
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophosphorylation of aliphatic alkynes catalyzed by CuNPs/ZnO for the synthesis of vinyl phosphonates. A DFT study on the reaction mechanism†‡
Leandro Fortunato ,
Yanina Moglie,
Viviana Dorn* and
Gabriel Radivoy*
,
Yanina Moglie,
Viviana Dorn* and
Gabriel Radivoy*
INQUISUR, Departamento de Química, Universidad Nacional del Sur (UNS)-CONICET, Av. Alem 1253, 8000 Bahía Blanca, Argentina. E-mail: gradivoy@criba.edu.ar
First published on 28th March 2017
Abstract
The reaction mechanism of the CuNPs/ZnO-catalyzed hydrophosphorylation of aliphatic alkynes for the synthesis of vinyl phosphonates has been investigated. Based on experimental observations and theoretical calculations through DFT studies, a reaction mechanism is proposed, in which a crucial role of both the solvent and the catalyst support is suggested. DFT studies and experimental data, are consistent with a reaction mechanism based on a copper-catalyzed anti-Markovnikov hydrophosphorylation process that favours the formation of the vinyl phosphonate E isomer.
Introduction
Vinyl phosphonates are well-known constituents of the family of phosphorus containing organic compounds. They are of extensive importance in synthetic organic chemistry both as intermediates and as final products,1 and have different applications as monomers and co-monomers in polymeric materials.2 At present, the main methods for the synthesis of vinyl phosphonates are based on expensive and/or toxic homogeneous Pd3 or Ni4 catalysts, most of them requiring the use of phosphine ligands and/or severe reaction conditions. Recently, the silver-catalyzed phosphorylation of styrenes by using AgNO3/K2O2S8/TEMPO has been reported.5 On the other hand, copper-based catalysts for the synthesis of vinyl phosphonates through hydrophosphorylation of alkynes have remained almost unexplored to date.6In the last years, we have actively been working in the development of new and mild methodologies based on the use of bare or supported copper nanoparticles (CuNPs) for their application in the construction of C–C and C–heteroatom bonds.7 In a recent publication,8 we informed the direct synthesis of vinyl phosphonates starting from aliphatic alkynes and commercial diethyl phosphite, catalyzed by CuNPs supported on ZnO. The reactions were carried out in acetonitrile (ACN) as solvent, under air, in the absence of any additive or ligand, and under mild reaction conditions. Notably, the use of ZnO as support and ACN as solvent was mandatory for the reaction to take place.
On the other hand, there are few papers in the scientific literature about DFT computational studies regarding the mechanism involved in the hydrophosphorylation of alkynes. Beletskaya et al.9a have investigated the Markovnikov-type regioselective hydrophosphorylation of terminal alkynes employing a nickel catalyst and phosphine ligands in THF, as well as the mechanism involved in this alkyne hydrophosphorylation reaction by means of DFT methods. After that work, the same group published an in-depth theoretical study about the pathway involved in the key step of the hydrophosphorylation reaction, i.e. the alkyne insertion into the metal-phosphorus or metal-hydrogen bonds.9b More recently, Zhao and coworkers6a reported a very interesting mechanistic study about the copper-catalyzed (CuI) phosphorylation of terminal alkynes in the presence of a base (NEt3) and DMSO as the solvent, although in this case the starting alkyne is converted into the corresponding copper acetylide under the reaction conditions.
With regard to the phosphorylating species acting in these reactions, it is known that H-phosphonates may exist in two tautomeric forms being the (RO)2P(O)H tautomer more stable than the (RO)2POH one.9d–e On the other hand, it has been suggested that the presence of a Lewis acid/base such as ZnO, could promote the formation of (RO)2POH tautomer.10 Unveiling the mechanistic nature of the hydrophosphorylation reaction is crucial for the rational design of new and selective catalysts. Prompted by our continuing interest in the use of computational methods for the elucidation of organic reaction mechanisms,11 we carried out a theoretical study based on DFT methods in order to shed light into the mechanism involved in the CuNPs/ZnO-catalyzed hydrophosphorylation of alkynes for the synthesis of vinyl phosphonates.
Results and discussion
As we reported in our previous work,8 a distinctive feature of this CuNPs/ZnO catalyst was the observed selectivity dependent on the alkyne nature, aromatic alkynes giving β-ketophosphonates whereas aliphatic ones leading to the corresponding vinyl phosphonates as the main reaction products. The reaction conditions are shown in Scheme 1. | ||
| Scheme 1 Synthesis of vinyl phosphonates from aliphatic alkynes. | ||
Based on some additional experiments, we proposed a plausible mechanistic pathway for the formation of the β-ketophosphonate products through a radical oxyphosphorylation process. However, a full explanation for the hydrophosphorylation of the aliphatic alkynes leading to the corresponding anti-Markovnikov vinyl phosphonates was not possible from the experimental results. Even though, on the basis of the experimental observations we could disregard both the participation of radical species (hydrophosphorylation also works in the presence of TEMPO) and copper acetylides as reaction intermediates (deuterated-vinyl phosphonate product was obtained when starting from 1-deuterio-oct-1-yne).
It is worthy of note that the nature of the solvent and the catalyst support showed to be crucial for the outcome of the reaction and, consequently, need to be considered in a mechanistic proposal. Thus, the use of ZnO as the catalyst support and acetonitrile as the solvent proved to be essential for the hydrophosphorylation to take place. As above mentioned, Lewis acid (Zn2+) and/or Lewis basic (O2−) sites on ZnO could be promoting the tautomerism of the dialkyl phosphite (Fig. 1). In fact, other supports tested such as PVP, CeO2, cellulose, Celite, MWNT and MCM-41 gave low conversions into the desired vinyl phosphonate products. On the other hand a dramatic fall in conversion values was observed when a solvent different from acetonitrile was used (MeOH, H2O, DMSO, THF, toluene, dichloromethane).
 | ||
| Fig. 1 Tautomeric forms of H-phosphonate. | ||
In view of these observations, we propose that the reaction is likely to occur via a copper-catalyzed anti-Markovnikov hydrophosphorylation process (Scheme 2), leading to the corresponding E vinyl phosphonates through the addition of the (EtO)2(HO)P: nucleophile (N, Fig. 1) to the carbon–carbon triple bond. Thus, we assume that ZnO could be playing a non-innocent role in the catalytic system, probably through the P–H bond activation, with the solvent acting as a ligand for copper. As shown in Scheme 2, the protonated ZnO species would be responsible for the proton transfer to give the final product and the Cu/ZnO catalyst to restart the catalytic cycle. In Scheme 2 the structures for the proposed intermediates (IN) and the transition states (TS) are shown.
 | ||
| Scheme 2 Proposed mechanistic pathway for the synthesis of (E) vinyl phosphonates from aliphatic alkynes catalysed by CuNPs/ZnO. | ||
With the aim to explain these experimental results and find a close understanding of the reaction mechanism, we performed a computational analysis with the Gaussian 09 (ref. 12) software package. For this purpose, we simplified the reactive system and theoretically studied the process by using ethynylcyclohexane and dimethyl phosphite as model starting materials. The density functional theory (DFT)13 calculations were performed with the B3LYP14 functional, applying the D2 Grimme's dispersion corrections15 as implemented in G09 Rev. C.01, which is known to be an appropriate methodology for the mechanistic studies on Cu-catalyzed reactions,9b,6a and the 6-31+G* basis set for all atoms. The energies in solution were obtained with the Tomasi's polarized continuum model (PCM)16 using acetonitrile as implemented in Gaussian 09.
As we assumed that the reaction would start when the alkyne is activated by the copper catalyst, it was necessary to establish each catalyst ligand and how these ligands were attached to the metal acting as stabilizers. We modeled different structures for the active copper catalyst with the aim to find the most representative structure for the catalyst acting in our mechanistic proposal. With regard to the zinc oxide, small clusters of (ZnO)n, n = 1–7 have been modeled through DFT methods,17 and we adopted a dimeric species with a square planar geometry as the most simple and stable structure for model this oxide.18 So, we assumed that copper would be attached to ZnO dimer by the oxygen atoms and with the solvent (acetonitrile) acting as ligands. As can be seen from Fig. 2, the formation of all the proposed structures I–VI could take place exothermically, while, the catalyst I showed to be the simplest one and thermochemically more favored, since it occur with an exothermicity of −63.5 kcal mol−1. When this catalyst was modeled using DMSO instead of acetonitrile as ligand, the process showed to be markedly less exothermic (−46.6 kcal mol−1).
 | ||
| Fig. 2 Geometries and formation energies for the different copper catalysts modeled (B3LYP-D/6-31+G*). | ||
Once the catalyst was modeled, we start the computational study of the proposed mechanism. We considered that, in the first stage, the reaction mechanism would involve the formation of an alkynyl copper complex with an endothermicity of 0.44 kcal mol−1, through a π-coordination between the alkyne and the copper catalyst (IN1, Scheme 2).
With regard to the phosphorylating species, as commented above, despite being the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O form more stable (8.4 kcal) than the P–OH one,19 coordination of ZnO with the P center favours the formation of the P–OH tautomer (N, Fig. 1), thus being 8.1 kcal mol−1 more stable than the PO form. As shown in Scheme 2, this P–OH phosphonate tautomer, would be the active phosphorylating agent, which through a nucleophilic attack over IN1, would lead to the intermediate IN2 (−19.3 kcal mol−1), the driving force for the tautomerization process being also assisted by the formation of P–Cu bond (IN2, 2.3 Å). To locate the transition state (TS1) for this reaction pathway in a more simple way, we modeled a reactive-like structure that we called reactive-complex (RC) and a product-like structure called product-complex (PC). The formation of the product-complex is exothermic (−7.0 kcal mol−1) and takes place with a very low activation energy of 0.21 kcal mol−1 (Fig. 3).
O form more stable (8.4 kcal) than the P–OH one,19 coordination of ZnO with the P center favours the formation of the P–OH tautomer (N, Fig. 1), thus being 8.1 kcal mol−1 more stable than the PO form. As shown in Scheme 2, this P–OH phosphonate tautomer, would be the active phosphorylating agent, which through a nucleophilic attack over IN1, would lead to the intermediate IN2 (−19.3 kcal mol−1), the driving force for the tautomerization process being also assisted by the formation of P–Cu bond (IN2, 2.3 Å). To locate the transition state (TS1) for this reaction pathway in a more simple way, we modeled a reactive-like structure that we called reactive-complex (RC) and a product-like structure called product-complex (PC). The formation of the product-complex is exothermic (−7.0 kcal mol−1) and takes place with a very low activation energy of 0.21 kcal mol−1 (Fig. 3).
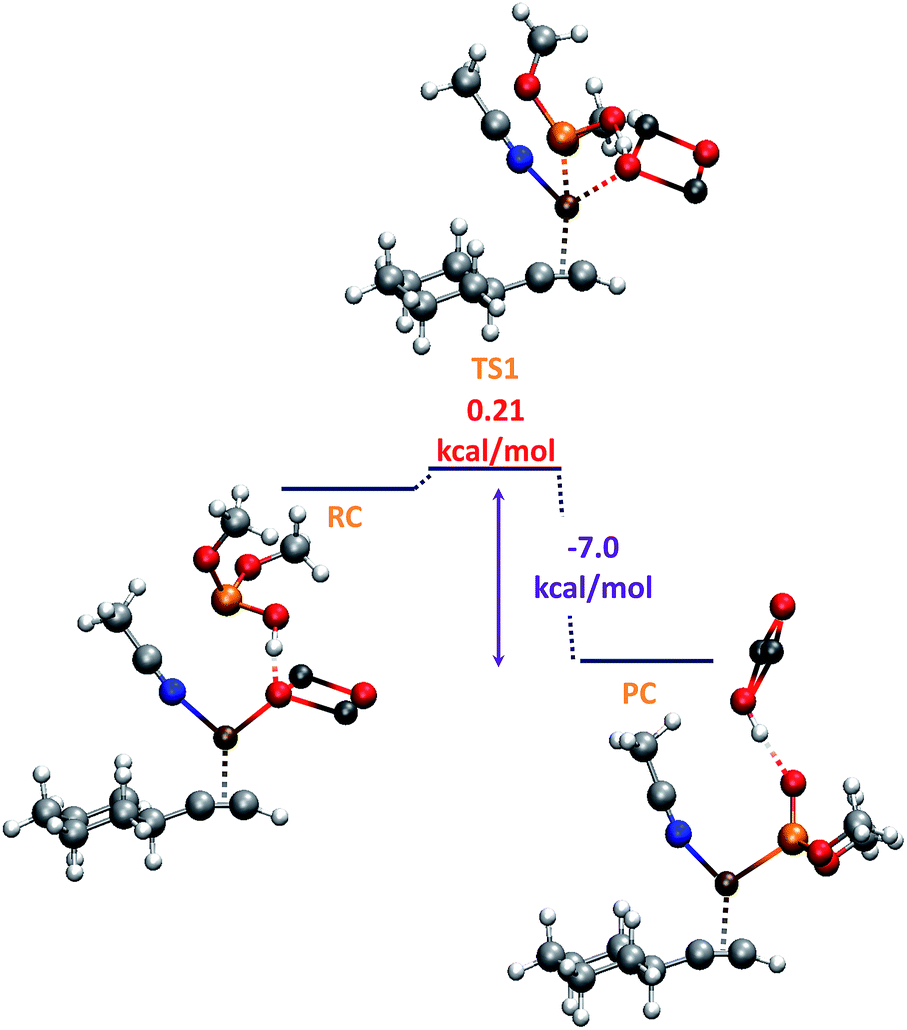 | ||
| Fig. 3 B3LYP-D/6-31+G* potential energy profile for initial RC and PC complexes. | ||
In a subsequent step, through a nucleophilic addition, the P–C bond is formed with a high activation energy of 33.3 kcal mol−1, exothermically leading to the five membered cyclic intermediate IN3 (−21.3 kcal mol−1), which shows a strong coordination between copper and the oxygen atom of PO group, with a O–Cu distance of 2.5 Å (showed as a red dashed line in Fig. 4). When this step was modeled by using DMSO as ligand and solvent, the process showed to be less exothermic (−17.1 kcal mol−1) and with a higher activation energy (38.3 kcal mol−1).
 | ||
| Fig. 4 B3LYP-D/6-31+G* potential energy profile for the nucleophilic addition and protonation steps in the formation of E vinyl phosphonate and MK charges (in blue). | ||
The next step involves the ZnO–H approach to intermediate IN3, through a strong interaction between zinc and the oxygen atom of the PO group, rendering the intermediate IN4 with an exothermicity of 26.0 kcal mol−1 (Fig. 4).
The proton transfer from the ZnO–H to give the intermediate IN5 is kinetically and thermodynamically unfavourable, being the most energy-demanding reaction step, it is slightly exothermic (−0.09 kcal mol−1) and occur with an activation energy of 33.5 kcal mol−1. As can be seen from Fig. 4, a stabilizing electrostatic interaction (red dashed line) is observed both in IN4 and TS3 structures, between the H atom of acetonitrile and the O atom of zinc oxide, located at a distance of 1.9 Å. This stabilizing interaction is evidenced by the Merz-Singh–Kollman (MK) charges20 which have a larger value for the H atom of acetonitrile closest to the O atom of zinc oxide, both in IN4 and TS3 (Fig. 4, charge values indicated in blue). This step was also the most energy-demanding when it was modeled using DMSO, but the process showed to be endothermic (+4.5 kcal mol−1) and with a higher activation energy (39.9 kcal mol−1).
Finally, the active copper catalyst is regenerated leading to the E vinyl phosphonate product with an endothermicity of 5.4 kcal mol−1; here again when the reaction was modeled with DMSO as ligand and solvent this step showed to be endothermic by 16.6 kcal mol−1.
On the other hand, we experimentally observed the formation of 5–30% of the Z vinyl phosphonate in all the hydrophosphorylation reactions. Taking into account the aforementioned mechanistic considerations, the formation of the Z isomer could proceed through the mechanism shown in Scheme 3.
 | ||
| Scheme 3 Proposed mechanistic pathway for the formation of (Z) vinyl phosphonate by-product. | ||
The key difference with the mechanism proposed in Scheme 2 for the E isomer formation, is that in this case, an external nucleophilic attack by a non-coordinated phosphorus nucleophile onto the alkynyl copper complex (IN1) to give intermediate IN6 would take place, via a C–P bond9a formation step. The DFT calculations about this process showed that it is 5 kcal mol−1 less exothermic than the formation of IN3 (E isomer). The P–C distance is 1.77 Å in the intermediate IN3 (E isomer) and 1.81 Å in the intermediate IN6 (Z isomer), and IN6 is 4.1 kcal mol−1 less stable than IN3. This could be probably due to the lack of extra-stabilizing factors such as the formation of a P–Cu bond (present in IN2), and the possibility of a strong coordination between copper and the oxygen atom of PO group in IN3, which is avoided for IN6 intermediate due to the trans orientation of these two groups (Fig. 5).
 | ||
| Fig. 5 B3LYP-D/6-31+G* geometry of intermediates IN3 (E isomer) and IN6 (Z isomer). | ||
The formation of IN7 through an interaction between Zn and the oxygen atom of the PO group (Fig. 6), is more exothermic (−30.9 kcal mol−1) than that of IN4 (E isomer, −26.0 kcal mol−1). This result is in line with the fact that IN6 (precursor of intermediate IN7) is less stable than IN3 (precursor of intermediate IN4). Finally, the proton transfer from ZnO–H to give IN8, is the most energy-demanding reaction step, and occurs with an activation energy of 40.0 kcal mol−1, i.e. 6.5 kcal mol−1 higher than that of the same proton transfer step for the case of the E isomer (formation of intermediate IN5). Besides, the formation of IN8 is endothermic (+4.6 kcal mol−1) whereas the formation of IN5 is slightly exothermic (−0.09 kcal mol−1). In part, this could be due to the lack of stabilizing electrostatic interactions, contrary to that observed for IN4 and TS3 (Fig. 4) which lead to the E vinyl phosphonate product (compare MK charges in Fig. 4 with those shown in Fig. 6). Finally, the Z vinyl phosphonate is formed 6 kcal mol−1 more endothermically (+11.5 kcal mol−1) than its E isomer, being the full process exothermic in 50.7 kcal mol−1, i.e. 10 kcal mol−1 less exothermic than that of the formation of the E isomer.
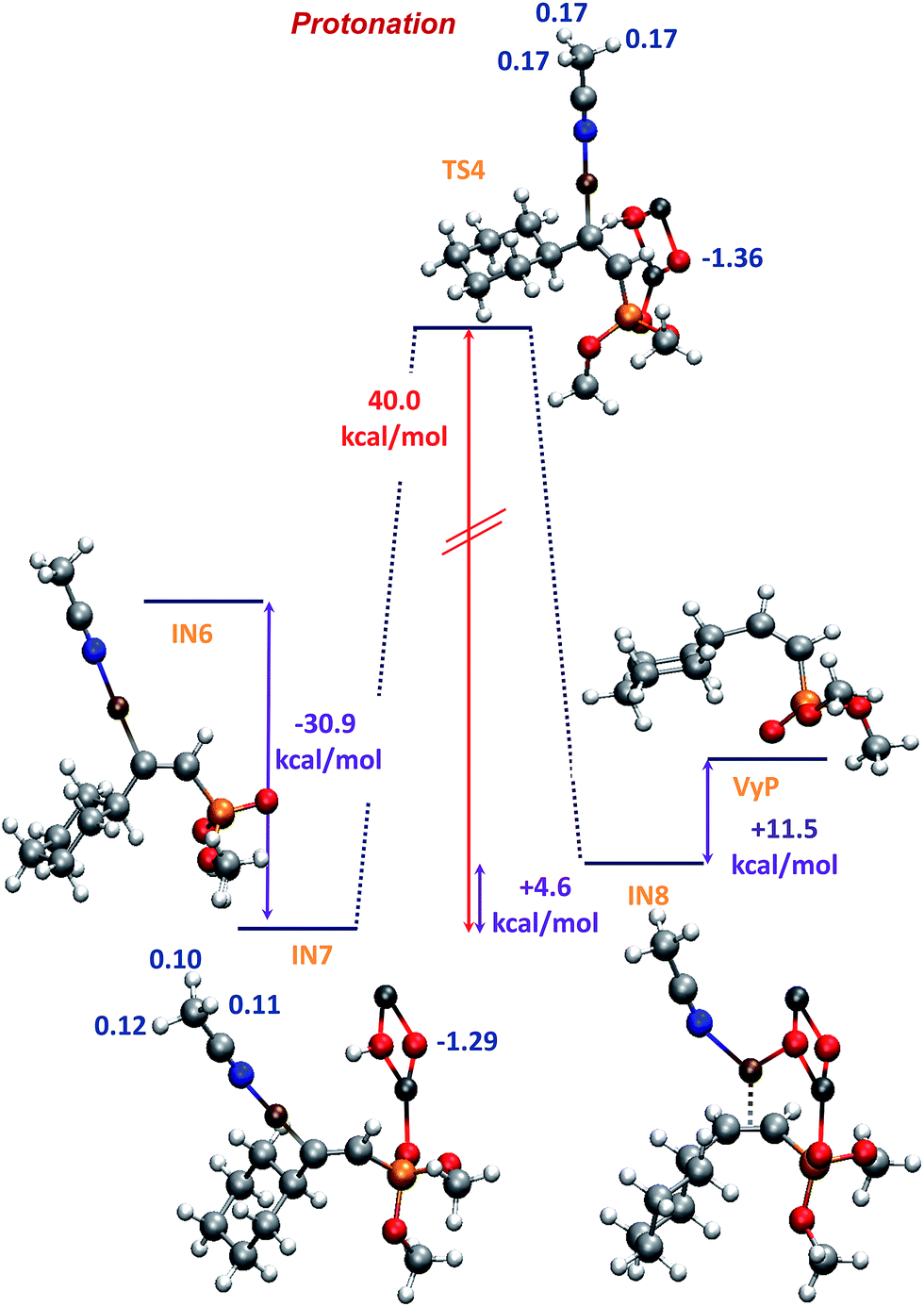 | ||
| Fig. 6 B3LYP-D/6-31+G* potential energy profile for the protonation step corresponding to Z vinyl phosphonate and MK charges (in blue). | ||
Conclusions
In the present study we addressed the mechanism of the copper-catalyzed hydrophosphorylation of aliphatic alkynes by dialkyl phosphites for the synthesis of vinyl phosphonates. Based on experimental observations and theoretical calculations through DFT studies, we have proposed a plausible reaction mechanism that reveals a crucial role for both acetonitrile solvent, acting as ligand, and ZnO support, activating the P–H bond and acting as a proton transfer agent in different steps of the reaction. The DFT results are in agree with the experimentally observed stereoselectivity, since a strong coordination between metals (Cu or Zn) and the oxygen atom of the PO group is observed for the formation of the E isomer, thus suggesting that the participation of stable cyclic structures as intermediates would be very likely to occur in this copper-catalyzed hydrophosphorylation. On the other hand, the modeled mechanism using DMSO as solvent and ligand, showed to be energetically less favourable than the same mechanism modeled with acetonitrile.
Taking into account that most of the existing methods for the hydrophosphorylation of alkynes are based on the use of expensive and/or toxic palladium catalysts, the mechanistic data obtained in this work is of major relevance for the rationale design of new efficient and economical copper-based catalytic systems for P–C (vinilyc) bond construction.
Acknowledgements
This work was generously supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), and Secretaría General de Ciencia y Técnica – Universidad Nacional del Sur (SGCyT-UNS) from Argentina. L. F. thanks the CONICET for a postdoctoral fellowship.Notes and references
- (a) X. Y. Jiao and W. G. Bentrude, J. Org. Chem., 2003, 68, 3303 CrossRef CAS PubMed; (b) M. Maffei, Curr. Org. Synth., 2004, 1, 355 CrossRef CAS; (c) M. N. Noshi, A. El-awa, E. Torres and P. L. Fuchs, J. Am. Chem. Soc., 2007, 129, 11242 CrossRef CAS PubMed. For review articles, see: (d) T. Minami and J. Motoyoshiya, Synthesis, 1992, 333 CrossRef CAS; (e) P. Adler, A. Fadel and N. Rabasso, Tetrahedron, 2014, 70, 4437 CrossRef CAS.
- (a) G. J. Schlichting, J. L. Horan, J. D. Jessop, S. D. Nelson, S. Seifert, Y. Yang and A. M. Herring, Macromolecules, 2012, 45, 3874 CrossRef CAS; (b) J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508 CrossRef CAS PubMed; (c) Q. Wu and R. A. Weiss, Polymer, 2007, 48, 7558 CrossRef CAS.
- (a) M. Kalek, A. Ziadi and J. Stawinski, Org. Lett., 2008, 10, 4637 CrossRef CAS PubMed; (b) N. S. Gulykina, T. M. Dolgina, G. N. Bondarenko and I. P. Beletskaya, Russ. J. Org. Chem., 2003, 39, 847 CrossRef. For review articles about the hydrophosphorylation of alkynes see: (c) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS PubMed; (d) L. Coudray and J. L. Montchamp, Eur. J. Org. Chem., 2008, 3601 CrossRef CAS PubMed; (e) V. P. Ananikov, L. L. Khemchyan and I. P. Beletskaya, Synlett, 2009, 2375 CrossRef CAS.
- (a) G. Trostyanskaya, D. Y. Titskiy, E. A. Anufrieva, A. A. Borisenkko, M. A. Kazankova and I. P. Beletskaya, Russ. Chem. Bull., 2001, 50, 2095 CrossRef; (b) L. B. Han, C. Zhang, H. Yazawa and S. Shimada, J. Am. Chem. Soc., 2004, 126, 5080 CrossRef CAS PubMed; (c) L. Liu, Y. Lv, Y. Wu, X. Gao, Z. Zeng, Y. Gao, G. Tang and Y. Zhao, RSC Adv., 2014, 4, 2322 RSC; (d) Y. Wu, L. Liu, K. Yan, P. Xu, Y. Gao and Y. Zhao, J. Org. Chem., 2014, 79, 8118 CrossRef CAS PubMed.
- Q. Gui, L. Hu, X. Chen, J. Liu and Z. Tan, Chem. Commun., 2015, 51, 13922 RSC.
- (a) L. Liu, Y. Wu, Z. Wang, J. Zhu and Y. Zhao, J. Org. Chem., 2014, 79, 6816 CrossRef CAS PubMed; (b) I. G. Trostyanskaya and I. P. Beletskaya, Tetrahedron, 2014, 70, 2556 CrossRef CAS.
- For some recent works, see: (a) F. Nador, M. A. Volpe, F. Alonso, A. Feldhoff, A. Kirschning and G. Radivoy, Appl. Catal., A, 2013, 455, 39 CrossRef CAS; (b) F. Alonso, Y. Moglie, G. Radivoy and M. Yus, J. Org. Chem., 2013, 78, 5031 CrossRef CAS PubMed; (c) F. Nador, M. A. Volpe, F. Alonso and G. Radivoy, Tetrahedron, 2014, 70, 6082 CrossRef CAS.
- V. Gutierrez, E. Mascaró, F. Alonso, Y. Moglie and G. Radivoy, RSC Adv., 2015, 5, 65739 RSC.
- (a) V. P. Ananikov, L. L. Khemchyan, I. P. Beletskaya and Z. A. Starikovac, Adv. Synth. Catal., 2010, 352, 2979 CrossRef CAS; (b) V. P. Ananikov and I. P. Beletskaya, Chem.–Asian J., 2011, 6, 1423 CrossRef CAS PubMed; (c) J. Stawinski and A. Kraszewski, Acc. Chem. Res., 2002, 35, 952 CrossRef CAS PubMed; (d) J.-N. Li, L. Liu, Y. Fu and Q.-X. Guo, Tetrahedron, 2006, 62, 4453 CrossRef CAS; (e) J. P. Guthrie, Can. J. Chem., 1979, 57, 236 CrossRef CAS.
- M. Hosseini-Sarvari and M. Tavakolian, New J. Chem., 2012, 36, 1014 RSC.
- (a) V. B. Dorn, M. A. Badajoz, M. T. Lockhart, A. B. Chopa and A. B. Pierini, J. Organomet. Chem., 2008, 693, 2458 CrossRef CAS; (b) M. E. Budén, V. B. Dorn, M. Gamba, A. B. Pierini and R. A. Rossi, J. Org. Chem., 2010, 75, 2206 CrossRef PubMed; (c) F. Nador, Y. Moglie, A. Ciolino, A. Pierini, V. Dorn, M. Yus, F. Alonso and G. Radivoy, Tetrahedron Lett., 2012, 53, 3156 CrossRef CAS; (d) V. B. Dorn, G. F. Silbestri, M. T. Lockhart, A. B. Chopa and A. B. Pierini, New J. Chem., 2013, 37, 1150 RSC; (e) M. J. Lo Fiego, V. B. Dorn, M. A. Badajoz, M. T. Lockhart and A. B. Chopa, RSC Adv., 2014, 4, 49079 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- W. Kohn and I. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (c) E. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef.
- (a) S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef CAS PubMed; (b) S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- (a) S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS; (b) S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239 CrossRef CAS; (c) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS.
- X. Cheng, F. Li and Y. Zhao, J. Mol. Struct.: THEOCHEM, 2009, 894, 121 CrossRef CAS.
- The calculated (B3LYP-D/6-31+G*) formation energy of the dimer (ZnO)2 is −58.4 kcal mol−1.
- B. G. Janesco, H. C. Fischer, M. J. Bridle and J.-L. Montchamp, J. Org. Chem., 2015, 80, 10025 CrossRef PubMed.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129 CrossRef CAS.
Footnotes |
| † In honour of Prof. Adriana Pierini. |
| ‡ Electronic supplementary information (ESI) available: Computational procedure, xyz coordinates and total energies in atomic units for all of the calculated structures. See DOI: 10.1039/c7ra01037k |
| This journal is © The Royal Society of Chemistry 2017 |