
Stimuli-responsive polymers that change their properties according to external environment provide a great opportunity for the development of functional materials.1–4 Various types of stimuli-responsive polymers have been exploited for the design of functional materials such as biological interfaces, drug delivery systems, and logic gates.2,5–8 Poly(2-isopropyl-2-oxazoline) (PiPOx) is a well-known thermoresponsive polymer that exhibits hydrophilic–hydrophobic thermal transition via the lower critical solution temperature.9 Because PiPOxs have a lower critical solution temperature, around human body temperature, and excellent biocompatibility, they have been receiving significant attention for biomedical applications.10–13 Macrocyclic polymers (i.e., polymers with a closed topology) have no chain ends,14 and the structural uniqueness endows them with properties distinguishable from those of linear polymers. Namely, macrocyclic polymers have smaller hydrodynamic volume, lower solution viscosity, higher thermal stability, and higher glass transition temperature as compared to linear polymers of similar molecular weight.15–17 Moreover, macrocyclic polymers often show improved chemical stability due to the absence of chain ends,18 and the closed topology offers additional advantages for their application in the biomedical field.19–22 For example, Grayson et al. reported a cyclic poly(ethylene imine) (PEI) as a carrier for in vitro gene transfection, which exhibited higher transfection efficacy and lower cytotoxicity than linear PEI.20 In addition, some macrocyclic polymers displayed longer circulation times than linear polymers. Such unique properties of macrocyclic polymers are highly beneficial for drug delivery and other biomedical applications.23 However, because of their synthetic difficulty, macrocyclic polymers have not been sufficiently investigated. Ring-closure reactions between the end groups of linear polymers are frequently used for the synthesis of macrocyclic polymers;24,25 namely, the click reaction between azide and alkyne groups, which affords a triazole heterocycle in high yields and under very mild conditions,26,27 proved to be very useful.28–32 In this study, we have synthesized dual stimuli-responsive macrocyclic PiPOxs via click chemistry. Clickable PiPOxs ending with alkyne groups have been prepared via cationic ring-opening polymerization of oxazoline monomers.33–37 The click reaction with azobenzene derivatives having two azide groups afforded macrocyclic PiPOxs with a photoresponsive azobenzene group in the macrocyclic ring. The dual stimuli-responsive properties of the macrocyclic PiPOx were compared with those of the linear PiPOx.Experimental section
Materials and measurements
All commercially available reagents were reagent grade and used without further purification. Dichloromethane, N,N-dimethylformamide (DMF), carbon tetrachloride, tetrahydrofuran (THF), and acetonitrile were freshly distilled before each use. Recycling preparative size-exclusion chromatography (SEC) was performed on a LC-9201 (JAI, Tokyo, Japan) instrument equipped with JAIGEL-1H, JAIGEL-2H, and JAIGEL-3H columns using CHCl3 as the eluent. Electronic absorption spectra were measured using a V-660 (JASCO, Tokyo, Japan) spectrophotometer equipped with a thermostatic cell holder coupled with a controller (ETCS-761, JASCO, Tokyo, Japan). 1H NMR spectra were recorded using a Bruker DPX 400 (400 MHz) spectrometer in CDCl3 or CD2Cl2. Analytical SEC was performed on a JASCO HPLC equipped with HF-403HQ and HF-404HQ columns (Shodex, Tokyo, Japan) using THF as the eluent. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) measurements were performed on a Bruker model LRF20 using dithranol (1,8,9-trihydroxyanthracene) as the matrix. FT-IR spectra were recorded on Bruker Vertex 70 FT-IR spectrometer. UV irradiation experiments were performed using a UV hand lamp (Vilber Lourmat, France) equipped with 4 W UV discharge tubes. For the UV irradiation, quartz cells or NMR tubes were placed at a distance of 5 mm from the lamp.Determination of thermal transition temperature
The transmittance of the solution at 800 nm was measured using a spectrophotometer. The heating rate of the sample cells was adjusted to 1.0 °C min−1. The thermal transition temperature was taken as the temperature at which the transmittance reached 50% in the resulting transmittance versus temperature curves.Synthesis
The synthetic routes to azobenzene-containing linear and cyclic poly(2-isopropyl-2-oxazoline)s (l-Az-PiPOx and c-Az-PiPOx, respectively) are outlined in Scheme 1. 4,4′-Bis(azidomethyl)azobenzene and 2-isopropyl-2-oxazoline (iPOx) were synthesized according to literature procedures.38–40 | ||
| Scheme 1 Synthesis of l-Az-PiPOx and c-Az-PiPOx. | ||
Prop-PiPOx2k-Prop. 1.27 g of propargyl p-toluenesulfonate (1.04 mL, 6.02 mmol), 11.4 g of iPOx (12.0 mL, 100.7 mmol) and 0.835 g of N-methylpropargylamine (1.02 mL, 12.08 mmol) in acetonitrile (15.0 mL) were used to prepare Prop-PiPOx2k-Prop as white powder (7.8 g, 68%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 4.23–4.12 (m; initiation terminal N–CH2–C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ), 3.48–3.42 (broad d; –CH2–CH2– on the polymer backbone), 2.88–2.60 (broad t; –CH– on the polymer side chain, α-terminal –CCH, ω-terminal –N–CH2–CCH), 2.33 (broad s; terminal –N–CH3), 1.06 (strong broad s; –CH– on the polymer side chain).
), 3.48–3.42 (broad d; –CH2–CH2– on the polymer backbone), 2.88–2.60 (broad t; –CH– on the polymer side chain, α-terminal –CCH, ω-terminal –N–CH2–CCH), 2.33 (broad s; terminal –N–CH3), 1.06 (strong broad s; –CH– on the polymer side chain).
Prop-PiPOx4k-Prop. 0.79 g of propargyl p-toluenesulfonate (0.65 mL, 3.76 mmol), 14.3 g of iPOx (15.0 mL, 126.4 mmol) and 0.401 g of N-methylpropargylamine (0.49 mL, 5.80 mmol) in acetonitrile (20 mL) were used to prepare Prop-PiPOx4k-Prop as white powder (12.3 g, 86%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 4.24–4.13 (m; initiation terminal N–CH2–C
), 3.50–3.43 (broad d; –CH2–CH2– on the polymer backbone), 2.89–2.69 (broad t; –CH– on the polymer side chain, α-terminal –CCH, ω-terminal –N–CH2–CCH), 2.32 (broad s; terminal –N–CH3), 1.08 (strong broad s; –CH– on the polymer side chain).
PiPOx1k-Prop. 1.93 g of methyl p-toluenesulfonate (1.56 mL, 10.36 mmol), 9.5 g of iPOx (10.0 mL, 83.9 mmol) and 1.43 g of N-methylpropargylamine (1.75 mL, 20.7 mmol) in acetonitrile (12.5 mL) were used to prepare PiPOx1k-Prop mixture solution. The solution was concentrated under reduced pressure and 2.0 g of residue was purified by recycling preparative SEC using CHCl3 as the eluent. After purification, 1.4 g of PiPOx1k-Prop was recovered as a white powder upon lyophilisation (yield = 70%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 3.50–3.43 (broad d; –CH2–CH2– on the polymer backbone), 3.04 (broad s; terminal N–CH3), 2.90–2.55 (broad m; –CH– on the polymer side chain, terminal –N–CH2–C
CH), 2.32 (broad s; terminal –N–CH3), 1.08 (strong broad s; –CH– on the polymer side chain).
PiPOx2k-Prop. 0.55 g of propargyl p-toluenesulfonate (0.45 mL, 2.97 mmol), 5.7 g of iPOx (6.0 mL, 50.4 mmol) and 0.246 g of N-methylpropargylamine (0.30 mL, 3.6 mmol) in acetonitrile (9.0 mL) were used to prepare PiPOx2k-Prop as white powder (4.2 g, 74%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 3.50–3.43 (broad d; –CH2–CH2– on the polymer backbone), 3.04 (broad s; terminal N–CH3), 2.90–2.59 (broad m; –CH– on the polymer side chain, terminal –N–CH2–C
CH), 2.32 (broad s; terminal –N–CH3), 1.08 (strong broad s; –CH– on the polymer side chain).
c-Az-PiPOx2k. 500.0 mg of Prop-PiPOx2k-Prop (0.25 mmol), 74.5 mg of 4,4′-bis(azidomethyl)azobenzene (0.26 mmol), 399.0 mg of copper(II) sulfate (2.5 mmol), 495.3 mg of sodium ascorbate (2.5 mmol), and 600 mL of THF/H2O (99/1) were used to afford c-Az-PiPOx2k as a bright orange powder (160 mg, 32%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 7.92–6.82 (m; N–CH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the triazole ring, –CH on the azobenzene group), 5.61 (s; N–CH2–C between the triazole ring and the azobenzene group), 4.60 (broad s; N–CH2–C beside the triazole ring), 3.69 (broad s; N–CH2–C opposite side of the triazole ring), 3.47–3.41 (broad d; –CH2–CH2– on the polymer backbone), 2.88–2.50 (broad m; –CH– on the polymer side chain), 2.26 (broad s; terminal –N–CH3), 1.06 (strong broad s; –CH– on the polymer side chain).
N in the triazole ring, –CH on the azobenzene group), 5.61 (s; N–CH2–C between the triazole ring and the azobenzene group), 4.60 (broad s; N–CH2–C beside the triazole ring), 3.69 (broad s; N–CH2–C opposite side of the triazole ring), 3.47–3.41 (broad d; –CH2–CH2– on the polymer backbone), 2.88–2.50 (broad m; –CH– on the polymer side chain), 2.26 (broad s; terminal –N–CH3), 1.06 (strong broad s; –CH– on the polymer side chain).
c-Az-PiPOx4k. 500.0 mg of Prop-PiPOx4k-Prop (0.13 mmol), 37.3 mg of 4,4′-bis(azidomethyl)azobenzene (0.13 mmol), 199.5 mg of copper(II) sulfate (1.3 mmol), 247.6 mg of sodium ascorbate (2.5 mmol), and 600 mL of THF/H2O (99/1) were used to afford c-Az-PiPOx4k as a bright orange powder (160 mg, 32%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 8.01–6.81 (m; N–CH
N in the triazole ring, –CH on the azobenzene group), 5.61 (s; N–CH2–C between the triazole ring and the azobenzene group), 4.61 (broad s; N–CH2–C beside the triazole ring), 3.70 (broad s; N–CH2–C opposite side of the triazole ring), 3.49–3.43 (broad d; –CH2–CH2– on the polymer backbone), 2.89–2.51 (broad m; –CH– on the polymer side chain), 2.27 (broad s; terminal –N–CH3), 1.07 (strong broad s; –CH– on the polymer side chain).
l-Az-PiPOx2k. 500 mg of PiPOx1k-Prop (0.5 mmol), 73.1 mg of 4,4′-bis(azidomethyl)azobenzene (0.25 mmol), 399.0 mg of copper(II) sulfate (2.5 mmol), 495.3 mg of sodium ascorbate (2.5 mmol), and 60 mL of THF/H2O (9/1) were used to afford l-Az-PiPOx2k as a bright orange powder (210 mg, 42%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 7.92–6.83 (m; N–CH
N in the triazole ring, –CH on the azobenzene group), 5.60 (s; N–CH2–C between the triazole ring and the azobenzene), 3.70 (broad s; N–CH2–C beside the triazole ring), 3.49–3.42 (broad d; –CH2–CH2– on the polymer backbone), 3.04 (broad s; terminal N–CH3), 2.90–2.53 (broad m; –CH– on the polymer side chain), 2.27 (broad s; terminal –N–CH3), 1.07 (strong broad s; –CH– on the polymer side chain).
l-Az-PiPOx4k. 500 mg of PiPOx2k-Prop (0.5 mmol), 36.5 mg of 4,4′-bis(azidomethyl)azobenzene (0.125 mmol), 399.0 mg of copper(II) sulfate (2.5 mmol), 495.3 mg of sodium ascorbate (2.5 mmol), and 60 mL of THF/H2O (9/1) were used to afford l-Az-PiPOx4k as a bright orange powder (205 mg, 41%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ (ppm): 7.92–6.83 (m; N–CH
N in the triazole ring, –CH on the azobenzene group), 5.60 (s; N–CH2–C between the triazole ring and the azobenzene), 3.70 (broad s; N–CH2–C beside the triazole ring), 3.49–3.43 (broad d; –CH2–CH2– on the polymer backbone), 3.07 (broad s; terminal N–CH3), 2.90–2.52 (broad m; –CH– on the polymer side chain), 2.27 (broad s; terminal –N–CH3), 1.08 (strong broad s; –CH– on the polymer side chain).
Results and discussion
Synthesis and characterization
The synthetic routes to l-Az-PiPOxs and c-Az-PiPOxs are summarized in Scheme 1. Firstly, propargyl-bearing telechelic PiPOxs (Prop-PiPOx-Prop) were prepared for the synthesis of c-Az-PiPOxs. In order to introduce propargyl groups at both initiation and termination ends, propargyl p-toluenesulfonate and N-methylpropargylamine were used as the initiator and terminating agent, respectively. The ratio of initiator to monomer was adjusted to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 and 1:34 to obtain Prop-PiPOx-Prop with number average molecular weights (Mn) approximately 2.0 and 4.0 kDa, respectively. For the synthesis of l-Az-PiPOxs, the propargyl group was introduced only at the termination end. Methyl p-toluenesulfonate and N-methylpropargylamine were used as the initiator and terminating agent, respectively, to afford PiPOx-Prop. In order to obtain l-Az-PiPOxs with Mn similar to that of c-Az-PiPOxs, the initiator to monomer ratio was adjusted to 1:8 or 1:17. Next, the Cu(I)-catalysed cycloaddition (click reaction) between the propargyl groups in Prop-PiPOx-Prop and PiPOx-Prop and the azide groups in 4,4′-bis(azidomethyl)azobenzene were performed to obtain l-Az-PiPOxs and c-Az-PiPOxs, respectively. To minimize the formation of dimeric or trimeric compounds, the synthesis of c-Az-PiPOxs was performed under diluted conditions. It should be noted that l-Az-PiPOxs and c-Az-PiPOxs were purified by recycling preparative SEC to remove dimeric and trimeric compounds as well as low molecular weight impurities.
:17 and 1:34 to obtain Prop-PiPOx-Prop with number average molecular weights (Mn) approximately 2.0 and 4.0 kDa, respectively. For the synthesis of l-Az-PiPOxs, the propargyl group was introduced only at the termination end. Methyl p-toluenesulfonate and N-methylpropargylamine were used as the initiator and terminating agent, respectively, to afford PiPOx-Prop. In order to obtain l-Az-PiPOxs with Mn similar to that of c-Az-PiPOxs, the initiator to monomer ratio was adjusted to 1:8 or 1:17. Next, the Cu(I)-catalysed cycloaddition (click reaction) between the propargyl groups in Prop-PiPOx-Prop and PiPOx-Prop and the azide groups in 4,4′-bis(azidomethyl)azobenzene were performed to obtain l-Az-PiPOxs and c-Az-PiPOxs, respectively. To minimize the formation of dimeric or trimeric compounds, the synthesis of c-Az-PiPOxs was performed under diluted conditions. It should be noted that l-Az-PiPOxs and c-Az-PiPOxs were purified by recycling preparative SEC to remove dimeric and trimeric compounds as well as low molecular weight impurities.
The synthesized Az-PiPOxs were characterized by 1HNMR, MALDI-TOF-MS, and FT-IR measurements. The 1HNMR spectra of both c-Az-PiPOxs and l-Az-PiPOxs showed the proton signals of the azobenzene groups at 7.9–6.8 ppm, indicating the successful incorporation of the azobenzene units into the PiPOx chains through the click reaction. The 1HNMR spectra suggested that the azobenzene groups were predominantly in the trans configuration (Fig. 1 and S5†).
 | ||
| Fig. 1 1H NMR spectra of c-Az-PiPOx2k (top) and l-Az-PiPOx2k (bottom) in CD2Cl2. | ||
The absolute molecular weights of Az-PiPOxs were measured by MALDI-TOF-MS analysis (Fig. 2 and S8†). In both cases of c-Az-PiPOxs and l-Az-PiPOxs, a series of peaks at approximately 113.16 Da intervals was observed, and the obtained m/z values showed less than 0.1% deviation from the calculated values. The Mn obtained form MALDI-TOF-MS (Mn-MS) of c-Az-PiPOx2k, l-Az-PiPOx2k, c-Az-PiPOx4k, and l-Az-PiPOx4k were estimated to be 4.18, 4.04, 2.72 and 2.79 kDa, respectively.
 | ||
| Fig. 2 MALDI-TOF-MS spectra of c-Az-PiPOx2k (red line) and l-Az-PiPOx2k (blue line). | ||
FT-IR measurements were performed to monitor the cycloaddition reaction between 4,4′-bis(azidomethyl)azobenzene and the terminal alkyne groups of PiPOxs. As shown in Fig. 3, the FT-IR spectrum of 4,4′-bis(azidomethyl)azobenzene displays a sharp absorption peak at 2090 cm−1, which originates from the azide groups. This peak completely disappeared upon formation of c-Az-PiPOx2k and l-Az-PiPOx2k, indicating complete conversion during the cycloaddition reaction. The same aspect was observed for c-Az-PiPOx4k and l-Az-PiPOx4k (Fig. S9†).
 | ||
| Fig. 3 FT-IR spectra of 4,4′-bis(azidomethyl)azobenzene, l-Az-PiPOx2k and c-Az-PiPOx2k. | ||
Analytical SEC was performed for Az-PiPOxs (Fig. 4 and S12†) to estimate Mn values (Mn-sec) and dispersity indices (Đ). The retention peaks of c-Az-PiPOx2k, l-Az-PiPOx2k, c-Az-PiPOx4k, and l-Az-PiPOx4k were appeared at 20.43, 20.65, 21.20 and 20.85 min, respectively. The Mn-sec values of c-Az-PiPOx2k, l-Az-PiPOx2k, c-Az-PiPOx4k, and l-Az-PiPOx4k were estimated to be 1.84, 2.53, 3.29, and 3.71 kDa, respectively, based on polystyrene standards. It is noteworthy that the Mn-sec values of l-Az-PiPOx2k and l-Az-PiPOx4k were only 260 and 330 Da deviated from the Mn-MS values of them, whereas the Mn-sec values of c-Az-PiPOx2k and c-Az-PiPOx4k were much smaller than the Mn-MS values of them (Table 1). In the SEC experiment, the retention time depends on the hydrodynamic volume of the polymer molecules. Generally, the hydrodynamic volume of macrocyclic polymers is smaller than that of linear polymers with equivalent molecular weight. Thus, the large differences of Mn-sec values between c-Az-PiPOxs and l-Az-PiPOxs would be another evidence of macrocycle formation. Because the polymerization of iPOx proceeded through a living mechanism, PiPOx-Prop and Prop-PiPOx-Prop showed very narrow molecular weight distributions. After the click reaction with 4,4′-bis(azidomethyl)azobenzene, the resulting c-Az-PiPOx2k, l-Az-PiPOx2k, c-Az-PiPOx4k, and l-Az-PiPOx4k were exhibited Đ values of 1.06, 1.06, 1.07, and 1.09. In the preparation procedure of Az-PiPOxs, by-products and unreacted polymers were removed by recycling preparative SEC. Hence, very narrow molecular weight distributions could be obtained for both l-Az-PiPOxs and c-Az-PiPOxs.
 | ||
| Fig. 4 SEC traces of l-Az-PiPOx2k (blue line) and c-Az-PiPOx2k (red line). | ||
| Mn,theoa | SECb | MSc | DPc | |||
|---|---|---|---|---|---|---|
| Mn | Đ | Mn | Đ | |||
| a Determined from the initiator/monomer ratio and the degree of conversion.b Determined by SEC analysis in THF using polystyrene standard.c Determined by MALDI-TOF-MS analysis. | ||||||
| PiPOx1k-Prop | 1000 | 1020 | 1.12 | 1180 | 1.02 | 10 |
| PiPOx2k-Prop | 2000 | 2050 | 1.07 | 1990 | 1.01 | 17 |
| Prop-PiPOx2k-Prop | 2000 | 2540 | 1.09 | 2240 | 1.01 | 20 |
| Prop-PiPOx4k-Prop | 4000 | 4550 | 1.06 | 3890 | 1.01 | 34 |
| l-Az-PiPOx2k | 2290 | 2530 | 1.07 | 2790 | 1.00 | 22 |
| c-Az-PiPOx2k | 2290 | 1840 | 1.06 | 2720 | 1.01 | 21 |
| l-Az-PiPOx4k | 4290 | 3710 | 1.09 | 4040 | 1.01 | 31 |
| c-Az-PiPOx4k | 4290 | 3290 | 1.07 | 4180 | 1.01 | 33 |
Photoisomerization of the azobenzene moiety
In order to monitor the photoisomerization of the azobenzene moiety, a solution of c-Az-PiPOx2k in phosphate buffer (10 mM PBS and 150 mM NaCl, pH 7.4) was prepared and its change in UV-Vis absorption upon UV irradiation was measured. Upon 365 nm UV irradiation, the absorption spectrum of the c-Az-PiPOx2k solution (0.2 g L−1) showed a gradual decrease of the peak at 329 nm and an increase of the band at 263 and 432 nm with clear isosbestic points at 282 and 390 nm (Fig. 5a), indicating trans-to-cis photoisomerization of the azobenzene moiety. The spectral shift was saturated after 15 min of 365 nm irradiation. Opposite changes were observed after 5 min of 254 nm UV irradiation (Fig. 5b). The cis-to-trans photoisomerization was faster than the trans-to-cis photoisomerization, presumably because the trans configuration is more stable. In order to confirm the reversibility of the photoisomerization, this experiment was repeated five times: as shown in Fig. 5c, the photoisomerization of the azobenzene moiety in c-Az-PiPOx2k was fully reversible under 365 and 254 nm UV irradiation.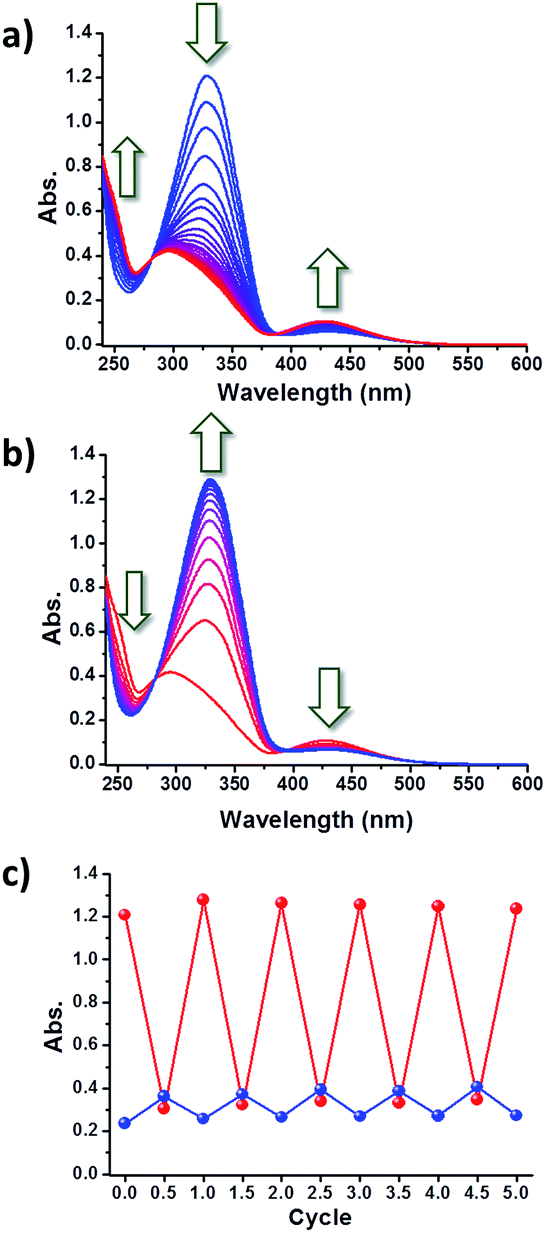 | ||
| Fig. 5 UV-Vis absorption spectral changes of c-Az-PiPOx2k in 10 mM PBS (0.2 g L−1) upon (a) 365 and (b) 254 nm irradiation. (c) Absorbance changes at 263 (blue) and 329 nm (red) upon 365 and 254 nm irradiation for 5 cycles. | ||
The photoisomerization of c-Az-PiPOx2k and 4,4′-bis(azidomethyl)azobenzene was monitored by 1H NMR spectroscopy. Solutions of c-Az-PiPOx2k and 4,4′-bis(azidomethyl)azobenzene in CD2Cl2 were irradiated with 365 nm UV light for 2 h to reach steady state conditions, and the 1H NMR spectra were recorded (Fig. 6). The cis ratio of c-Az-PiPOx2k was estimated to be slightly lower (80%) than that of 4,4′-bis(azidomethyl)azobenzene (89%).
 | ||
| Fig. 6 1HNMR spectra of 4,4′-bis(azidomethyl)azobenzene in the trans (a) and cis (b) configuration, and of c-Az-PiPOx2k in the trans (c) and cis configuration (d), in CD2Cl2. The peaks under the blue and red circles are attributed to the trans and cis forms of the azobenzene moieties, respectively. | ||
Thermoresponsiveness of Az-PiPOxs
The thermal transition temperatures of Az-PiPOxs were determined by temperature-dependent transmittance measurements. The transmittance of the solutions at 800 nm was monitored during temperature elevation of 1 °C min−1 rate, and the thermal transition temperatures were defined as the temperature at which 50% of the transmittance change occurred. Fig. 7 shows temperature-dependent transmittance changes of c-Az-PiPOxs and l-Az-PiPOxs at the concentration of 2.0 g L−1. The thermal transition temperatures of l-Az-PiPOx2k, c-Az-PiPOx2k, l-Az-PiPOx4k and c-Az-PiPOx4k were determined to be 39.4, 33.8, 39.9 and 35.5 °C, respectively. It is well known that the thermal transition temperature of PiPOx decreases by increasing molecular weight. As expected, Az-PiPOx2ks show higher thermal transition temperature than Az-PiPOx4ks. On the other hand, the thermal transition temperatures of l-Az-PiPOxs were higher than those of c-Az-PiPOxs. Because the chemical structures of l-Az-PiPOxs and c-Az-PiPOxs are not identical, the direct comparison of thermal transition temperatures may not be greatly meaningful. However, the absence of chain terminals in c-Az-PiPOxs may influence on hydrophilicity of total polymers. When compared the optical transmittance change curves, l-Az-PiPOxs showed sharper transition than c-Az-PiPOxs. Such aspect has been reported previously in several poly(N-isopropyl acrylamide)-based systems.30,41 It has been expected that the cyclic topology of thermoresponsive polymers can be affected the phase transition by the existence of repulsive force to prohibit intermolecular aggregation and steric constraints to induce intramolecular chain interactions. | ||
| Fig. 7 Temperature dependent transmittance changes of (a) Az-PiPOx2ks and (b) Az-PiPOx4ks. Black and blue lines are corresponding to c-Az-PiPOxs and l-Az-PiPOxs, respectively. Solid and dashed lines are corresponding to trans and cis configurations of each polymers, respectively. | ||
To examine concentration dependency of thermal transition temperature, solutions of each Az-PiPOxs with concentration of 4.0, 2.0, 1.0 and 0.5 g L−1 in 10 mM PBS (NaCl 150 mM, pH 7.4) were prepared and optical transmittance changes were recorded. As increasing concentration of all Az-PiPOxs, the thermal transition temperatures were gradually decreased. Fig. 8 shows concentration dependent thermal transition temperature changes of Az-PiPOxs. All Az-PiPOxs showed linear correlation between thermal transition temperatures and logarithmic concentration. The steepness of slopes were varied by molecular weight and molecular topology. The slopes of Az-PiPOx2ks were steeper than those of Az-PiPOx4ks, indicating that lower molecular weight polymers have greater concentration dependency. On the other hand, the slopes of c-Az-PiPOxs were steeper than those of l-Az-PiPOxs, indicating cyclic polymers have higher concentration dependency than linear polymers.
 | ||
| Fig. 8 Concentration dependent thermal transition temperature changes of (a) Az-PiPOx2ks and (b) Az-PiPOx4ks. | ||
The azobenzene moieties of l-Az-PiPOxs and c-Az-PiPOxs were photoisomerized to the cis form by 365 nm UV irradiation. After saturation of the photoisomerization, the thermal transition temperatures of l-Az-PiPOx2k, c-Az-PiPOx2k, l-Az-PiPOx4k, and c-Az-PiPOx4k at 2.0 g L−1 were increased about 42.9, 39.1, 42.7 and 38.6 °C, respectively (Fig. 7 and 8). This observation can be explained by the polarization and morphological change of the azobenzene moiety: cis-azobenzene has a higher polarity than the trans isomer because the lone pair electrons are constrained to be on the same side. One of the unique aspects of the thermoresponsiveness of l-Az-PiPOxs and c-Az-PiPOxs is the difference in thermal transition temperature between trans and cis isomers. In our previous study, an increase in thermal transition temperature of only 0.9 °C was observed upon trans-to-cis photoisomerization of a PiPOx polymer with azobenzene moieties at the α and ω terminal ends.5 In contrast, in this work, the cis and trans forms of both l-Az-PiPOxs and c-Az-PiPOxs exhibited significantly different thermal transition temperatures, where the differences of l-Az-PiPOx2k, c-Az-PiPOx2k, l-Az-PiPOx4k, and c-Az-PiPOx4k were 3.5, 5.3, 3.8, and 3.1 °C, respectively. This unique property can be attributed to morphological changes of the polymers by photoisomerization. In our previous study, the photoisomerization of the azobenzene moieties cannot induce morphological changes of whole of polymer because the azobenzene moieties are located at the terminal ends of the polymeric structure. In contrast, the morphologies of l-Az-PiPOxs and c-Az-PiPOxs can be drastically changed because the photoisomerizable unit located in the middle of polymer chains. Moreover, the morphological aspect of c-Az-PiPOxs would be greatly influenced by the photoisomerization due to the closed topology.
The large change in thermal transition temperature would be very useful for the design of thermo- and photo-responsive materials. Namely, Az-PiPOxs are highly attractive for application in the biomedical field because its thermal transition proceeds near the body temperature body temperature. As an example, we measured the phase transition of c-Az-PiPOx2k at 2.0 g L−1 concentration at body temperature. The solution of trans-c-Az-PiPOx2k became turbid when the temperature was raised to 36 °C. Under these conditions, the solution was continuously irradiated with 365 nm UV light and became transparent as a result of the trans-to-cis photoisomerization process (Fig. 9). Alternatively, the clear solution became turbid by continuous irradiation with 254 nm UV light. Thus, the hydrophilic–hydrophobic transition of Az-PiPOxs could be successfully controlled by photoirradiation.
 | ||
| Fig. 9 Turbidity change of c-Az-PiPOx2k at 36 °C by UV irradiation. | ||
Conclusions
In summary, dual stimuli-responsive linear and macrocyclic poly(2-isopropyl oxazoline) polymers containing a photoresponsive azobenzene group were prepared via living cationic polymerization followed by click reaction. In each polymer, the azobenzene moiety exhibited photoisomerization upon UV irradiation, and hydrophilic–hydrophobic phase transition was achieved upon temperature elevation. The photoisomerization of the azobenzene moiety resulted in the change of the thermal transition temperature of the polymers. cis isomers exhibited a higher thermal transition temperature than trans isomers. Thus, the phase transition can be controlled by 365 and 254 nm UV irradiation. Such unique property would make it an attractive material for various applications.Acknowledgements
This work was supported by the Mid-Career Researcher Program (2014R1A2A1A10051083) funded by the National Research Foundation (NRF) of Korea.Notes and references
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- J. H. Kim, Y. Jung, D. Lee and W. D. Jang, Adv. Mater., 2016, 28, 3499–3503 CrossRef CAS PubMed.
- J.-H. Kim, E. Koo, S.-Y. Ju and W.-D. Jang, Macromolecules, 2015, 48, 4951–4956 CrossRef CAS.
- C. de las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- F. D. Jochum, F. R. Forst and P. Theato, Macromol. Rapid Commun., 2010, 31, 1456–1461 CrossRef CAS PubMed.
- S. Masuda, T. Shimizu, M. Yamato and T. Okano, Adv. Drug Delivery Rev., 2008, 60, 277–285 CrossRef CAS PubMed.
- H. Uyama and S. Kobayashi, Chem. Lett., 1992, 1643–1646 CrossRef CAS.
- A. L. Demirel, M. Meyer and H. Schlaad, Angew. Chem., Int. Ed., 2007, 46, 8622–8624 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- R. Victor, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef PubMed.
- J. Zhao, R. Hoogenboom, G. Van Assche and B. Van Mele, Macromolecules, 2010, 43, 6853–6860 CrossRef CAS.
- Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097 CrossRef CAS.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- X. Zhu, N. Zhou, Z. Zhang, B. Sun, Y. Yang, J. Zhu and X. Zhu, Angew. Chem., Int. Ed., 2011, 50, 6615–6618 CrossRef CAS PubMed.
- K. Endo, in New Frontiers in Polymer Synthesis, Springer, 2008, pp. 121–183 Search PubMed.
- M. Trabi and D. J. Craik, Trends Biochem. Sci., 2002, 27, 132–138 CrossRef CAS PubMed.
- H. Wei, D. S. Chu, J. Zhao, J. A. Pahang and S. H. Pun, ACS Macro Lett., 2013, 2, 1047–1050 CrossRef CAS PubMed.
- M. A. Cortez, W. T. Godbey, Y. Fang, M. E. Payne, B. J. Cafferty, K. A. Kosakowska and S. M. Grayson, J. Am. Chem. Soc., 2015, 137, 6541–6549 CrossRef CAS PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS PubMed.
- X. Y. Tu, M. Z. Liu and H. Wei, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1447–1458 CrossRef CAS.
- B. Chen, K. Jerger, J. M. Fréchet and F. C. Szoka, J. Controlled Release, 2009, 140, 203–209 CrossRef CAS PubMed.
- K. Pangilinan and R. Advincula, Polym. Int., 2014, 63, 803–813 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- D. M. Eugene and S. M. Grayson, Macromolecules, 2008, 41, 5082–5084 CrossRef CAS.
- D. Pasini, Molecules, 2013, 18, 9512–9530 CrossRef CAS PubMed.
- X.-P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- Y. Satokawa, T. Shikata, F. Tanaka, X.-P. Qiu and F. M. Winnik, Macromolecules, 2009, 42, 1400–1403 CrossRef CAS.
- Y.-Y. Yuan, J.-Z. Du and J. Wang, Chem. Commun., 2012, 48, 570–572 RSC.
- R. Hoogenboom, F. Wiesbrock, H. Huang, M. A. Leenen, H. M. Thijs, S. F. van Nispen, M. van der Loop, C.-A. Fustin, A. M. Jonas and J.-F. Gohy, Macromolecules, 2006, 39, 4719–4725 CrossRef CAS.
- A. Makino and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1251–1270 CrossRef CAS.
- J.-S. Park and K. Kataoka, Macromolecules, 2006, 39, 6622–6630 CrossRef CAS.
- J.-H. Kim, E. Lee, J.-S. Park, K. Kataoka and W.-D. Jang, Chem. Commun., 2012, 48, 3662–3664 RSC.
- J.-H. Kim, D. Yim and W.-D. Jang, Chem. Commun., 2016, 52, 4152–4155 RSC.
- W. A. Velema, M. van der Toorn, W. Szymanski and B. L. Feringa, J. Med. Chem., 2013, 56, 4456–4464 CrossRef CAS PubMed.
- T. Ogoshi, K. Yoshikoshi, T. Aoki and T. A. Yamagishi, Chem. Commun., 2013, 49, 8785–8787 RSC.
- M. Meyer and H. Schlaad, Macromolecules, 2006, 39, 3967–3970 CrossRef CAS.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, MALDI-TOF-MS and IR spectra, SEI profiles. See DOI: 10.1039/c7ra01042g |
| This journal is © The Royal Society of Chemistry 2017 |